

Abstract
Background
Intrachromosomal amplification of chromosome 21 (iAMP21) results from breakage-fusion-bridge cycles and chromothripsis is a distinct marker of a subgroup of B cell acute lymphoblastic leukemia (B-ALL) cases associated with a poor prognosis. iAMP21 accounts for 2% of pediatric B-ALL and occurs predominantly in older children or adolescents. ETV6-RUNX1 fusion, resulting from t(12;21)(p13;q22), is associated with an excellent outcome in younger children with B-ALL. Coexistence of iAMP21 with ETV6-RUNX1 fusion is extremely rare with limited clinical information available.
Results
We report the case of an 18-year old Caucasian man diagnosed with ETV6-RUNX1 fusion positive B-ALL. He was treated with intensive chemotherapy and achieved remission for 6 months before relapse, 15 months after the initial diagnosis. G-band karyotyping and Fluorescence in situ hybridization (FISH) analyses performed on bone marrow revealed complex abnormalities: 41,X,-Y,der(3)t(3;20)(p11.2;q11.2),-4,t(5;22)(q32;q11.2),del(9)(p13),dic(9;17)(p13;p11.2),t(12;21)(p13;q22),der(14)t(14;17)(p11.2;q11.2),der(17;22)(q11.2;q11.2),-20,add(21)(q22),-22[4]/46,XY[15] with an iAMP21 and an ETV6-RUNX1. Additional molecular studies confirmed ETV6-RUNX1 fusion and with a TP53 mutation. High-resolution single nucleotide polymorphism microarray (SNP array) revealed the iAMP21 to be chromothripsis of 21q and subsequent metaphase FISH further delineated complex genomic aberrations. Although the patient received intensive chemotherapy with allogenic stem cell transplant, he died 26 months after initial diagnosis. We searched the literature and identified six cases showing coexisting iAMP21 and ETV6-RUNX1. The median age for these six patients was 10 years (range, 2–18) and males predominated. The median overall survival (OS) was 28 months.
Conclusions
Patients with B-ALL associated with both iAMP21 and ETV6-RUNX1 tend to be older children or adolescents and have a poor prognosis.
Keywords: B-ALL, iAMP21, RUNX1 amplification, ETV6-RUNX1 fusion, SNP microarray
Background
The latest revision to the World Health Organization (WHO) classification of B-cell lymphoblastic leukemia/lymphoma (B-ALL) has added B-ALL with intrachromosomal amplification of chromosome 21 (iAMP21) as an entity in the group of B-ALL with recurrent genetic abnormalities [1]. iAMP21 is a distinct marker that can be readily detected by metaphase FISH [2] and is caused by breakage-fusion-bridge cycles and chromothripsis, which is a phenomenon reported in cancer genomes, resulted from tens to hundreds of genomic rearrangements occur in a cellular crisis. Chromthripsis can involve one or more chromosomes, often with massive copy number aberrations [3]. Recent study suggested that hyperploidy and telomere attrition could be triggering events for chromothripsis and are frequently associated with TP53 mutation [4].
B-ALL associated with iAMP21 is a poor prognostic subgroup that represents 2% of pediatric B-ALL cases. The median age of patients is 9 years old and there is a prevalence of males. Patients with iAMP21 often show low platelet and low white blood cell counts (WBC) [5–8]. These patients have a relapse rate that is three times higher than other B-ALL patients are and therefore patients often require intensified therapy, particularly in older children or adolescents with B-ALL [9].
The t(12;21)(p13;q22) which results in the formation of the ETV6-RUNX1 fusion gene accounts for about 25% of pediatric B-ALL. Patients with B-ALL associated with ETV6-RUNX1 tend to be younger children and patients have a favorable outcome [10]. iAMP21 has been reported rarely in B-ALL associated with ETV6-RUNX1 [11].
In this study, we describe a patient with B-ALL associated with both iAMP21 with ETV6-RUNX1 that we have characterized extensively by using molecular and cytogenetic methods. We also reviewed the literature and identified six similar cases [7, 12]. This combination of molecular alterations in B-ALL tends to occur in older male patients who have a poor prognosis.
