

Abstract
Telomere length (TL) is a widely used marker of biological aging and is associated with an increased risk of morbidity and mortality. Recently, there has been evidence for an association between socioeconomic status (SES) and TL, particularly with measures of education and childhood SES. Individual differences in TL are also influenced by genetic factors, with heritability estimates from twin and sibling studies ranging from 34 to 82 percent. Yet unknown is the additive heritability of TL due to measured genetic variations and the extent that heritability is modified by SES. Data from the Health and Retirement Study, a nationally-representative cohort of older adults (mean 69 years), were used to provide the first estimates of molecular-based heritability of TL using genome-wide complex trait analysis (GCTA). We found that additive genetic variance contributed 28 percent (p=0.012) of total phenotypic variance of TL in the European American sample (n=3290). Estimation using the GCTA and KING Robust relationship inference methods did not differ significantly in this sample. None of the variance from the gene-by-SES interactions examined contributed significantly to the total TL variance. Estimation of heritability and genetic interaction with SES in the African American sample (n=442) was too unstable to provide reliable estimates.
Keywords: telomere length, heritability, GCTA, socioeconomic status
INTRODUCTION
Over the last decade telomere length (TL), the repeated DNA that creates protective caps at the ends of chromosomes, has become an innovative and controversial biomarker of aging and of general health status. Studies have shown associations between shorter TL and mortality and various types of age-related diseases such as cardiovascular disease (e.g., stroke, heart attack), cancer, and diabetes as well as osteoporosis, cognitive function, dementia, depression, and inflammatory diseases such as arthritis (Aubert and Lansdorp 2008; Cawthon et al. 2003; Demissie et al. 2006; Weischer et al. 2012; Zee et al. 2010). Many have suggested that TL measurement is a new tool for aging and health status characterization with multiple novel applications (Blasco 2005; Harari et al. 2013; Vera and Blasco 2012), the largest being that health can be more accurately assessed long before disease is evident.
Recently, there has been evidence for an association between socioeconomic status (SES) and TL, particularly with measures of education, childhood SES, and adult SES (Needham 2012; Prescott et al. 2012; Needham et al. 2013; Needham et al. 2014; Mitchell et al. 2014; Savolainen et al. 2014). These studies report that lower SES levels are associated with shorter TL implying that the stress of living in a more disadvantaged circumstance ages people faster than at higher SES levels. This fits with a much larger literature that shows that a lower SES level, especially during childhood, degrades health over the life course (Cohen et al. 2010). If TL can be used as an indicator of advanced aging or weathering (Geronimus et al. 2006), it could provide more a direct measure of health for researchers.
In a parallel literature, twin and genetic association studies have provided evidence that genes may regulate TL. Genetic association studies have found several single nucleotide polymorphisms (SNPs) and genes that appear to be related to TL (Codd et al. 2010; Levy et al. 2010; Mirabello et al. 2010; Rafnar et al. 2009; Soerensen et al. 2012). Evidence of genetic influence on TL has also been shown through studies of heritability, defined as the proportion of the variance in a phenotype (the observable characteristic of interest, in this case TL) that is due to genetic variation. Heritability estimates range from 0 to 1, with higher heritabilities indicating that a phenotype is more strongly patterned by genetic factors. Initial twin and sibling studies all supported a moderate to strong genetic contribution to TL, but showed divergent estimates of TL heritability ranging from 34 to 82 percent (Andrew et al. 2006; Bischoff et al. 2005; Slagboom, Droog, and Boomsma 1994; Vasa-Nicotera et al. 2005). A recent meta-analysis that investigated TL heritability in six large, independent family-based cohorts supports high TL heritability (0.70; 95 percent CI 0.64–0.76) (Broer et al. 2013).
Family-based estimates of heritability, however, must be interpreted with caution because they may capture non-additive as well as additive genetic effects. For example, twin studies will include the effects of gene-by-gene interactions (GxG; epistasis) as identical twins share virtually 100% of their DNA sequence, and thus they share all genetic effects, including both additive and non-additive (Plomin et al. 2013). Non-additive genetic effects include any non-linear effects that contribute to the genetic variance of a trait, such as GxG, gene-by-environment interactions (GxE), or genetic dominance (where the presence of one allele contributes more strongly to phenotype patterning than the presence of the other allele). Additive genetic effects include only those that are linearly associated with the trait of interest (that is, the individual effects of each allele can be summed to obtain the total genetic effect). Heritabilities that include only additive genetic effects are referred to as “narrow-sense heritabilities,” while those that also include non-additive genetic effects are referred to as “broad-sense heritabilities.”
