

Abstract
Nucleotide analogues have been well accepted as therapeutic agents active against a number of viruses. However, their use as antiviral agents is limited by the need for phosphorylation by endogenous enzymes, and if the analogue is orally administered, by low bioavailability due to the presence of an ionizable diacid group. To circumvent these limitations, a number of prodrug approaches have been proposed. The ideal prodrug achieves delivery of a parent drug by attachment of a non-toxic moiety that is stable during transport and delivery, but is readily cleaved to release the parent drug once at the target. Here, a brief overview of several promising prodrug strategies currently under development is given.
Keywords: antiviral agents, oral bioavailability, prodrugs, pronucleotides
1. Introduction
Of the approximately 40 antiviral drugs formally licensed for use, half are nucleoside or nucleotide analogues [1]. Nucleoside drugs per se must usually be phosphorylated to the 5′-mono-, 5′-di-, and finally, 5′-triphosphate by intracellular or viral kinases [2] in order to inhibit their therapeutic targets. This requirement limits efficacy, as phosphorylation to the monophosphate by endogenous kinases is slow and typically is the rate-limiting step in human cells [3,4].
The administration of a nucleoside drug as its monophosphate (NMP) is a well-known approach to overcoming this obstacle [3,5]. However, this entails a penalty in the form of decreased membrane permeability. Nucleotide analogues contain an ionizable –O-P(O)(OH)2 group that exists chiefly as a dianion at physiological pH, resulting in low oral bioavailability [5]. In addition, if a NMP succeeds in crossing the intestinal membrane, it then becomes a potential substrate for phosphohydrolases (phosphatases and 5′-nucleotidases), which remove the phosphate group [6]. The use of a nucleoside phosphonate –CH2-P(O)(OH)2 circumvents dephosphorylation, but decreased transport remains an obstacle.
Formulation strategies [7–11] to overcome these limitations are beyond the scope of this short review, which has as its focus an alternative approach: prodrug modification of nucleotide drugs. Promoieties can be attached at a number of positions on an NMP or nucleotide analogue [12,13]. However, the introduction of promoieties at the phosphorus (–[O,CH2]-P(O)(X)(Y) where X,Y = OR, OR′, NHR″) directly addresses the problem of blocking P-OH ionization in vivo. The attachment of a well-designed promoiety increases delivery of the drug to its target, provided that its biochemical and physical properties – including lipophilicity, site-specificity and chemical stability – are conducive to this end [5,13,14].
A prodrug must be stable under delivery conditions [3,5], but it must be capable of conversion to its active parent drug in vivo [5], at a rate consistent with pharmacological efficacy. The prodrug and metabolized promoiety/promoieties should have low acute and chronic toxicity [5]. Control of these and other crucial properties, such as aqueous solubility and lipophilicity, remains a key challenge in the development of an effective prodrug.
Esterification with pivaloyloxymethyl (POM), p-acyloxybenzyl (PAOB), or isopropyloxy–carbonyloxymethyl (POC) groups has been reviewed extensively [3,5,6,13,15,16] and will not be addressed here. Also, of recent interest, but omitted from this discussion is the approach of Hostetler et al. to improve the oral bioavailability of certain antiviral phosphonate drugs by esterification with an ether lipid ester that mimics the natural lipid lysophosphatidylcholine, thus potentially delivering the prodrug within the cell intact [4,17,18]. Our review will examine the prodrug approaches represented by the structures in Figure 1.
Figure 1.
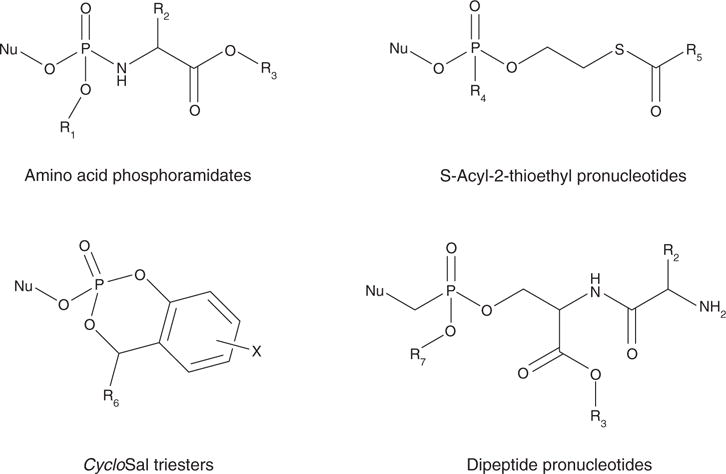
General structures exemplifying the antiviral prodrug approaches discussed in this review.
2. Phosphoramidate ‘ProTide’ approach
McGuigan has introduced prodrugs (‘ProTides’) based on an amino acid ester promoiety, attached to the drug (as a aryl monophosphate or phosphonate) via a P-N bond, applying this approach to: 4′-azidouridine [19], 4′-azidoadenosine [20], 2′,3′-dideoxyuridine (ddU) [21], carbocyclic L-d4A (L-Cd4A) [22], stauvidine (d4T) [23], 9-[2-(phosphonomethoxy) ethyl]adenine (PMEA) [24], 3′-azidothymidine (AZT) [25], abacavir (ABC) [26] and tenofovir (PMPA, 9-[(R)-2-(phosphonomethoxy)propyl]adenine) [27].
The original approach involved preparation of simple alkyloxy phosphoramidates (Figure 2A(1)), but has evolved into aryloxy phosphoramide pronucleotides with distinct structure–activity relationship detail [28]. Phosphorodiamidates (Figure 2A(2)) were also prepared, but no biological benefit versus the phosphoramidates was observed and synthetic yields were lower [28]. Interestingly, analogues linked through an oxygen resulted in a significant decrease in antiviral activity [29], possibly because the nucleoside monophosphate was not released from the diester intermediate [28]. Diaryl pronucleotides (Figure 2A(3)) were not active in kinase-deficient cells [30], due to poor intracellular delivery of the NMP or possibly chemical instability of the diaryl masking groups. Overall, aryloxy phosphoramidates (Figure 2A(4)) appear to hold the most promise for delivery of the therapeutic agent.
Figure 2.
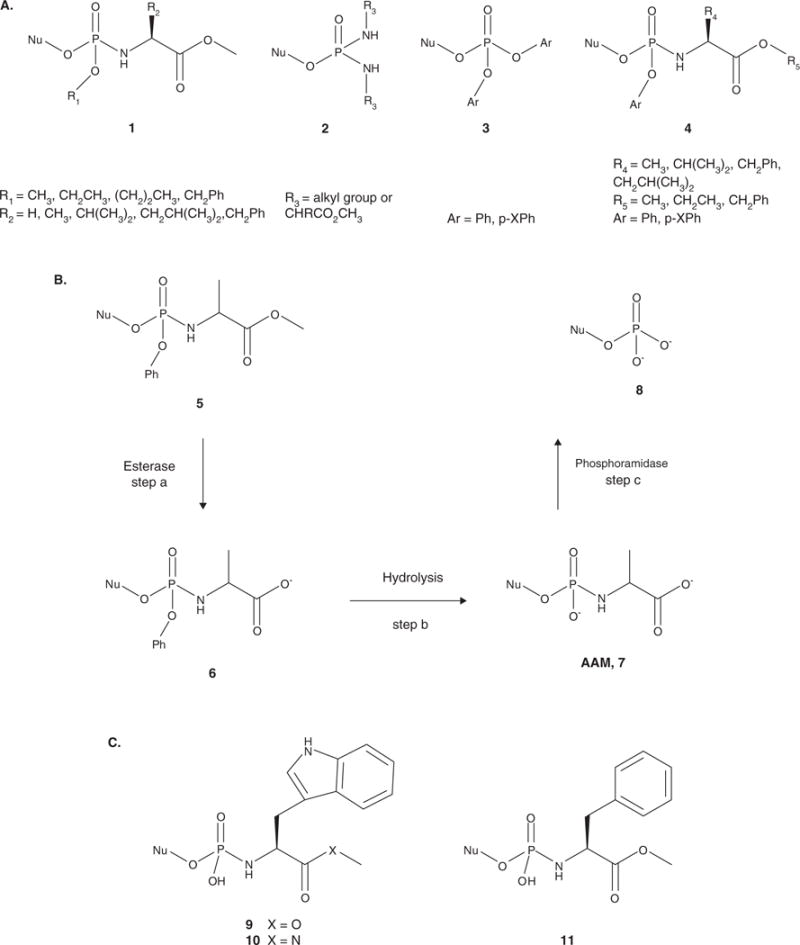
A. General structures of pronucleotides investigated by McGuigan and coworkers. The alkyloxy phosphoramidates (1) were initially evaluated. Phosphorodiamidates (2) were also tested, but showed no biological advantage over the phosphoramidates. The diaryl triesters (3) lost antiviral activity in kinase-deficient cells. However, the aryloxy phosphoramidates (4) proved to be the most successful pronucleotides. B. Activation pathway of the aryloxy phosphoramidate pronucleotides. Step a involves cleavage of the ester group by an esterase to form intermediate 6. Hydrolysis of the aryl group facilitated by intramolecular cyclization of the amino acid moiety followed by hydrolysis produces the amino acyl metabolite (AAM, 7). Cleavage of the P-N bond by a phosphoramidase yields the 5′-nucleoside monophosphate 8 (step c). C. Structures of the amino acid phosphoramidate monoesters: tryptophan methyl ester (9), tryptophan methyl amide (10) and phenylalanine methyl ester (11). The L-Val, L-Leu and L-Ala methyl ester derivatives were also synthesized.