Results
The patient was an 18-year old Caucasian man who presented initially with pancytopenia. A complete blood count showed: WBC 2.0 × 109/L, platelets 88 ×109/L and hemoglobin 8.3 g/dL. Bone marrow examination showed 61% blasts and the patient was diagnosed with a B-ALL at another institution (Table 1). FISH studies performed on the bone marrow aspirate smears showed ETV6-RUNX1 fusion in 28% of interphases with no evidence of BCR-ABL1 or MLL gene rearrangements. No concurrent chromosome data were available from the initial bone marrow studies. The patient did not have central nervous system (CNS) involvement and he was treated with the intrathecal cytarabine, daunorubicin, vincristine, intrathecal methotrexate, PEG asparaginase and prednisone (CALGB 10403 regimen) elsewhere. The patient did not respond well initially although he eventually achieved remission for 6 months after a second round of chemotherapy. The patient then began to show minimal residual disease by flow cytometry immunophenotypic analysis 8 months after the initial diagnosis, and eventually relapsed 15 months after the diagnosis. The patient was transferred to our institution at this time (Table 1).
Table 1.
Clinical and laboratory data of the patient
| Date | 10/9/2013 | 6/24/2014 | 12/9/2014 | 12/31/2014 | 6/8/2015 | 8/19/2015 |
|---|---|---|---|---|---|---|
| Significant event | Initial diagnosis | MRD Positive | 1st
Relapse |
Persistent disease | Post-ASCTa | 2nd
Relapse |
| BM Blast (%) (0–5) | 61 | NA | 5 | 54 | 1 | 25 |
| WBC (x109/L) | 2.0 | 5.5 | 4.6 | 2.8 | 5.6 | 2.2 |
| PLT (x103/μL) | 88 | 383 | 202 | 79 | 133 | 79 |
| HB (g/dL) | 8.3 | 10.8 | 13.3 | 12.9 | 9.1 | 11.4 |
| Flow-cytometry analysis | Positiveb | Positive | Positivec | Positived | NA | Positive |
| G-Band karyotyping | NAe | NA | 46,XY | Complex | 46,XY | 46,XY |
| ETV6-RUNX1/iAMP21 FISH | Positive | Negative | Borderline Negative | Positive | NA | NA |
| ETV6-RUNX1 by PCR | NA | NA | NA | Positive | NA | NA |
| Treatment | CALGBf | Post- CALGB | Blinatumomab | Hyper-CVADg + Inotuzumab | 50 days Post-ASCT | EPOCHh + Rituxan |
a Post ASCT post allogeneic stem cell transplantation
bCD19lo, CD22+, cytoplasmic CD79a+, HLA-DR+, aberrant CD13 and CD33 expression, CD3-, CD10-, CD20-, surface Ig-, CD34-, CD38-,MPO-
cCD10+, CD13+, CD19lo, CD33+/lo, CD9-, CD20-, CD34-, surface Ig-
dCD10+, CD13+, CD19+, CD22+,CD33+ (subset), CD45 dim, CD38 dim, CD58, cytoplasmic CD79a, CD81, TdT+, cytoplasmic CD3-, CD15-, CD20-, CD25-, CD34-, CD66c-, CD117-, CRLF2-, cytoplasmic IgM-, MPO-
e NA not available
fCALGB: IT Cytarabine, Daunorubicin, Vincristine, IT Methotrexate, PEG Asparaginase and Prednisone
gHyper-CVAD: Cyclophosphamide, Vincristine, Doxorubicin Hydrochloride, Dexamethasone
hEPOCH: Etoposide, Vincristine, Cyclophosphamide, Doxorubicin Hydrochloride
