

Abstract
Background
Duchenne and Becker Muscular Dystrophy (DMD and BMD, respectively), are common forms of inherited muscle disease. Information regarding the epidemiology of these conditions, including genotype, is still sparse.
Objective
To establish the prevalence and genetic profile of DMD and BMD in Puerto Rico.
Methods
We collected data from medical records in all Muscular Dystrophy Association (MDA) clinics in Puerto Rico in order to estimate the prevalence of DMD and BMD and to describe the genotypic profile of these patients. Patients selected for data analysis matched “definite”, “probable” and “possible” case definitions as established by MD STARnet.
Results
A total of 141 patients matched the inclusion criteria, with 64.5% and 35.5% being categorized into DMD and BMD, respectively. DMD and BMD prevalence in Puerto Rico was estimated at 5.18 and 2.84 per 100,000 males, respectively. Deletion was the most common form of mutation (66.7%) in the dystrophin gene, with exons in segment 45 to 47 being the most frequently affected.
Conclusions
This is the first report of the prevalence and genetic profile characteristics of DMD and BMD in Puerto Rico. Prevalence of DMD was similar to that reported worldwide, while prevalence of BMD was higher. Genetic profile was consistent with that reported in the literature.
Keywords: Duchenne muscular dystrophy, Becker muscular dystrophy, dystrophinopathy, muscular dystrophy, Puerto Rico
Introduction
Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD) are X-linked recessive allelic disorders caused by mutations in the dystrophin (DMD) gene (1). These occur almost exclusively in males and are included in the conditions commonly referred to as dystrophinopathies.
The DMD gene is the largest protein-coding gene in humans, localized at Xp21.2 with a size of 2.4 Mb(2–4). RNA transcribed from the dystrophin gene is expressed predominantly in skeletal and cardiac muscle, with lower expression in brain tissue (5). This gene encodes for dystrophin, a cytoskeletal protein that bridges the inner cytoskeleton and the extracellular matrix of muscle fibers(6). Due to its large size, the rate of DMD gene mutations is high (7). Rearrangements such as gene deletions and duplications are the most common type of aberration found in the DMD gene, but 20% to 35% of DMD and BMD cases result from point mutations(8,9).
A recently published review of the epidemiology of DMD and BMD by Mah et al estimated the worldwide prevalence to be 4.78 and 1.53 per 100,000 males, respectively (10). However, there was a lack of studies from Latin America in the literature review that served as the basis to calculate these estimates.
This is the first publication that reports estimates for the prevalence and genotype description of DMD and BMD patients in Puerto Rico.
Materials and Methods
This is a retrospective chart-review study encompassing data from 141 patients attending the Muscular Dystrophy Association (MDA) neuromuscular clinics in Puerto Rico (4 clinics in total). These clinics are the only facilities in Puerto Rico dedicated to the care of patients with muscular dystrophy, and the community is well aware that they are sponsored by the Muscular Dystrophy Association. Thus, we estimate that over 90% of DMD and BMD patients in Puerto Rico are treated in these four clinics. The study protocol was approved by the Institutional Review Board (IRB) of the University of Puerto Rico Medical Sciences Campus (IRB ID: 0890113), and authorized by the Muscular Dystrophy Association Office in Puerto Rico.
Inclusion criteria followed case definitions as established by the Muscular Dystrophy Surveillance Tracking and Research Network (11). “Definite”, “probable” and “possible” case definitions were included for data analysis. “Definite” cases have symptoms referable to DMD or BMD and either (1) a documented DMD gene mutation, (2) muscle biopsy evidencing abnormal dystrophin without an alternative explanation, or (3) creatine kinase level at least 10 times normal, pedigree compatible with X-linked recessive inheritance, and an affected family member who meets criterion 1 or 2. “Probable” cases have symptoms referable to DMD or BMD, a creatine kinase level at least 10 times normal, family history consistent with an X-linked muscular dystrophy, and either (1) no DMD gene mutation analysis or muscle biopsy, (2) an inconclusive muscle biopsy, or (3) a negative DMD gene mutation analysis. “Possible” cases have elevated creatine kinase and documented clinical symptoms referable to DMD or BMD. A neuromuscular specialist in each clinic evaluated patients and conferred clinical diagnosis of either DMD or BMD following previously published diagnosis criteria(12), if applicable.