Aryloxy phosphoramidates were designed to release the NMP intracellularly via both chemical and enzymatic mechanisms (Figure 2B). The first step in the activation process is cleavage of the amino acid ester by a carboxyesterase [28] to afford 6 (Figure 2B), although a thorough investigation by Venkatachalam and coworkers found that activation by a lipase or protease is also possible and that these enzymes have different specificities for the substituent on the aryl group, the amino acid and the stereochemistry at the phosphorus [31]. Subsequent nucleophilic attack at the phosphorus by the carboxyl group releases the aryloxy group, forming a transient cyclic diester, which is hydrolyzed to form the amino acyl metabolite (AAM, Figure 2B(7)) [28]. In the final step, the amino acid moiety is cleaved by a phosphoramidase to release the nucleoside monophosphate (Figure 2B(8)) and an amino acid [28].
McGuigan and coworkers have thoroughly studied the aryloxy phosphoramidates of d4T and have been able to gain extensive structure–activity relationship insight. In general, the methyl, ethyl and benzyl esters lead to potent activity, while bulkier esters (t-butyl and isopropyl) are significantly less active than the methyl ester [32], most likely due to the increased stability to enzymatic hydrolysis [20]. A quantitative structure–activity relationship (QSAR) study on the variation of amino acid esters further described the most potent esters as those with considerable lipophilicity slightly removed from the ester bond [33]. In a separate report, it was found that the conversion of AAM to NMP was inhibited when benzyl alcohol was released [34]. This inhibition was not observed when ethanol or methanol was released [34].
Although there are some exceptions depending on the nature of the drug used, the most successful pronucleotides contain L-alanine as the amino acid [34,35]. Exchange of L-alanine for either glycine or L-leucine reduced the antiviral activity 70- and 13-fold, respectively [36]. When L-valine was used, the antiviral activity was reduced 147-fold, and furthermore, 100% of the intact prodrug was recovered when it was exposed to pig liver carboxyesterases [34]. When D-alanine was substituted for L-alanine, the potency was decreased 35-fold [34]. The exact reason for the preference for alanine remains unknown. When the achiral amino acid analogue α,α-dimethylglycine was prepared, the antiviral activity was only reduced three-fold [36], which illustrates the fact that natural amino acids are not essential for activity. However, when such amino acids were used, a preference for α-amino acids was observed [34]. The β-amino acid phosphoramidates showed efficient ester cleavage, but no phenyl loss was detected, and the AAM was not observed [34]. This suggests a possible entropy barrier that increases with chain length.
Studies to determine the optimal aryloxy group were also performed [37]. The greatest activity was achieved when the aryl group had a p-Cl substituent, and generally for aryl groups that function as mildly electron-withdrawing, lipophilic substituents [37]. The potential for toxicity of the released phenol was not discussed. A naphthyl group was also reported to be an effective aryl moiety for delivering anticancer agents [38], and this activity is most likely transferable to antiviral agents.
To obtain successful intracellular delivery of NMP, the pronucleotides need to be resistant to hydrolysis during the absorption and distribution process. The chemical stability of the pronucleotides was studied, and all exhibited satisfying stability over the range of pHs studied (2.0 – 7.4) [39]. The aryloxy phosphoramidates were significantly less resistant to decomposition in plasma or cell extracts, indicative of the need for enzymatic activation [39]. After overnight exposure to pig liver carboxyesterases, CEM cell extract, human serum and mouse serum, AAM was formed from a majority of the L-amino acid-containing pronucleotides [34]. When carbocyclic adenosine phosphoramidates were evaluated in intestinal and liver S9 homogenates, some of the most antivirally potent analogues exhibited complete decomposition over 1 h in intestinal homogenate [22], but isopropyl and t-butyl esters on the amino acid increased intestinal stability [22]. Similarly, D-alanine and glycine exhibited the highest intestinal stability [22], which highlights the complexity in obtaining a structure–activity relationship. Although the usage of this aryloxy phosphoramidate prodrug approach with nucleotide analogues containing a phosphonate may be more difficult due to decreased chemical stability [40], an example of successful application to tenofovir has been described [27].
The pharmacokinetics and oral bioavailability of aryloxy phosphoramidates, specifically abacavir phosphoramidates, were examined [35]. When the abacavir methyl alaninyl–phosphoramidate was administered intravenously, the pronucleotide was rapidly cleared from the plasma with a half-life of 7 min [35]. Similar results were observed following oral administration [35]. However, the major metabolite observed was the AAM [35]. Total exposure to the pronucleotide and its active metabolites was reported to approach that estimated for a similar dose of the parent drug, abacavir, resulting in an overall bioavailability of 50% [35]. The epithelial permeability of a series of d4T aryloxy phosphoramidates was evaluated in Caco-2 and MDCK monolayers [41]. The pronucleotides exhibited relatively low permeability, which may be partially explained by their susceptibility to first-pass metabolism in the intestinal epithelial cells and by being substrates of P-gp [41]. In general, this work exemplifies the difficulty in delivering the NMP to the target while avoiding significant metabolism during absorption and distribution. To obtain optimal antiviral activity of each pronucleotide, the fine tuning of each element (amino acid, ester, and aryl moiety) is required.
3. Monoester prodrugs
Amino acid phosphoramidate monoesters designed to release the NMP after a single activation by an endogenous phosphoramidase have been described by Wagner, who has applied this approach to AZT [42,43] and ddA [44], as well as anticancer drugs [45].
After the delivery of AZT monophosphate by a glycoslyated carrier attached through lysine was reported [46], Wagner and coworkers proposed that NMP could be efficiently delivered by non-polar amino acid phosphoramidate monoesters and that the aryl group was not necessary. Furthermore, these phosphoramidate monoesters were stable in cell culture and rat and human plasma [42]. A series of these compounds were synthesized containing an amino acid (tryptophan methyl ester [Figure 2C(9)] or phenylalanine methyl ester [Figure 2C(11)]) connected via a P-N bond to the NMP with the other P-OH left as a free acid or esterified to a simple alkyl group [47]. The tryptophan monoester (Figure 2C(9)) exhibited the best antiviral activity, with an eight-fold increase over AZT with no cytotoxicity observed at the levels tested [47]. Further studies have been done to investigate the activation pathway of these pronucleotides and optimize their structures.
The effect of changing the amino acid was studied in peripheral blood mononuclear cells (PBMC) [42]. The best antiviral activity was obtained with the L-alanine methyl ester [42], consistent with McGuigan’s ProTides. Furthermore, enhanced activity was observed with the L-tryptophan derivative (Figure 2C(9)) compared with the L-phenylalanine (Figure 2C(11)), L-valine and L-leucine derivatives [42]. When evaluated in CEM cells, the L-alanine and L-phenylalanine derivatives exhibited antiviral activity comparable to AZT [42]. This suggests that a simple structure–activity relationship does not exist. In order to avoid the polar carboxylate formed after interaction of the pronucleotides with carboxyesterases, the amino acid methyl ester was substituted by a methyl amide [42]. The authors reported that this exchange had little effect on the antiviral activity of the tryptophan derivatives, while the phenylalanine methyl amide derivatives exhibited increased potency [42]. However, the methyl amide derivatives exhibited greater in vitro and in vivo stability [48]. The antiviral activity did not exhibit a strong dependence on the amino acid stereochemistry [42], but the inclusion of the D-isomer versus the L-isomer led to decreased volumes of distribution [48]. Overall, the L-tryptophan methyl amide derivative (Figure 2C(10)) was selected for further studies.
To better understand the differences in potency, Wagner and coworkers investigated the ability of the pronucleotide to deliver NMP intracellularly [42]. The antiviral activity is strongly related to the intracellular levels of nucleoside triphosphate. In both PBMCs and CEM cells, AZT was able to produce higher levels of AZT triphosphate than the pronucleotides [42]. However, when evaluated in CEM cells, the intracellular levels of the tryptophan methyl ester (Figure 2C(9)) and phenylalanine methyl ester (Figure 2C(11)) pronucleotides did not plateau [49]. Therefore, the differences in potency may be derived from the ability of a phosphoramidase to cleave the P-N bond and release the NMP.
The oral bioavailability, disposition and stability of the AZT phosphoramidate monoesters were evaluated in rats [50]. The phosphoramidate monoesters were stable in tissue homogenates, intestinal contents, and rat and human plasma [48,50]. The tryptophan methyl amide derivative (Figure 2C(10)) exhibited the best pharmacokinetic parameters. However, in simulated gastric fluids at pH 2.0, the pronucleotide exhibited a significantly reduced half-life of 5 h, but greater stability as the pH increased [50]. These results are consistent with greater chemical hydrolysis of P-N bonds at lower pH [51]. The pronucleotide was not detected in plasma or urine, which was confirmed in an in situ single pass perfusion study where little or no absorption of the pronucleotide in the 120 min perfusion period was detected [50]. AZT was observed in plasma and urine samples accounting for 29.5% of the dose, while 54.3% of the dose was recovered 4 h post-dosing (intravenously) as intact pronucleotide in the bile [50]. These results offer some possible explanations for the zero oral bioavailability of the pronucleotide. Not only will the pronucleotide exist as a charged species at physiological pH, but pre-systemic hydrolysis of the P-N bond would lead to a dianionic monophosphate. This fact, combined with its molecular weight, makes biliary excretion difficult to avoid [50].
The phosphoramidate monoester pronucleotide approach was applied to AZT to create a pronucleotide with increased antiviral activity and decreased cytotoxicity. Unfortunately, decomposition of the phosphoramidate monoester in simulated gastric fluid was observed, and the pronucleotide exhibited little or no bioavailability when evaluated in rats.
4. SATE pronucleotides
S-Acyl-2-thioethyl (SATE) protecting groups for nucleotide drugs have been introduced for the delivery of a number of NMP including d4T [52], PMEA [53], elvucitabine (β-L-FD4C) [54], acyclovir [55,56], AZT [57–59] and cytarabine (Ara-C) [60].
