

Abstract
Soybean lipoxygenase (SLO) serves as a prototype for fundamental understanding of hydrogen tunneling in enzymes. Its reactivity depends on the active site structure around a mononuclear, nonheme iron center. The available crystal structures indicate five-coordinate iron, while magnetic circular dichroism experiments suggest significant populations of both five-coordinate (5C) and six-coordinate (6C) iron in ferrous SLO. Quantum mechanical calculations of gas phase models produce only 6C geometries. Herein mixed quantum mechanical/molecular mechanical (QM/MM) calculations are employed to identify and characterize the 5C and 6C geometries. These calculations highlight the importance of the protein environment, particularly two Gln residues in a hydrogen-bonding network with Asn694, the ligand that can dissociate. This hydrogen-bonding network is similar in both geometries, but twisting of a dihedral angle in Asn694 moves its oxygen away from the iron in the 5C geometry. These insights are important for future simulations of SLO.
Soybean lipoxygenase (SLO) is a member of an important family of enzymes that catalyze the oxidation of unsaturated fatty acids.1−4 SLO also serves as a valuable prototype for understanding fundamental aspects of hydrogen tunneling in enzymes. The significance of hydrogen tunneling in native SLO is illustrated by the large hydrogen/deuterium kinetic isotope effect of ∼80 at room temperature for the enzyme-catalyzed proton-coupled electron transfer (PCET) reaction with the linoleic acid substrate, in conjunction with even larger kinetic isotope effects of up to ∼700 for mutant SLO enzymes.5−7 These experimental results have inspired a variety of theoretical studies.7−16 The reactivity of SLO depends strongly on the active site structure, which is comprised of an iron center surrounded by six nearby residues, including three histidines, one asparagine, one isoleucine, and a water or hydroxide group. Many experimental techniques have been used to determine the structure of the active site in the relevant states of the enzyme.17−24
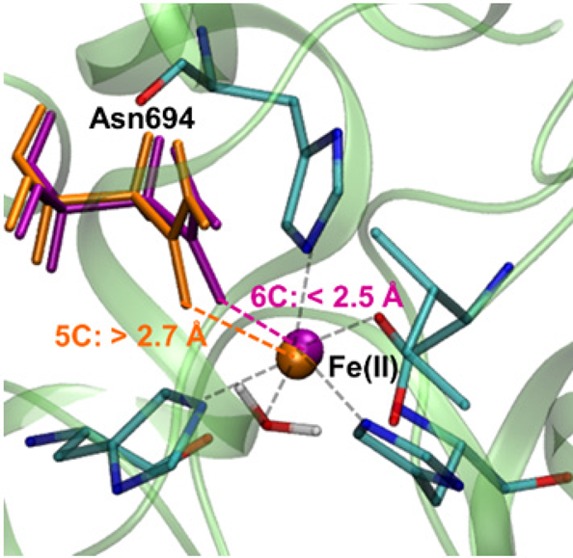
A key question that has been debated in the literature is whether the iron center in SLO is directly coordinated to five or six ligands in the ferrous state. The crystal structure of the presumed ferrous state indicates that the iron is coordinated to five ligands because the sixth closest ligand, Asn694, is 2.85 Å from the iron.20,23,24 X-ray absorption fine structure (EXAFS) studies also suggest that this distance is greater than 2.5 Å for the ferrous state.19 Moreover, magnetic circular dichroism (MCD) experiments and the analysis of the relationship between the d-d transition energies and the geometry of the Fe(II) center in ferrous SLO9,17,18,21,22 indicate that in the solution phase, the ferrous state of apo SLO exists as an approximately 40:60 mixture of five-coordinate (5C) and six-coordinate (6C) iron.9,17,18,21,22 Despite the experimental evidence for the existence of the 5C geometry of the ferrous state of apo SLO, previous density functional theory (DFT) geometry optimizations with the B3LYP functional for small model compounds mimicking the active site resulted in only the 6C geometry without imposing geometrical constraints.9 More recent quantum mechanical/molecular mechanical (QM/MM) calculations using the ONIOM approach25 identified both 5C and 6C geometries and concluded that a key difference between these geometries is the orientation and hydrogen-bonding interactions of the water ligand. This previous work proposed that the crystal structure is a mixture of the 5C and 6C geometries based on the significantly longer distance between the Fe and the oxygen of Asn694 in the optimized 5C geometry.
