

ABSTRACT
The voltage-dependent K+ channel Kv1.3 (also known as KCNA3), which plays crucial roles in leukocytes, physically interacts with KCNE4. This interaction inhibits the K+ currents because the channel is retained within intracellular compartments. Thus, KCNE subunits are regulators of K+ channels in the immune system. Although the canonical interactions of KCNE subunits with Kv7 channels are under intensive investigation, the molecular determinants governing the important Kv1.3– KCNE4 association in the immune system are unknown. Our results suggest that the tertiary structure of the C-terminal domain of Kv1.3 is necessary and sufficient for such an interaction. However, this element is apparently not involved in modulating Kv1.3 gating. Furthermore, the KCNE4-dependent intracellular retention of the channel, which negatively affects the activity of Kv1.3, is mediated by two independent and additive mechanisms. First, KCNE4 masks the YMVIEE signature at the C-terminus of Kv1.3, which is crucial for the surface targeting of the channel. Second, we identify a potent endoplasmic reticulum retention motif in KCNE4 that further limits cell surface expression. Our results define specific molecular determinants that play crucial roles in the physiological function of Kv1.3 in leukocytes.
KEY WORDS: Potassium channels, Trafficking, Channelosome, Intracellular retention
Summary: Kv1.3 and KCNE4 form functional channels in leukocytes. KCNE4 associates with the C-terminal domain of Kv1.3 providing two independent and additive mechanisms for the intracellular retention of the complex.
INTRODUCTION
Voltage-dependent K+ (Kv) channels are crucial for the cardiac action potential and propagation of the nerve impulse. In addition, Kv channels play important roles in many cellular processes such as maintenance of the resting membrane potential, regulation of cell volume and proliferation (Hille, 2001). Kv currents present substantial variability and functional versatility in tissues, which is achieved by heterooligomerization and by post-translational modifications that regulate their activity. The Kv1.3 channel (also known as KCNA3), which plays important roles in the immune system response, is not an exception. Kv1.3 controls leukocyte physiology by modulating the membrane potential and driving force for Ca2+ entry through Ca2+ release-activated Ca2+ channels (ICRAC). Indeed, pharmacological blockade of Kv1.3 inhibits the immune response in vivo, and aberrant surface expression of Kv1.3 is linked to the development of autoimmune diseases (Chandy et al., 2004). For example, T-effector memory cells from patients with multiple sclerosis and other autoimmune dysfunctions present an elevated number of Kv1.3 channels at the cell membrane (Varga et al., 2010). In addition, systemic lupus erythematosus cells display an abnormal surface distribution of channels (Nicolaou et al., 2007). By contrast, immunosuppression can be related to an impairment of the cell surface expression of Kv1.3 (Villalonga et al., 2010). In summary, the pharmacological regulation of Kv1.3 activity and localization is of enormous clinical interest.
Kv1.3 heterooligomeric associations also modulate physiological responses by governing both the number and the spatial distribution of surface channels (Vicente et al., 2008). Assembly with Kv1.5 (also known as KCNA5) modulates channel behavior, and heterotetrameric channels with different stoichiometries might form in mononuclear phagocytes, such as macrophages and dendritic cells, thereby fine-tuning cellular responses (Felipe et al., 2010). In addition, co-assembly with accessory subunits, such as Kvβ (also known as KCNAB) or KCNE subunits, generates further functional diversity that affects both channel gating kinetics and trafficking (Sole et al., 2009; Vicente et al., 2005). In this context, KCNE4 efficiently regulates Kv1.3 activity by promoting its retention within the endoplasmic reticulum (ER), thereby impairing its surface expression (Sole et al., 2009). Specifically, KCNE4, which is present in the immune system, might act as a very powerful dominant-negative regulatory subunit of Kv1.3 channels in leukocytes (Sole and Felipe, 2010). Although the T1 domain of the channel, located at the N-terminus, is the molecular determinant involved in the interaction with Kvβ subunits (Gulbis et al., 2000), nothing is known about the domains involved in this physiologically relevant Kv1.3–KCNE4 association, which, by controlling channel surface expression, could have an enormous influence on the immunological response.
KCNE subunits are single-transmembrane-domain proteins that canonically associate with Kv7 channels (McCrossan and Abbott, 2004). KCNE4 is the most divergent member of the KCNE family (KCNE1–KCNE5) and plays a dominant-negative role for numerous K+ channels, such as Kv7.1 (KCNQ1), Kv1.1 (KCNA1), Kv1.3, Kv4.2 (KCND2) and KCa1.1 (KCNMA1) (McCrossan and Abbott, 2004). Although residues from the S6 domain of Kv7.1 are necessary for KCNE4-dependent inhibition of channel gating, the physical association itself involves the Kv7.1 channel C-terminus (Vanoye et al., 2009). Structural features are quite different among different K+ channel families, and surprisingly, no studies have addressed the interactions of KCNE4 with other K+ channels. Because KCNE4 exerts important influences over Kv1.3 that could modulate the immune response (Sole et al., 2009), we aimed to decipher the molecular determinants in Kv1.3 that are involved in the association with KCNE4. Our data suggest that the tertiary topology of the C-terminal domain of Kv1.3 is the structural element responsible for the interaction with KCNE4. Although this domain is sufficient to transfer the physical association capability to other Kv channels, such as Kv1.5, it is not involved in the control of the Kv1.3 gating. A major consequence of this association is the decrease in functional channels at the surface owing to massive intracellular retention. In this context, we demonstrate that KCNE4 masks the recently identified forward di-acidic trafficking motif at the C-terminus of Kv1.3, thereby impairing the plasma membrane targeting. In addition, an ER retention motif in KCNE4 further participates in the intracellular retention of the Kv1.3–KCNE4 complex, which, in turn, can alter the immunological response.
RESULTS
Kv1.3 and KCNE4 associate in CY15 dendritic cells
Kv1.3 and KCNE4 are present in leukocytes, and KCNE4 modulates the surface expression of the functional complex at the plasma membrane (Sole et al., 2009). Therefore, we first investigated whether a functional heterooligomeric complex is present in immune system cells. Leukocytes, which express a limited repertoire of K+ channels, exhibit important differences among cell lineages. Therefore, we analyzed the voltage-dependent K+ currents in both Jurkat T-cells and CY15 dendritic cells (Fig. 1A–D). Depolarizing pulses elicited K+ currents that resembled those from Kv1.3 channels in Jurkat T-cells and CY15 cells (Fig. 1A,B), although both cell types slightly differed in the slow C-type inactivation. To elucidate whether these macroscopic currents were indeed conducted by Kv1.3, biophysical and pharmacological properties, such as the cumulative inactivation and the inhibition by margatoxin (MgTx), were analyzed. Cumulative inactivation was obtained by applying a train of 15 depolarizing voltage steps of 250 ms from −80 to +60 mV once every second (Fig. 1C,D). We found that K+ currents were sensitive to MgTx in both cell lines (Fig. 1E). Thus K+ currents in T-lymphocytes and dendritic cells are indeed mostly conducted by Kv1.3. However, Kv currents in CY15 dendritic cells showed less cumulative inactivation than in T-lymphocytes (34±2% and 66±8%, mean±s.e.m., respectively). In addition, 1 and 10 nM MgTx inhibited Kv currents in Jurkat lymphocytes (72±7% and 83±4%, respectively) more potently than in CY15 cells (17±8% and 49±7%, respectively). Steady-state activation indicated that whereas Jurkat T-cells displayed a similar V0.5 (voltage for half maximal activation) to HEK-293 cells transfected with Kv1.3 (−11.9±1.9 and −15.1±2.3 mV, mean±s.e.m., respectively), the half-activation voltage shifted to depolarizing potentials in CY15 cells (0.20±3.3 mV) (Fig. 1F). Other mononuclear phagocytes, such as macrophages, exhibit similar biophysical and pharmacological alterations owing to the presence of heterotetrameric Kv1.3–Kv1.5 channels (Villalonga et al., 2007). Indeed, unlike Jurkat T-lymphocytes, but similar to human dendritic cells (Zsiros et al., 2009), mouse CY15 cells express Kv1.5 (Fig. 1G). Furthermore, although Jurkat and CY15 cells both had substantial levels of Kv1.3, the amount of KCNE4 was much higher in dendritic cells (Fig. 1G). Further immunocytochemistry studies showed that Kv1.3 and KCNE4 colocalized in Jurkat T-cells to a lesser extent. Furthermore, unlike in T-cells, Kv1.3 was mostly retained intracellularly in CY15 cells (Fig. 1H). Our results suggest that, unlike in Jurkat cells, when the expression of KCNE4 is abundant in dendritic cells, Kv1.3 remains mostly intracellularly, further supporting observations from HEK-293 cells and Raw 264.7 macrophages (Sole et al., 2009). To gain insight regarding whether a Kv1.3–KCNE4 association might participate in this phenotype, we performed co-immunoprecipitation studies that demonstrated, for the first time, that Kv1.3 and KCNE4 interact in CY15 dendritic cells (Fig. 1I).
Fig. 1.