The SATE approach utilizes both enzymatic and chemical mechanisms to activate the pronucleotide and release the NMP (Figure 3A) [61]. Removal of the S-acyl-2-thioethyl protecting group and release of the monophosphate is initiated by esterase-mediated hydrolysis of the acyl group, which produces a reactive thiol group [61]. Nucleophilic attack on the α-carbon results in a reactive 2-mercaptoethyl ester that decomposes spontaneously to release the diester (Figure 3A(14)) and ethylene sulfide (episulfide) [61]. If the second ester is also a SATE group, the process is then repeated, resulting in release of the NMP [61]. It has been proposed that if SATE mixed pronucleotides are used, the aryl group in SATE phosphotriesters is cleaved by a type 1 phosphodiesterase and a phosphoramidase cleaves the protecting group bound via a P-N bond [61]. However, the cleavage of the aryl ester or phosphoramidate could be concomitant with cleavage of the SATE group [61].
Figure 3.
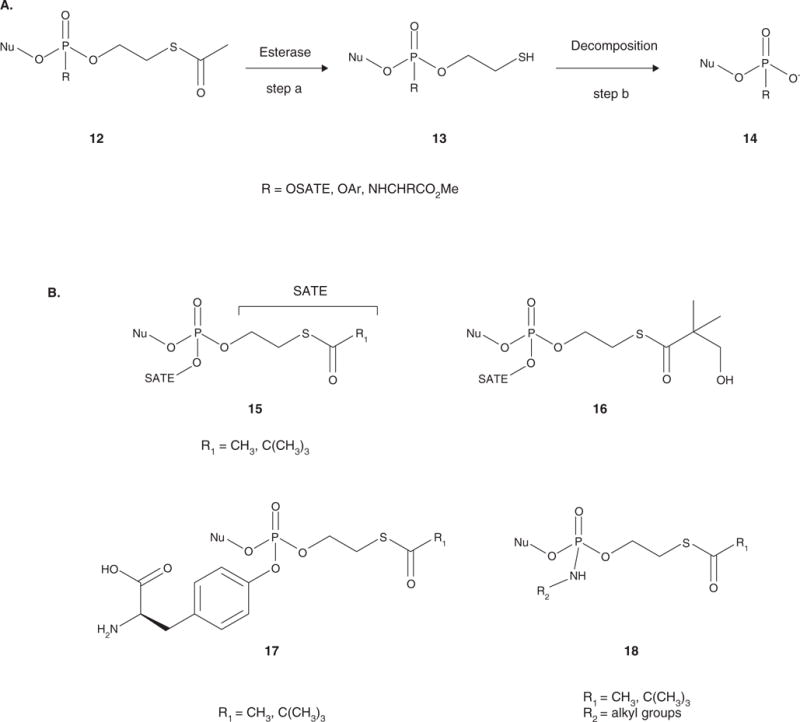
A. Activation of the SATE pronucleotides. The acyl group is cleaved to release the intermediate, which contains a reactive thiol group 13 (step a). Intramolecular cyclization to form an ethylene sulfide results in spontaneous decomposition to the diester or phosphoramidate 14 (step b). If the pronucleotide contains a second SATE moiety, the process is repeated until the 5′-nucleoside monophosphate is released. If the second group is an aryloxy group or a phosphoramidate, hydrolysis can either proceed sequentially or concomitantly to release the 5′-nucleoside monophosphate. B. General structures of the SATE pronucleotides: bis(SATE) triesters (15), bis(hydroxyl-tBuSATE) triesters (16), aryl SATE phosphotriesters (17), and SATE phosphoramidate diesters (18).
Although the release of the NMP is essential, a further point to consider is promoiety toxicity. In the activation process, carboxylic acids and episulfide were released [61]. The body can metabolize the carboxylic acids, and no cytotoxicity was reported for the episulfide [61]. When the SATE promoiety and its metabolites were evaluated in various cell lines, no additional cytotoxicity was observed [61]. In vivo toxicity studies in cynomolgus monkeys showed neither clinical symptoms nor behavior problems indicative of toxicity [61].
In all cases, application of the bis(SATE) phosphotriester (Figure 3B(15)) approach has led to increased in vitro antiviral activity compared to the parent nucleoside and efficient delivery of the NMP. Bis(MeSATE) d4T monophosphate was 10- to 17-fold more potent than d4T in wild-type CEM cells and PBMCs, and furthermore, antiviral activity was retained in thymidine kinase-deficient cells [52]. A slight increase in cytotoxicity was also observed for the pronucleotides [52]. Administration of the bis(tBuSATE) Ara-C phosphotriester resulted in significant antiviral activity in a resistant cell line due to lack of the appropriate kinase [60].
Stability and transport across a Caco-2 monolayer was used to determine the optimal R1 group (Figure 3B) [53]. Transport of the pronucleotides across Caco-2 monolayers resulted in intact pronucleotide in the basolateral compartment only when the bis(tBuSATE) phosphotriester was evaluated, whereas no intact pronucleotide or metabolites were observed for the bis(MeSATE) phosphotriester [53]. Although very low amounts of the intact pronucleotide were detected, the greatest uptake in the Caco-2 monolayer was observed for the bis(tBuSATE) phosphotriester, which could be due to the increased enzymatic stability or lipophilicity of the pivaloyl group [53]. The bis(tBuSATE) phosphotriester was extensively metabolized to the monoester during transport in vivo, resulting in no intact pronucleotide observed in the plasma [62]. Some bis(tBuSATE) phosphotriesters exhibit poor aqueous solubility and had to be administered with 5% dimethyl sulfoxide (DMSO), which affected the esterase activity and significantly increased the half-lifes of the pronucleotides [62].
In an attempt to obtain more favorable stability and bioavailability, a SATE group bearing a functionalized acyl moiety was investigated (Figure 3B(16)) [57,59,62]. The addition of one hydroxyl group led to antiviral activity comparable to the nucleoside and increased activity compared to the bis(tBuSATE) phosphotriester [62]. The addition of a hydroxyl group to the bis(tBuSATE) promoiety decreased the hydrolysis rate and increased the half-life and aqueous solubility of the pronucleotide [62]. Furthermore, intact pronucleotide was observed when the bis(hydroxyl-tBuSATE) phosphotriester was evaluated in a Caco-2 monolayer [62].
Several variations of the SATE pronucleotide approach have been described. Initial attempts at the intracellular delivery of NMP with SATE esters involved the use of two SATE groups [52,55,60,62]. Although these bis(SATE) pronucleotides (Figure 3B(15 and 16)) exhibited increased antiviral activity and decreased cytotoxicity, the removal of the second SATE group proved difficult and proceeded much more slowly due to the negative charge at the phosphate [61]. Therefore, Gosselin and Imbach evaluated two different kinds of SATE mixed esters: aryl SATE phosphotriesters (Figure 3B(17)) [64,65] and SATE phosphoramidate diesters (Figure 3B(18)) [64,67].
A phenyl (tBuSATE) mixed phosphotriester was synthesized, but the NMP was not released from the prodrug when studied in cell extract [63]. Several derivatives of L-tyrosine SATE phosphotriesters (Figure 3B(17)) were investigated for stability and antiviral activity [64,65]. Since the presence of the free carboxylic acid on the tyrosine residue resulted in decreased antiviral activity, most likely due to decreased membrane permeability, the moiety was modified to contain polar, but not anionic, functionalities [66]. The pronucleotide with the shortest half-life exhibited the best antiviral activity [64]. The resulting mixed SATE phosphotriesters exhibited enhanced antiviral activity in CEM kinase-deficient cells, illustrating the successful delivery of the NMP intracellularly [64].
Another solution to the slow activation of the bis(SATE) phosphotriesters was the SATE phosphoramidate diesters (Figure 3B(18)) modeled after the phosphoramidates of McGuigan and Wagner. Initial studies with various alkylamines illustrated that the rate limiting hydrolysis of the phosphoramidate was dependent on the basicity and bulk of the amine [65,67]. However, when the pKa of the amine was appropriate (approx. 5 – 11.2), the phosphoramidate diesters effectively delivered NMP intracellularly [67]. Phosphoramidate diesters of AZT were as potent as AZT in wild-type CEM cells and retained significant antiviral activity in kinase-deficient cells [58]. Interestingly, the isopropylamino derivative exhibited the greatest potency, which illustrates the flexibility in the amine moiety and demonstrates that an α-amino acid is not a structural requirement [58].
Périgaud and coworkers have recently investigated the use of aryl SATE phosphotriesters [57,59]. Building on the ability of (hydroxyl-tBu)SATE to increase the solubility and stability of the prodrug, functionalization of the acyl moiety with polar groups was applied to the phenyl SATE mixed phosphotriester analogues. Additionally, an analogue with a valine replacing the acyl group was studied, but the compound was unstable in cell extract and did not maintain antiviral activity in a thymidine kinase (TK) deficient cell line [57,59]. Introduction of one hydroxyl group on the acyl moiety resulted in greater stability in cell extracts compared to the tBuSATE analogue [57]. The derivative containing two hydroxyl groups showed decreased enzymatic stability compared to the (hydroxyl-tBu)SATE analogue, but it resulted in a significant loss in activity in a TK-deficient cell line [57]. The monohydroxylated prodrug showed anti-HIV activity in the micromolar range comparable to the tBuSATE analogue, and the solubility of the pronucleotide was greatly increased [57], which demonstrates the necessity of obtaining optimized pharmacokinetic properties to achieve effective prodrugs.
Application of the SATE pronucleotide approach to numerous nucleoside monophosphates and nucleotide analogues has resulted in increased antiviral activity in vitro. However, when evaluated for transport across Caco-2 monolayers and for bioavailability, no intact prodrug was observed illustrating premature hydrolysis. As pointed out by Gosselin, Imbach, and Périgaud, these facts illustrate the necessary balance that needs to be achieved between lipophilicity, solubility and enzymatic stability [61].
5. CycloSal prodrugs
Meier et al. have shown that salicyl alcohol is an effective bifunctional masking unit for nucleotides, that is cleaved by a pH-dependent mechanism to deliver the active drug [68]. They have illustrated the utility of this approach using various nucleoside and nucleotide analogues including acyclovir [69], 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA, adefovir) [70], 2′,3′-dideoxyadenosine (ddA) [71], 2′,3′-dideoxy-2′,3′-didehydroadenosine (d4A) [71], 5-[(E)-2-bromovinyl]-2′-deoxyuridine (BVdU or brivudin) [72,73], 2′,3′-dideoxy-2′,3′-didehydrothymidine (d4T) [74–79] and carbocyclic 3′-azidothymidine analogues [80].