Herein we use different quantum mechanical/molecular mechanical (QM/MM) methods to address this issue concerning the structure of the active site for apo SLO in the ferrous state. We also identify two different minima corresponding to the 5C and 6C geometries. Our analysis highlights the importance of including the protein environment and of including critical residues that hydrogen bond to Asn694 in the QM region. We also investigate several types of density functionals and find that the results are predominantly consistent among them. The results provide insight into the delicate structural balance within the active site of SLO and characterize two qualitatively different thermally accessible conformations that may be relevant to catalytic activity. In addition, this study points out potential pitfalls in these types of QM/MM calculations, particularly the importance of including the protein environment and searching for qualitatively distinct local minima. Moreover, our results and conclusions differ from those of the previous QM/MM study25 in several key aspects that will be discussed below.
Figure 1 depicts the gas phase models used to mimic the active site of ferrous SLO. The initial coordinates were obtained from a crystal structure of SLO (PDB ID 3PZW).20,23,24 In this initial geometry, the Fe–O694 distance is 2.85 Å, suggesting that Asn694 is not coordinated to the iron. Here Fe–O694 is simplified notation to denote the distance between Fe and OD1(Asn694). We performed DFT geometry optimizations for the two different model systems depicted in Figure 1. All calculations herein were performed for the high-spin state with S = 2, on the basis of experimental studies.26 The smaller model, denoted Mod-S, included only Fe and the six closest residues (His499, His504, His690, Ile839, Asn694, and a bound water molecule). The larger model, denoted Mod-L, also included Gln495 and Gln697, which form a hydrogen-bonded network with Asn694. The larger model replaced the three His residues with NH3 and the Ile residue with formate to be the same as the model used in previous studies by Solomon and co-workers.9 We used a range of functionals, including B3LYP,27,28 B3LYP-D3,29 B3P86,28,30 M06-L,31 and in some cases M06-2x,32 for these geometry optimizations. The Fe–O694 distance was 2.17–2.21 Å for the smaller model and 2.22–2.26 Å for the larger model (Table S1). Thus, both of these model systems resulted in the 6C geometry, illustrating that gas phase models without the protein environment are not sufficient to obtain the 5C geometry. Note that applying a large number of angle restraints determined from the crystal structure was found to lead to a longer Fe–O694 distance,9 but this procedure depends on prior knowledge of the desired structure.
Figure 1.
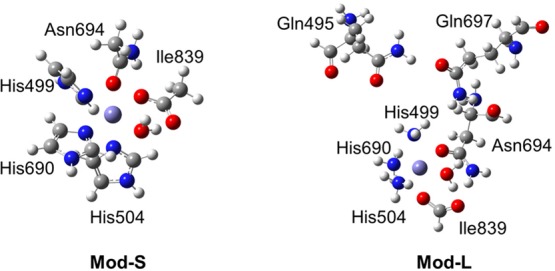
Depiction of the two gas phase model systems studied in this paper. The Mod-S model includes the Fe center and His499, His504, His690, Ile839, Asn694, and a bound water molecule. The Mod-L model also includes Gln495 and Gln697 and replaces the His residues by NH3 and the Ile residue by formate.
To explore the impact of the protein environment on the active site geometry in ferrous SLO, we conducted QM/MM geometry optimizations including the entire protein environment, as depicted in Figure 2. These calculations were performed using the Qsite module in the Schrödinger package.33 We investigated two different QM regions (Figure 2). The first QM region, denoted QM-S, included only Fe and the six closest residues, and the second QM region, denoted QM-L, also included Gln495 and Gln697. The calculations were performed with and without solvent. The geometry optimizations with the QM-L quantum region resulted in two different minima corresponding to the 5C geometry, with an Fe–O694 distance greater than 2.6 Å, and the 6C geometry, with a shorter distance of 2.3–2.4 Å (Table 1). For the QM-S quantum region, the initial geometry optimizations resulted in the 6C geometry. However, subsequent geometry optimizations with the QM-S quantum region starting from a 5C geometry obtained with the QM-L quantum region resulted in the 5C geometry (Table S2). These results indicate that the inclusion of the two hydrogen-bonding Gln residues in the QM region facilitates the identification of minimum energy structures corresponding to the 5C geometry, but such minima are also present on the potential energy surface using the smaller QM region that excludes these two Gln residues.