A complete record review of all active patients in the MDA clinics from the first to the most recent visit to the clinics was performed for all patients. Data collection included age at time of data collection and age at diagnosis (defined as age at which the physician documented a DMD or BMD diagnosis). Four cases were diagnosed when they were less than 1 year old. For statistical analysis, age at diagnosis for these cases was coded as 1 year old. In addition, data from confirmatory studies was collected, including results from muscle biopsy, creatine kinase (CK), DNA studies performed by certified clinical laboratories and/or family history of DMD or BMD. Details obtained from the DNA study report included type of mutation and the affected segment of the dystrophin gene.
This data was submitted and accepted for inclusion in the Leiden Open Variation Database for DMD (7,13).
Statistical Analysis
Statistical analysis was done using SPSS version 12 on Windows XP operating system, and R version 3.2.4(14), R Commander version 2.2-3(15) and the EZR plug-in for R Commander(16) on OS X 10.11.4. Frequency distributions and Mann-Whitney tests were used for data analysis. Denominators for the estimation of disease prevalence were obtained from population estimates of the male population in for Puerto Rico for the year 2012 (1,757,189 males for 2012) available online from the United States Census Bureau (17).
Results
141 patients (all male) met the inclusion criteria for the study. Table 1 displays general characteristics of the entire sample. 91 patients (64.5%) were classified as DMD while 50 (35.5%) were classified as BMD. Data about age at diagnosis, age at the time of the study (“actual age”) and CK levels were available for 111 patients (87.7%), 140 patients (99.3%) and 111 patients (79.3%), respectively. Median age at diagnosis was 6 years old. 84 patients (59.2%) had a molecular study available for analysis, while 37 (26.1%) had a muscle biopsy result on record. Based on 2012 census data (17) , and considering that the data was collected from all four major muscular dystrophy clinics on the island, the minimum prevalence of DMD and BMD in Puerto Rico was estimated as 8.02 per 100,000 males (141 DMD/BMD males/1,757,189 total males for 2012 in Puerto Rico). The minimum prevalence of DMD and BMD in Puerto Rico is 5.18 per 100,000 males (91 DMD males/1,757,189 total males for 2012 in Puerto Rico) and 2.84 (50 BMD males/1,757,189 total males for 2012 in Puerto Rico) per 100,000 males, respectively. Table 2 details the case definitions per DMD and BMD diagnosis using the Muscular Dystrophy Surveillance Tracking and Research Network criteria established above. The majority of patients were classified as a “Definite” case.
Table 1.
General and clinical characteristics of patients with DMD/BMD. Muscular Dystrophy Association Clinics, Puerto Rico. 2012.
| Variable | Median (range), or number (percent) |
|---|---|
| Total cases | 141 |
| Actual age (median) n = 140 | 19 (4 to 72) |
| Age of diagnosis (median) n = 111 | 6 (1 to 59) |
| CK at diagnosis, units per liter (median) n = 102 | 11250 (63 to 86700) |
| Number of patients with Duchenne Muscular Dystrophy | 91 (64.5%) |
| Number of patients with Becker Muscular Dystrophy | 50 (35.5%) |
| Positive DNA study | 84 (59.2%) |
| Positive muscle biopsy | 37 (26.1%) |
| Positive family history | 49 (34.5%) |
Table 2.