At time of relapse, the complete blood count showed: WBC 2.8 × 109/L, platelets 79 ×109/L and hemoglobin 12.9 g/dL. Bone marrow examination showed 54% blasts. Conventional cytogenetic analysis on the relapsed bone marrow showed a complex karyotype of 41,X,-Y,-3,-4,del(5)(p14),der(5)t(5;22) (q22;q11.2),del(10)(q24q25),-12,-14,-17,add(17)(p11.2),-20,+add(21)(p11.2),der(21)add(21)(p11.2)hsr(?21),der(21)t(12;21)(p13;q22),-22,add(22)(p11.2),+der(?)t(?;5)(?;?)t(?;22)(?;?),+mar[4]/46,XY[15] as initially reported. A nuclear fusion of ETV6-RUNX1 signal with RUNX1 amplification were observed in 27.5% of the interphases (Fig. 1). High-resolution SNP microarray revealed losses of chromosomes Yq, 3p, 4, 9p, 17p and 20p, as well as chromothripsis-like pattern of chromosome 21q (Fig. 2). Subsequent metaphase FISH analysis on the G-banded chromosomes targeting ETV6-RUNX1, DS523/D5S721/EGR1, CSF1R, CDKN2A/CEP9, TP53/CEP17 and DS20S108 along with whole chromosome painting (WCP) for chromosomes 17 and 22 (Figs. 3 and 4) showed: 1) a der(3)t(3;20) (p11.2;q11.2)(D20S108+); 2) a der(5)t(5;22)(q32;q11.2)(WCP22+); 3) a del(9)(p13) (CDKN2A-,D9Z1+), a dic(9;17)(p13;p11.2)(CDKN2A-,D9Z1+;D17Z1+,TP53-,WCP17+); 4) a t(12;21)((p13;(q22)RUNX1+; ETV6+,RUNX1+) and add(21)(RUNX1+++++); 5) a der(14)t(14;17)(p11.2;q11.2)(WCP17+); 6) a der(17)t(17;22) (TP53+,D17Z1+,WCP17+,WCP22+); 7) der((22)t(5;22)(CSF1R+,WCP22+) (Table 2). By integrating all the SNP array and chromosome and/or metaphase FISH, the above karyotype was further refined to 41,X,-Y,der(3)t(3;20)(p11.2;q11.2),-4,t(5;22)(q32;q11.2), del(9)(p13),dic(9;17)>(p13;p11.2),t(12;21)(p13;q22),der(14)t(14;17)(p11.2;q11.2), der(17;22)(q11.2;q11.2),-20,add(21)(q22),-22[4]/46,XY[15] (Figs. 3 and 4). In addition, sequencing analysis revealed a 10 base pair deletion-insertion mutation in exon 4 of TP53 (NM_000546(TP53): c.310_321delinsGT p.Q104fs), resulting in a loss of TP53 function. While this specific mutation is not previously reported in the catalogue of somatic mutations in cancer (COSMIC), this region in exon 4 is known to be involved by similar deleterious (frameshift and truncating) mutations. The patient was treated with blinatumomab and the hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone)/inotuzumab regimen, but only a partial remission was achieved. Due to persistent disease, the patient eventually received a matched unrelated donor allogeneic stem cell transplant (ASCT) 19 months after the initial diagnosis and 6 months after relapse. Unfortunately, the post-transplant course was complicated by liver veno-occlusive disease and relapse of B-ALL. Despite further therapy with R-EPOCH (rituximab, etoposide, vincristine, cyclophosphamide, and doxorubicin) and the patient died 26 months after initial diagnosis.
Fig. 1.
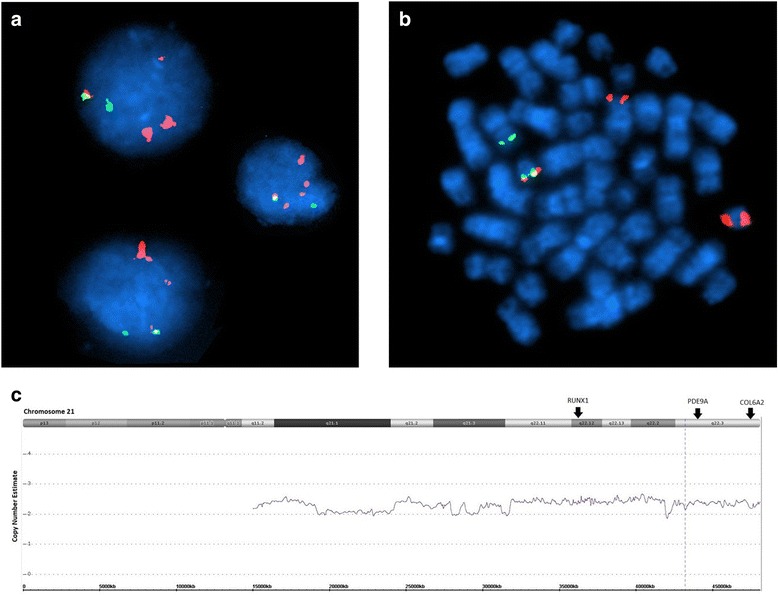
Interphase FISH, metaphase FISH, and SNP microarray analyses. a Interphase FISH showed iAMP21 and ETV6-RUNX1 fusion. b Metaphase FISH indicated a derivative chromosome 21 with ETV6-RUNX1 fusion, an iAMP21, and a derivative 12 with a single RUNX1 signal. c SNP microarray showing the chromothripsis-like pattern of chromosome 21q11.2-21q22.3 (15,006,457 – 48,097,372)
Fig. 2.