While the meta-analysis of family-based cohorts included a wider spectrum of related individuals (Broer et al. 2013), the heritability of TL in unrelated individuals has not been estimated. New techniques, such as genome-wide complex trait analysis (GCTA), can be used to estimate the total amount of phenotypic variance that can be explained by genotyped SNPs in unrelated individuals (Yang et al. 2011). That is, instead of using familial relationships to estimate heritabilities, the GCTA method estimates heritability using genetic variations that are measured across the entire genome through array-based genotyping (genotyping “chips”). This approach estimates heritability due only to additive genetic effects (narrow-sense heritability), and may underestimate the total heritability of the trait if there are genetic variations that contribute to phenotypic variance but are not well-captured by the SNPs on the genotyping array utilized. The GCTA method, implemented in the GCTA software, has been successfully applied to estimate molecular-based heritability for multiple traits in unrelated individuals including height, intelligence, personality, type I diabetes, and even economic and political preferences (Benjamin et al. 2012; Deary et al. 2012; Lee et al. 2011; Vinkhuyzen et al. 2012; Yang et al. 2010). Vinkhuyzen et al. (2013) provides an excellent review of the concepts of heritability and its estimation in families and unrelated individuals using whole-genome analysis methods. Taken together, this research suggests that there are both genetic and non-genetic mechanisms that influence TL. To date only one study has attempted to combine both literatures to show that the effect of SES or social disadvantage appears to be moderated by genes (Mitchell et al. 2014). However, this study was on young children (age 9) from a highly select sample. Further, it used candidate markers from the serotonergic and dopaminergic pathways. The aim of the present study is to provide the first estimates of molecular-based heritability of TL by using a multi-ethnic, nationally-representative cohort of older adults and to examine whether heritability varies by lifecourse SES. Thus, it first provides a lower bound of the effects of genetics on TL by estimating the narrow-sense, additive heritability due to measured genetic variation. Second, it examines whether heritability of TL differs by SES level. That is, on average, do genes influence TL more or less for people in higher SES levels compared to lower SES levels? Third, it compares heritability estimates for TL obtained when relatedness among study participants is calculating using a method that does not account for admixture (GCTA) to a method that does (kinship-based inference for GWAS (KING)) (Manichaikul et al. 2010). By more fully examining the interaction between SES-related disadvantage and genome-wide variations, we hope to help to elucidate the pathway by which SES-related health disparities emerge. It is well-known that persons with low SES experience poorer health and wellbeing relative to higher SES persons; however, what remains unknown is why the effect is heterogeneous among low SES groups (Adler and Rehkopf 2008). One potential explanation for the heterogeneity in outcomes may be biological, both through direct effects of biological differences and differential responsiveness to environmental influences (Mitchell et al. 2013).
MATERIALS AND METHODS
Sample
Telomere length, genotype, and survey data are linked from the Health and Retirement Study (HRS) (Juster and Suzman, 1995; Sonnega et al. 2014). The HRS is a nationally representative, prospective panel study of approximately 20,000 community-dwelling adults in the contiguous United States over 50 years of age with oversamples of African-Americans and Hispanics (Heeringa and Connor 1995). The study collects information about income, work, assets, pension plans, health insurance, disability, physical health and functioning, cognitive functioning, and health care expenditures. It is designed to provide reliable data on the decisions, choices, and behaviors of people as they age and respond to changes in public policy, the economy, and health (Sonnega et al. 2014). The study is funded by the National Institutes of Aging and conducted by the Institute for Social Research at the University of Michigan (U01AG009740).
The HRS sample for this study consisted of a random one half of the entire HRS sample that was preselected to have an Enhanced Face-to-Face Interview (EFTF) in 2008. In addition to the core interview, the EFTF interview includes a set of physical performance tests, anthropometric measurements, blood and saliva samples, and self-administered questionnaire on psychosocial topics. Approximately fifty percent of households with at least one living respondent were selected for the EFTF interview across all primary sampling units (PSUs). The sample was selected at the household-level to ensure that the same request was made to both members of a household. Exclusion criteria for the EFTF interview and saliva collection included 1) needing to be interviewed by proxy, b) residing in a nursing home, or c) preferring to be interviewed by telephone. Consent to DNA collection was administered in person at the time of the interview. The HRS study protocol, including DNA collection, was approved by the University of Michigan Institutional Review Board.
TL Assessment and Genotyping
The 2008 Telomere Data include average TL data from HRS respondents who consented and provided a saliva sample during the 2008 interview wave (N=6,221). Respondents who provided a saliva sample did not significantly differ from those who were asked but did not consent across age, sex, or SES variables (education, income, wealth). A saliva sample was obtained directly using an Oragene Collection Kit. The saliva consent rate in 2008 was 85 percent. Among respondents who consented, 99 percent successfully completed a usable saliva sample. After collection, saliva samples were immediately sent to a central laboratory where the DNA was extracted and stored frozen at −80 C until being plated and shipped for genetic analyses.
Average TL was assayed using quantitative PCR (qPCR) by comparing telomere sequence copy number in each participant’s sample (T) to a single-copy gene copy number (S) (Cawthon 2002). The resulting T/S ratio is proportional to mean TL (Aviv et al. 2009; Cawthon 2002). The coefficient of variation (CV) for each sample was calculated based on two or three pairs of T and S runs – three runs for plates 2–9, 11 and 13 and based on two runs for plates 1, 10, 13–64. Comparison of CVs of 10 quality control DNA samples from 10 plates each with 3 runs showed that 3 runs compared to 2 runs did not reduce CVs. Therefore, all subsequent plates were assayed with 2 runs. Samples that had smaller than 12.5 percent CV were considered acceptable and samples with greater than 12.5 percent CV were reassayed. A total of 5916 participant samples from the HRS cohort were tested. Of these participant samples, 88.9 percent passed all CV-related QC criteria. After repeated testing, the final success rate was 98.2 percent (N=5808). Assays and QC were performed by Telome Health (Telomere Diagnostics, http://www.telomehealth.com/). T/S ratio scores ranged from 0.2 to 21.1. To remove outliers, 54 samples (< 1 percent) with T/S ratio 3.5 or greater were not included in the analyses. T/S ratio were transformed with the natural logarithm prior to analysis to improve normality.