Kv1.3 and KCNE4 form functional channels in leukocytes. Endogenous expression of Kv1.3 and KCNE4 was analyzed in human Jurkat T-lymphocytes and mouse CY15 dendritic cells. Endogenous voltage-dependent K+ currents were elicited in Jurkat (A) and CY15 cells (B). Cells were held at −60 mV, and pulse potentials were applied as indicated. Cumulative inactivation of K+ currents was elicited in Jurkat (C) and CY15 cells (D) by a train of 15 depolarizing 250 ms pulses ranging from −80 mV to +60 mV once every 1 s. (E) K+ currents elicited in Jurkat (left axis) and CY15 cells (right axis) at the peak current density (+80 mV) in the presence or the absence of 1 and 10 nM MgTx. Black bars, Jurkat T-cells; white bars, CY15 dendritic cells. Values are shown as the mean±s.e.m. (n=6–8 cells/group). (F) Steady-state activation of outward K+ currents. Gray circles, HEK-293 cells transfected with Kv1.3; black circles, Jurkat cells; white circles, CY15 cells. Values are shown as the mean±s.e.m. (n=4–6 independent cells). (G) Protein expression of Kv1.3, Kv1.5 and KCNE4 in leukocytes as determined by western blotting. HEK-293 cells were used as a negative control. Notably, although Jurkat and CY15 dendritic cells express both Kv1.3 and KCNE4, the abundance of KCNE4 and Kv1.5 is much lower in T-cells and was barely detected. (H) Representative confocal images of Kv1.3 and KCNE4 in Jurkat T-lymphocytes and CY15 dendritic cells. Scale bars: 10 µm. (I) KCNE4 co-immunoprecipitates with Kv1.3 in dendritic cells. Western blots from Kv1.3 and KCNE4 co-immunoprecipitation. Lysates were immunoprecipitated for Kv1.3 (IP: Kv1.3). Upper panel: Kv1.3 immunoblot (IB: Kv1.3). Lower panel: KCNE4 immunoblot (IB: KCNE4). SM, starting material (input); IP+, immunoprecipitation in the presence of the anti-Kv1.3 antibody; IP−, immunoprecipitation in the absence of the anti-Kv1.3 antibody; SN+, supernatant from the IP+; SN−, supernatant from the IP−.
Our data indicate that Kv1.3 and KCNE4 form oligomeric channels in leukocytes. This association alters the channel behavior, thereby leading to physiological consequences. To explore the molecular determinants involved in the Kv1.3–KCNE4 interaction, we further validated and extended our results in HEK-293 cells (Fig. 2). As previously reported (Sole et al., 2009), KCNE4 inhibits Kv1.3 currents when expressed in HEK-293 cells, and this is associated with an intracellular retention of the channel. Although Kv1.3 was efficiently targeted to the membrane surface, KCNE4, similar to Kv1.5, exhibited a marked intracellular phenotype (Fig. 2A–C). As mentioned above, the presence of KCNE4 caused the intracellular retention of Kv1.3 (Fig. 2D). However, although Kv1.5 and KCNE4 shared an intracellular location, their colocalization was clearly minor (Fig. 2E). Kv1.5 and KCNE4 are present in macrophages and dendritic cells, and whether these two subunits associate is an open debate. Grunnet et al. (2003) have demonstrated that KCNE4 does not modulate Kv1.5, whereas Abbott and co-workers (Crump et al., 2016) found a Kv1.5–KCNE4 association relevant to heart physiology. To elucidate this issue, we elicited voltage-dependent K+ currents in HEK-293 cells transfected with Kv1.3 and Kv1.5 in the presence or the absence of KCNE4 (Fig. 2F–I). In line with what we and others previously found (Grunnet et al., 2003), the presence of KCNE4 inhibited Kv1.3 but did not modulate Kv1.5 (Fig. 2J). To further explore any putative association, we assessed fluorescence resonance energy transfer (FRET) for Kv1.3 and Kv1.5 with KCNE4 in HEK-293 cells under the same conditions (Fig. 2K). Our results show that although Kv1.3 and KCNE4 triggered significant positive values, indicating a molecular association, the co-expression of Kv1.5 and KCNE4 produced no significant energy transfer, further supporting the electrophysiological (Fig. 2H,I) and confocal (Fig. 2E) studies. Our results indicate that, in our conditions, as in similar previous observations in oocytes (Grunnet et al., 2003), there is no association between KCNE4 and Kv1.5 in HEK-293 cells. This was further confirmed by co-immunoprecipitation experiments (see below).
Fig. 2.
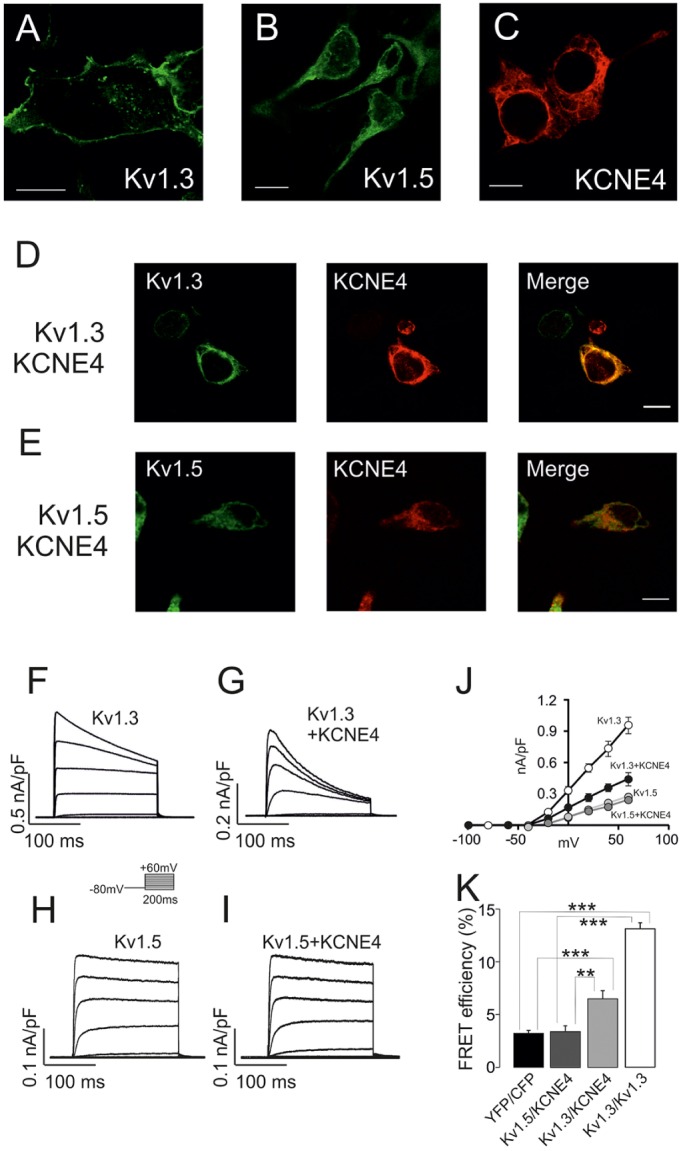
Kv1.3, but not Kv1.5, associates with KCNE4 in HEK-293 cells. KCNE4 modulates Kv1.3 trafficking and activity. HEK-293 cells were transfected with Kv1.3–YFP or Kv1.5–YFP in the presence or absence of KCNE4–CFP. Confocal images of (A) Kv1.3–YFP, (B) Kv1.5–YFP and (C) KCNE4–CFP. (D) HEK-293 cells co-transfected with Kv1.3 and KCNE4. (E) Kv1.5 and KCNE4. Color code: green, channels; red, KCNE4; yellow in merge panels shows colocalization. Scale bars: 10 µm. Voltage-dependent K+ currents were elicited in HEK-293 cells transfected with Kv1.3 (F,G) and Kv1.5 (H,I) in the absence (F,H) or the presence (G,I) of KCNE4. Cells were held at −80 mV, and pulse potentials were applied as indicated. (J) Current density versus voltage plot of outward K+ currents. White circles, Kv1.3; black circles, Kv1.3+KCNE4; light gray circles, Kv1.5; dark gray circles, Kv1.5+KCNE4. Values are shown as the mean±s.e.m. (n=6–10 independent cells). (K) Molecular association of Kv1.3 and Kv1.5 with KCNE4 as measured by FRET efficiency (%). HEK-293 cells were transfected with Kv1.3–YFP and Kv1.5–YFP in the presence of KCNE4–CFP. YFP–CFP and Kv1.3–YFP or Kv1.3–CFP were used as negative and positive controls, respectively. Values are shown as the mean±s.e.m. (n>25 independent cells). **P<0.01; ***P<0.001 (Student's t-test).
The C-terminus of Kv1.3 is required for KCNE4 association
Our data show that KCNE4 controls the channel behavior of Kv1.3 but not of Kv1.5. However, the structural motifs involved in this association are completely unknown. We first checked whether the intracellular N- and C-terminal domains of Kv1.3 are involved in the KCNE4 association. To that end, we constructed mutant channels lacking the N-terminus (Kv1.3ΔN) or the C-terminus (Kv1.3ΔC) of Kv1.3 (Fig. S1A,B). Because KCNE4 alters the cellular distribution of Kv1.3 in HEK-293 (Fig. 2D) and dendritic cells (Fig. 1H), we wondered whether the trafficking of Kv1.3 mutants was altered in the presence of KCNE4. Unlike wild-type Kv1.3 (Kv1.3 wt; Fig. 2A), Kv1.3ΔN and Kv1.3ΔC showed a markedly intracellular distribution (Fig. S1A,B) in the absence of KCNE4. Whereas KCNE4 triggered an important intracellular retention of Kv1.3 wt (Fig. 3A–C), the distribution of Kv1.3ΔN (Fig. 3D–F) and Kv1.3ΔC (Fig. 3G–I) was not apparently altered. However, an extensive pixel-by-pixel analysis illustrated that, although there was not a significant decrease in the colocalization of Kv1.3ΔN versus Kv1.3 wt with KCNE4, Kv1.3ΔC–KCNE4 colocalization decreased by ∼26% (P<0.001 versus Kv1.3, n=25) (Fig. 3J). To further decipher which Kv1.3 domain was involved in the KCNE4 association, co-immunoprecipitation experiments were performed in HEK-293 cells co-transfected with KCNE4 in the presence of Kv1.3, Kv1.3ΔN and Kv1.3ΔC (Fig. 3K,L). KCNE4 co-immunoprecipitated with Kv1.3 (Fig. 3K, bottom panel) and with Kv1.3ΔN (Fig. 3L, bottom panel) but not with Kv1.3ΔC (Fig. 3L, bottom panel). These results suggest that the C-terminal domain of Kv1.3 is required for interaction with KCNE4.