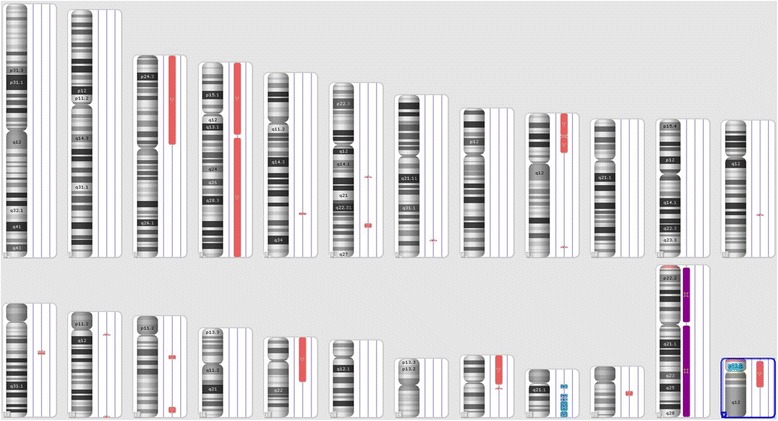
Chromosome view of SNP microarray analysis showing multiple copy number losses
Fig. 3.
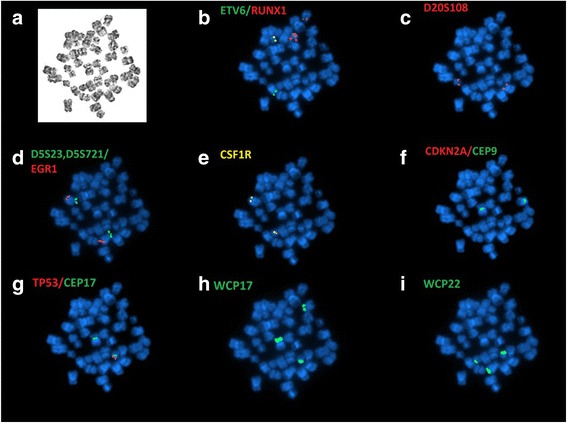
Sequential G-banding and metaphase FISH was performed to refine initial karyotyping result. a G-banded metaphase. b Metaphase FISH indicated ETV6 (green) and RUNX1 (red) fusion as well as RUNX1 amplification. c No deletions for D20S108/20q12 probe in red, one signal on a normal chromosome 20 and the other signal on the derivative chromosome 3. d No deletions for D5S23/D5S721(5p15.2) in green and EGR1 (5q31) in red). e No rearrangement for CSF1R/ 5q33–34, however, one copy was translocated to chromosome 22. f Homozygous deletion of CDKN2A (9p21) in red; centromere 9 in green. g Hemizygous deletion of TP53 (17p13.1) in red; centromeric 17 in green. h Whole chromosome painting (WCP) for 17 (green) stained three different chromosomes, indicated translocations. i WCP for 22 (green) stained three different chromosomes, indicated translocations
Fig. 4.
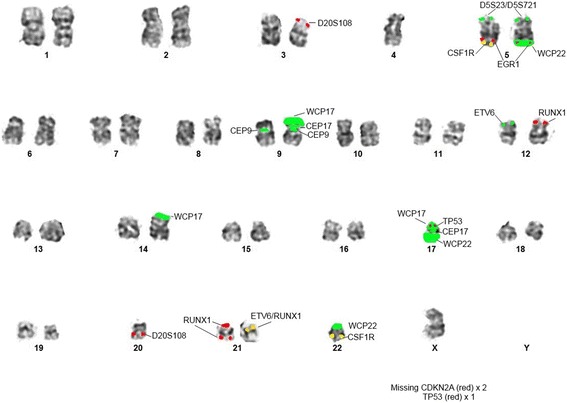
Refined karyotype of abnormal metaphase displayed in Fig. 3a with co-localized FISH signals indicated a hypodiploid clone with 1) a der(3)t(3;20)(p11.2;q11.2)(D20S108+); 2) a der(5)t(5;22)(q32;q11.2)(WCP22+); 3) a del(9)(p13)(CDKN2A-), a dic(9;17)(p13;p11.2)(D9Z1+,CDKN2A-;D17Z1+,TP53-,WCP17+); 4) a t(12;21)(p13;q22)(RUNX1+; ETV6+,RUNX1+) and add(21)(RUNX1+++++); 5) a der(14)t(14;17)(p11.2;q11.2)(WCP17+); 6) a der(17)t(17;22) (TP53+,D17Z1+,WCP17+,WCP22+); 7) der(22)t(5;22)(CSF1R+,WCP22+)
Table 2.