Genotyping of common variants was obtained using the llumina HumanOmni2.5 BeadChip, which measures ~2.4 million SNPs. Samples were excluded if they had a call rate <98 percent, or were a first- or second- degree relative with another HRS participant in the sample. SNPs were excluded if they had a call rate <98 percent, had >4 discordant calls in study duplicates, had >1 Mendelian error in control samples, or were out of Hardy-Weinberg equilibrium in European American or African American sub-samples (p<10−4). Genotyping was conducted by the NIH Center for Inherited Disease Research (CIDR) at Johns Hopkins University. Genotyping quality control and final preparation of the data were performed by the Genetics Coordinating Center at the University of Washington. The genotyping was funded as a separate award from the National Institute on Aging (RC2 AG036495).
From the full HRS genetic sample, we used a combination of self-reported race and principal components analysis of genome-wide genotype data using the SNPRelate package in R to select a sub-sample of non-Hispanic White participants (European Americans) and a sub-sample of African American participants for analysis. In each ethnic group separately, the GCTA program was used to obtain a relationship matrix (GCTA relationship matrix) among all participants. Following that, a subset of unrelated participants was selected so that all participant pairs had relatedness less than 0.025 (N=8,401 European Americans; N=1,386 African Americans). A secondary relationship matrix was estimated in the full HRS genetic sample by KING robust method (Manichaikul et al. 2010). The same subset of samples were extracted to constitute the corresponding KING relationship matrix for the analysis conducted here. Also, GCTA was used to estimate two sets of ethnic group specific principal components separately from the GCTA matrix and the KING matrix for use as the corresponding analysis covariates. The final analytic samples were created by combining the unrelated genetic sample with the sample with available telomere data as diagramed in Figure 1.
Figure 1.
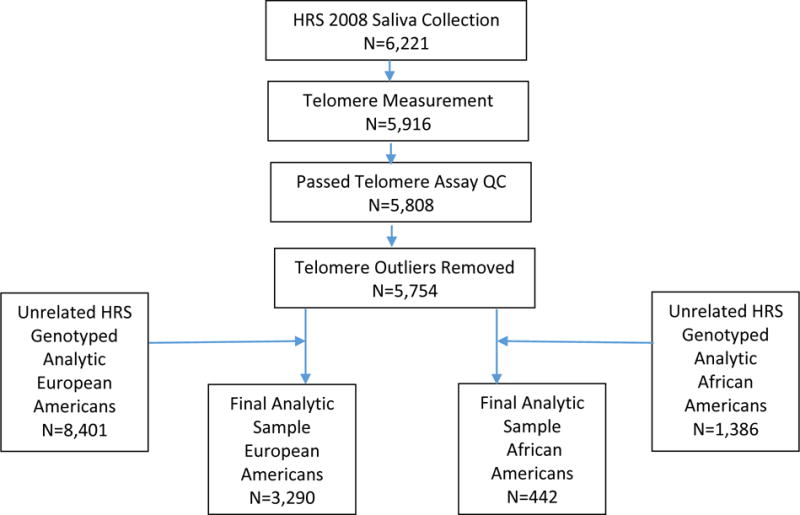
Sample inclusion from saliva collection to telomere heritability analysis
Socioeconomic Measures
Information on SES in childhood, and educational attainment, income and wealth in adulthood was collected prior to and concurrent with DNA collection and TL assessment.
Maternal and Paternal Education
Childhood SES was represented by parental education which was collected at the baseline interview. Because of differences in question administration over time, maternal education was coded as a dichotomous variable indicating whether mothers had ≥ 8 years of education. Paternal education was coded analogously. Respondents were only included in the analyses if they did not have missing data on the parental education measures. Respondents with and without data on these measures did not differ statistically by age, gender, or other measures of SES.
Education
Years of education was asked at baseline and originally reported as a continuous variable with a maximum of 17. For the purposes of these analyses, education was represented as a dichotomous variable indicating whether the respondent had at least some college education.
Income and Wealth
The income measure used was reported household income at the time of DNA collection. Income was dichotomized at the median (< $38,900, and $38,900 or more). Since some HRS respondents are retired at the time of interview, household wealth was chosen in addition to household income as a supplemental measure of adult economic circumstances. Wealth was measured using an assets-less-debts approach by subtracting debt from the sum of net worth as reported at the baseline interview – items such as value of the home, checking and savings accounts, individual retirement accounts, certificates of deposit, bonds, and shares of stocks or mutual funds. For comparability, wealth was also dichotomized at the median (< $188,000, and $188,000 or more). Income and wealth data were drawn from the RAND HRS data files – version N which includes summary measures for income and wealth compiled from an extensive battery of HRS questions and imputed values where income and wealth variables are missing (Chien et al. 2014). Because of the multitude of variables that contribute to the calculated household income and wealth variables produced by RAND, approximately half of all respondents in 2008 have at least one imputed value contributing to these totals.