Fig. 3.

KCNE4 associates with the Kv1.3 C-terminal domain. HEK-293 cells were transfected with Kv1.3–YFP channels and KCNE4. (A–I) Confocal images from HEK-293 cells transfected with wt and mutant Kv1.3–YFP channels and KCNE4–CFP. (A,D,G) Kv1.3–YFP, Kv1.3ΔN–YFP and Kv1.3ΔC–YFP channels in green. (B,E,H) Cellular distribution of KCNE4–CFP in red. (C,F,I) Merged images show colocalization in yellow. Scale bars: 10 µm. Note that in all cases, Kv1.3 is mostly intracellular. Representative cartoons of different Kv1.3 channels are shown at the left of their respective images. The red dotted ellipse highlights the deleted domain. (J) Histogram representing the relative colocalization between Kv1.3 channels and KCNE4. Results in arbitrary units (A.U.) are the mean±s.e.m. of a pixel-by-pixel analysis on 25–40 cells. ***P<0.001 vs Kv1.3 (Student's t-test). (K,L) Representative western blots from Kv1.3–YFP, Kv1.3ΔN–YFP and Kv1.3ΔC–YFP co-immunoprecipitation. (K) Kv1.3–YFP immunoprecipitation against GFP (IP: Kv1.3). Upper panel: GFP immunoblot (IB: Kv1.3). Lower panel: KCNE4–HA immunoblot (IB: KCNE4). Kv1.3–YFP and KCNE4–HA are indicated with arrows. (L) Immunoprecipitation against GFP for Kv1.3ΔN–YFP (left side) and Kv1.3ΔC–YFP (right side). The top panel corresponds to the immunoblotting against GFP (IB: Kv1.3), in which Kv1.3ΔN–YFP and Kv1.3ΔC–YFP are indicated by arrows. The bottom panel represents immunoblotting against KCNE4–HA (IB: KCNE4). KCNE4–HA is indicated with an arrow. SM, starting material (input); IP+, immunoprecipitation in the presence of the anti-GFP antibody; IP−, immunoprecipitation in the absence of the anti-GFP antibody; SN+, supernatant from the IP+; SN−, supernatant from the IP−. Note that the anti-GFP antibody used recognizes YFP.
The removal of important intracellular domains, such as the N- and C-termini, triggers major structural changes and impairs the trafficking of Kv1.3 (Martinez-Marmol et al., 2013). Therefore, we further analyzed the involvement of the Kv1.3 C-terminal domain in the KCNE4 interaction by using chimeric Kv1.3–Kv1.5 channels. We used Kv1.5 because this channel, which is also expressed in macrophages and dendritic cells, is not associated with KCNE4 (Grunnet et al., 2003; Vicente et al., 2006; Zsiros et al., 2009, and see Fig. 2). Chimeric channels were obtained by replacing the N- and C-terminal domains of Kv1.5 with those of Kv1.3 (Kv1.3NKv1.5 and Kv1.3CKv1.5, respectively) and vice versa. We first studied the cellular distribution of the chimeras (Fig. S1C–F). Similar to Kv1.3 wt (Fig. 4A–C), the targeting of the Kv1.3NKv1.5 chimera to the plasma membrane was impaired by the presence of KCNE4 (Fig. 4D–F). KCNE4 and Kv1.3CKv1.5 both showed intracellular retention, but their distribution patterns were different (Fig. 4G–I). A pixel-by-pixel analysis (Fig. 4S) showed that Kv1.3CKv1.5 colocalization with KCNE4 was reduced by more than 50% compared to Kv1.3 wt (P<0.001, Student's t-test, n=30).
Fig. 4.

The C-terminus of Kv1.3, but not Kv1.5, is necessary and sufficient for the association with KCNE4. Confocal images from HEK-293 cells transfected with Kv1.3–YFP (A–C), Kv1.3NKv1.5–YFP (D–F) and Kv1.3CKv1.5–YFP (G–I) in the presence of KCNE4–CFP. All Kv1.3 channels presented an intracellular distribution, but the colocalization of Kv1.3CKv1.5 with KCNE4–CFP is 40% lower than that with Kv1.3 or Kv1.3NKv1.5. HEK-293 cells were also transfected with Kv1.5–YFP (J–L), Kv1.5NKv1.3 (M–O) and Kv1.5CKv1.3 (P–R) in the presence of KCNE4. Although all Kv1.5 channels were distributed intracellularly, notable colocalization was only observed between KCNE4 and Kv1.5CKv1.3. Cartoons on top of panels represent chimeric channels with the N- and C-terminal domains, and six transmembrane domains (boxes). Blue, Kv1.3 domains; green, Kv1.5 domains. Color code in confocal images: green, channels; red, KCNE4; yellow, colocalization in merge panels. Scale bars: 10 μm. (S) Histogram representing the relative colocalization between Kv1.3 and Kv1.5 channels and chimeras and KCNE4. Results are mean±s.e.m. (n=25–30 independent cells). Black columns represent Kv1.3 and Kv1.3–Kv1.5 chimeras (1.3N1.5 and 1.3C1.5). White columns represent Kv1.5 and Kv1.5–Kv1.3 chimeras (1.5N1.3 and 1.5C1.3). ***P<0.001 (Student's t-test). (T) Western blots for Kv1.3–YFP and Kv1.5–YFP with KCNE4–HA co-immunoprecipitation. Left panels: Kv1.3–YFP immunoprecipitation against GFP (IP: GFP). Right panels: Kv1.5–YFP immunoprecipitation against GFP (IP: GFP). Upper panels: Kv1.3–YFP and Kv1.5–YFP immunoblots (IB: GFP). Lower panels: KCNE4–HA immunoblots (IB: KCNE4). YFP channels and KCNE4–HA are marked with arrows. (U) Western blots for Kv1.3–Kv1.5–YFP and Kv1.5–Kv1.3–YFP chimeras with KCNE4–HA co-immunoprecipitation. Left panels: immunoprecipitation against GFP of Kv1.3NKv1.5–YFP and Kv1.3CKv1.5–YFP (IP: GFP). Right panels: immunoprecipitation against GFP for Kv1.5NKv1.3–YFP and Kv1.5CKv1.3–YFP (IP: GFP). Top panels correspond to the immunoblotting against Kv1.3 and Kv1.5 chimeras (IB: GFP), where channels are denoted by an arrow. Bottom panels correspond to the immunoblotting against KCNE4–HA (IB: KCNE4). KCNE4–HA is marked with an arrow, and co-immunoprecipitates with Kv1.3NKv1.5–YFP and Kv1.5CKv1.3–YFP. SM, starting material (input); IP+, immunoprecipitation in the presence of the anti-GFP antibody; IP−, immunoprecipitation in the absence of the anti-GFP antibody; SN+, supernatant from the IP+; SN−, supernatant from the IP−. Note that the anti-GFP antibody used recognizes YFP.
Although the C-terminus of Kv1.3 is required for the KCNE4 interaction, we next wanted to know whether this domain was sufficient for the association with the regulatory subunit. We generated Kv1.5 chimeras where the N- or C-terminus was replaced with the corresponding domain from Kv1.3 (Kv1.5NKv1.3 and Kv1.5CKv1.3, respectively). Kv1.5 and both the Kv1.5–Kv1.3 chimeras presented a low level of cell surface targeting both with and without KCNE4 (Fig. 4J–R; Fig. S1E,F, respectively). However, Kv1.5CKv1.3 showed higher colocalization with KCNE4 than Kv1.5NKv1.3 or Kv1.5 wt (P<0.001 vs Kv1.5+KCNE4, n=30) (Fig. 4S). This higher colocalization suggests that the C-terminus of Kv1.3 is both necessary and sufficient for the interaction with KCNE4.
To further address the role of the Kv1.3 C-terminus in the interaction with KCNE4, we performed co-immunoprecipitation experiments. Unlike Kv1.3 (Fig. 4T, left panels), Kv1.5 did not co-immunoprecipitate with KCNE4 (Fig. 4T, right panels). Although Kv1.3NKv1.5 co-immunoprecipitated with KCNE4, no significant co-immunoprecipitation of Kv1.3CKv1.5 with KCNE4 was observed (Fig. 4U, left panels). Furthermore, KCNE4 co-immunoprecipitated with Kv1.5CKv1.3 but not with either Kv1.5NKv1.3 or Kv1.5 (Fig. 4T,U, right panels). Thus, both the intracellular localization and the immunoprecipitation results support a role for the C-terminal domain of Kv1.3 in the association with KCNE4.
The C-terminal domain of Kv1.3 is not involved in modulating the Kv1.3 current density
Evidence collected during almost two decades suggests that structural domains implicated in the canonical interactions of Kv7 channels with KCNE peptides are not related to the modulation of gating, although this area is still under intensive investigation (Liin et al., 2015; Wrobel et al., 2012). Because Kv1.3 is structurally different from Kv7 channels, we investigated whether the C-terminus-based association between Kv1.3 and KCNE4 modulated the channel current density. To that end, we performed patch-clamp experiments using HEK-293 cells transfected with Kv1.3, Kv1.5 and the Kv1.3–Kv1.5 chimeras in the presence and the absence of KCNE4 (Fig. S2). KCNE4 inhibited Kv1.3 and Kv1.3NKv1.5 currents by ∼50%. However, as previously described (Grunnet et al., 2003), K+ currents from Kv1.5 were not affected (see also Fig. 2J–I). Similarly, the Kv1.3CKv1.5 chimera and the other Kv1.5 constructs containing Kv1.3 intracellular domains (Kv1.5NKv1.3 and Kv1.5CKv1.3) were also not inhibited by the presence of KCNE4. Considering that Kv1.5CKv1.3 efficiently co-immunoprecipitated with KCNE4 (Fig. 4U), our data suggest that the C-terminal domain of the Kv1.3 physically interacts with KCNE4 but does not cause an alteration of the current density. Therefore, similar to Kv7 channels, additional research is required to fully understand the mechanism underlying channel modulation.