Figure 2.
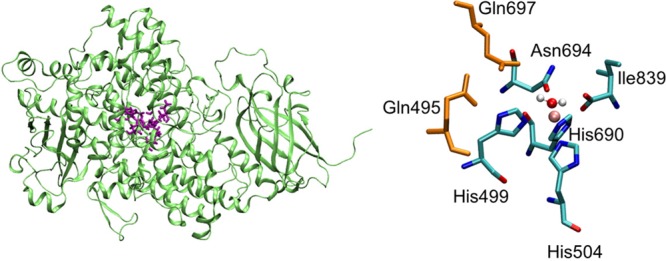
SLO apo enzyme with QM region depicted in purple (left), and a magnified view of the QM region with the iron in pink and the two Gln residues in orange (right).
Table 1. Fe–O694 Distance Determined by QM/MM Geometry Optimization of Ferrous SLO with QM-L Region and Various Functionalsa.
| without
solvent |
with solvent |
||||
|---|---|---|---|---|---|
| Min-5C | Min-6C | energy difference (kcal/mol)b | Min-5C | Min-6C | |
| B3LYP | 3.09 | 2.36 | 4.3 | 2.85 | 2.36 |
| B3LYP-D3 | 3.35 | 2.36 | –1.8 | 2.65 | 2.36 |
| B3P86 | 3.24 | 2.36 | 4.7 | 2.93 | 2.36 |
| M06-L | 3.14 | 2.37 | 0.3 | 2.71 | 2.37 |
Distances are given in Å, and energies are given in kcal/mol. The initial coordinates were chosen to correspond to 5C or 6C geometries for the corresponding geometry optimizations. The QM region and the MM region including all residues within 20 Å of the Fe center were optimized simultaneously.
The energy difference is the energy of the 6C minimum subtracted from the energy of the 5C minimum without solvent. Note that this energy difference corresponds to these two specific minima, but there are many related minima of each type with different energies on the potential energy surface. Because this energy difference does not include the effects of conformational sampling, it cannot be used to determine the relative populations of these two states. This energy difference is only given for the system without solvent because for the solvated system, the 5C and 6C systems have a slightly different number of solvent molecules due to the solvation procedure. Other local minima have different energies and span a range.
Table 1 presents the Fe–O694 distances for the two geometries identified with the QM-L quantum region using four different functionals both with and without solvent. Table S3 presents the Fe–O694 distances for the two geometries obtained by optimizing only the QM region. Minima corresponding to both the 5C and the 6C geometries were found for all functionals studied. Inclusion of the solvent environment in these geometry optimizations did not significantly impact the active site geometry. The energy differences between the 5C and 6C minima are also given in Table 1. Because these energy differences correspond to two specific local minima on the potential energy surface and do not include the effects of conformational sampling, these energies cannot be associated with the free energy difference used to determine the relative thermal populations of these two states. Nevertheless, the observation of both 5C and 6C geometries with reasonably similar energies, considering the large size of the system, is consistent with the experimental MCD measurements indicating a significant population of both 5C and 6C species.22
Previous experimental work discussed the potential significance of hydrogen-bonding interactions involving Gln495, Gln697, and Asn694. In particular, the possibility of a hydrogen bond between NE2 of Gln697 and OD1 of Asn694, denoted HB3 in Figure 3A, was discussed.6,21,23 However, our QM/MM geometry optimizations resulted in the formation of the HB2 hydrogen bond rather than the HB3 hydrogen bond (Figure 3B). To further investigate this issue, we analyzed four different crystal structures of SLO (Table S4). The N–O distances associated with the HB2 and HB3 hydrogen bonds are similar in all of these crystal structures, ranging from 3.00 to 3.20 Å, suggesting the possibility of forming either hydrogen bond. As illustrated by the portion of the crystal structure 3PZW shown in Figure 3A, however, the positions of the heavy atoms are not conducive to the HB3 hydrogen bond because the atoms comprising the C–N–O angle, labeled α in Figure 3A, are nearly linear. Specifically, this angle ranges from 154°–162° in the four crystal structures. As a result, the protons on the NE2 of Gln697 are not located in an appropriate position to form the HB3 hydrogen bond. In contrast, the crystal structure heavy atom positions are consistent with the formation of the HB2 hydrogen bond.