Analysis
Using genome-wide complex trait analysis (GCTA), we estimated the variance in TL explained by the additive effects of all measured SNPs across the genome in unrelated individuals separately by ethnic group and controlling for population structure (Yang et al. 2011). Unlike classical biometric models of heritability that rely on expected genetic relatedness of family members, this method estimates genetic relatedness from the SNP data in individuals that are unrelated through the use of measured genetic variations.
The effects of the SNPs were modeled as random effects using a linear mixed modeling approach, and the proportion of TL variance explained by all of the SNPs across the genome simultaneously was estimated by a restricted maximum likelihood approach that utilizes genetic relationships estimated for all pairs of individuals. All common genotyped autosomal SNPs with minor allele frequency >1 percent (N=1,486,411 for Whites and N=1,894,712 for AA) were used to infer the degree of genetic relatedness (kinship coefficient) among all pairs of individuals in the sample and thus create relationship matrices separately for each ethnic group. We then estimated the variance in TL explained by the genome-wide SNPs (molecular-based heritabilities) after adjusting for each SES measure as a covariate. Next, we included gene-by-environment (GxE) interactions in the heritability estimation, using each SES measure as a separate environment (Zheng et al. 2013, -gXe option). All heritabilities were calculated separately by ethnic group. All models were adjusted for age, sex, 10 ethnic group-specific principal components, and telomere plate (contributes approximately 3 percent of variance in TL in this sample). Finally, we partitioned the variance in TL explained by the SNPs on each of the autosomal chromosomes to determine the molecular-based heritability attributable to each chromosome separately.
For all analyses, we conservatively used the GCTA-based relationship matrix to exclude respondents with estimated relatedness greater than 0.025 from the analyses. In addition, in order to investigate whether systematic inflation in the degree of estimated relatedness among individuals of the same racial group had any impact on the heritabilities, we conducted each set of analyses by estimating the relatedness of the individuals using the GCTA and KING Robust relationship matrices separately (Manichaikul et al. 2010). The KING method was developed principally to remove potential bias due to population admixture which can make ethnic minorities appear more related than they are.
RESULTS
The final sample consisted of 3732 respondents (N=3290 European Americans, N=442 African Americans). Descriptive statistics are provided in Table 1. European Americans had a mean TL of 1.30 (SD=0.33, min=0.23, max=3.50) and mean age of 69.3 years (SD=10.1), while African Americans had a mean TL of 1.44 (SD=0.40, min=0.38, max=3.38) and mean age of 67.1 years (SD=9.9). The African American sample had a larger percentage of women (64.7%) than the European American sample (58.8%) and had a larger percentage of participants with lower childhood and adult measures of SES.
Table 1.
Descriptive Statistics for HRS European Americans and African Americans
| European Americans (N=3290) |
African Americans (N=442) |
|
|---|---|---|
| Mean (SD) | Mean (SD) | |
| Telomere length (T:S ratio) | 1.30 (0.33) | 1.44 (0.40) |
| Ln(telomere length) | 0.23 (0.25) | 0.33 (0.28) |
| Age, years | 69.3 (10.1) | 67.1 (9.90) |
| N (%) | N (%) | |
|
| ||
| Female | 1935 (58.8) | 246 (64.7) |
| Mother Education <8 years | 406 (12.3) | 135 (30.5) |
| Father Education <8 years | 590 (17.9) | 184 (41.6) |
| Education < High School | 2230 (67.8) | 344 (77.8) |
| Income < $38,900 | 1366 (41.5) | 278 (62.9) |
| Wealth < $188,000 | 1290 (39.2) | 342 (77.4) |
HRS: Health and Retirement Study; Ln: natural log
GCTA was used to estimate the heritability and contribution of genome-wide GxE variance to TL while adjusting for potential confounders: age, sex, top 10 PCs, and telomere plate number. For TL, additive genetic variance contributed 28 percent (p=0.012) of total phenotypic variance in the European American sample using the KING approach to relationship inference. The estimated heritability using the GCTA approach to relationship inference was 31 percent (p=0.011). The heritability estimates using these different approaches to relationship inference did not differ substantially in this cohort (Table 2).
Table 2.
Comparison of Heritability of Telomere Length Among European Americans Using GCTA and KING Robust Relationship Inference Methods
| N | h2 | SE (h2) | p value | |
|---|---|---|---|---|
| KING Robust | 3290 | 0.28 | 0.13 | 0.012 |
| GCTA | 3290 | 0.31 | 0.14 | 0.011 |
KING: kinship-based inference for GWAS; GCTA: genome-wide complex trait analysis; h2: heritability; SE: standard error.