Their original goal in creating these pronucleotides was to find a masking unit that would deliver the nucleotide analogue exclusively by a chemical mechanism. Initial attempts using bis(alkyl), bis(phenyl), or bis(benzyl) nucleotide triesters proved unsuccessful at releasing the NMP by a purely chemical mechanism [6]. The charge formed once one ester was cleaved led to a stable compound resistant to further chemical hydrolysis [16]. However, Meier and coworkers found that they could successfully mask the phosphate with phenyl and benzyl esters of salicyl alcohol, while the nucleoside was attached by esterification via the 5′-hydroxyl group (Figure 4A(19)) [81]. These esters are distinct enough to achieve differentiated chemical hydrolysis independent of any enzymatic activity [82]. This principle was validated by comparing the half-lifes of the triesters in phosphate buffer at pH 7.3 and cell extracts with fetal calf serum, which showed similar half-lifes for the triesters in both media [83].
Figure 4.
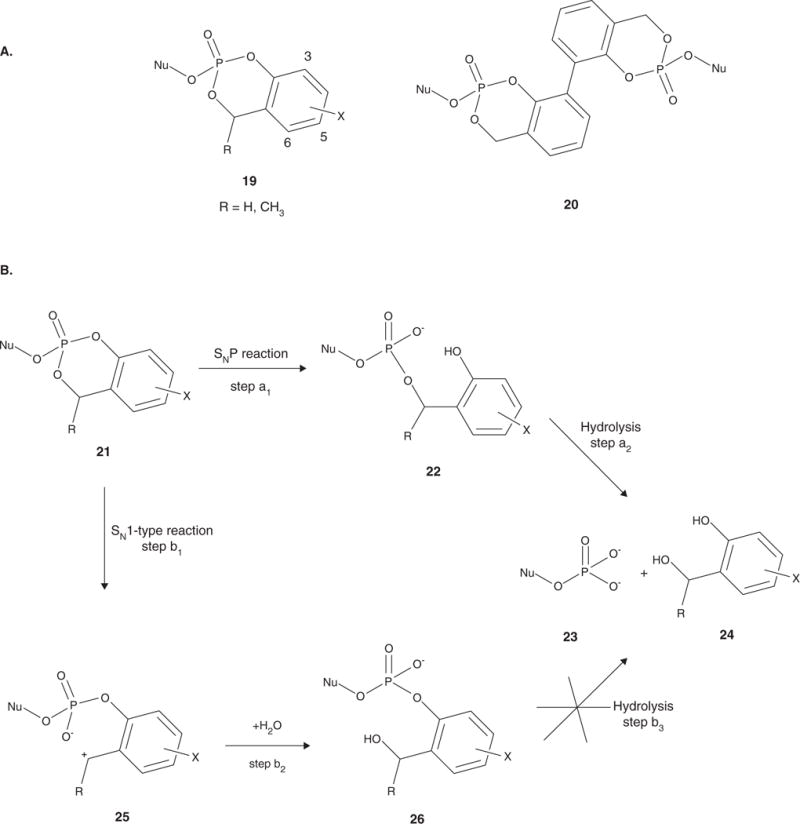
A. The triester pronucleotides studied by Meier and coworkers. The cycloSal pronucleotides (19) led to increased intracellular levels of NMP. The bis-cycloSal pronucleotides (20) are able to deliver two molecules of drug per biomolecule administered. B. Activation pathway for the cycloSal pronucleotides. Cleavage of the phenyl ester is initiated by nucleophilic attack at the phosphorus to produce the diester 22 (step a1). Cleavage of the benzyl ester yields the nucleotide analogue 23 and salicyl alcohol 24 (step a2). The benzyl ester of 21 can also first be hydrolyzed resulting in the charged intermediate 25 (step b1), which then reacts with water to produce 26 (step b2). Hydrolysis of 26 does not occur (step b3), and the nucleotide is not released.
The successful cycloSal pronucleotide releases the nucleotide (Figure 4B(23)) and the salicyl alcohol (Figure 4B(24)) intracellularly [81]. The nucleotide is released through a cascade, which is initiated by cleavage of the phenyl ester via nucleophilic attack on the phosphorus atom by hydroxide to form the diester (Figure 4B(22)) (step a1). The ring, now activated by the strong electron-donating hydroxyl group, allows for cleavage of the benzyl ester to yield the nucleotide analogue (Figure 4B(23)) and salicyl alcohol (Figure 4B(24)) via a SN1-type reaction at the benzyl position (step a2). It is also possible that hydrolysis of the benzyl ester can occur to first produce a charged intermediate (Figure 4B(25)) (step b1), which then reacts with water to yield 26 (Figure 2B) (step b2). Further hydrolysis of 26 (Figure 2B) does not occur, thus preventing the release of the nucleotide analogue. However, in hydrolysis studies, the major products were the NMP and salicyl alcohol. When evaluated for cytotoxicity in mice, salicyl alcohol showed no toxicity [82].
As the cycloSal pronucleotides were designed to release the active drug via a chemical cascade mechanism, the stability and hydrolysis pathways of these pronucleotides can be fine-tuned by varying the substituents on the aromatic ring. Acceptor substituents in the 5- or 6-position decrease stability, while donor substituents at the 3- or 5-position increase the stability of the triesters (Figure 4A(19)) [82]. Bulky substituents (tert-butyl groups) at the 3- and/or 5-position increase the amount of the phenyl phosphate diester (Figure 4B(25)) observed. When substitution was made at the benzyl position, the half-life decreased drastically compared to the unsubstituted analogue and the major product was the diester (Figure 4B(25)) in hydrolysis studies [82]. However, the addition of a donor substituent at the 6-position caused the major hydrolysis product to be the desired diester (Figure 4B(22)) [82].
Although there is a benefit – lack of dependence on enzyme expression differences in tissues, individuals and species – to the use of a pronucleotide activated by a chemical mechanism, the possibility of extracellular release of the active drug or efflux of the pronucleotide, due to the establishment of a concentration equilibrium across the cell membrane, cannot be ignored. To remedy these potential problems, Meier et al. designed a way to trap the pronucleotide inside the cell [77,79,84]. In theory, the attachment of a moiety to the aromatic ring that can be enzymatically activated to release a more polar group will prevent penetration of the cellular membrane by the compound and trap the pronucleotide [75]. The initial attempts included the use of an esterase to release an alcohol or carboxylic acid [85]. The released alcohol group was not polar enough to prevent efflux of the pronucleotide, and when a two, three, or four carbon linker was used to attach a methyl or benzyl ester to the aromatic ring, the compounds did not show good esterase affinity [78]. The feasibility of acetoxymethyl (AM) and pivaloyloxymethyl (POM) esters as enzyme-cleavable triggers was demonstrated by their significantly decreased stability in cell extracts versus plasma, illustrating selective activation of these compounds intracellularly [73]. Attempts at intracellular trapping of the pronucleotide with an amino acid ester trigger moiety resulted in a large differential between the buffer and cell extract stability and sustained antiviral activity in kinase-deficient cells [84]. Although a strongly polar group was required to trap the pronucleotide in the cell, Meier and coworkers found that the release of the NMP from the carboxylic acid intermediate was unsatisfactorily slow, and therefore, they sought a trigger to accelerate the release of the NMP [77].
They found that the incorporation of an aldehyde, a strong electron-withdrawing group, in the aromatic ring increased the release of the NMP [77,79]. When a diacetoxymethyl (di-AM) group was attached to the cycloSal ring at the 5-position, cleavage of the di-AM group resulted in the rapid formation of the benzyl phosphodiester intermediate and, subsequently, the release of the NMP, but the loss of antiviral activity in TK-deficient cells was 26-fold, illustrating only a partial delivery of the prodrug [74,75]. The attachment of 5-(1-acetoxyvinyl) to 3-alkyl-cycloSal-d4TMP showed the best potential for ‘lock-in’ of the pronucleotide, as these analogues showed increased chemical stability, the acetoxyvinyl groups were rapidly cleaved in cell extracts and the 5-(1-acetoxyvinyl)-3-alkyl-cycloSal-d4TMPs retained antiviral activity in thymidine kinase-deficient cells [77].
One potential limitation of the cycloSal approach is interaction between the pronucleotides and human acetylcholinesterase (AChE) or butyrylcholinesterase (BChE) [81,82]. None of the tested pronucleotides inhibit the essential human AChE [86], but inhibition of BChE has been observed and a structure–activity relationship has been established [87]. Studies have shown that inhibitory activity of the pronucleotide varies greatly with the NMP or nucleotide analogue used, the stereochemistry at the phosphorus and the substituents on the aromatic ring. Increasing the size of the pronucleotide by adding bulky substituents to the aromatic ring decreases the inhibitory activity of the triester. In one example, the IC50 value was decreased four-fold when a cyclohexyl group was added at the 5-position [88]. However, antiviral data was not reported for this compound and the fact that bulky substituents at that position increase the formation of the undesired diester (Figure 4B(26)) was not addressed. The best results (50-fold reduction in BChE inhibition) were observed when a bis-cycloSal moiety (Figure 4A(20)) was used [89].
Recently, the development of bis-cycloSal pronucleotides was reported (Figure 4A(20)) [89]. These pronucleotides deliver two molecules of active drug for each biomolecule that is delivered. The hydrolysis pathway of these pronucleotides was studied in detail. The bis-cycloSal pronucleotides were found to be more stable than the monomers. Interestingly, no hydrolysis of the benzyl ester was observed. Rather, the pronucleotides were hydrolyzed exclusively to the NMP [89]. These pronucleotides exhibited reduced BChE inhibition [88,89] and maintained antiviral activity comparable to that of the nucleoside in wild-type CEM cells while displaying significantly enhanced antiviral activity in kinase-deficient cells [89]. The pronucleotides displayed considerably higher cytotoxicity than the nucleoside.