Figure 3.

(A) Active site obtained from the crystal structure 3PZW, including definitions of the potential hydrogen bonds involving Gln495, Gln697, and Asn694 and the C–N–O angle α. (B) Superimposition of the 5C and 6C structures of ferrous SLO determined by QM/MM geometry optimization using the QM-L quantum region and the B3LYP functional. The Fe–O694 distance is 2.85 Å in the 5C structure (dark colors, black dashed line) and is 2.36 Å in the 6C structure (light colors, gray dashed line). The two hydrogen bonds HB1 and HB2 are depicted by dashed lines, and the associated distances and angles in the optimized geometries are given in Table S5.
To compare the 5C and 6C structures, we superimposed the corresponding optimized structures and found that the hydrogen-bonding network comprised of HB1 and HB2 is present in both structures, as depicted in Figure 3B and quantified by the hydrogen-bonding distances given in Table S5. Previous work suggested that differences in this hydrogen-bonding network could be responsible for the distinct 5C and 6C geometries.9 However, these QM/MM calculations indicate that this hydrogen-bonding network is similar for both geometries (Table S5). The main difference between the 5C and 6C structures is the dihedral angle within the side chain of Asn694. The twisting of this angle moves the O694 away from the iron, leading to the 5C geometry.
Although these results are similar to those of a previous study25 using the ONIOM method in that both studies identified 5C and 6C geometries, several key differences have been identified. First, a significant conclusion of the previous study is that the water ligand adopts different conformations with distinct hydrogen-bonding interactions in the 5C and 6C geometries. According to these previous calculations, the ligated water molecule is hydrogen bonded to OT1(Ile839) and OD1(Asn694) in the 5C geometry; the ligated water molecule is also hydrogen bonded to OT1(Ile839) in the 6C geometry, but one of the OH bonds is pointing toward NE2(His504) and thus cannot form a hydrogen bond with OD1(Asn694) in the 6C geometry. The difference in coordination of the iron center is attributed to this difference in the ligated water conformation. In contrast, in the present work the ligated water maintains the same orientation and hydrogen-bonding interactions in both the 5C and 6C geometries. In both cases, the water is hydrogen bonded to OT1(Ile839), and its oxygen is ∼2.70 Å from OD1(Asn694), but the angle is not sufficiently linear to be characterized as a hydrogen bond (Figure 4). Additional minima with alternative water conformations cannot be ruled out, however, and it is not possible to determine the conformation of the water molecule experimentally. The previous work showed that the calculated CD spectra of the 5C and 6C geometries with different water conformations are in qualitative agreement with the experimental data,25 but other geometries may also be in qualitative agreement with the experimental CD spectra. The present work shows that different conformations of the ligated water molecule are not required to obtain the 5C and 6C coordinated iron center. Thus, the conformation of the ligated water molecule does not appear to determine the coordination of the iron center.
Figure 4.