All analyses were adjusted for age, sex, PCs (10), and telomere plate
Adjustment for one’s own education level, maternal education, paternal education, income or wealth did not substantively impact the estimate of TL heritability. Heritability estimates for TL ranged from 0.27 to 0.28 regardless of the adjustment covariate, and p-values assessing the significance of TL heritability were all <0.05. Models that included assessment of the GxE interactions between the SNPs and each social factor maintained relatively consistent heritability estimates for TL as well (ranging from 0.17 when the model included an interaction between genetics and wealth to 0.28 when the model included an interaction between genetics and maternal education). The proportion of TL variance attributable to GxE interactions was generally small, with most interactions accounting for less than 5% of the trait variance, with the exception of the gene-by-wealth interaction (18%). However, p-values for the GxE interaction terms showed that none of the GxE interactions significantly contributed to the total TL variance (p-values for the interaction term ranged from 0.257 to 0.500), indicating that interaction between the genetic and socioeconomic factors is not a substantial contributor to the variability of TL (Table 3).
Table 3.
Estimation of Telomere Heritability Adjusting for SES Covariates vs. Explicitly Modeling GxE Interactions in European Americans (N=3290)
| h2 | SE (h2) | p value | GxE h2 | GxE SE (h2) | GxE p value | |
|---|---|---|---|---|---|---|
| KING Robust | 0.28 | 0.13 | 0.012 | |||
| Education Adjustment | 0.28 | 0.13 | 0.012 | |||
| Education Interaction | 0.25 | 0.13 | 0.05 | 0.24 | 0.420 | |
| Maternal Education Adjustment | 0.28 | 0.13 | 0.012 | |||
| Maternal Education Interaction | 0.28 | 0.28 | 0.00 | 0.31 | 0.500 | |
| Paternal Education Adjustment | 0.28 | 0.13 | 0.002 | |||
| Paternal Education Interaction | 0.26 | 0.22 | 0.03 | 0.26 | 0.449 | |
| Income Adjustment | 0.27 | 0.13 | 0.015 | |||
| Income Interaction | 0.27 | 0.17 | 0.00 | 0.22 | 0.500 | |
| Wealth Adjustment | 0.27 | 0.13 | 0.014 | |||
| Wealth Interaction | 0.18 | 0.18 | 0.18 | 0.25 | 0.257 |
SES: socioeconomic status; GxE: gene-by-environment interaction; h2: heritability; SE: standard error; KING: kinship-based inference for GWAS
All analyses were adjusted for age, sex, PCs (10), and telomere plate
When we partitioned the genetic variance of TL across the chromosomes, we found that it was distributed fairly evenly across the genome. The proportion of total TL variance accounted for by the genetic variation on each of the 22 autosomal chromosomes, separately, ranged from <0.01 to 0.088. The estimates of variance explained did not differ significantly by chromosome (p=0.369), indicating that excess heritability was not observed for chromosomes containing genes with large regulatory effects on TL and telomerase such as Chromosome e (TERC) and Chromosome 5 (TERT) (Armanios and Blackburn 2010). Not unexpectedly, chromosome size was correlated with the amount of TL variance explained (r=0.31), with larger chromosomes explaining more of the genetic variance of TL than smaller chromosomes.
We attempted to run the same set of analyses in the African American sub-sample, but the sample size was too small to achieve meaningful estimates of heritability.
DISCUSSION
In this study, we utilized a fairly new approach to estimating heritability of a biological indicator in a sample of unrelated adults. We examined the effect of assumptions about the population structure and relatedness of the sample on that estimate. In addition, we used two methods to examine the contribution of GxE interaction on the genome-wide variance of TL. These results from a nationally-representative population-based sample show that common SNPs in total explain approximately 28 percent of the phenotypic variance in TL among European Americans.
The GCTA estimate of TL heritability is much lower (28–31 percent) than the estimate from the twin-based meta-analysis (70 percent) (Broer et al. 2013). Twin and family-based estimates of heritability have repeatedly been found to be higher than molecular-based estimates in unrelated individuals, so this pattern is not surprising (Plomin and Deary 2015; Trzaskowski, Dale, and Plomin 2013). However, while some processes like height, weight, and IQ have shown more similar (although still lower) estimates of heritability, behavioral data has shown substantial differences in the heritability estimates. Certainly, heritability estimates from measured genetic data are lower bounds because they utilize only the information from genetic variations that are included on the genotyping array or are strongly correlated with measured SNPs; thus, these estimates may not capture all the possible additive effects. However, the more likely explanation for the substantially lower heritability estimated by the GCTA method is that this method does not capture non-additive genetic effects due to GxG interactions and GxE interactions (Trzaskowski et al. 2013). Thus, it may be possible that GxG and GxE interactions explain as much as half of the heritability of TL. Further work should attempt to tease out these multiplicative relationships.
One potentially important GxE relationship is with SES. Given the relationship between SES and TL in both children and adults (Cohen et al. 2010; Mitchell et al. 2014; Needham 2012; Needham 2013), there was reason to hypothesize that SES would explain part of the variation in TL and/or moderate the effect of genetics on TL. However, we found that neither the childhood nor adult SES measures used in this study contributed to the variance explained. That is, adjusting for SES did not change the amount of TL variance explained by genes. This implies that SES is not a mediating variable through which these genes operate to influence TL. This is not to say the SES does not have an effect on TL—just that the genetic markers used here don’t operate through SES. We also found no evidence that the genetic contribution to TL was moderated by SES, since none of the GxE contributions were significant.