The structural conformation of the Kv1.3 C-terminal domain is responsible for the association with KCNE4
To gain further insights about the C-terminal Kv1.3 motif implicated in the KCNE4 interaction, we generated Kv1.3 mutants with stop codons sequentially introduced (Fig. 5). Fig. 5A shows a schematic representation of the truncated Kv1.3 channels. Fig. 5B shows the C-terminus of Kv1.3, highlighting the mutations and the main forward trafficking and associative motifs. As previously described (Martinez-Marmol et al., 2013), the progressive removal of residues from the Kv1.3 C-terminus yielded proteins that gradually decreased in size and that displayed altered glycosylation (Fig. 5C,D). Co-immunoprecipitation analysis of the channels in the presence of KCNE4 demonstrated that the association of KCNE4 with Kv1.3 decreased concomitantly with increasingly severe truncations of the C-terminal domain (Fig. 5C,D). Our results indicate that no specific region of the Kv1.3 C-terminus is involved in the KCNE4 interaction. Instead, the shorter the C-terminus, the greater the reduction in KCNE4 co-immunoprecipitation that was observed (Fig. 5E). The correlation was notable (r2=0.8175), yielding a Pearson coefficient of 0.9041, which is statistically significant (P=0.0133). Therefore, our data suggest that rather than a specific motif, the conformational tertiary structure of the Kv1.3 C-terminal domain is responsible for the KCNE4 association.
Fig. 5.

The structure of the Kv1.3 C-terminal domain mediates the association with KCNE4. HEK-293 cells were transfected with Kv1.3 wt, various Kv1.3 truncated mutants and KCNE4. (A) Scheme representing Kv1.3 wt and the different C-terminus truncation mutants. Bars indicate the N-terminus in white (Nt), the S1–S6 transmembrane domains in black and the C-terminal domain in gray (Ct). (B) Schematic diagram of the Kv1.3 C-terminal domain highlighting some important forward trafficking signatures and the distal PDZ motif. The residues in black indicate the truncations. A unique change (A to S) between human and rodents is indicated in gray. The S6 transmembrane domain is represented as a gray barrel. The HRETE, ExExE, YMVIEE and the PDZ motifs are indicated by gray boxes. The SwissProt number, as well as the number of residues in the C-terminus, for rat, mouse and humans is also indicated. (C) Cell lysates. Immunoprecipitation of Kv1.3–YFP C-terminus mutants with anti-GFP antibody. Top panels show the immunoblot against GFP (Kv1.3). Bottom panels show the immunoblot against KCNE4 (arrow). (D) Co-immunoprecipitation (Co-IP). Cell lysates (as in C) were immunoprecipitated with anti-GFP antibody (IP Kv1.3) and immunoblotted against Kv1.3 (IB: Kv1.3, top panel) and KCNE4 (IP: KCNE4, bottom panel). Note that the anti-GFP antibody used recognizes YFP. (E) Plot of the remaining number of C-terminal residues in the Kv1.3 C-terminus truncated mutants versus the co-immunoprecipitation of KCNE4. Values (mean±s.e.m., n=4) are calculated as the logarithm (log) of co-immunoprecipitation in arbitrary units (AU) standardized to the maximum level (scored as 1) of the co-immunoprecipitation of KCNE4 with Kv1.3 wt from D. Values followed a notable linear correlation (r2=0.8175) which yielded a statistical significant (P=0.0133) Pearson’s coefficient (0.9041).
Structural motifs responsible for the KCNE4-mediated Kv1.3 ER retention
The binding of KCNE4 to Kv1.3 triggers massive ER retention that, in turn, downregulates cell surface expression (Sole et al., 2009). Our data indicate that KCNE4 interacts with Kv1.3 through the structural conformation of its C-terminal domain. This region contains several forward trafficking elements, including the potent YMVIEE motif (Fig. 5B), which is crucial in the COPII-dependent forward trafficking of Kv1.3 to the cell surface (Martinez-Marmol et al., 2013). Therefore, we wondered whether the KCNE4 interaction impairs these events. In fact, KCNE4 hindered the interaction of Sec24D, an element of the COPII machinery, with Kv1.3 (Fig. 6). In the presence of Sar1(H79G), which leads to the retention of the channel in the ER due to constitutively active GTPase activity, Kv1.3 co-immunoprecipitated with Sec24D. KCNE4, which also causes localization of Kv1.3 to the ER, impeded this association (Fig. 6A,B). To further analyze this mechanism, we performed FRET experiments addressing the potential interaction between Kv1.3 and Sec24D (Fig. 6C–G). Kv1.3 colocalized with KCNE4 and Sec24D in discrete locations. Staining for the three proteins identified two (positive and negative) FRET region of interest (ROI) populations, increasing the variability of the results (Fig. 6G). However, elevated levels of KCNE4 correlated with negative FRET values for Kv1.3 and Sec24D in discrete ROIs (Fig. 6H). This indicates that the Kv1.3–Sec24D interaction decreased when KCNE4 was abundant. Our results suggest that KCNE4 and Sec24 competed for the association with Kv1.3, further supporting the idea that the C-terminal domain of the channel is involved in that interaction.
Fig. 6.

KCNE4 interacts with the C-terminal domain of Kv1.3, thereby impairing the association with the COPII anterograde trafficking mechanism. HEK-293 cells were transfected with Kv1.3 in the presence or the absence of KCNE4, Sec24D–YFP and Sar1(H79G)–HA. (A,B) Co-immunoprecipitation of Kv1.3 and Sec24D in the presence or the absence of KCNE4. (A) Expression of Sec24D, Kv1.3 and KCNE4 in starting materials (cell lysates). (B) Co-immunoprecipitations. Samples were immunoprecipitated (+IP) against Sec24D–YFP and immunoblotted (IB) against GFP (Sec24D), Kv1.3 and KCNE4. Sar1(H79G) and KCNE4 expression led to the retention of Kv1.3 in the ER. Note that KCNE4 notably impaired the association between Kv1.3 and Sec24D. In addition, KCNE4 showed a slight co-immunoprecipitation with Sec24. Note that the anti-GFP antibody used recognizes YFP. (C–H) Molecular interaction between Kv1.3 and Sec24D in the presence or the absence of KCNE4. (C–F) A representative FRET experiment for Kv1.3 and Sec24D in the presence of KCNE4. (C) Sec24D–YFP (red) in the acceptor panel. (D) Kv1.3–CFP (green) in the donor panel. (E) Colocalization is shown in yellow. (F) Magnification of the boxed area in E. Circles highlight some ROIs. Scale bar: 10 µm. (G) FRET efficiency of the Kv1.3–Sec24D interaction in the presence or the absence of Sar1(H79G) and KCNE4. YFP and CFP, and Kv1.3–CFP and Kv1.3–YFP pairs were used as negative and positive controls, respectively. Note that although FRET values between Kv1.3 and Sec24D were positive, the presence of KCNE4 triggered a larger variability. (H) Relative KCNE4 expression in arbitrary units (AU) in FRET-positive or FRET-negative ROIs. Considering the FRET between Kv1.3 and Sec24D, two types of ROI were observed. When ROI triggered clearly positive FRET, KCNE4 expression was low. However, in ROIs with no relevant FRET values, KCNE4 expression was higher. Values are shown as the mean±s.e.m. of n>30 cells. *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
To gain insights into whether the KCNE4 masking of the YMVIEE motif at the C-terminal domain of Kv1.3 was entirely responsible for the observed ER retention, we analyzed the intracellular localization of a Kv1.3 mutant, in which residues E483 and E484 had both been replaced with I (hereafter referred to as E483/484I), and which exhibits poor cell membrane targeting due to an impaired association with COPII (Martinez-Marmol et al., 2013). KCNE4 was co-expressed with Kv1.3 wt and Kv1.3 (E483/484I) channels (Fig. 7). Whereas KCNE4 retained the Kv1.3 wt intracellularly (Fig. 7A–G), KCNE4 did not alter the intracellular expression pattern of Kv1.3 (E483/484I) (Fig. 7H–N). However, the surface expression analysis indicated that KCNE4 triggered a stronger ER retention of both Kv1.3 and Kv1.3 (E483/484I) channels than upon mutating the Kv1.3 forward trafficking motif alone (Fig. 7O). Therefore, other ER retention motifs (ERRMs) within KCNE4 might be involved in preventing forward trafficking of Kv1.3. Basic elements, such as RxR and KKxx, are important ERRMs (Gao et al., 2014; Zerangue et al., 1999), and KCNE4 does contain a positively charged signature (K110SKRREKKSS119) within its C-terminal domain (Fig. 8A). Thus, we investigated whether this motif further enhances ER retention of the Kv1.3–KCNE4 channelosome. We mutated several basic residues of the ERRM signature to alanine residues in order to disrupt putative canonical and recurrent signals (Fig. 8A). When expressed alone, the KCNE4 (KSAAREAKSS) mutant [denoted KCNE4(ERRM)] showed twice the cell surface targeting observed with the wt KCNE4 (Fig. 8B–G,P). In addition, when Kv1.3 was co-expressed with KCNE4(ERRM), the channel reached the membrane more efficiently than with KCNE4 wt (Fig. 8H–O). These data are summarized in Fig. 8P. Thus, it appears that KCNE4 retains the Kv1.3 channelosome within the cell by two independent, but complementary, mechanisms: (1) through a KCNE4 interaction with the C-terminus of Kv1.3, which hinders the di-acidic forward trafficking signature, and (2) by providing potent ERRMs to the complex.
Fig. 7.