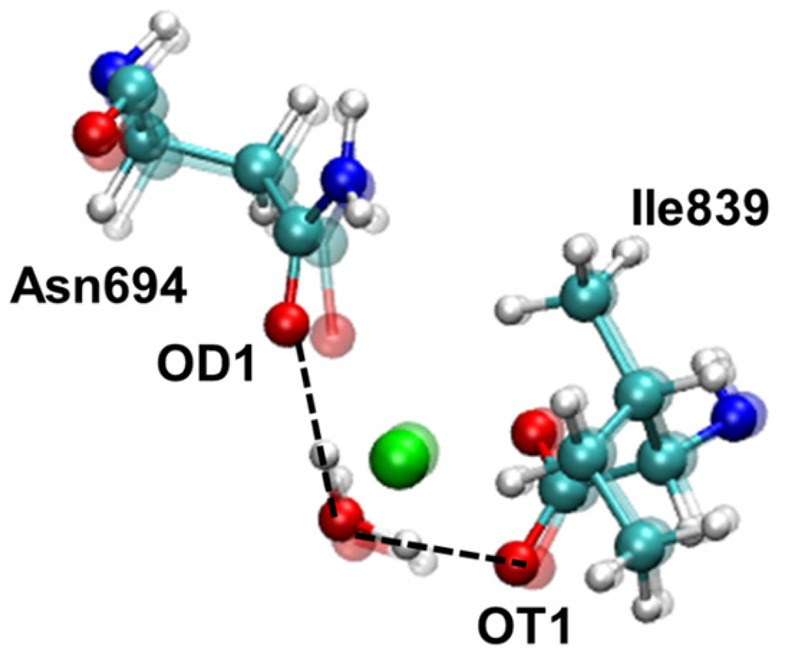
Orientation of the ligated water molecule depicted by superimposition of the 5C and 6C structures of ferrous SLO determined by QM/MM geometry optimization using the QM-L quantum region and the B3LYP functional. The distance between OD1(Asn694) and the ligated water O was 2.68 Å and 2.72 Å for the 5C and 6C structures, respectively. The angle formed by OD1(Asn694), the ligated water H, and the ligated water O was 135° and 123° for the 5C and 6C structures, respectively. The distance between OT1(Ile839) and the ligated water O was 2.52 Å for both the 5C and 6C geometries. The angle formed by OT1(Ile839), the ligated water H, and the ligated water O was 165° for both the 5C and 6C structures.
Another key difference between the present study and the previous study is the calculated Fe–O694 distance for the 5C geometry. The previous study25 calculated an Fe–O694 distance of 3.46 Å for the 5C geometry, which is significantly longer than the crystal structure distance of 2.85 Å in the 3PZW structure,20,23,24 the refinement of structures 1YGE(20) and 1F8N.20,23 According to our calculations that include the larger QM region and solvent, the Fe–O694 distance for the 5C geometry is 2.85 Å with the B3LYP functional and ranges from 2.65–2.93 Å with other functionals. On the basis of the longer Fe–O694 distances calculated with the ONIOM approach, the previous study proposed that the crystal structure is a mixture of the 5C and 6C geometries, supporting this hypothesis by pointing out that the average of the distances obtained for the 5C and 6C geometries is similar to the distance in the crystal structure. In contrast, on the basis of the agreement between our calculated Fe–O694 distance for the 5C geometry and the crystal structure distance, our calculations suggest that the crystal structure corresponds to the pure 5C geometry. Examination of the crystallographic data indicates that the crystal structure was solved at a high resolution of 1.4 Å, and the active site is extremely well-defined with no possibility of an alternative conformation for Asn694.20,23,24 Thus, the previous hypothesis that the crystal structure is a mixture of 5C and 6C geometries is not consistent with the experimental crystallographic data, which definitively specifies a 5C geometry. Finally, in contrast to the previous work, the present study includes Gln495 and Gln697 in the QM region and analyzes the associated hydrogen-bonding interactions, which have been shown experimentally through mutation of these Gln residues to impact the coordination of the iron center as well as catalysis.34
Herein computational methods were employed to identify and characterize the distinct geometries associated with five-coordinate and six-coordinate iron in ferrous SLO. The identification of both 5C and 6C geometries is consistent with experimental MCD data suggesting significant populations of both species. In the 5C geometry, twisting of a dihedral angle within the side chain of Gln694 moves the O694 away from the iron, thereby breaking the Fe–O694 metal–ligand bond. The hydrogen-bonding network involving two Gln residues and the Asn694 ligand is similar for both the 5C and 6C geometries. Moreover, this network involves the hydrogen bond between the amide nitrogen of Gln697 and the backbone oxygen of Asn694 rather than the side chain oxygen of Asn694 as suggested previously. DFT geometry optimizations of gas phase model systems of the active site produced only the 6C geometry. Thus, these calculations highlight the importance of including the protein environment, as well as searching for local minima. The insights provided by these calculations about the geometry around the Fe center in SLO will be important for future calculations of the PCET reaction catalyzed by this enzyme.