Distribution by Case Definitions for DMD and BMD patients based on Muscular Dystrophy Surveillance Tracking and Research Network criteria.
| DMD Number (percent) |
BMD Number (percent) |
|
|---|---|---|
| Definite | 76 (83.5) | 35 (70.0) |
| Probable | 6 (6.6) | 5 (10.0) |
| Possible | 9 (9.9) | 10 (20.0) |
| Total | 91 (100.0) | 50 (100.0) |
The genetic profile of patients having results of a molecular study in the medical record is displayed in Table 3. The majority of patients with a molecular study have evidence of a deletion, comprising 66.7%. Figure 1 shows exon mutations over a representation of the dystrophin exon sequence. The segment that was most frequently affected was segment 45 to 47.
Table 3.
Gene mutations in patients with DMD/BMD. Muscular Dystrophy Association Clinics, Puerto Rico. 2012 (n = 141)
| Type of Mutation | Number (percent) |
|---|---|
| Deletion | 56 (66.7%) |
| Duplication | 2 (2.4%) |
| Insertion | 1 (1.2%) |
| Other | 6 (7.1%) |
| No mutation | 19 (22.6%) |
Figure 1.
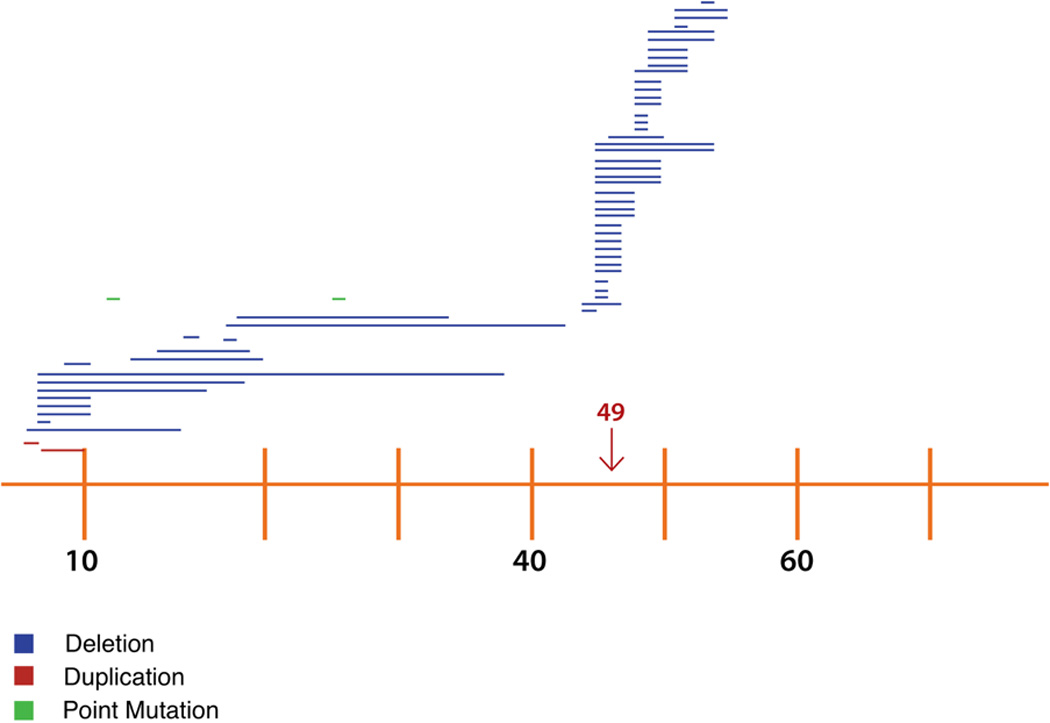
Distribution of mutations along the DMD gene.
1 unit on the X axis represents 1 exon on the DMD gene, increasing sequentially from left to right.
Age and CK profiles of DMD and BMD patients are shown in Table 4. CK distribution for DMD and BMD patients is presented in Figure 2. Differences in age at the time of the study, as well as age at diagnosis were statistically significant (p = 0.001 and p = 0.002 respectively) suggesting that BMD patients were diagnosed later in life. CK levels at the time of diagnosis also demonstrated a statistically significant difference (p = 0.001) with patients diagnosed with DMD having much higher levels at the time of diagnosis.