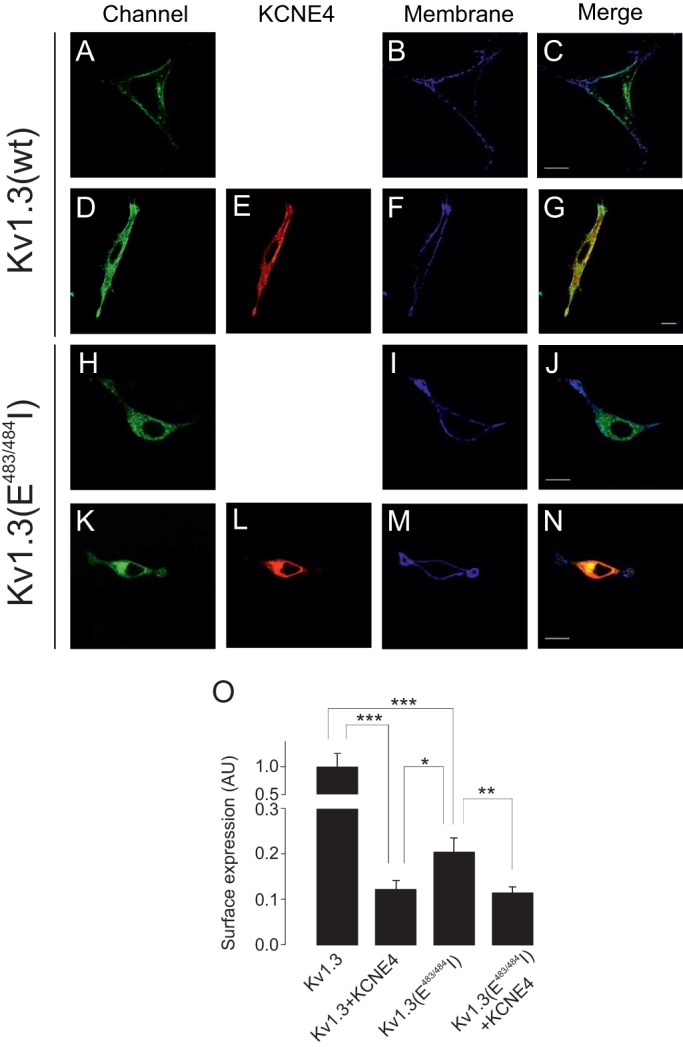
The KCNE4 interaction further aggravated the intracellular retention of the Kv1.3(E483/484I) mutant. HEK-293 cells were transfected with Kv1.3wt, Kv1.3(E483/484I) and KCNE4. Membrane surface labeling was performed using WGA as described in the Materials and Methods. (A–G) Representative confocal images of Kv1.3 wt in the absence (A–C) or the presence (D–G) of KCNE4. (H–N) Representative confocal images of Kv1.3(E483/484I) in the absence (H–J) or the presence (K–N) of KCNE4. (A,D,H,K) Kv1.3 channels in green. (E,L) KCNE4 in red. (B,F,I,M) WGA membrane staining in blue. (C,G,J,N) Color code in merged panels: Kv1.3 channels and KCNE4 colocalization, yellow; channels and membrane, cyan; triple colocalization, white. Scale bars: 10 µm. (O) Kv1.3 membrane surface expression in arbitrary units (AU) in a pixel-by-pixel analysis. Values are shown as the mean±s.e.m. of n>25 cells. *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
Fig. 8.

The disruption of the ERRM of KCNE4 only partially counteracted the intracellular retention of Kv1.3. HEK-293 cells were transfected with Kv1.3–YFP, KCNE4–CFP and the KCNE4(ERRM)–CFP mutant. (A) Cartoon illustrating the ERRM in the C-terminal domain of KCNE4. Positive residues (R and K) in the wild-type ERRM (brown box) were mutated to alanine residues to disrupt the motif. N-term, N-terminal extracellular domain; C-term, C-terminal intracellular domain; TMD, transmembrane domain. (B–G) Confocal images of KCNE4 wt (B–D) and the KCNE4(ERRM) mutant (E–G). (B,E) KCNE4. (C,F) Membrane staining with WGA. (D,G) Merge channel. (H–O) Representative images of Kv1.3 in the presence of KCNE4 (H–K) and KCNE4(ERRM) (L–O). (H,L) Kv1.3 in green; (I,M) KCNE4 in red; (J,N) membrane in blue; (K,O) merge. Note that although Kv1.3 colocalization with KCNE4 showed marked intracellular retention in all cases, the channel reached the surface better in the presence of KCNE4(ERRM). Scale bars: 10 µm. (P) Pixel-by-pixel analysis of the relative membrane surface expression, in arbitrary units (AU), of KCNE4 (black bars) and Kv1.3 (white bars). Note that KCNE4(ERRM) showed double the KCNE4 membrane surface expression. Furthermore, Kv1.3 reached the cell surface more efficiently in the presence of KCNE4(ERRM) than with KCNE4. Values are shown as the mean±s.e.m. (n>25 cells). *P<0.05; **P<0.01; ***P<0.001 (Student's t-test).
DISCUSSION
KCNE4 is a regulatory subunit that modulates the cell surface abundance and the spatial localization of Kv1.3 channels. Both proteins are present in leukocytes and form heterooligomeric channels (Sole and Felipe, 2010; Sole et al., 2009, 2013). In the present work, we analyzed the molecular determinants in Kv1.3 that participate in the Kv1.3–KCNE4 interaction. Our data suggest that the tertiary structural conformation of the C-terminal domain of Kv1.3 is responsible for the association with KCNE4. However, other domains, which are yet to be characterized, are responsible for the gating modulation. Furthermore, we also demonstrated that two independent, but complementary, mechanisms collectively participate in the intracellular retention of the Kv1.3–KCNE4 channelosome. Overall, our results shed light onto the mechanisms that control Kv1.3 surface expression, which is closely linked to immune cell activation. In addition, our data illustrate how differing tissue-specific oligomeric associations configure both functional and trafficking heterogeneity.
Regulatory subunits, such as KCNE and Kvβ subunits, modulate Kv subcellular distribution (Kanda and Abbott, 2012; Roura-Ferrer et al., 2010; Shi et al., 1996). For instance, Kvβ2.1 (also known as KCAB2) stabilizes Kv1 channels at the cell surface (Shi et al., 1996), and KCNE1 participates in the Kv7.1 plasma membrane location (Kanda and Abbott, 2012; Roura-Ferrer et al., 2010). In contrast, KCNE4 impairs Kv1.3 surface targeting, triggering dramatic consequences for the channel function (Sole et al., 2009). Kv1.3 is crucial for the immune response, and an alteration in its membrane abundance can be responsible for both physiological changes and pathological conditions. For instance, the number of Kv1.3 channels increases during T-lymphocyte activation (Varga et al., 2010). Similarly, macrophages elevate Kv1.3 at the plasma membrane during activation and proliferation (Vicente et al., 2003). Furthermore, Kv1.3 channels are targeted to raft microdomains under specific insults (Vicente et al., 2008). However, exacerbated surface expression or altered spatial localization is related to autoimmune diseases (Chandy et al., 2004; Nicolaou et al., 2007; Varga et al., 2010). In contrast, immunosuppression decreases Kv1.3, triggering similar effects to those of channel blockers (Chandy et al., 2004; Villalonga et al., 2010). Therefore, the control of functional Kv1.3 channels at the cell surface is crucial for appropriate cell responses. In addition, KCNE4 expression is tightly regulated in leukocytes (Sole et al., 2013). In this scenario, the association with KCNE4, by controlling the number and spatial localization of surface Kv1.3, would be essential. Kv1.3 is considered a multi-pharmacological target because channel alterations are present in the onset of numerous pathologies, including sensory discrimination, autoimmune diseases, type II diabetes, obesity and cancer (Perez-Verdaguer et al., 2016b). Considering the variety of functions in which Kv1.3 participates (i.e. activation, proliferation and apoptosis), it is tempting to speculate that this diversity is achieved by a variety of complementary subunit associations that govern channel localization and function.
Our present data suggest that it is the folded structural conformation of the Kv1.3 C-terminus, rather than specific signatures, that is important for the KCNE4 association. Although KCNE peptides bind to Kv7 isoforms through interactions with the C-terminal domain (Wrobel et al., 2012; Zheng et al., 2010), nothing was known previously about their association with other Kv channels. Structural elements are quite different among Kv families (Hille, 2001). For example, the Kv7 family as well as the EAG superfamily contain tetramerization domains at their C-termini (Hausammann and Grutter, 2013; Jenke et al., 2003; Schwake et al., 2006), whereas Kv1–Kv6 channels contain their tetramerization domains, named T1, at the N-terminus (Li et al., 1992). The T1 domain and nearby structures participate in associations with Kvβ subunits and caveolin (Gulbis et al., 2000; Perez-Verdaguer et al., 2016a). However, the C-terminus of Kv1 channels does contain important elements for their trafficking and localization. A PDZ domain, which interacts with PSD proteins, such as PSD95 or SAP-97, is positioned at the very distal part of the channel (Szilagyi et al., 2013; Tiffany et al., 2000). In addition, forward trafficking motifs, such as VxxSL, HRETE or YMVIEE, are also located within this intracellular region (Li et al., 2000; Martinez-Marmol et al., 2013; Zhu et al., 2007). However, there was little information about the interactions of this C-terminal domain with ancillary partners (Misonou and Trimmer, 2004). The COPII machinery is responsible for a di-acidic interaction within the C-terminal domain of the Kv1.3 channel (Martinez-Marmol et al., 2013; Spear et al., 2015). Our new data suggest that the interaction between KCNE4 and the Kv1.3 C-terminal domain masks this motif and triggers a marked ER retention, which impairs membrane targeting. Furthermore, this association does not function alone, as the strong intracellular phenotype is further ensured by a strong ERRM in KCNE4.