There were also no substantial differences in models using SES as a covariate adjustment or those where SES was explicitly modeled as a GxE interaction for the TL phenotype. We interpret this finding to mean that the additive effect of common SNPs is relatively constant across the SES gradient. We suggest that what we have measured here may in fact be the basic genetic architecture for the biological processes that affect TL, but not necessarily the genetic determinants of TL change or the potential responsiveness of TL due to the environment. Of course this is speculative without additional examinations in other samples and without additional SES measures.
In contrasting our results on the effects of SES on TL with the broader SES and TL literature it is important to remember while adults of low socioeconomic status generally have shorter telomeres than those of higher status, associations with specific measures of SES are more nuanced. For example, while some studies have found TL is longer for college-educated adults (Needham 2013), others have that education was not associated with TL (Carroll 2013). Adults living in low income or poverty generally have shorter telomere lengths (Chae et al. 2014; Geronimus et al. 2010; Yen and Lung 2013) but that is not always true (Needham et al. 2013). Finally, occupational characteristics show no relationship with TL (Fujishiro et al. 2014). Potentially more interesting is to note that in examining TL differences in children by parental SES (education and income), children of lower SES already appear to have shorter TL (Needham et al. 2012; Mitchell et al. 2014). However, most other child TL literature focuses not on SES specifically, but broader stressful-life events or environments—all of which seem to show negative correlations with child telomere length (e.g. a child’s risk level as defined by the Child Welfare System (Asok et al. 2013), exposure to institutionalized care (Drury et al. 2012), family violence and instability (Drury et al. 2014; Mitchell et al. 2014), and neighborhood disorder and poverty (Theall et al. 2013)).
A key assumption underlying many existing methods of inferring relatedness is that genotypes for all individuals are representative of a common set of allele frequencies. However, deviations from this assumption are to be expected in representative, population-based samples with population substructure and admixture. The methods used to infer relationships among individuals can have an effect on the algorithms used downstream from high-throughput genotyping such as GWAS and molecular-based heritability estimation. In contrast to many relationship inference tools, the KING Robust method does not use the underlying assumption of homogeneity and is therefore a more robust inference in the presence of population substructure (Manichaikul et al. 2010). While the differences in the heritability estimates of TL using the GCTA-generated and KING Robust relationship matrices were not substantively different with this phenotype in this European American cohort, it remains important to compare different relationship inference methods especially when population homogeneity cannot be assumed (such as in African American and other admixed populations). Further analyses should also consider the REAP (relatedness estimation in admixed populations) method, especially where both population substructure and admixed ancestry are present (Thornton et al. 2012).
This study represents significant progress in the estimation of genetic contribution to TL. However, there are several limitations to this work. First, while we were able to estimate heritability of TL in the European American sub-sample, the size of the African American sample was too small to even generate an estimate of the total additive genetic variance. Hopefully in the future, other studies with TL measured on large African American samples can be combined to examine heritability and the contribution of SES differences on TL variance in African Americans and other minority populations. Second, we acknowledge that this set of results may be specific to this cohort and the SES measures that we used. Previous analyses that used different environmental variables found similarly robust heritabilities of biological phenotypes suggesting that the bias due to environmental confounding is minimal (Conley et al. 2014). Third, a larger sample size would facilitate more precise investigation of the differences in heritability across strata of SES, as well as provide more power for testing the significance of interactions. Finally, we were not able to study the effect of SES on the change in TL over time, due to the cross-sectional nature of this study. Ultimately the theory underlying the importance of TL is that experienced stress, in this case due to lower SES, degrades TL faster over time than one might expect due to aging. Unfortunately, there are no large enough samples that include 1) longitudinal data on TL, 2) SES measures across the lifecourse, and 3) genome-wide data in a sample large enough to estimate the heritability of change in TL. Additionally, it is reasonable to assume that the estimate of measured heritability (i.e. narrow, or additive, heritability) for change in TL is actually quite small if most of the effect of excessive telomere erosion is due to stress (or even gene-stressful environment interactions) because those effects are not included in the estimate of molecular heritability.
The results of this study suggest that common SNPs assayed on large-scale genotyping arrays account for just under one third of the heritability of TL. While several genes and SNPs associated with TL have been identified through genetic association studies, these SNPs combined only account for a small fraction of the estimated genetic variance. This suggests that: 1) GWAS of TL with much larger sample sizes will be required to identify additional SNPs that contribute to TL, and 2) multiplicative genetic effects including GxG and GxE may be an equally fruitful avenue, although again larger samples may be requisite.