Acknowledgments
We are grateful to Judith Klinman and Adam Offenbacher for inspiring us to study this aspect of SLO and for helpful discussions. This work was funded by the National Institutes of Health Grant GM056207.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b01626.
Details of computational methods, geometrical parameters and coordinates of optimized gas-phase model systems, key distances obtained with QM-S quantum region, key distances obtained with QM-L region optimizing only the QM region, analysis of hydrogen-bonding interactions in available crystal structures, hydrogen-bond distances and angles for optimized geometries, and coordinates of QM/MM-optimized geometries for QM-L quantum region with B3LYP functional. (PDF)
QM/MM optimized 5C geometry with B3LYP, QM-L region, without solvent. (PDB)
QM/MM optimized 6C geometry with B3LYP, QM-L region, without solvent. (PDB)
QM/MM optimized 5C geometry with B3LYP, QM-L region, with solvent. (PDB)
QM/MM optimized 6C geometry with B3LYP, QM-L region, with solvent. (PDB)
The authors declare no competing financial interest.
Supplementary Material
References
- Gardner H. W. Recent Investigations into the Lipoxygenase Pathway of Plants. Biochim. Biophys. Acta, Lipids Lipid Metab. 1991, 1084, 221–239. 10.1016/0005-2760(91)90063-N. [DOI] [PubMed] [Google Scholar]
- Siedow J. N. Plant Lipoxygenase - Structure and Function. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 145–188. 10.1146/annurev.pp.42.060191.001045. [DOI] [Google Scholar]
- Yamamoto S. Mammalian Lipoxygenases - Molecular-Structures and Functions. Biochim. Biophys. Acta, Lipids Lipid Metab. 1992, 1128, 117–131. 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]
- Fordhutchinson A. W.; Gresser M.; Young R. N. 5-Lipoxygenase. Annu. Rev. Biochem. 1994, 63, 383–417. 10.1146/annurev.bi.63.070194.002123. [DOI] [PubMed] [Google Scholar]
- Knapp M. J.; Rickert K.; Klinman J. P. Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 2002, 124, 3865–3874. 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- Meyer M. P.; Tomchick D. R.; Klinman J. P. Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1146–1151. 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S. S.; Sharma S. C.; Scouras A. D.; Soudackov A. V.; Carr C. A. M.; Hammes-Schiffer S.; Alber T.; Klinman J. P. Extremely Elevated Room-Temperature Kinetic Isotope Effects Quantify the Critical Role of Barrier Width in Enzymatic C-H Activation. J. Am. Chem. Soc. 2014, 136, 8157–8160. 10.1021/ja502726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov A. M.; Ulstrup J. Proton and hydrogen atom tunnelling in hydrolytic and redox enzyme catalysis. Can. J. Chem. 1999, 77, 1085–1096. 10.1139/v99-099. [DOI] [Google Scholar]
- Lehnert N.; Solomon E. I. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J. Biol. Inorg. Chem. 2003, 8, 294–305. 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase. J. Am. Chem. Soc. 2004, 126, 5763–5775. 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M.; Siegbahn P. E. M.; Warshel A. Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase. J. Am. Chem. Soc. 2004, 126, 2820–2828. 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- Siebrand W.; Smedarchina Z. Temperature dependence of kinetic isotope effects for enzymatic carbon-hydrogen bond cleavage. J. Phys. Chem. B 2004, 108, 4185–4195. 10.1021/jp030575t. [DOI] [PubMed] [Google Scholar]