KCNE peptides control the surface abundance of Kv7 channels. Whereas KCNE1 increases the Kv7.1 membrane staining, KCNE4, similar to our observations for Kv1.3, reduces Kv7.1 surface expression and lipid raft localization (Roura-Ferrer et al., 2010). The balance between forward trafficking and ER retention signals is crucial for the membrane targeting and function (Misonou and Trimmer, 2004). In two-pore-domain background K+ channels (K2P channels), the influence of protein–protein interactions on sorting decisions is crucial. The adaptor protein 14-3-3 interacts with TASK-1 and TASK-3 (also known as KCNK3 and KCNK9, respectively) to mask a retention signal in the C-terminus of the channels, which is important for the COPI binding (Mathie et al., 2010). In fact, 14-3-3 promotes plasma membrane expression of several ion channels, such as Cav2.2 (also known as CACNA1B) (Liu et al., 2015). The effect of KCNE4 would be the opposite; by interacting with the C-terminus of Kv1.3, the ancillary peptide would hide a di-acidic forward trafficking motif important for COPII anterograde traffic. By contrast, the adaptor protein p11 (also known as protein S100-A10), which interacts with TASK-1, carries a di-lysine retention signal that retains the channel at the ER (Mathie et al., 2010). KCNE4 association also provides a strong ERRM to Kv1.3. Unlike TASK channels, the KCNE4 mechanism can be considered to be redundant and effective. Several forward trafficking signals have been identified in the C-terminal domain of Kv1.3. Thus, the HRETE motif, its alternative ExExE extension and the YMVIEE cluster containing a di-acid motif promote anterograde transport through the COPII-mediated early secretory pathway (Martinez-Marmol et al., 2013; Spear et al., 2015). However, the interaction of KCNE4 with the fully folded C-terminal domain fine-tunes cell surface expression by both masking forward trafficking signals and providing an ER retention signal.
Furthermore, our results suggest that motifs involved in the association are not responsible for the gating control, as KCNE regulatory subunits can bind to channels without modifying their kinetics (McCrossan and Abbott, 2004; Wrobel et al., 2012). In this context, KCNE4, although it associates with Kv7.4 (also known as KCNQ4), does not modulate the channel activity. However, the transfer of the S6 domain from Kv7.1 to Kv7.4 leads to inhibition, whereas transferring the S6 domain from Kv7.4 to Kv7.1 prevents inhibition by KCNE4 (Vanoye et al., 2009). This effect is due to a dipeptide domain (KT, amino acids 326–327) preceding the S6 segment. Neither Kv1.3 nor Kv1.5 contains such an element. Although Kv1.1–Kv1.6 channels possess a lysine residue in this position, this residue cannot support the KCNE4 modulation because it is specific for only Kv1.3 and Kv1.1 (Grunnet et al., 2003; Sole et al., 2009). The C-terminus and S5–S6 domains of Kv7.1 are required for modulation by KCNE1 and KCNE3 (Melman et al., 2004). Evidence supports the idea that the C-terminus of Kv7.1 acts by anchoring and correctly positioning the regulatory subunit (Manderfield et al., 2009; Vanoye et al., 2009). Furthermore, multiple interactions have been described between the KCNE and the gating machinery of Kv7.1, such as residues S1, S4, S5 and S6 (Chouabe et al., 2000; Franqueza et al., 1999; Melman et al., 2004; Nakajo and Kubo, 2007; Panaghie et al., 2006; Xu et al., 2008). Our data indicate that, similar to the Kv7.1–KCNE interaction, the C-terminus of Kv1.3 plays a role in anchoring KCNE4 to the complex, but that additional interactions modulate channel gating.
Our results are physiologically relevant because KCNE4 and Kv1.3 associate in antigen-presenting cells. Kv1.3 channels participate in the immune response and, through a variable KCNE4 interaction, dendritic cells and macrophages might be able to precisely modulate their cell physiology. This work highlights some of the mechanisms by which interacting proteins might govern the intracellular trafficking and surface abundance of ion channels involved in the immune response.
MATERIALS AND METHODS
Expression plasmids, chimeric channels and site-directed mutagenesis
Rat (r)Kv1.3 in pRcCMV was provided by Todd C. Holmes (New York University, NY). Mouse (m)KCNE4 in pSGEM was from Michael Sanguinetti (University of Utah, Salt Lake City, UT). piRES-EGFP-hKCNE4-HA was obtained from Alfred L. George (Northwestern University Feinberg School of Medicine, Chicago, IL). Human (h)Kv1.5 has been extensively analyzed in our laboratory. rKv1.3 and hKv1.5 were subcloned into pEYFP-C1 and pECFP-C1 (Clontech) to preserve normal channel behavior. mKCNE4 was introduced in pECFP-N1 (Clontech). rKv1.3–YFP, mKCNE4–CFP and hKCNE4–HA constructs were previously described (Sole et al., 2009). Kv1.3–CFP, Kv1.5–YFP, Kv1.3–Kv1.5 chimeras and Kv1.3 mutants and truncations have been analyzed previously (Martinez-Marmol et al., 2013). Kv1.3–YFP and KCNE4–CFP mutants were generated using the QuikChange and QuikChange multi-site-directed mutagenesis kits (Stratagene). All constructs and mutants were verified using automated DNA sequencing. The pEYFP-Sec24D and the HA-Sar1(H79G) were from Theresa H. Ward (The London School of Hygiene & Tropical Medicine, London, UK) and Rainer Pepperkok (EMBL, Heidelberg, Germany), respectively.
Cell culture
HEK-293 cells, Jurkat human T-lymphocytes and CY15 mouse dendritic cells were cultured in Dulbecco's modified Eagle's medium (DMEM) and RPMI culture medium, respectively (LONZA), supplemented with 10% fetal bovine serum (FBS), 10,000 U/ml penicillin, 100 µg/ml streptomycin and 2 mM L-glutamine (GIBCO).
For patch-clamp experiments, trypsinized HEK-293 cells from a confluent 100-mm dish were electroporated with 1 µg of DNA using a Bio-Rad Genepulser Xcell (Bio-Rad) with a 0.2-cm-gap cuvette and a single 110 V and 25 ms pulse. Transfected cells were then plated on a 50-mm dish.
For confocal imaging and co-immunoprecipitation experiments, cells were seeded (70–80% confluence), in six-well dishes containing poly-lysine-coated coverslips or in 100-mm dishes, respectively, 24 h before transfection with selected cDNAs. Metafectene PRO (Biontex) was used for transfection according to the supplier's instructions. The amount of transfected DNA was 500 ng per well of a six-well dish and 4 µg for a 100-mm dish. Next, 4–6 h after transfection, the mixture was removed from the dishes and replaced with fresh culture medium. All the experiments were performed 24 h after transfection.
Electrophysiology
Transfected HEK-293 cells were trypsinized 24 h after electroporation and re-plated on 35-mm glass-bottom dishes coated with Matrigel (BD Biosciences). After 2–4 h, cells were extensively washed with whole-cell external recording solution, containing the following (in mM): 150 NaCl, 5 KCl, 10 CaCl2, 2 MgCl2, 10 glucose and 10 HEPES, pH 7.4. Transfected HEK-293 cells were selected using an Olympus FV1000 confocal microscope equipped with spectral detectors and SIM scanner. Whole-cell K+ currents in HEK-293, Jurkat and CY15 cells were recorded at room temperature using an Axopatch 200B and an EPC-10 (HEKA), respectively, and the appropriate software was used for data recording and analysis. Ionic currents were capacitance- and series-resistance-compensated by 80–90%, sampled at 10 kHz (Digidata 1440; Molecular Devices) and filtered at 2.9 kHz. Patch electrodes of 2–4 MΩ were fabricated in a P-97 puller (Sutter Instruments Co.) from borosilicate glass (outer diameter 1.2 mm and inner diameter 0.94 mm; Clark Electromedical Instruments Co.). Electrodes for HEK-293 cells were filled with a solution containing the following (in mM): 4 NaCl, 150 KCl, 1 MgCl2, 0.5 EGTA, 5 ATP(K) and 10 HEPES, pH 7.4. HEK-293 cells were clamped to a holding potential of −80 mV. Electrodes for CY15 and Jurkat cells were filled with a solution containing the following (in mM): 84 K-aspartate, 36 KCl, 10 KH2PO4, 6 K2ATP, 5 HEPES, 5 EGTA, 3 MgCl2, pH 7.2. The extracellular solution contained (in mM): 136 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES and 10 D-glucose, pH 7.4. CY15 and Jurkat cells were clamped to a holding potential of −60 mV. To evoke voltage-gated currents, all cells were stimulated with 250-ms square pulses ranging from −80 to +80 mV in 10 mV steps. The normalized G/Gmax versus the voltage curve was fitted using Boltzmann's equation: G/Gmax=1/(1+exp(V0.5−V/k)), where V0.5 is the voltage at which the current is half-activated, and k is the slope factor of the activation curve. Cumulative inactivation of K+ currents was elicited in Jurkat and CY15 cells by a train of 15 depolarizing 250-ms pulses from −80 mV to +60 mV once every 1 s. All recordings were routinely subtracted for leak currents.
Protein extraction, co-immunoprecipitation and western blotting
Cells were washed twice in cold PBS and lysed on ice with lysis solution (1% Triton X-100, 10% glycerol, 50 mM HEPES, 150 mM NaCl, pH 7.2) supplemented with 1 µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml pepstatin and 1 mM phenylmethylsulfonyl fluoride as protease inhibitors. Homogenates were centrifuged at 12,000 g for 10 min, and the supernatant was collected. Protein content was determined using the Bio-Rad Protein Assay (Bio-Rad).