Table 4.
Age and CK profiles of DMD and BMD patients. In parenthesis is the number of patients from either DMD or BMD with available data for each variable.
| DMD | BMD | p-value (Mann-Whitney test) |
|
|---|---|---|---|
| Age at time of study median (n) | 18.0 (90) | 20.0 (50) | 0.001 |
| Age at Diagnosis median (n) | 5.5 (74) | 9.0 (37) | 0.002 |
| CK at Diagnosis (U/L) median (n) | 13759.5 (68) | 6476 (34) | 0.001 |
Figure 2.
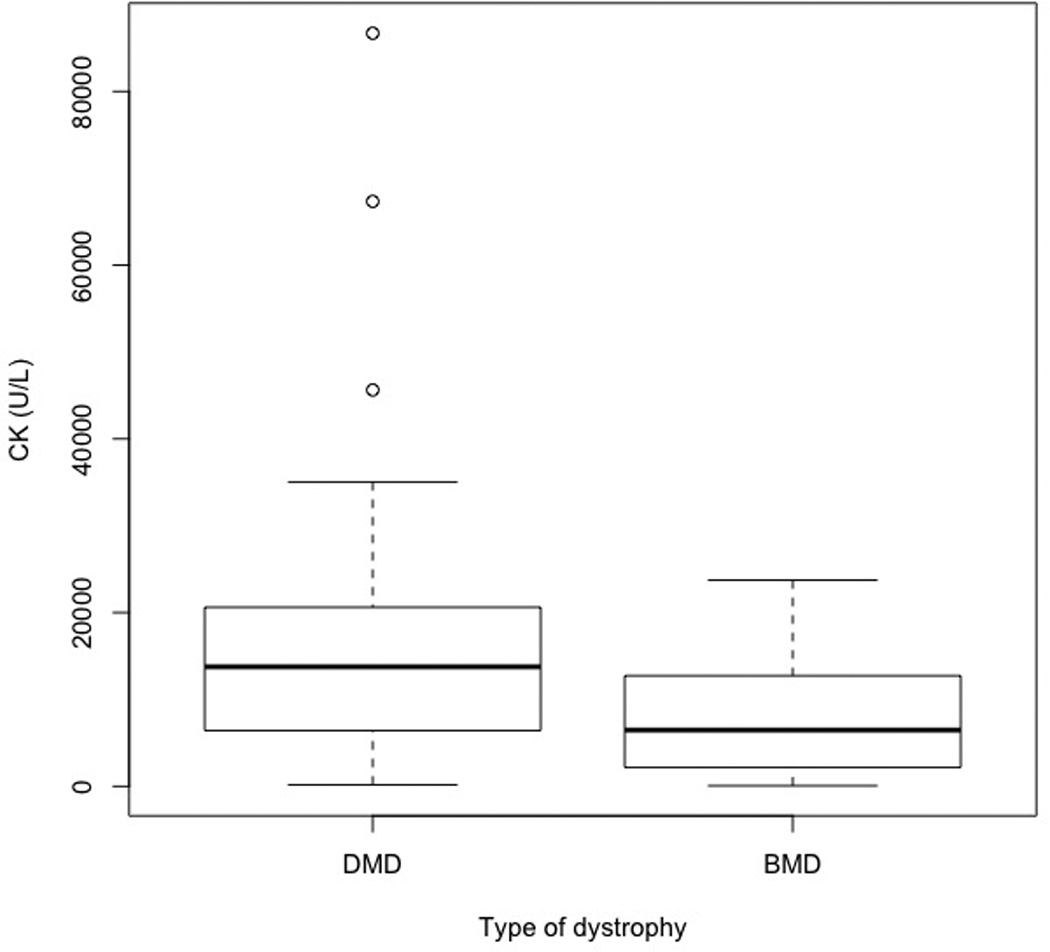
Creatine kinase levels at time of diagnosis by type of muscular dystrophy
Discussion
To our knowledge, this is the first study describing prevalence and genotypic characteristics of patients with DMD and BMD in Puerto Rico. After retrospective analysis of medical record data, one hundred forty one patients met the inclusion criteria for diagnosis of these conditions. Of these, 91 patients were diagnosed with DMD, 50 were diagnosed with BMD, and 19 patients were still pending final characterization of their conditions. As stated above, the minimum prevalence for both DMD and BMD in Puerto Rico is estimated as 8.02 per 100,000 males, while estimated individual prevalence of DMD and BMD is 5.18 and 2.84 per 100,00 males, respectively. As a reminder, the values reported by Mah in 2014 for estimated worldwide prevalence of DMD and BMD were 4.78 and 1.53 per 100,000 males, respectively (10).