G-band, FISH, and SNP-array results comparison
| Chromosome | G-band | FISH | SNP-array |
|---|---|---|---|
| Y | -Y | NDa | Yp11.31q11.23(2,650,140-28,799,937)x0-1 |
| 3 | der(3)t(3;20)(p11.2;q11.2) | D20S108+ | 3p26.3p11.2(61,891-87,302,938)x1-2 |
| 4 | −4 | ND | 4q13.3q35.2(73,857,745-190,957,473)x1 ~ 2 |
| 5 | t(5;22)(q22;q11.2) | wcp22+;wcp22+,CSF1R+ | |
| 9 | del(9)(p13); dic(9;17)(p13;p11.2) |
CDKN2A-,D9Z1+ D9Z1+,CDKN2A-;D17Z1+,TP53-,WCP17+ |
9p24.3p21.3(203,861-21,608,158)x1 ~ 2, 9p21.3(21,617,251-23,426,271)x1 ~ 2, 9p21.3p13.1(23,436,107-38,787,480)x1 ~ 2, |
| 12 | t(12;21) | RUNX1+;ETV6+,RUNX1+ | |
| 13 | NLb | ND | 13q14.2(47,925,533-48,966,146)x1 ~ 2, 13q14.2(48,984,342-49,177,989)x1 ~ 2, 13q14.2q14.3(49,182,831-51,194,835)x1 ~ 2 |
| 14 | der(14;17)(p11.2;q11.2) | wcp17+ | |
| 17 | der(17)t(17;22) | TP53+,D17Z1+,WCP17+,WCP22+ | 17p13.3q21.32(525-45,059,684)x1 ~ 2 |
| 20 | −20 | disomy for 20q12(D20S108) | 20p13q11.21(61,568-29,497,009)x1 ~ 2 |
| 21 | add(21)(q22) | RUNX1+++++ | 21q11.2q21.1(15,006,457-19,142,414)x2 ~ 3, 21q21.2q21.3 (24,806,259-27,905,650)x2 ~ 3, 21q21.3(28,498,600-28,835,696)x2 ~ 3, 21q21.3(29,805,880-31,211,807)x2 ~ 3, 21q22.11q22.2(31,795,184-41,909,261)x2 ~ 3, 21q22.2q22.3(42,364,149-48,097,372)x2 ~ 3 |
| 22 | -22, t(5;22) | t(5;22)(CSF1R+,WCP22+) |
aND not done
bNL normal
Discussion
We report the case of an 18-year-old with B-ALL associated with iAMP21 and ETV6-RUNX1. The patient had a very poor outcome despite intensified chemotherapy and allogeneic stem cell transplant. We also searched the literature and identified six additional cases of B-ALL with co-existing iAMP21 and the ETV6-RUNX1 [7, 11–14] (Table 3). The median age of these seven patients was 10 years old (range, 2–18) and the median WBC count was 9.1 ×109/L (range, 0.7–34.2 ×109/L). Six of seven (85.7%) cases had karyotypic information with 3 showing an apparently normal karyotype at the diagnosis, likely the result of limited blasts dividing in short-term culture. The remaining 3 cases showed iAMP21 which presented as either the “der(21)” or an “add(21)”; 2 of these cases also had highly complex karyotypes including the current patient. Four of 7 cases had detailed ETV6/RUNX1 FISH data (Table 3). Case 1 showed the ETV6-RUNX1 fusion amplification as a sole finding. Patients 2 and 3 apparently showed the ETV6-RUNX1 fusion as the primary clone and iAMP21 as apparent evidence of a clonal evolution. Interestingly, very similar to the findings as observed in our case (case 7 in Table 3), patient 4 had the ETV6-RUNX1 fusion only with a normal karyotype at the diagnosis, and had the additional iAMP21 in the relapsed B-ALL. These findings further indicate that iAMP21 is likely a secondary event that results in disease progression. OS information is available for 3 of 7 (42.9%) patients; OS was 34, 28 and 24 months in patients 1, 4 and 7, respectively (Table 3). Patient 1 had a better OS, likely attributable to younger age at the diagnosis. The overall poor prognosis observed in these patients suggests that the adverse clinical impact of iAMP21 overrides the presumably better prognosis associated with ETV6-RUNX1 in B-ALL.