References
- Adler NE, Rehkopf DH. US disparities in health: descriptions, causes, and mechanisms. Annu Rev Public Health. 2008;29:235–252. doi: 10.1146/annurev.publhealth.29.020907.090852. [DOI] [PubMed] [Google Scholar]
- Andrew T, Aviv A, Falchi M, Surdulescu GL, Gardner JP, Lu X, Kimura M, et al. Mapping genetic loci that determine leukocyte telomere length in a large sample of unselected female sibling pairs. Am J Hum Genet. 2006;78(3):480–6. doi: 10.1086/500052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nature Reviews Genetics. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008;88(2):557–79. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- Aviv A, Chen W, Gardner JP, Kimura M, Brimacombe M, Cao X, Srinivasan SR, Berenson GS. Leukocyte telomere dynamics: longitudinal findings among young adults in the bogalusa heart study. Am J Epidemiol. 2009;169(3):323–29. doi: 10.1093/aje/kwn338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DJ, Cesarini D, van der Loos MJ, Dawes CT, Koellinger PD, Magnusson PK, Chabris CF, et al. The genetic architecture of economic and political preferences. Proc Natl Acad Sci U S A. 2012;109(21):8026–31. doi: 10.1073/pnas.1120666109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff C, Graakjaer J, Petersen HC, Hjelmborg J, Vaupel JW, Bohr V, Koelvraa S, Christensen K. The heritability of telomere length among the elderly and oldest-old. Twin Res Hum Genet. 2005;8(5):433–9. doi: 10.1375/183242705774310141. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- Broer L, Codd V, Nyholt DR, Deelen J, Mangino M, Willemsen G, Albrecht E, et al. Meta-analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet. 2013;21(10):1163–68. doi: 10.1038/ejhg.2012.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JE, Diez Roux AV, Fitzpatrick AL, Seeman T. Low social support is associated with shorter leukocyte telomere length in late life: multi-ethnic study of atherosclerosis. Psychosom Med. 2013;75(2):171–177. doi: 10.1097/PSY.0b013e31828233bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM. Telomere measurement by quantitative pcr. Nucleic Acids Res. 2002;30(10):e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon R, Smith K, O’Brien E, Sivatchenko A, Kerber R. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361:393–395. doi: 10.1016/S0140-6736(03)12384-7. [DOI] [PubMed] [Google Scholar]
- Chien S, Campbell N, Hayden O, Hurd M, Main R, Mallett J, Martin C, et al. Rand hrs data documentation, version n. Social Security Administration and National Institute on Aging; 2014. [Google Scholar]
- Codd V, Mangino M, van der Harst P, Braund PS, Kaiser M, Beveridge AJ, Rafelt S, et al. Common variants near terc are associated with mean telomere length. Nat Genet. 2010;42(3):197–9. doi: 10.1038/ng.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45(4):422–7. doi: 10.1038/ng.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Chen E, Matthews KA. Childhood socioeconomic status and adult health. Ann N Y Acad Sci. 2010;1186:37–55. doi: 10.1111/j.1749-6632.2009.05334.x. [DOI] [PubMed] [Google Scholar]
- Conley D, Siegal ML, Domingue BW, Mullan Harris K, McQueen MB, Boardman JD. Testing the key assumption of heritability estimates based on genome-wide genetic relatedness. J Hum Genet. 2014;59(6):342–5. doi: 10.1038/jhg.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deary IJ, Yang J, Davies G, Harris SE, Tenesa A, Liewald D, Luciano M, et al. Genetic contributions to stability and change in intelligence from childhood to old age. Nature. 2012;482(7384):212–5. doi: 10.1038/nature10781. [DOI] [PubMed] [Google Scholar]
- Demissie S, Levy D, Benjamin EJ, Cupples LA, Gardner JP, Herbert A, et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell. 2006;5:325–330. doi: 10.1111/j.1474-9726.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- Geronimus AT, Hicken M, Keene D, Bound J. Weathering and Age Patterns of Allostatic Load Scores among Blacks and Whites in the United States. Am J Public Health. 2006 May;96(5):826–33. doi: 10.2105/AJPH.2004.060749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari Y, Romano GH, Ungar L, Kupiec M. Nature Vs Nurture: Interplay Between The Genetic Control Of Telomere Length And Environmental Factors. Cell Cycle. 2013;12(22):3465–70. doi: 10.4161/cc.26625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeringa SC, Connor JF. HRS Documentation Report DR-002. University of Michigan; Ann Arbor, MI: 1995. Technical description of the health and retirement study sample design. [Google Scholar]
- Juster FT, Suzman R. An overview of the health and retirement study. J Hum Resources. 1995;30:S7–S56. [Google Scholar]
- Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88(3):294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Neuhausen SL, Hunt SC, Kimura M, Hwang SJ, Chen W, Bis JC, et al. Genome-wide association identifies obfc1 as a locus involved in human leukocyte telomere biology. Proc Natl Acad Sci U S A. 2010;107(20):9293–8. doi: 10.1073/pnas.0911494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust Relationship Inference in Genome-Wide Association Studies. Bioinformatics. 2010;26(22):2867–73. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Yu K, Kraft P, De Vivo I, Hunter DJ, Prescott J, Wong JY, et al. The association of telomere length and genetic variation in telomere biology genes. Hum Mutat. 2010;31(9):1050–8. doi: 10.1002/humu.21314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, McLanahan S, Brooks-Gunn J, Garfinkel I, Hobcraft J, Notterman D. Genetic differential sensitivity to social environments: Implications for research. Am J Public Health. 2013;103(S1):S102–S110. doi: 10.2105/AJPH.2013.301382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks-Gunn J, Garfinkel I, Notterman D. Social disadvantage, genetic sensitivity, and children’s telomere length. Proc Natl Acad Sci U S A. 2014;111(16):5944–9. doi: 10.1073/pnas.1404293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham BL. Socioeconomic status and cell aging in children. Soc Sci Med. 2012;74(12):1248–51. doi: 10.1016/j.socscimed.2012.02.019. [DOI] [PubMed] [Google Scholar]