It is noteworthy that the estimated minimum prevalence of BMD in Puerto Rico is 8% higher than that reported as the worldwide prevalence. Since there is no cure for this condition, a higher prevalence can be explained by an increased incidence, a lower mortality, or a combination of these two factors. Longer life expectancy (lower mortality) due to appropriate management of the disease and its associated complications might hence increase prevalence. Access to adequate diagnostic facilities may also result in a higher frequency of diagnosis, with a subsequent impact on the report of new cases of the disease. The island’s small size might also allow for less genetic variation and perpetuation of mutations resulting in more new cases of the disease.
Patients diagnosed with DMD were diagnosed at an earlier age than those with BMD (5.5 years old vs 9.0 years old, respectively). This may be explained by BMD patients presenting symptoms later in life. The difference in age at the time of the study (BMD patients older than DMD patients) might be due to better survival of BMD patients.
Molecular genetic testing of DMD can establish the diagnosis of either DMD or BMD without muscle biopsy in most individuals. Thus, the current guidelines for diagnosis establish that deletion/duplication DMD gene analysis should be done first and, on those whose result was non-informative, then DMD gene sequencing analysis should follow. If no DMD pathogenic variant is identified after performing both types of genetic analysis described above, then skeletal muscle biopsy is indicated for western blot and immunohistochemistry studies of dystrophin (18).
Approximately half (59.2%) of patients diagnosed with DMD or BMD in our sample had a molecular (DNA) study available as part of their diagnosis. Although genetic testing is an important diagnostic tool for this condition, these were not commonly used until 1990 (10). Since the average age for our study population is 20.47 years old, this could partially explain the low number of patients having a result for a diagnostic DNA study. Although not all research articles about this condition report the usage of molecular studies as a requisite for diagnosis, there are some that do. In Canada, 74% of patients had genetic testing performed for their diagnosis (19). A recent study in DMD patients in the USA cites that 94.7% of their reported population had genetic testing available for analysis (20). Genetic testing can be costly for patients, and it is not usually covered by medical insurance, which could explain the low number of DNA studies available for analysis in our clinics. As detailed above, a DMD gene sequencing analysis should follow if the initial DMD gene analysis is non-informative. Not all patients have the economic means to more testing, raising the possibility of patients not being properly diagnosed if their initial testing is unremarkable. This reveals a health disparity issue amongst our patient population, primarily regarding awareness of diagnostic guidelines and/or coverage of expenditures associated to genetic diagnostic tests in individuals who meet clinical criteria.
Of those patients who underwent a genetic test, 58 patients had deletions or duplications, whereas only 7 of the remaining 26 patients (~27%) with uninformative results on the DMD rearrangement analysis were able to undergo reflex testing for DMD sequence variations. There is a large variability on the genomic mechanisms leading to DMD or BMD. Deletions of one or more exons account for up to 60-70% of pathogenic variants, duplications account for up to 10%, and the smaller genomic sequence alterations account for approximately 25-30%(9,21–24). Large rearrangements involving multiple exons cluster in two recombination hot spots. The proximal hot spot comprises exons 2-20 (30%) and the distal hot spot comprises exons 44-53 (70%) (25). Duplications preferentially cluster at the proximal hot spot with duplication of exon 2 being the most common (26)
In contrast to the expected rates, amongst those 58 patients with large pathogenic DMD rearrangements identified, 56 had deletions of one or multiple exons (~96.6%), whereas duplications were only seen in 2 patients (~3.4%). Both patients with duplications had involvement of exons within the proximal hot spot but only one of them affected exon 2. As observed in other populations, 31% of our patients had pathogenic rearrangements involving the proximal cluster whereas 69% involved the distal cluster.