The cycloSal approach has effectively achieved delivery of an NMP through a pH-dependent mechanism triggered by enzymatic release of the polar cycloSal phosphotriester intracellularly. In a majority of these cases, salicyl alcohol is successful in masking the phosphate and releasing the active drug in an enzyme-independent manner. These pronucleotides increase antiviral activity in vitro compared to the parent nucleoside, especially in kinase-deficient cells, while often demonstrating increased cytotoxicity, although Meier and coworkers reported that the salicyl alcohol is non-toxic. Modification of the salicyl alcohol aromatic ring substituents can increase the stability and decrease the BChE inhibitory activity of the pronucleotide. With the further refinement of their intracellular trapping strategy, Meier and coworkers have demonstrated a versatile method to deliver nucleoside monophosphates or nucleotide analogues.
6. New amino acid and peptide conjugates as pronucleotides
In the context of bioterrorism, the development of an orally available therapy [18] effective against such viral pathogens as variola virus (smallpox) is of urgent concern. Cidofovir is an FDA-approved, nucleoside monophosphonate derivative known to be effective against variola and related viruses, but has inadequate oral bioavailability and thus has become an important target of contemporary prodrug strategies.
McKenna and coworkers have recently studied the use of biologically benign amino acids and peptides conjugated to cidofovir or cyclic cidofovir via a phosphonate ester with the serine side chain hydroxyl group to increase oral bioavailability of the drugs [12,90–93].
Enhanced oral bioavailability resulting from conjugation of L-valine to acyclovir by esterification of the hydroxyl group [5,94] has stimulated interest in amino acids or dipeptides as promoieties. A dipeptide promoiety might interact with a nutritional peptide transporter, such as the human oligopeptide transporter 1 (hPEPT1), in the gastrointestinal tract [95]. In this approach, an amino acid or dipeptide was attached via an ethylene glycol (EG) linker (Figure 5(27)) or directly via esterification of a serine side chain hydroxyl group (Figure 5(28 and 29)). It was hypothesized that the use of a naturally occurring phosphoserine bond might allow for activation by a phosphoserine/phosphothreonine specific protein phosphatase or a dual-specificity protein phosphatase [96,97]. The other phosphonic acid –OH group was masked by intramolecular esterification to afford a cyclic cidofovir (cHPMPC) derivative (Figure 5(29)) or via esterification by an ethyl group (Figure 5(28)).
Figure 5.
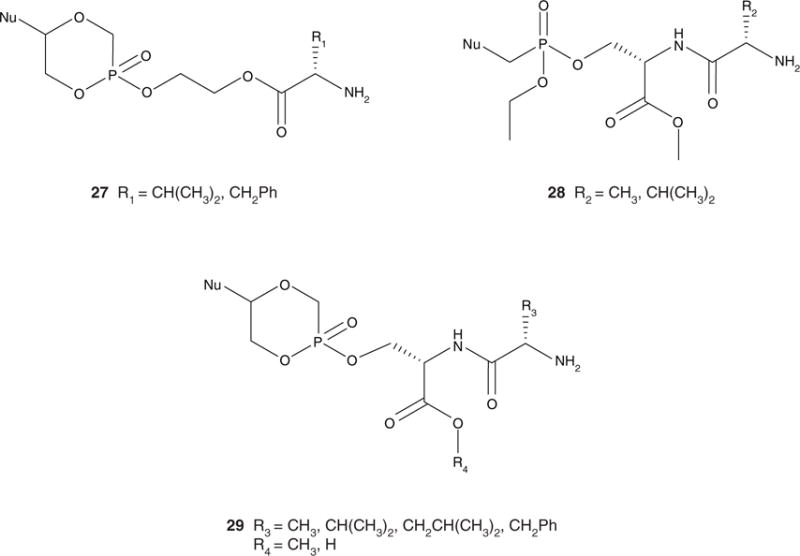
General structures of the ethylene glycol-linked amino acid (27) and serine phosphoester dipeptide pronucleotides (28 and 29).
Pronucleotides containing an amino acid–EG–cHPMPC conjugate made by POH–HOC esterification were synthesized (Figure 5(27)) [90]. While the analogue was stable in moderately acidic and neutral buffers, it was rapidly metabolized to cHPMPC by liver, intestinal and cellular enzymes [90]. The L-valine pronucleotide exhibited a four-fold increase in antiviral activity compared to ganciclovir in a human cytomegalovirus (HCMV) plaque reduction assay and showed no cytotoxicity in KB cells [90]. However, the EG-linked amino acid conjugates did not exhibit increased bioavailability compared to the parent compound after direct injection into the gastrointestinal tract of rats [90].
Dipeptide diesters of acyclic cidofovir (HPMPC) were also synthesized (Figure 5(28)) [91], incorporating a dipeptide esterified to the phosphonate via the serine side chain hydroxyl group and an ethyl ester masking the other P-OH group. Although these conjugates exhibited enhanced bioavailability, the ethyl group was not cleaved in an in vivo rat model. The P-OEt monoester metabolite was detected in plasma, and this compound did not exhibit significant activity in an in vitro vaccinia plaque reduction assay [91].
Dipeptide monoesters of cHPMPC containing a variable N-terminal amino acid and serine esterified to cHPMPC via the side chain hydroxyl group of serine have been synthesized (Figure 5(29)) [12,92,98]. It should be noted that in addition to elegantly masking the P-OH by the internal esterification, the cHPMPC structure is associated with decreased nephrotoxicity compared to HPMPC [99]. When exposed to intracellular cyclic CMP phosphodiesterase, cHPMPC generates the parent drug [99]. Similar stability profiles were observed for the dipeptide pronucleotides and the EG-linked pronucleotides. An eight-fold increase in oral bioavailability compared to the parent drug was observed for the L-Val-L-Ser(OMe) cHPMPC pronucleotide in a rat model. Exchange of the L-valine for other hydrophobic amino acid residues, such as Ala, Phe and Leu, resulted in enhanced oral bioavailability compared to the parent compound, while the free carboxylic acid analogue, L-Val-L-Ser(OH) cHPMPC, exhibited the lowest bioavailability of all compounds tested [98]. To investigate the effect of varying the amino acid stereochemistry on bioavailability, a series of prodrugs were synthesized [93,98]. The use of D-amino acids has the potential to provide additional enzymatic stability without necessarily incurring toxicity [100]. The best bioavailability was achieved with the pronucleotides containing amino acids with D-stereochemistry in the N-terminal residue [98]. It was hypothesized that the increased enzymatic stability afforded by the D-isomers allows for more intact pronucleotide to be transported into the plasma. These modifications reveal that the observed enhanced transport might be due to a mechanism other than hPEPT1 [93,98].
Dipeptide conjugates of cHPMPC evaluated for in vitro antiviral activity in a plaque reduction assay demonstrated activity against HCMV in the submicromolar range, which was 10-fold lower than ganciclovir, the positive control [92]. These conjugates exhibited little to no cytotoxicity at concentrations up to 100 μM in human foreskin fibroblast (HFF) and KB cells [92]. In vivo antiviral experiments are needed to understand the full potential of these conjugates as prodrugs.
7. Conclusion
Nucleosides and nucleotide analogues represent an important tool in the treatment of viruses and cancer. However, their therapeutic use is hindered by several factors. The first phosphorylation step of nucleosides is often the rate-determining step in obtaining the active nucleoside triphosphate, and viruses can also develop resistance to nucleoside drugs through kinase deficiencies, as well as by modifying nucleoside transport. Nucleotide analogues usually exist as dianions at physiological pH, and, therefore, transverse of cellular membranes is often limited. Over the past decade, several creative prodrug strategies have been developed to overcome these deficiencies. Further optimization of the specifics to obtain a balance between transport, toxicity and activation should lead to the development of pronucleotides that can more effectively achieve oral absorption, bypass pre-systemic metabolism and deliver nucleotide analogues to exert their therapeutic action.
8. Expert opinion
The treatment of viral infections by nucleotide analogues is well recognized. Although their value has been proven, their therapeutic utility is often limited by low bioavailability. As a result, the development of orally available prodrug modifications of such drugs is highly desirable. Several developing approaches aimed at improving the oral absorption of nucleotide drugs have been reviewed here. All of these approaches show promise at some level but, in our opinion, the efficient development of prodrug strategies in general is impeded by the lack of comprehensive, reliable models for predicting oral bioavailability, pharmacokinetics and toxicity based on the molecular structure of a prodrug.
The pronucleotide approaches discussed here have been developed using ‘chemist’s intuition’ to optimize strategies for masking charge to achieve oral absorption and for activation in vivo. Absorption is traditionally estimated empirically by methods such as lipid/water solubility and membrane permeability in an animal model. Despite the rudimentary nature of these predictive tools, the application of chemical ingenuity has been remarkably successful in affording useful prodrug modifications. Yet the uncertainty of achieving a beneficial outcome in a given case points to a lack of sophistication in current models for prodrug behavior in vivo, and thus improvement of these models is a key long-term objective for drug delivery research.
When designing therapeutic agents with enhanced oral absorption, the fundamental processes of absorption, distribution, metabolism, excretion and transport can be addressed only individually and semi-empirically. For a prodrug to be successful, a complex series of conditions need to be met. The prodrug must have robust stability in the gut, be substantially transported intact into the plasma, and thence to the target cell. Preferably at the target, and certainly not prematurely, the prodrug must be converted to its active form at an efficacious rate. The prodrug and promoiety metabolites must not display excess toxicity. The ideal prodrug will satisfy all these criteria, and other desiderata. Unfortunately, today’s state of the art does not allow us to optimize all of these properties in a rationalized, unified manner based on prediction from a particular molecular structure. Physiologically based computer models are now emerging to guide candidate drug selection prior to clinical studies [101]. However, it is clear that improved rational prodrug design will also depend on further advances in understanding transport at the molecular level, especially the expression of active transporters, their specificity and mechanisms of action, in addition to more predictable in vivo activation. Accurate in silico modeling of potential human toxicity remains a yet more difficult challenge.