For co-immunoprecipitation, 1 mg of protein was brought up to 500 µl of lysis buffer for immunoprecipitation (in NaCl 150 mM, HEPES 50 mM, Titron X-100 1%, pH 7.4, supplemented with protease inhibitors). Samples were precleared with 50 µl of protein-G–Sepharose beads for 2 h at 4°C with gentle mixing. Next, each sample was incubated in a small chromatography column (BioRad Micro Bio-Spin Chromatography Columns), which contained 2.5 µg of the desired antibody previously crosslinked to protein-A or -G–Sepharose beads, for 1 h at room temperature with gentle mixing. Columns were centrifuged for 30 s at 1000 g. The supernatant was kept and stored at −20°C. Columns were washed four times with 500 µl of lysis buffer and centrifugations of 30 s at 1000 g. Finally, for elution, the columns were incubated with 100 µl of 0.2 M glycine pH 2.5 and spun for 30 s at 1000 g. The eluted proteins were prepared by adding 20 µl of SDS Laemmli SDS loading buffer (5×) and 5 µl of 1 M Tris-HCl pH 10, and the preparations were boiled and separated on 10% SDS-PAGE gels. Next, they were transferred to nitrocellulose membranes (Immobilon-P; Millipore) and blocked in 0.05% Tween-20 in PBS supplemented with 5% dried milk before immunoreaction. Filters were immunoblotted with antibodies against HA (1:1000, cat. no. H 6908; Sigma), GFP (1:1000, cat. no. 11 814 460 001; Roche), Kv1.3 (1:200, cat. no. 75-009; NeuroMab), Kv1.5 (1:500, cat. no. APC-004; Alomone) and KCNE4 (1:500, cat. no. 18289-1-AP; Proteintech). Anti-β-actin antibody was used as a loading control (1:50,000, cat. no. A5441; Sigma).
Irreversible crosslinking of the antibody to the Sepharose beads was performed after an incubation of the antibody with protein-A or -G–Sepharose beads for 1 h at room temperature. The beads were then incubated with 500 µl of 5.2 mg/ml of dimethyl pimelimidate (Pierce) for 30 min at room temperature by gentle mixing. The beads were then washed four times with 500 µl of 1× TBS, four times with 500 µl of 0.2 M glycine pH 2.5 and three times more with 1× TBS. Once these steps were performed, the columns were incubated with the protein lysates in order to perform the immunoprecipitation as described above.
Immunocytochemistry
Jurkat and CY15 cells were cultured on 20×20 mm poly-lysine-coated glass coverslips for up to 24 h. Cells were fixed in methanol (−20°C) for 15 min at room temperature, washed twice in PBS without K+ (denoted PBS-K+, wash buffer) for 5 min to rehydrate cells, and blocked and permeabilized for 1 h with blocking buffer (PBS-K+ containing 0.05% Triton X-100, 5% non-fat milk, 10% bovine serum albumin). Cells were subsequently incubated with rabbit anti-KCNE4 (1:100, cat. no. 18289-1-AP; Proteintech) for 2 h, and unbound antibody was removed with wash buffer. Coverslips were exposed to Cy3-conjugated goat anti-rabbit-IgG secondary antibody (1:200, Molecular Probes) for 1 h. After washing to remove unbound secondary antibody, CY15 cells were blocked again for 1 h in blocking buffer. CY15 cells were incubated with anti-Kv1.3 (1:50, cat. no. 75-009; NeuroMab) at 4°C overnight, washed, and then exposed to Cy5-conjugated goat anti-mouse-IgG (1:200, Molecular Probes) for 1 h. After removing unbound secondary antibody, coverslips were mounted with Mowiol (Calbiochem) and examined with a confocal laser-scanning fluorescent microscope.
Confocal microscopy and image analysis
For confocal image acquisition, cells were seeded on poly-lysine-coated coverslips and transfected 24 h later. The next day, cells were quickly washed twice, fixed with 10% paraformaldehyde for 10 min, and washed three times for 5 min with PBS-K+. Finally, coverslips were mounted on microscope slides (Acefesa) with Mowiol. Preparations were dried at room temperature before imaging.
For membrane surface labeling under non-permeabilized conditions, wheat germ agglutinin conjugated to Texas Red (WGA–Texas Red®, Invitrogen) was used. Live cells (on ice) were quickly washed with PBS at 4°C and stained with a dilution of WGA–TexasRed (1:1500) in DMEM supplemented with 30 mM HEPES for 15 min at 4°C. Cells were washed twice and fixed with 4% paraformaldehyde in PBS for 10 min. Next, cells were washed and mounted as described above.
The fluorescence resonance energy transfer (FRET) was achieved by an acceptor photobleaching technique and was measured in discrete ROIs. Fluorescent proteins from fixed cells were excited with the 458 nm or the 514 nm lines using low excitation intensities. Next, 475- to 495-nm bandpass and >530-nm longpass emission filters were applied. The YFP was bleached using maximum laser power. We obtained ∼80% of acceptor intensity bleaching. After photobleaching, images of the donors and acceptors were taken. The FRET efficiency was calculated using the equation [(FCFPafter−FCFPbefore)/FCFPbefore]×100, where FCFPafter was the fluorescence of the donor after bleaching and FCFPbefore was the fluorescence before bleaching. The loss of fluorescence as a result of the scans was corrected by measuring the CFP intensity in an unbleached part of the cell.
All images were acquired with a Leica TCS SL laser-scanning confocal spectral microscope (Leica Microsystems), equipped with argon and helium-neon lasers. All the experiments were performed with a 63× oil-immersion objective lens, NA 1.32. All offline image analysis was performed using Image J and SigmaPlot software. A pixel-by-pixel colocalization analysis was performed using JACoP (Just Another Colocalization Plugin). Manders split coefficients, which are proportional to the amount of fluorescence of the colocalizing pixels in each color channel, were obtained. Thus, although the spatial resolution of the light microscope is limited by the wavelength of the light (less than 200 nm), our data were exhaustively corroborated by the rest of the above-mentioned techniques such as FRET, co-immunoprecipitation and electrophysiological recording.
Acknowledgements
Authors thank the Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB) for confocal microscopy experiments. The English editorial assistance of the American Journal Experts is also acknowledged.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
A.F. and M.M.T. conceived and supervised the present study. R.R.-M. provide reagents and tools. L.S., S.R.R., A.S.-A. and A.V.-G. performed the experiments and analyzed the data. A.F. and L.S. wrote the manuscript. All authors contributed to the interpretation and commented on the manuscript.
Funding
Supported by the Ministerio de Economía y Competitividad (MINECO), Spain (BFU2014-54928-R and BFU2015-70067-REDC) and Fondo Europeo de Desarrollo Regional (FEDER). This work was also supported by the National Institutes of Health (R01GM84136, R01GM084136S1 and R01GM109888-01 to M.M.T.). L.S., S.R.R. and A.V.-G. hold a fellowship from the Ministerio de Economía y Competitividad (MINECO) and A.S.-A. from Generalitat de Catalunya. R.M.-M. was supported by the Juan de la Cierva program (MINECO). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.191650.supplemental
References
- Chandy K. G., Wulff H., Beeton C., Pennington M., Gutman G. A. and Cahalan M. D. (2004). K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 25, 280-289. 10.1016/j.tips.2004.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C., Neyroud N., Richard P., Denjoy I., Hainque B., Romey G., Drici M.-D., Guicheney P. and Barhanin J. (2000). Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc. Res. 45, 971-980. 10.1016/S0008-6363(99)00411-3 [DOI] [PubMed] [Google Scholar]
- Crump S. M., Hu Z., Kant R., Levy D. I., Goldstein S. A. N. and Abbott G. W. (2016). Kcne4 deletion sex- and age-specifically impairs cardiac repolarization in mice. FASEB J. 30, 360-369. 10.1096/fj.15-278754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felipe A., Soler C. and Comes N. (2010). Kv1.5 in the immune system: the good, the bad, or the ugly? Front. Physiol. 1, 152 10.3389/fphys.2010.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L., Lin M., Splawski I., Keating M. T. and Sanguinetti M. C. (1999). Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274, 21063-21070. 10.1074/jbc.274.30.21063 [DOI] [PubMed] [Google Scholar]
- Gao C., Cai Y., Wang Y., Kang B.-H., Aniento F., Robinson D. G. and Jiang L. (2014). Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci. 19, 508-515. 10.1016/j.tplants.2014.04.004 [DOI] [PubMed] [Google Scholar]
- Grunnet M., Rasmussen H. B., Hay-Schmidt A., Rosenstierne M., Klaerke D. A., Olesen S.-P. and Jespersen T. (2003). KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels. Biophys. J. 85, 1525-1537. 10.1016/S0006-3495(03)74585-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis J. M., Zhou M., Mann S. and MacKinnon R. (2000). Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science 289, 123-127. 10.1126/science.289.5476.123 [DOI] [PubMed] [Google Scholar]
- Hausammann G. J. and Grutter M. G. (2013). Chimeric hERG channels containing a tetramerization domain are functional and stable. Biochemistry 52, 9237-9245. 10.1021/bi401100a [DOI] [PubMed] [Google Scholar]
- Hille B. (2001). Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer. [Google Scholar]
- Jenke M., Sanchez A., Monje F., Stuhmer W., Weseloh R. M. and Pardo L. A. (2003). C-terminal domains implicated in the functional surface expression of potassium channels. EMBO J. 22, 395-403. 10.1093/emboj/cdg035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda V. A. and Abbott G. W. (2012). KCNE regulation of K+ channel trafficking - a Sisyphean Task? Front. Physiol. 3, 231 10.3389/fphys.2012.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Jan Y. N. and Jan L. Y. (1992). Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science 257, 1225-1230. 10.1126/science.1519059 [DOI] [PubMed] [Google Scholar]