Limitations in this study are mostly linked to the nature of retrospective data collection in a healthcare facility. Data was obtained from four different clinics that had not implemented electronic medical record systems. Multiple physicians evaluated patients throughout the time period analyzed. In addition, there was no standardized form to record results from the evaluation of patients with DMD or BMD, which made data collection more difficult. Our sample size is small when compared to other studies; however, this is to be expected based on the population size of Puerto Rico. We believe that collecting data from all four MDA clinics on the island allowed us to collect data from most patients suffering from these conditions in the island.
Conclusion
This is the first report of the prevalence and genetic profile characteristics of DMD and BMD in Puerto Rico. The prevalence of these conditions in our population is similar to that reported worldwide, with a slightly higher prevalence of BMD patients. In addition, genetic characteristics were very similar to those reported in other parts of the world. There is a notable lack of genetic test data available in the population under study. This study also demonstrates the need for a standardized instrument to be used by clinicians following patients suffering from DMD and BM in Puerto Rico. The development of an island-wide registry for this population could be of benefit for future analysis, eliminating most of the limitations in the current study.
Acknowledgments
Research reported in this publication was supported in part by the National Institute of Minority Health and Health Disparities of the National Institutes of Health under award number G12MD007600. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest
The authors have no conflict of interest to report.
References
- 1.Emery AEH, Muntoni F, Quinlivan RCM. OUP Oxford. 2015. [cited 2015 Apr 15]. Duchenne Muscular Dystrophy [Internet] p. 320. Available from: https://books.google.com/books?id=qAi3BgAAQBAJ&pgis=1. [Google Scholar]
- 2.Hammonds RG. Protein sequence of DMD gene is related to actin-binding domain of alpha-actinin. [cited 2016 Feb 13];Cell [Internet] 1987 Oct 9;51(1):1. doi: 10.1016/0092-8674(87)90002-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3652206. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. [cited 2016 Feb 10];Cell [Internet] 1987 Dec 24;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3319190. [DOI] [PubMed] [Google Scholar]
- 4.Davison MD, Critchley DR. alpha-Actinins and the DMD protein contain spectrin-like repeats. [cited 2016 Feb 13];Cell [Internet] 1988 Jan 29;52(2):159–160. doi: 10.1016/0092-8674(88)90503-x. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3342446. [DOI] [PubMed] [Google Scholar]
- 5.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurology. 2003:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 6.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. [cited 2016 Feb 14];Nature [Internet] 1992 Feb 20;355(6362):696–702. doi: 10.1038/355696a0. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1741056. [DOI] [PubMed] [Google Scholar]
- 7.Aartsma-Rus A, Van Deutekom JCT, Fokkema IF, Van Ommen GJB, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle and Nerve. 2006:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 8.Buzin CH, Feng J, Yan J, Scaringe W, Liu Q, den Dunnen J, et al. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. [cited 2016 Feb 14];Hum Mutat [Internet] 2005 Feb;25(2):177–188. doi: 10.1002/humu.20132. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15643612. [DOI] [PubMed] [Google Scholar]
- 9.Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. [cited 2016 Feb 14];Am J Hum Genet [Internet] 2003 Apr;72(4):931–939. doi: 10.1086/374176. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1180355&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. [cited 2015 Apr 3];Neuromuscul Disord [Internet] 2014 Jun;24(6):482–491. doi: 10.1016/j.nmd.2014.03.008. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24780148. [DOI] [PubMed] [Google Scholar]