In summary, although great strides in design have been made by combining simple pharmacological principles, intuition and empirical data, the ideal prodrug will only be obtained efficiently with the development and optimization of an integrated, comprehensive model for transport, activation and toxicity. In the meantime, progress in this field will continue to be driven by the heuristic savoir-faire and synthetic skills of medicinal chemists.
Acknowledgments
CM thanks the NIH for grants U01 AI061457 and R44 AI056864 in support of his work. LP is a 2008 – 2009 WiSE Merit Fellow.
Footnotes
Declaration of interest
LP and CM are co-inventors on a patent related to a portion of the work discussed in this review.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30(2):115–33. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Rideout JL, Henry DW, Beacham LM III, editors. Nucleosides, nucleotides, and their biological applications. (Proceedings of the 5th International Round Table, October 20–22, 1982) Academic Press; New York: 1983. [Google Scholar]
- 3•.Ariza ME. Current prodrug strategies for the delivery of nucleotides into cells. Drug Des Rev Online. 2005;2(5):373–87. A thorough review of current prodrug strategies as utilized with nucleotides for all purposes. [Google Scholar]
- 4.Hostetler KY. Synthesis and antiviral evaluation of broad spectrum, orally active analogs of cidofovir and other acyclic nucleoside phosphonates. Adv Antivir Drug Des. 2007;5:167–84. [Google Scholar]
- 5.Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97(3):1109–34. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- 6.Wagner CR, Iyer VV, McIntee EJ. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med Res Rev. 2000;20(6):417–51. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 7.Hillaireau H, Le Doan T, Appel M, Couvreur P. Hybrid polymer nanocapsules enhance in vitro delivery of azidothymidine-triphosphate to macrophages. J Control Release. 2006;116(3):346–52. doi: 10.1016/j.jconrel.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Vinogradov SV, Zeman AD, Batrakova EV, Kabanov AV. Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J Control Release. 2005;107(1):143–57. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duzgunes N, Simoes S, Slepushkin V, et al. Delivery of antiviral agents in liposomes. Methods Enzymol. 2005;391:351–73. doi: 10.1016/S0076-6879(05)91020-3. [DOI] [PubMed] [Google Scholar]
- 10.Rossi L, Serafini S, Pierige F, et al. Erythrocyte-based drug delivery. Expert Opin Drug Deliv. 2005;2(2):311–22. doi: 10.1517/17425247.2.2.311. [DOI] [PubMed] [Google Scholar]
- 11.Aguzzi C, Cerezo P, Viseras C, Caramella C. Use of clays as drug delivery systems: possibilities and limitations. Appl Clay Sci. 2007;36(1–3):22–36. [Google Scholar]
- 12.McKenna CE, Kashemirov BA, Eriksson U, et al. Cidofovir peptide conjugates as prodrugs. J Organomet Chem. 2005;690(10):2673–8. [Google Scholar]
- 13.Anastasi C, Quelever G, Burlet S, et al. New antiviral nucleoside prodrugs await application. Curr Med Chem. 2003;10(18):1825–43. doi: 10.2174/0929867033457034. [DOI] [PubMed] [Google Scholar]
- 14.Van de Waterbeemd H, Lennernas H, Artursson P. Drug bioavailability: estimation of solubility, permeability, absorption and bioavailability. Methods Princ Med Chem. 2003;18 2003. [Google Scholar]
- 15.De Clercq E, Field HJ. Antiviral prodrugs – the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;147(1):1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schultz C. Prodrugs of biologically active phosphate esters. Biorg Med Chem. 2003;11(6):885–98. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- 17••.Painter GR, Hostetler KY. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol. 2004;22(8):423–7. doi: 10.1016/j.tibtech.2004.06.008. A comprehensive review of the synthesis, development and design rationale of the ether lipid ester pronucleotides. [DOI] [PubMed] [Google Scholar]
- 18•.Quenelle DC, Collins DJ, Wan WB, et al. Oral treatment of cowpox virus and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother. 2004;48(2):404–12. doi: 10.1128/AAC.48.2.404-412.2004. In vivo studies of the ether lipid esters against vaccinia and cowpox virus after oral administration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perrone P, Luoni GM, Kelleher MR, et al. Application of the phosphoramidate ProTide approach to 4′-azidouridine confers sub-micromolar potency versus Hepatitis C virus on an inactive nucleoside. J Med Chem. 2007;50(8):1840–9. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 20.Perrone P, Daverio F, Valente R, et al. First example of phosphoramidate approach applied to a 4′-substituted purine nucleoside (4′-azidoadenosine): conversion of an inactive nucleoside to a submicromolar compound versus Hepatitis C virus. J Med Chem. 2007;50(22):5463–70. doi: 10.1021/jm070362i. [DOI] [PubMed] [Google Scholar]
- 21.Mehellou Y, McGuigan C, Brancale A, Balzarini J. Design, synthesis, and anti-HIV activity of 2′,3′-didehydro-2′,3′-dideoxyuridine (d4U), 2′,3′-dideoxyuridine (ddU) phosphoramidate ‘ProTide’ derivatives. Bioorg Med Chem Lett. 2007;17(13):3666–9. doi: 10.1016/j.bmcl.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 22.McGuigan C, Hassan-Abdallah A, Srinivasan S, et al. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J Med Chem. 2006;49(24):7215–26. doi: 10.1021/jm060776w. [DOI] [PubMed] [Google Scholar]
- 23.Balzarini J, Karlsson A, Aquaro S, et al. Mechanism of anti-HIV action of masked alaninyl d4T-MP derivatives. Proc Natl Acad Sci USA. 1996;93(14):7295–9. doi: 10.1073/pnas.93.14.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballatore C, McGuigan C, De Clercq E, Balzarini J. Synthesis and evaluation of novel amidate prodrugs of PMEA and PMPA. Bioorg Med Chem Lett. 2001;11(8):1053–6. doi: 10.1016/s0960-894x(01)00128-7. [DOI] [PubMed] [Google Scholar]
- 25.McGuigan C, Pathirana RN, Mahmood N, et al. Aryl phosphates derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antiviral Res. 1992;17(4):311–21. doi: 10.1016/0166-3542(92)90026-2. [DOI] [PubMed] [Google Scholar]
- 26.Balzarini J, Aquaro S, Hassan-Abdallah A, et al. Improved antiviral activity of the aryloxymethoxyalaninyl phosphoramidate (APA) prodrug of abacavir (ABC) is due to the formation of markedly increased carbovir 5′-triphosphate metabolite levels. FEBS Lett. 2004;573(1–3):38–44. doi: 10.1016/j.febslet.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 27.Lee WA, He GX, Eisenberg E, et al. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother. 2005;49(5):1898–906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28••.Cahard D, McGuigan C, Balzarini J. Aryloxy phosphoramidate triesters as protides. Mini Rev Med Chem. 2004;4(4):371–81. doi: 10.2174/1389557043403936. A key review that describes the development of the aryloxy phosphoramidate pronucleotides and their use as antiviral agents. [DOI] [PubMed] [Google Scholar]
- 29.McGuigan C, Nickson C, O’Connor TJ, Kinchington D. Synthesis and anti-HIV activity of some novel lactyl and glycolyl phosphate derivatives. Antiviral Res. 1992;17(3):197–212. doi: 10.1016/0166-3542(92)90041-3. [DOI] [PubMed] [Google Scholar]
- 30.McGuigan C, Pathirana RN, Davies MPH, et al. Diaryl phosphate derivatives act as pro-drugs of AZT with reduced cytotoxicity compared to the parent nucleoside. Bioorg Med Chem Lett. 1994;4(3):427–30. [Google Scholar]
- 31.Venkatachalam TK, Samuel P, Qazi S, Uckun FM. Effect of change in nucleoside structure on the activation and antiviral activity of phosphoramidate derivatives. Biorg Med Chem. 2005;13(18):5408–23. doi: 10.1016/j.bmc.2005.04.083. [DOI] [PubMed] [Google Scholar]
- 32.McGuigan C, Sutton PW, Cahard D, et al. Synthesis, anti-human immunodeficiency virus activity and esterase lability of some novel carboxylic ester-modified phosphoramidate derivatives of stavudine (d4T) Antivir Chem Chemother. 1998;9(6):473–9. doi: 10.1177/095632029800900603. [DOI] [PubMed] [Google Scholar]
- 33.Knaggs MH, McGuigan C, Harris SA, et al. A QSAR study investigating the effect of l-alanine ester variation on the anti-HIV activity of some phosphoramidate derivatives of d4T. Bioorg Med Chem Lett. 2000;10(18):2075–8. doi: 10.1016/s0960-894x(00)00397-8. [DOI] [PubMed] [Google Scholar]
- 34•.Saboulard D, Naesens L, Cahard D, et al. Characterization of the activation pathway of phosphoramidate triester prodrugs of stavudine and zidovudine. Mol Pharmacol. 1999;56(4):693–704. An article describing the characterization of the activation pathway of the aryloxy phosphoramidate pronucleotides. [PubMed] [Google Scholar]