- Needham BL, Adler N, Gregorich S, Rehkopf D, Lin J, Blackburn EH, Epel ES. Socioeconomic status, health behavior, and leukocyte telomere length in the national health and nutrition examination survey, 1999–2002. Soc Sci Med. 2013;85:1–8. doi: 10.1016/j.socscimed.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham BL, Carroll JE, Roux AVD, Fitzpatrick AL, Moore K, Seeman TE. Neighborhood characteristics and leukocyte telomere length: the multi-ethnic study of atherosclerosis. Health & Place. 2014;28:167–172. doi: 10.1016/j.healthplace.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Deary IJ. Genetics and intelligence differences: five special findings. Mol Psychiatry. 2015;20(1):98–108. doi: 10.1038/mp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Haworth CM, Meaburn EL, Price TS, Davis OS. Common DNA markers can account for more than half of the genetic influence on cognitive abilities. Psychol Sci. 2013;24(4):562–8. doi: 10.1177/0956797612457952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott J, Du M, Wong JYY, Han J, De Vivo I. Paternal age at birth is associated with offspring leukocyte telomere length in the nurses’ health study. Hum Reprod. 2012;27(12):3622–3631. doi: 10.1093/humrep/des314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, Jakobsdottir M, et al. Sequence variants at the tert-clptm1l locus associate with many cancer types. Nat Genet. 2009;41(2):221–7. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen K, Eriksson JG, Kananen L, Kajantie E, Pesonen AK, Heinonen K, Räikkönen K. Associations between early life stress, self-reported traumatic experiences across the lifespan and leukocyte telomere length in elderly adults. Biol Psychol. 2014;97:35–42. doi: 10.1016/j.biopsycho.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Slagboom PE, Droog S, Boomsma DI. Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet. 1994;55(5):876–82. [PMC free article] [PubMed] [Google Scholar]
- Soerensen M, Thinggaard M, Nygaard M, Dato S, Tan Q, Hjelmborg J, Andersen-Ranberg K, et al. Genetic variation in tert and terc and human leukocyte telomere length and longevity: a cross-sectional and longitudinal analysis. Aging Cell. 2012;11(2):223–7. doi: 10.1111/j.1474-9726.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnega A, Faul JD, Ofstedal MB, Langa KM, Phillips JW, Weir DR. Cohort profile: the health and retirement study. Int J Epidemiol. 2014;43(2):576–85. doi: 10.1093/ije/dyu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton T, Tang H, Hoffmann TJ, Ochs-Balcom HM, Caan BJ, Risch N. Estimating kinship in admixed populations. Am J Hum Genet. 2012;91(1):122–38. doi: 10.1016/j.ajhg.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski M, Dale PS, Plomin R. No genetic influence for childhood behavior problems from DNA analysis. J Am Acad Child Adolesc Psychiatry. 2013;52(10):1048–56 e3. doi: 10.1016/j.jaac.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasa-Nicotera M, Brouilette S, Mangino M, Thompson JR, Braund P, Clemitson JR, Mason A, et al. Mapping of a major locus that determines telomere length in humans. Am J Hum Genet. 2005;76(1):147–51. doi: 10.1086/426734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera E, Blasco MA. Beyond average: potential for measurement of short telomeres. Aging. 2012;4(6):379–92. doi: 10.18632/aging.100462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkhuyzen AA, Pedersen NL, Yang J, Lee SH, Magnusson PK, Iacono WG, McGue M, et al. Common snps explain some of the variation in the personality dimensions of neuroticism and extraversion. Transl Psychiatry. 2012;2:e102. doi: 10.1038/tp.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkhuyzen NA, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation And Partition Of Heritability In Human Populations Using Whole-Genome Analysis Methods. Annu Rev Genet. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischer M, Bojesen SE, Cawthon RM, Freiberg JJ, Tybjaerg-Hansen A, Nordestgaard BG. Short telomere length, myocardial infarction, ischemic heart disease, and early death. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:822–829. doi: 10.1161/ATVBAHA.111.237271. [DOI] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, et al. Common snps explain a large proportion of the heritability for human height. Nat Genet. 42(7):565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. Gcta: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee RY, Castonguay AJ, Barton NS, Germer S, Martin M. Mean leukocyte telomere length shortening and type 2 diabetes mellitus: a case-control study. Transl Res. 2010;155:166–169. doi: 10.1016/j.trsl.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Zheng J-S, Arnett DK, Lee Y-C, Shen J, Parnell LD, Smith CE, Richardson K, Li D, Borecki IB, Ordovas JM, Lai CQ. Genome-wide contribution of genotype by environment interaction to variation of diabetes-related traits. PLoS ONE. 2013;8(10):e77442. doi: 10.1371/journal.pone.0077442. [DOI] [PMC free article] [PubMed] [Google Scholar]