- 11.Mathews KD, Cunniff C, Kantamneni JR, Ciafaloni E, Miller T, Matthews D, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): case definition in surveillance for childhood-onset Duchenne/Becker muscular dystrophy. [cited 2016 Feb 13];J Child Neurol [Internet] 2010 Sep;25(9):1098–1102. doi: 10.1177/0883073810371001. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3674568&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jennekens FG, ten Kate LP, de Visser M, Wintzen AR. Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy. [cited 2016 Feb 14];Neuromuscul Disord [Internet] 1991 Jan;1(6):389–391. doi: 10.1016/0960-8966(91)90001-9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1822350. [DOI] [PubMed] [Google Scholar]
- 13.Leiden Muscular Dystrophy pages © [Internet] Leiden (Netherlands): Center for Human and Clinical Genetics LUMC; 2003. [cited 2016 M 24]. A from http://www. dmd. n No Title. [Google Scholar]
- 14. [cited 2016 Mar 24];R: The R Project for Statistical Computing [Internet] Available from: https://www.r-project.org/
- 15. [cited 2016 Mar 24];R Commander: A Basic-Statistics GUI for R [Internet] Available from: http://socserv.mcmaster.ca/jfox/Misc/Rcmdr/
- 16.Kanda Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. [cited 2016 Feb 13];Bone Marrow Transplant [Internet]. Macmillan Publishers Limited. 2013 Mar;48(3):452–458. doi: 10.1038/bmt.2012.244. Available from: http://dx.doi.org/10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.US Census Bureau DIS. [cited 2015 May 13];International Programs, International Data Base. Available from: http://www.census.gov/population/international/data/idb/region.php?N= Results&T=10&A=separate&RT=0&Y=2012&R=-1&C=RQ.
- 18.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. The Lancet Neurology. 2010:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 19.Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years - four states, 2007. [cited 2015 Apr 3];MMWR Morb Mortal Wkly Rep [Internet] 2009 Oct 16;58(40):1119–1122. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19834452. [PubMed] [Google Scholar]
- 20.Wang RT, Silverstein Fadlon CA, Ulm JW, Jankovic I, Eskin A, Lu A, et al. Online self-report data for duchenne muscular dystrophy confirms natural history and can be used to assess for therapeutic benefits. [cited 2015 Mar 18];PLoS Curr [Internet] 2014 Jan;:6. doi: 10.1371/currents.md.e1e8f2be7c949f9ffe81ec6fca1cce6a. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4207635&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed]
- 21.Bennett RR, den Dunnen J, O’Brien KF, Darras BT, Kunkel LM. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet. 2001;2:17. doi: 10.1186/1471-2156-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mendell JR, Buzin CH, Feng J, Yan J, Serrano C, Sangani DS, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology. 2001;57(4):645–650. doi: 10.1212/wnl.57.4.645. [DOI] [PubMed] [Google Scholar]
- 23.Dolinsky LCB, De Moura-Neto RS, Falcão-Conceição DN. DGGE analysis as a tool to identify point mutations, de novo mutations and carriers of the dystrophin gene. Neuromuscul Disord. 2002;12(9):845–848. doi: 10.1016/s0960-8966(02)00069-x. [DOI] [PubMed] [Google Scholar]
- 24.White S, Kalf M, Liu Q, Villerius M, Engelsma D, Kriek M, et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71(2):365–374. doi: 10.1086/341942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. [cited 2015 Apr 15];Am J Hum Genet [Internet] 1989 Dec;45(6):835–847. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1683480&tool=pmcentrez&rendertype=abstract. [PMC free article] [PubMed] [Google Scholar]
- 26.White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers ALJ, Lalic T, et al. Duplications in the DMD gene. Hum Mutat. 2006;27(9):938–945. doi: 10.1002/humu.20367. [DOI] [PubMed] [Google Scholar]