Table 3.
Clinicopathologic features of iAMP21 and ETV6-RUNX1 fusion positive B-ALL cases
| Cases | Age(Yr)/Gender | WBC Count (109/L) |
BM Blast % |
Additional Abnormalities | Outcome | Treatment | References |
|---|---|---|---|---|---|---|---|
| 1 | 2/M | 9.1 | NAa | Karyotype: 46,XY. FISH: ETV6-RUNX1 (4–5 copies) fusion in 80% cells | EFSb: 22 months. OSc: 34 months |
ALL-IC-BFM | Case #4 Haltrich, 2013 [12] |
| 2 | 10/M | NA | NA | RUNX1 amplification with ETV6-RUNX1 fusion in 5.5% cells and without ETV6-RUNX1 fusion in 88.5% cells | NA | NA | Case #4 Ma, 2001 [14] |
| 3 | 2/F | 78 | 98 | ETV6-RUNX1 fusion with RUNX1 amplification in 56% cells and without RUNX1 amplification in 23% cells. Karyotype: 46,XX | NA | NA | Case #23 Mikhail, 2002 [13] |
| 4 | 7/M | 34.2 | NA | At diagnosis: ETV6 deletion with ETV6-RUNX1 fusion. Normal Karyotype. At relapse: RUNX1 x 4-5, ETV6 deletion, ETV6-RUNX1 fusion. Karyotype: 46,XY,der(21)add(21)(q22)[25] |
EFS: 28 months | ALL-BFM’95/ALL-REZ-BFM 2002 | Case #1 Haltrich, 2013 [12] |
| 5 | 11/M | 0.7 | NA | Karyotype: 46,XY,add(21)(q22) | NA | NA | Case #528 Harrison, 2014 [7] |
| 6 | 13/M | NA | NA | Karyotype: 44,XY,del(1)(p33),-4,i(9)(q10),-17, t(19;?)(q13.3;?),dup(21)(q?),+1 ~ 2mar[cp16] |
NA | NA | Case #530 Harrison, 2014 [7] |
| 7 | 18/M | 2.0 | 54 | At diagnosis: ETV6-RUNX1 fusion. No Karyotype. At relapse: RUNX1 amplification (5 copies)/ETV6-RUNX1 in 27% cells. Complex karyotype. |
OS: 26 months | Chemo ASCTd |
Current Study |
a NA not available
b EFS event-free survival
c OS overall survival
d ASCT allogeneic stem cell transplantation
In the literature, B-ALL associated with iAMP21 is more frequent than cases of B-ALL with concomitant iAMP21 and ETV6-RUNX1 fusion. Using an arbitrary age range for adolescents, we summarized 22 cases of B-ALL with iAMP21 for comparison. All these 22 patients had a median age of 15 years at time of diagnosis (range, 13–20) (Table 4) [8, 11, 12, 15, 16] and the male-to-female ratio was 1.75. Most patients had a low WBC count with a median of 3.4×9/L (range, 1–15.8). Three (13.6%) patients had RUNX1 amplification with a normal karyotype; five (22.7%) patients showed a deletion of chromosome 7 as an additional abnormality. Clinical follow up data were available in 20 (90.9%) patients showing a median OS of 29.5 months (range, 9–86 months). Comparing B-ALL with iAMP21 versus B-ALL patients with coexistent iAMP21 and ETV6-RUNX1, the iAMP21 only patients had a younger age at disease onset; 9 years old for iAMP21 versus 15 years old for coexistent iAMP21 and ETV6-RUNX1, p = 0.00. Patients with B-ALL and iAMP21 only also had a higher WBC count; 25×109/L for iAMP21 only patients versus 5×109/L for patients with both iAMP21 and ETV6-RUNX1, p = 0.01. However, OS was insignificant between these two groups. Although, the clinical data are limited, we believe that patients with B-ALL associated with iAMP21 and ETV6-RUNX1 can be included in the cytogenetic subgroup of “iAMP21”.
Table 4.