- 35.McGuigan C, Harris SA, Daluge SM, et al. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J Med Chem. 2005;48(10):3504–15. doi: 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- 36.McGuigan C, Tsang HW, Cahard D, et al. Phosphoramidate derivatives of d4T as inhibitors of HIV: the effect of amino acid variation. Antiviral Res. 1997;35(3):195–204. doi: 10.1016/s0166-3542(97)00029-6. [DOI] [PubMed] [Google Scholar]
- 37.Siddiqui AQ, Ballatore C, McGuigan C, et al. The presence of substituents on the aryl moiety of the aryl phosphoramidate derivative of d4T enhances anti-HIV efficacy in cell culture: a structure–activity relationship. J Med Chem. 1999;42(3):393–9. doi: 10.1021/jm9803931. [DOI] [PubMed] [Google Scholar]
- 38.Congiatu C, McGuigan C, Jiang WG, et al. Naphthyl phosphoramidate derivatives of BVdU as potential anticancer agents: design, synthesis and biological evaluation. Nucleosides Nucleotides Nucleic Acids. 2005;24(5–7):485–9. doi: 10.1081/ncn-200061774. [DOI] [PubMed] [Google Scholar]
- 39.Siccardi D, Gumbleton M, Omidi Y, McGuigan C. Stereospecific chemical and enzymatic stability of phosphoramidate triester prodrugs of d4T in vitro. Eur J Pharm Sci. 2004;22(1):25–31. doi: 10.1016/j.ejps.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Mucha A, Grembecka J, Cierpicki T, Kafarski P. Hydrolysis of the phosphonamidate bond in phosphono dipeptide analogs – the influence of the nature of the N-terminal functional group. Eur J Org Chem. 2003;2003(24):4797–803. [Google Scholar]
- 41.Siccardi D, Kandalaft LE, Gumbleton M, McGuigan C. Stereoselective and concentration-dependent polarized epithelial permeability of a series of phosphoramidate triester prodrugs of d4T: An in vitro study in Caco-2 and Madin-Darby canine kidney cell monolayers. J Pharmacol Exp Ther. 2003;307(3):1112–9. doi: 10.1124/jpet.103.056135. [DOI] [PubMed] [Google Scholar]
- 42.Chang SL, Griesgraber GW, Southern PJ, Wagner CR. Amino acid phosphoramidate monoesters of 3′-azido-3′-deoxythymidine: relationship between antiviral potency and intracellular metabolism. J Med Chem. 2001;44(2):223–31. doi: 10.1021/jm000260r. [DOI] [PubMed] [Google Scholar]
- 43.McIntee EJ, Remmel RP, Schinazi RF, et al. Probing the mechanism of action and decomposition of amino acid phosphomonoester amidates of antiviral nucleoside prodrugs. J Med Chem. 1997;40(21):3323–31. doi: 10.1021/jm960694f. [DOI] [PubMed] [Google Scholar]
- 44.Chang SL, Griesgraber G, Wagner CR. Comparison of the antiviral activity of hydrophobic amino acid phosphoramidate monoesters of 2′,3′-dideoxyadenosine (DDA) and 3′-azido-3′-deoxythymidine (AZT) Nucleosides, Nucleotides Nucleic Acids. 2001;20(8):1571–82. doi: 10.1081/NCN-100105248. [DOI] [PubMed] [Google Scholar]
- 45.Abraham TW, Kalman TI, McIntee EJ, Wagner CR. Synthesis and biological activity of aromatic amino acid phosphoramidates of 5-fluoro-2′-deoxyuridine and 1-beta-arabinofuranosylcytosine: Evidence of phosphoramidase activity. J Med Chem. 1996;39(23):4569–75. doi: 10.1021/jm9603680. [DOI] [PubMed] [Google Scholar]
- 46.Molema G, Jansen RW, Pauwels R, et al. Targeting of antiviral drugs to T4-lymphocytes. Anti-HIV activity of neoglycoprotein-AZTMP conjugates in vitro. Biochem Pharmacol. 1990;40(12):2603–10. doi: 10.1016/0006-2952(90)90577-8. [DOI] [PubMed] [Google Scholar]
- 47.Wagner CR, McIntee EJ, Schinazi RF, Abraham TW. Aromatic amino acid phosphoramidate di- and triesters of 3′-azido-3′-deoxythymidine (AZT) are non-toxic inhibitors of HIV-1 replication. Bioorg Med Chem Lett. 1995;5(16):1819–24. [Google Scholar]
- 48.Song H, Griesgraber George W, Wagner Carston R, Zimmerman Cheryl L. Pharmacokinetics of amino acid phosphoramidate monoesters of zidovudine in rats. Antimicrob Agents Chemother. 2002;46(5):1357–63. doi: 10.1128/AAC.46.5.1357-1363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Drontle DP, Wagner CR. Designing a pronucleotides stratagem: lessons from amino acid phosphoramidates of anticancer and antiviral pyrimidines. Mini Rev Med Chem. 2004;4(4):409–19. doi: 10.2174/1389557043403945. A review of the phosphoramidate monoester pronucleotides. [DOI] [PubMed] [Google Scholar]
- 50.Song H, Johns R, Griesgraber GW, et al. Disposition and oral bioavailability in rats of an antiviral and antitumor amino acid phosphoramidate prodrug of AZT-monophosphate. Pharm Res. 2003;20(3):448–51. doi: 10.1023/a:1022616523678. [DOI] [PubMed] [Google Scholar]
- 51.Huskey SEW, Luffer-Atlas D, Dean BJ, et al. Substance P receptor antagonist. I: Conversion of phosphoramidate prodrug after i.v. administration to rats and dogs. Drug Metab Dispos. 1999;27(11):1367–73. [PubMed] [Google Scholar]
- 52.Girardet JL, Perigaud C, Aubertin AM, et al. Increase of the anti-HIV activity of D4T in human T-cell culture by the use of the sate pronucleotide approach. Bioorg Med Chem Lett. 1995;5(24):2981–4. [Google Scholar]
- 53.Annaert P, Gosselin G, Pompon A, et al. Comparison of the disposition of ester prodrugs of the antiviral agent 9-(2-phosphonylmethoxyethyl)adenine [PMEA] in Caco-2 monolayers. Pharm Res. 1998;15(2):239–45. doi: 10.1023/a:1011914618109. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Carmichael E, Feng M, et al. Bis-S-acyl-2-thioethyl (SATE)-bearing 5′-monophosphate prodrug of beta-L-FD4C as potent anti-HBV agent. Bioorg Med Chem Lett. 1998;8(1):57–62. doi: 10.1016/s0960-894x(97)10178-0. [DOI] [PubMed] [Google Scholar]
- 55.Perigaud C, Gosselin G, Girardet JL, et al. The S-acyl-2-thioethyl pronucleotide approach applied to acyclovir. Part I. Synthesis and in vitro anti-hepatitis B virus activity of bis(S-acyl-2-thioethyl) phosphotriester derivatives of acyclovir. Antiviral Res. 1999;40(3):167–78. doi: 10.1016/s0166-3542(98)00059-x. [DOI] [PubMed] [Google Scholar]
- 56.Hantz O, Perigaud C, Borel C, et al. The SATE pronucleotide approach applied to acyclovir. Part II. Effects of bis(SATE) phosphotriester derivatives of acyclovir on duck hepatitis B virus replication in vitro and in vivo. Antiviral Res. 1999;40(3):179–87. doi: 10.1016/s0166-3542(98)00060-6. [DOI] [PubMed] [Google Scholar]
- 57.Villard AL, Coussot G, Lefebvre I, et al. Phenyl phosphotriester derivatives of AZT: variations upon the SATE moiety. Bioorg Med Chem. 2008;16(15):7321–9. doi: 10.1016/j.bmc.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 58.Egron D, Imbach JL, Gosselin G, et al. S-Acyl-2-thioethyl phosphoramidate diester derivatives as mono-nucleotide prodrugs. J Med Chem. 2003;46(21):4564–71. doi: 10.1021/jm0308444. [DOI] [PubMed] [Google Scholar]
- 59.Peyrottes S, Villard AL, Coussot G, et al. A step further in the SATE mononucleotide prodrug approach. Nucleic Acids Symp Ser. 2008;52(1):539–40. doi: 10.1093/nass/nrn273. [DOI] [PubMed] [Google Scholar]
- 60.Groschel B, Cinatl J, Perigaud C, et al. S-acyl-2-thioethyl (SATE) pronucleotides are potent inhibitors of HIV-1 replication in T-lymphoid cells cross-resistant to deoxycytidine and thymidine analogs. Antiviral Res. 2002;53(2):143–52. doi: 10.1016/s0166-3542(01)00205-4. [DOI] [PubMed] [Google Scholar]
- 61••.Peyrottes S, Egron D, Lefebvre I, et al. SATE pronucleotide approaches: an overview. Mini Rev Med Chem. 2004;4(4):395–408. doi: 10.2174/1389557043404007. An overview of the SATE approach including toxicity of the metabolites, stability studies and in vivo studies of the pronucleotides. [DOI] [PubMed] [Google Scholar]
- 62.Shafiee M, Deferme S, Villard AL, et al. New bis(SATE) prodrug of AZT 5′-monophosphate: in vitro anti-HIV activity, stability, and potential oral absorption. J Pharm Sci. 2001;90(4):448–63. doi: 10.1002/1520-6017(200104)90:4<448::aid-jps1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 63.Schlienger N, Beltran T, Perigaud C, et al. Rational design of a new series of mixed anti-HIV pronucleotides. Bioorg Med Chem Lett. 1998;8(21):3003–6. doi: 10.1016/S0960-894X(98)00535-6. [DOI] [PubMed] [Google Scholar]
- 64.Coussot G, Lefebvre I, Dimalta D, et al. SATE (aryl) phosphotriester series. II. Stability studies and physicochemical parameters. Nucleosides Nucleotides Nucleic Acids. 2003;22(5–8):907–9. doi: 10.1081/NCN-120022683. [DOI] [PubMed] [Google Scholar]
- 65••.Jochum A, Schlienger N, Egron D, et al. Biolabile constructs for pronucleotide design. J Organomet Chem. 2005;690(10):2614–25. A comprehensive review of the SATE pronucleotide approach discussing the activation of the prodrug and different promoieties. [Google Scholar]