- Tejero I.; Garcia-Viloca M.; Gonzalez-Lafont A.; Lluch J. M.; York D. M. Enzyme dynamics and tunneling enhanced by compression in the hydrogen abstraction catalyzed by soybean lipoxygenase-1. J. Phys. Chem. B 2006, 110, 24708–24719. 10.1021/jp066263i. [DOI] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase: Dynamical behavior and temperature dependence of kinetic isotope effects. J. Am. Chem. Soc. 2007, 129, 187–196. 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- Mavri J.; Liu H. B.; Olsson M. H. M.; Warshel A. Simulation of tunneling in enzyme catalysis by combining a biased propagation approach and the quantum classical path method: Application to lipoxygenase. J. Phys. Chem. B 2008, 112, 5950–5954. 10.1021/jp0758420. [DOI] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. Impact of Distal Mutation on Hydrogen Transfer Interface and Substrate Conformation in Soybean Lipoxygenase. J. Phys. Chem. B 2010, 114, 6653–6660. 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slappendel S.; Veldink G. A.; Vliegenthart J. F. G.; Aasa R.; Malmstrom B. G. EPR spectroscopy of soybean lipoxygenase-1. Determination of the zero-field splitting constants of high-spin Fe(III) signals from temperature and microwave frequency dependence. Biochim. Biophys. Acta, Protein Struct. 1980, 624, 30–39. 10.1016/0005-2795(80)90222-6. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gebhard M. S.; Solomon E. I. Spectroscopic Studies of the Nonheme Ferric Active-Site in Soybean Lipoxygenase - Magnetic Circular-Dichroism as a Probe of Electronic and Geometric Structure - Ligand-Field Origin of Zero-Field Splitting. J. Am. Chem. Soc. 1991, 113, 5162–5175. 10.1021/ja00014a004. [DOI] [Google Scholar]
- Scarrow R. C.; Trimitsis M. G.; Buck C. P.; Grove G. N.; Cowling R. A.; Nelson M. J. X-Ray Spectroscopy of the Iron Site in Soybean Lipoxygenase-1 - Changes in Coordination Upon Oxidation or Addition of Methanol. Biochemistry 1994, 33, 15023–15035. 10.1021/bi00254a011. [DOI] [PubMed] [Google Scholar]
- Minor W.; Steczko J.; Stec B.; Otwinowski Z.; Bolin J. T.; Walter R.; Axelrod B. Crystal structure of soybean lipoxygenase L-l at 1.4 angstrom resolution. Biochemistry 1996, 35, 10687–10701. 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Zhou J.; Neese F.; Pavel E. G. New insights from spectroscopy into the structure/function relationships of lipoxygenases. Chem. Biol. 1997, 4, 795–808. 10.1016/S1074-5521(97)90113-7. [DOI] [PubMed] [Google Scholar]
- Holman T. R.; Zhou J.; Solomon E. I. Spectroscopic and functional characterization of a ligand coordination mutant of soybean lipoxygenase-1: First coordination sphere analogue of human 15-lipoxygenase. J. Am. Chem. Soc. 1998, 120, 12564–12572. 10.1021/ja982844c. [DOI] [Google Scholar]
- Tomchick D. R.; Phan P.; Cymborowski M.; Minor W.; Holman T. R. Structural and functional characterization of second-coordination sphere mutants of soybean lipoxpgenase-1. Biochemistry 2001, 40, 7509–7517. 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- Chruszcz M.; Wlodawer A.; Minor W. Determination of protein structures - a series of fortunate events. Biophys. J. 2008, 95, 1–9. 10.1529/biophysj.108.131789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. What is the Real Nature of Ferrous Soybean Lipoxygenase-1? A New Two-Conformation Model Based on Combined ONIOM(DFT:MM) and Multireference Configuration Interaction Characterization. J. Phys. Chem. Lett. 2010, 1, 901–906. 10.1021/jz1001456. [DOI] [Google Scholar]
- Dunham W. R.; Carroll R. T.; Thompson J. F.; Sands R. H.; Funk M. O. The Initial Characterization of the Iron Environment in Lipoxygenase by Mossbauer-Spectroscopy. Eur. J. Biochem. 1990, 190, 611–617. 10.1111/j.1432-1033.1990.tb15616.x. [DOI] [PubMed] [Google Scholar]
- Lee C. T.; Yang W. T.; Parr R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry.III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- QSite, version 6.2; Schrödinger, LLC: New York, 2014. [Google Scholar]
- Schenk G.; Neidig M. L.; Zhou J.; Holman T. R.; Solomon E. I. Spectroscopic characterization of soybean lipoxygenase-1 mutants: the role of second coordination sphere residues in the regulation of enzyme activity. Biochemistry 2003, 42, 7294–302. 10.1021/bi027380g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.