Clinicopathologic features of iAMP21 positive adolescent B-ALL without ETV6-RUNX1 fusion
| Cases | Age(Yr)/Gender | WBC Count (109/L) | Additional Abnormalities | RUNX1 Copies/Cell | Outcome | References |
|---|---|---|---|---|---|---|
| 1 | 15/M | 3.1 | −8,der(16)t(1;16),−21 | 5–6 | CRb1 × 9 months | Johnson, 2015 [11] |
| 2 | 17/M | 2.4 | Normal Karyotype | >5 | Relapsed 58 months after diagnosis following SCT, CR2 × 3 y | Johnson, 2015 [11] |
| 3 | 20/M | 2.2 | −13,−14,+21 | 6–7 | CR1 × 30 months | Johnson, 2015 [11] |
| 4 | 19/M | 5.3 | del(7)(q11.2) | 5–9 | CR alive for 2 years | Knez, 2015 [15] |
| 5 | 13/M | 3.7 | None | 5–10 | Unknown | Knez, 2015 [15] |
| 6 | 15/M | 2.1 | None | 4–8 | Relapsed after 7 years of CR | Knez, 2015 [15] |
| 7 | 13/F | 3.0 | +X | >5 | CR and alive for 3 years | Knez, 2015 [15] |
| 8 | 15/M | 15.8 | del(7)(q31) | 8 | Relapse 2.5 y from diagnosis, CR2 at last chemo block | Haltrich, 2013 [12] |
| 9 | 14/M | 2.2 | None | 5–10 | CR 29 months | Reichard, 2011 [8] |
| 10 | 15/M | 2.0 | Not Done | 6–8 | CR 51 months | Reichard, 2011 [8] |
| 11 | 13/F | 2.8 | None | 5–10 | CR 18 months | Soulier, 2003 [16] |
| 12 | 17/M | 1.0 | add(1)(q25) | 8 | CR 19 months | Soulier, 2003 [16] |
| 13 | 19/F | 10.1 | del(7)(p14p21) | 6–8 | CR 21 months | Soulier, 2003 [16] |
| 14 | 14/M | 2.2 | inv(7)(p?15q?21) | 5–7 | CR 23 months | Soulier, 2003 [16] |
| 15 | 13/F | 3.8 | del(7)(q22q35),del(11)(p12) | 5 | CR 61 months | Soulier, 2003 [16] |
| 16 | 15/F | 9.9 | None | 4 | CR 86 months | Soulier, 2003 [16] |
| 17 | 15/M | 4.3 | -Y | 4–5 | CR 32 months | Soulier, 2003 [16] |
| 18 | 15/F | NAa | add(1)(p?),del(6)(q25) | >4 | CR 13 months | Soulier, 2003 [16] |
| 19 | 13/M | 7.6 | i(9)(q10),−16 | 4 | CR 18 months | Soulier, 2003 [16] |
| 20 | 13/F | 6.6 | add(4)(q31),del(7)(q3?2) | 5 | CR 10 months | Soulier, 2003 [16] |
| 21 | 14/M | 14.5 | Normal Karyotype | 6–15 | CR 48 months | Soulier, 2003 [16] |
| 22 | 15/F | NA | Normal Karyotype | 15–20 | Relapsed | Soulier, 2003 [16] |
a NA not available
b CR complete remission
In addition to the co-existing of ETV6-RUNX1 fusion and iAMP21, our patient also showed TP53 deletion with a concomitant TP53 mutation. TP53 deletion is frequently observed in B-ALL, particularly in those with hypodiploidy or familial Li Fraumeni syndrome or cancer predisposition syndrome [17]. Sequencing methods allow identification and better characterization of TP53 mutation in 90% hypodiploid childhood ALL that is important for prognostic assessment [18, 19]. The concomitant TP53 mutation with deletion could result in “two hits” for TP53 loss of function and could result in poorer prognosis in our patient [20]. In addition, the null-function of TP53 or other tumor suppressor gene, such as the homozygous CDKN2A deletions observed in our patient, can also promote the chromothripsis of 21q at the genomic level [21]. Hetero- or homozygous deletions of CDKN2A are recurrent findings in pediatric ALL. However, they are often considered as secondary events in childhood ALL and increase the likelihood of relapse [22, 23]. In our patient, the CDKN2A homozygous deletions were likely following the ETV6-RUNX1 fusions, to drive disease progression together with the iAMP21.