- Li D., Takimoto K. and Levitan E. S. (2000). Surface expression of Kv1 channels is governed by a C-terminal motif. J. Biol. Chem. 275, 11597-11602. 10.1074/jbc.275.16.11597 [DOI] [PubMed] [Google Scholar]
- Liin S. I., Barro-Soria R. and Larsson H. P. (2015). The KCNQ1 channel - remarkable flexibility in gating allows for functional versatility. J. Physiol. 593, 2605-2615. 10.1113/jphysiol.2014.287607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Zhou Q., Zhou J., Sun H., Wang Y., Zou X., Feng L., Hou Z., Zhou A., Zhou Y. et al. (2015). 14-3-3tau promotes surface expression of Cav2.2 (alpha1B) Ca2+ channels. J. Biol. Chem. 290, 2689-2698. 10.1074/jbc.M114.567800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield L. J., Daniels M. A., Vanoye C. G. and George A. L. Jr (2009). KCNE4 domains required for inhibition of KCNQ1. J. Physiol. 587, 303-314. 10.1113/jphysiol.2008.161281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Marmol R., Perez-Verdaguer M., Roig S. R., Vallejo-Gracia A., Gotsi P., Serrano-Albarras A., Bahamonde M. I., Ferrer-Montiel A., Fernandez-Ballester G., Comes N. et al. (2013). A non-canonical di-acidic signal at the C-terminus of Kv1.3 determines anterograde trafficking and surface expression. J. Cell Sci. 126, 5681-5691. 10.1242/jcs.134825 [DOI] [PubMed] [Google Scholar]
- Mathie A., Rees K. A., El Hachmane M. F. and Veale E. L. (2010). Trafficking of neuronal two pore domain potassium channels. Curr. Neuropharmacol. 8, 276-286. 10.2174/157015910792246146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan Z. A. and Abbott G. W. (2004). The MinK-related peptides. Neuropharmacology 47, 787-821. 10.1016/j.neuropharm.2004.06.018 [DOI] [PubMed] [Google Scholar]
- Melman Y. F., Um S. Y., Krumerman A., Kagan A. and McDonald T. V. (2004). KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 42, 927-937. 10.1016/j.neuron.2004.06.001 [DOI] [PubMed] [Google Scholar]
- Misonou H. and Trimmer J. S. (2004). Determinants of voltage-gated potassium channel surface expression and localization in Mammalian neurons. Crit. Rev. Biochem. Mol. Biol. 39, 125-145. 10.1080/10409230490475417 [DOI] [PubMed] [Google Scholar]
- Nakajo K. and Kubo Y. (2007). KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 130, 269-281. 10.1085/jgp.200709805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou S. A., Szigligeti P., Neumeier L., Lee S. M., Duncan H. J., Kant S. K., Mongey A. B., Filipovich A. H. and Conforti L. (2007). Altered dynamics of Kv1.3 channel compartmentalization in the immunological synapse in systemic lupus erythematosus. J. Immunol. 179, 346-356. 10.4049/jimmunol.179.1.346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G., Tai K.-K. and Abbott G. W. (2006). Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J. Physiol. 570, 455-467. 10.1113/jphysiol.2005.100644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Verdaguer M., Capera J., Martinez-Marmol R., Camps M., Comes N., Tamkun M. M. and Felipe A. (2016a). Caveolin interaction governs Kv1.3 lipid raft targeting. Sci. Rep. 6, 22453 10.1038/srep22453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Verdaguer M., Capera J., Serrano-Novillo C., Estadella I., Sastre D. and Felipe A. (2016b). The voltage-gated potassium channel Kv1.3 is a promising multitherapeutic target against human pathologies. Expert Opin. Ther. Targets 20, 577-591. 10.1517/14728222.2016.1112792 [DOI] [PubMed] [Google Scholar]
- Roura-Ferrer M., Sole L., Oliveras A., Dahan R., Bielanska J., Villarroel A., Comes N. and Felipe A. (2010). Impact of KCNE subunits on KCNQ1 (Kv7.1) channel membrane surface targeting. J. Cell Physiol. 225, 692-700. 10.1002/jcp.22265 [DOI] [PubMed] [Google Scholar]
- Schwake M., Athanasiadu D., Beimgraben C., Blanz J., Beck C., Jentsch T. J., Saftig P. and Friedrich T. (2006). Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J. Neurosci. 26, 3757-3766. 10.1523/JNEUROSCI.5017-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Nakahira K., Hammond S., Rhodes K. J., Schechter L. E. and Trimmer J. S. (1996). Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 16, 843-852. 10.1016/S0896-6273(00)80104-X [DOI] [PubMed] [Google Scholar]
- Sole L. and Felipe A. (2010). Does a physiological role for KCNE subunits exist in the immune system? Commun. Integr. Biol. 3, 166-168. 10.4161/cib.3.2.10602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole L., Roura-Ferrer M., Perez-Verdaguer M., Oliveras A., Calvo M., Fernandez-Fernandez J. M. and Felipe A. (2009). KCNE4 suppresses Kv1.3 currents by modulating trafficking, surface expression and channel gating. J. Cell Sci. 122, 3738-3748. 10.1242/jcs.056689 [DOI] [PubMed] [Google Scholar]
- Sole L., Vallejo-Gracia A., Roig S. R., Serrano-Albarras A., Marruecos L., Manils J., Gomez D., Soler C. and Felipe A. (2013). KCNE gene expression is dependent on the proliferation and mode of activation of leukocytes. Channels 7, 85-96. 10.4161/chan.23258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear J. M., Koborssy D. A., Schwartz A. B., Johnson A. J., Audhya A., Fadool D. A. and Stagg S. M. (2015). Kv1.3 contains an alternative C-terminal ER exit motif and is recruited into COPII vesicles by Sec24a. BMC Biochem. 16, 483 10.1186/s12858-015-0045-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi O., Boratko A., Panyi G. and Hajdu P. (2013). The role of PSD-95 in the rearrangement of Kv1.3 channels to the immunological synapse. Pflugers Arch. 465, 1341-1353. 10.1007/s00424-013-1256-6 [DOI] [PubMed] [Google Scholar]
- Tiffany A. M., Manganas L. N., Kim E., Hsueh Y.-P., Sheng M. and Trimmer J. S. (2000). PSD-95 and SAP97 exhibit distinct mechanisms for regulating K(+) channel surface expression and clustering. J. Cell Biol. 148, 147-158. 10.1083/jcb.148.1.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye C. G., Welch R. C., Daniels M. A., Manderfield L. J., Tapper A. R., Sanders C. R. and George A. L. Jr (2009). Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE subunits. J. Gen. Physiol. 134, 207-217. 10.1085/jgp.200910234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z., Hajdu P. and Panyi G. (2010). Ion channels in T lymphocytes: an update on facts, mechanisms and therapeutic targeting in autoimmune diseases. Immunol. Lett. 130, 19-25. 10.1016/j.imlet.2009.12.015 [DOI] [PubMed] [Google Scholar]
- Vicente R., Escalada A., Coma M., Fuster G., Sanchez-Tillo E., Lopez-Iglesias C., Soler C., Solsona C., Celada A. and Felipe A. (2003). Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J. Biol. Chem. 278, 46307-46320. 10.1074/jbc.M304388200 [DOI] [PubMed] [Google Scholar]
- Vicente R., Escalada A., Soler C., Grande M., Celada A., Tamkun M. M., Solsona C. and Felipe A. (2005). Pattern of Kv beta subunit expression in macrophages depends upon proliferation and the mode of activation. J. Immunol. 174, 4736-4744. 10.4049/jimmunol.174.8.4736 [DOI] [PubMed] [Google Scholar]
- Vicente R., Escalada A., Villalonga N., Texido L., Roura-Ferrer M., Martin-Satue M., Lopez-Iglesias C., Soler C., Solsona C., Tamkun M. M. et al. (2006). Association of Kv1.5 and Kv1.3 contributes to the major voltage-dependent K+ channel in macrophages. J. Biol. Chem. 281, 37675-37685. 10.1074/jbc.M605617200 [DOI] [PubMed] [Google Scholar]
- Vicente R., Villalonga N., Calvo M., Escalada A., Solsona C., Soler C., Tamkun M. M. and Felipe A. (2008). Kv1.5 association modifies Kv1.3 traffic and membrane localization. J. Biol. Chem. 283, 8756-8764. 10.1074/jbc.M708223200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalonga N., Escalada A., Vicente R., Sanchez-Tillo E., Celada A., Solsona C. and Felipe A. (2007). Kv1.3/Kv1.5 heteromeric channels compromise pharmacological responses in macrophages. Biochem. Biophys. Res. Commun. 352, 913-918. 10.1016/j.bbrc.2006.11.120 [DOI] [PubMed] [Google Scholar]
- Villalonga N., David M., Bielanska J., Vicente R., Comes N., Valenzuela C. and Felipe A. (2010). Immunomodulation of voltage-dependent K+ channels in macrophages: molecular and biophysical consequences. J. Gen. Physiol. 135, 135-147. 10.1085/jgp.200910334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel E., Tapken D. and Seebohm G. (2012). The KCNE tango - how KCNE1 interacts with Kv7.1. Front. Pharmacol. 3, 142 10.3389/fphar.2012.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Jiang M., Hsu K.-L., Zhang M. and Tseng G.-N. (2008). KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol. 131, 589-603. 10.1085/jgp.200809976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N., Schwappach B., Jan Y. N. and Jan L. Y. (1999). A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22, 537-548. 10.1016/S0896-6273(00)80708-4 [DOI] [PubMed] [Google Scholar]
- Zheng R., Thompson K., Obeng-Gyimah E., Alessi D., Chen J., Cheng H. and McDonald T. V. (2010). Analysis of the interactions between the C-terminal cytoplasmic domains of KCNQ1 and KCNE1 channel subunits. Biochem. J. 428, 75-84. 10.1042/BJ20090977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Gomez B., Watanabe I. and Thornhill W. B. (2007). Kv1 potassium channel C-terminus constant HRETE region: arginine substitution affects surface protein level and conductance level of subfamily members differentially. Mol. Membr. Biol. 24, 194-205. 10.1080/09687860601066309 [DOI] [PubMed] [Google Scholar]
- Zsiros E., Kis-Toth K., Hajdu P., Gaspar R., Bielanska J., Felipe A., Rajnavolgyi E. and Panyi G. (2009). Developmental switch of the expression of ion channels in human dendritic cells. J. Immunol. 183, 4483-4492. 10.4049/jimmunol.0803003 [DOI] [PubMed] [Google Scholar]