- 66.Peyrottes S, Gosselin G, Aubertin AM, Perigaud C. SATE (aryl) phosphotriester series. I. Synthesis and biological evaluation. Nucleosides Nucleotides Nucleic Acids. 2003;22(5–8):903–5. doi: 10.1081/NCN-120022682. [DOI] [PubMed] [Google Scholar]
- 67.Beltran T, Egron D, Pompon A, et al. Rational design of a new series of pronucleotide. Bioorg Med Chem Lett. 2001;11(13):1775–7. doi: 10.1016/s0960-894x(01)00299-2. [DOI] [PubMed] [Google Scholar]
- 68.Meier C. cycloSal-pronucleotides – design of chemical Trojan horses. Mini Rev Med Chem. 2002;2(3):219–34. doi: 10.2174/1389557023406205. [DOI] [PubMed] [Google Scholar]
- 69.Meerbach A, Klocking R, Meier C, et al. Inhibitory effect of cycloSaligenyl-nucleoside monophosphates (cycloSal-NMP) of acyclic nucleoside analogues on HSV-1 and EBV. Antiviral Res. 2000;45(1):69–77. doi: 10.1016/s0166-3542(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 70.Goerbig U, Balzarini J, Meier C. New cycloAMB-nucleoside phosphonate prodrugs. Nucleosides Nucleotides Nucleic Acids. 2007;26(6–7):831–4. doi: 10.1080/15257770701503894. [DOI] [PubMed] [Google Scholar]
- 71.Meier C, Knispel T, De Clercq E, Balzarini J. CycloSal-pronucleotides of 2′,3′-dideoxyadenosine and 2′,3′-dideoxy-2′, 3′-didehydroadenosine: Synthesis and antiviral evaluation of a highly efficient nucleotide delivery system. J Med Chem. 1999;42(9):1604–14. doi: 10.1021/jm981096z. [DOI] [PubMed] [Google Scholar]
- 72.Meier C, Meerbach A, Wutzler P. CycloSal-pronucleotides of brivudine monophosphate – highly active antiviral agents. Curr Med Chem Anti-Infect Agents. 2005;4(4):317–35. [Google Scholar]
- 73.Jessen HJ, Tonn V, Meier C. Intracellular trapping of cycloSal-pronucleotides by enzymatic cleavage. Nucleosides Nucleotides Nucleic Acids. 2007;26(6–7):827–30. doi: 10.1080/15257770701503886. [DOI] [PubMed] [Google Scholar]
- 74.Gisch N, Balzarini J, Meier C. 5-diacetoxymethyl-cycloSal-d4TMP – A prototype of enzymatically activated cycloSal-pronucleotides. Nucleosides Nucleotides Nucleic Acids. 2007;26(6–7):861–4. doi: 10.1080/15257770701504025. [DOI] [PubMed] [Google Scholar]
- 75.Gisch N, Balzarini J, Meier C. Enzymatically activated cycloSal-d4T-monophosphates: the third generation of cycloSal-pronucleotides. J Med Chem. 2007;50(7):1658–67. doi: 10.1021/jm0613267. [DOI] [PubMed] [Google Scholar]
- 76.Meier C, Ducho C, Jessen H, et al. Second-generation cycloSal-d4TMP pro-nucleotides bearing esterase-cleavable sites – the ‘trapping’ concept. Eur J Org Chem. 2006;2006(1):197–206. [Google Scholar]
- 77•.Gisch N, Pertenbreiter F, Balzarini J, Meier C. 5-(1-Acetoxyvinyl)-cycloSaligenyl-2′,3′-dideoxy-2′,3′-didehydrothymidine monophosphates, a second type of new, enzymatically activated cycloSaligenyl pronucleotides. J Med Chem. 2008;51(24):8115–23. doi: 10.1021/jm801197f. A report on the synthesis, stability and antiviral activity of the most recent generation of lock-in cycloSal pronucleotides. [DOI] [PubMed] [Google Scholar]
- 78.Vukadinovic-Tenter D, Balzarini J, Meier C. New developments of the ‘lock-in’ modified cycloSal-d4TMPs. Nucleosides Nucleotides Nucleic Acids. 2007;26(10–12):1325–8. doi: 10.1080/15257770701530707. [DOI] [PubMed] [Google Scholar]
- 79.Gisch N, Balzarini J, Meier C. Studies on enzyme-cleavable dialkoxymethyl-cycloSaligenyl-2′,3′-dideoxy-2′,3′-didehydrothymidine monophosphates. J Med Chem. 2008;51(21):6752–60. doi: 10.1021/jm800853p. [DOI] [PubMed] [Google Scholar]
- 80.Ludek OR, Balzarini J, Meier C. Synthesis and antiviral evaluation of carbocyclic 3′-azidothymidine (AZT) analogs and their cycloSal-phosphate triesters. Eur J Org Chem. 2006;2006(4):932–40. [Google Scholar]
- 81.Meier C, Balzarini J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antiviral Res. 2006;71(2–3):282–92. doi: 10.1016/j.antiviral.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 82.Meier C. CycloSal phosphates as chemical Trojan horses for intracellular nucleotide and glycosyl-monophosphate delivery – chemistry meets biology. Eur J Org Chem. 2006;2006(5):1081–102. [Google Scholar]
- 83.Meier C, Lorey M, De Clercq E, Balzarini J. cycloSal-2′,3′-dideoxy-2′,3′-didehydrothymidine monophosphate (cycloSal-d4TMP): synthesis and antiviral evaluation of a new d4TMP delivery system. J Med Chem. 1998;41(9):1417–27. doi: 10.1021/jm970664s. [DOI] [PubMed] [Google Scholar]
- 84.Jessen HJ, Balzarini J, Meier C. Intracellular trapping of cycloSal-pronucleotides: Modification of prodrugs with amino acid esters. J Med Chem. 2008;51(20):6592–8. doi: 10.1021/jm800815b. [DOI] [PubMed] [Google Scholar]
- 85.Meier C, Ruppel MFH, Vukadinovic D, Balzarini J. ‘Lock-in’-cycloSal-pronucleotides – a new generation of chemical Trojan horses? Mini Rev Med Chem. 2004;4(4):383–94. doi: 10.2174/1389557043403972. [DOI] [PubMed] [Google Scholar]
- 86.Ducho C, Balzarini J, Meier C. Non-inhibition of acetylcholinesterase by cycloSal nucleotides. Nucleosides Nucleotides Nucleic Acids. 2003;22(5–8):841–3. doi: 10.1081/NCN-120022667. [DOI] [PubMed] [Google Scholar]
- 87.Meier C, Ducho C, Goerbig U, et al. Interaction of cycloSal-pronucleotides with cholinesterases from different origins. A structure–activity relationship. J Med Chem. 2004;47(11):2839–52. doi: 10.1021/jm031032a. [DOI] [PubMed] [Google Scholar]
- 88.Ducho C, Jessel S, Gisch N, et al. Novel cycloSal nucleotides with reduced inhibitory potency toward human butyrylcholinesterase. Nucleosides Nucleotides Nucleic Acids. 2005;24(5–7):519–22. doi: 10.1081/ncn-200061791. [DOI] [PubMed] [Google Scholar]
- 89.Ducho C, Goerbig U, Jessel S, et al. Bis-cycloSal-d4T-monophosphates: Drugs that deliver two molecules of bioactive nucleotides. J Med Chem. 2007;50(6):1335–46. doi: 10.1021/jm0611713. [DOI] [PubMed] [Google Scholar]
- 90.Eriksson U, Hilfinger JM, Kim JS, et al. Synthesis and biological activation of an ethylene glycol-linked amino acid conjugate of cyclic cidofovir. Bioorg Med Chem Lett. 2007;17(3):583–6. doi: 10.1016/j.bmcl.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peterson LW, Kashemirov BA, Saejueng K, et al. Novel synthetic approaches to cidofovir and foscarnet prodrugs. Antiviral Res. 2007;74(3):A74. [Google Scholar]
- 92.Eriksson U, Peterson LW, Kashemirov BA, et al. Serine peptide phosphoester prodrugs of cyclic cidofovir: Synthesis, transport, and antiviral activity. Mol Pharm. 2008;5(4):598–609. doi: 10.1021/mp8000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peterson LW, Kashemirov BA, Sala-Rabanal M, et al. Synthesis and transport studies on serine side-chain-linked peptidomimetic prodrugs of cyclic cidofovir. Abstracts of Papers, 235th ACS National Meeting; New Orleans, LA, USA. April 6–10, 2008; 2008. MEDI-183. [Google Scholar]
- 94.Soul-Lawton J, Seaber E, On N, et al. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob Agents Chemother. 1995;39(12):2759–64. doi: 10.1128/aac.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han HK, Amidon GL. Targeted prodrug design to optimize drug delivery. AAPS PharmSci. 2000;2(1):E6. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Savle PS, Shelton TE, Meadows CA, et al. N-(Cyclohexanecarboxyl)-O-phospho-L-serine, a minimal substrate for the dual-specificity protein phosphatase IphP. Arch Biochem Biophys. 2000;376(2):439–48. doi: 10.1006/abbi.2000.1750. [DOI] [PubMed] [Google Scholar]
- 97.Pestell KE, Ducruet AP, Wipf P, Lazo JS. Small molecule inhibitors of dual specificity protein phosphatases. Oncogene. 2000;19(56):6607–12. doi: 10.1038/sj.onc.1204084. [DOI] [PubMed] [Google Scholar]
- 98.Peterson LW, Kashemirov BA, Blazewska KM, et al. Synthesis and structure-activity aspects of some cyclic cidofovir peptidomimetic prodrugs. Antiviral Res. 2007;74(3):A33. [Google Scholar]
- 99.Mendel DB, Cihlar T, Moon K, Chen MS. Conversion of 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl] cytosine to cidofovir by an intracellular cyclic CMP phosphodiesterase. Antimicrob Agents Chemother. 1997;41(3):641–6. doi: 10.1128/aac.41.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Williams RE, Lock EA. Sodium benzoate attenuates D-serine induced nephrotoxicity in the rat. Toxicology. 2005;207(1):35–48. doi: 10.1016/j.tox.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 101.Parrott N, Jones H, Paquereau N, Lave T. Application of full physiological models for pharmaceutical drug candidate selection and extrapolation of pharmacokinetics to man. Basic Clin Pharmacol Toxicol. 2005;96(3):193–9. doi: 10.1111/j.1742-7843.2005.pto960308.x. [DOI] [PubMed] [Google Scholar]