iAMP21 is also a chromothripsis phenomenal, resulted in the remodeling chromosome 21 in a nonrandom fashion, leading to a stable derivative of chromosome 21 with leukemic potential [6]. Recent studies have provided new insight about the mechanistic events and consequences of chromothripsis [4, 24, 25]. These non-random genomic instabilities of chromosome 21q could be an initial leukemic event [26] in B-ALL pathogenesis, although it was a secondary event in our patient. The additional segmental copy number aberrations involving other parts of the genome, often reflected by complex karyotypes, are likely a secondary event in pathogenesis. High-resolution microarray based testing integrated with traditional chromosome/FISH analysis, particularly the metaphase FISH as performed in our patient, would allow the refinement of the heterogeneous karyotypic findings in iAMP21 B-ALL cases. The clinically critical regions of iAMP21 likely within the 21q22.2-22q22.3 region encoding for 19 to 32 Mb [26–31] in size. These genomic complexities likely contribute to the tumor progression and the poor response to therapy in this subset of the B-ALL patients.
Conclusions
Our results suggest that the coexistence of iAMP21 and ETV6-RUNX1 fusion B-ALL is associated with relatively older age, male predominance, and a very poor prognosis. The presence of ETV6-RUNX1 does not appear to modify the poor prognosis imparted by iAMP21 in B-ALL. Older children with an ETV6-RUNX1 fusion positive B-ALL should be monitored closely for the development of iAMP21, particularly when a relapse of B-ALL is suspected. Patients with B-ALL associated with both iAMP21 and ETV6-RUNX1 fit best in the poor prognosis cytogenetic subgroup of “iAMP21”. Integrated genomic testing including high-resolution microarray and metaphase FISH is needed to refine the extremely complex genomic rearrangements.
Methods
Flow-cytometry immunophenotypic analyses
Eight- color flow cytometric immunophenotypic analysis was performed according to standard procedures. The panel included antibodies directed against: CD3, CD4, CD5, CD7, CD9, CD10, CD13, CD19, CD20, CD22, CD25, CD33, CD34, CD38, CD52, CD79a, CD117, BCL-2, HLA-DR, myeloperoxidase, IgM (cytoplasmic), kappa and lambda light chains (Becton-Dickinson Biosciences, San Jose, CA, USA), TdT (Supertechs Inc, Bethesda, MD, USA).
Cytogenetic and FISH analysis
Twenty-four and/or forty-eight hour unstimulated bone marrow cultures were setup for conventional cytogenetic analysis. Using a Leica-microscope imaging system (Leica Microsystems Inc., Chicago, IL) 20 metaphases were examined and karyotypes were prepared according to International System for Human Cytogenetic Nomenclature (ISCN 2013).
FISH studies were performed on cultured bone marrow metaphases and interphases using the probe sets targeting ETV6/RUNX1, BCR/ABL1(ES), MLL, CDKN2A/CEP9, D5S23/D5S721/EGR1, TP53/CEP17, D20S108 (Abbott Molecular, Inc. Abbott Park, IL); and CSF1R break-apart (5q32), WCP17, WCP22 (Cytocell Ltd, OGT, UK). A G-banded slide was destained in methanol and hybridized with all the FISH probes above subsequently, according to standard lab procedures. FISH images were then captured in Cytovision and 200 cells were scored by two technologists when applicable.
SNP microarrays
SNP microarray study was performed using the Affymetrix CytoScan HD array (Affymetrix, Inc. Santa Clara, CA) which contains 2.5 million markers, including 750,000 SNPs and 1.7 million non-polymorphic probes, with extensive coverage over 18,500 RefSeq genes, known cancer genes and 12,000 OMIM genes. In brief, 250 ng of genomic DNA for each NK cell line were hybridized to a CytoScan HD array according to the manufacturer’s protocols. Array data for copy number alterations (CNAs) and copy-neutral loss of heterozygosity (cnLOH) were analyzed using Affymetrix Chromosome Analysis Suite v.3.1 (ChAS) software and the Nexus copy number 7.5 (BioDiscovery Inc, El Segundo, CA) with a reference framework of NA33 (hg19). Regions of copy number alterations larger than 50 markers/400 kb for gain or 20 markers/100 kb for loss and copy neutral loss of heterozygosity (LOH) larger than 3 Mb are recorded. All CNAs were compared with known public databases of normal genomic variants (DGV).
Molecular study
Nanofluidics-based qualitative multi-parametric reverse-transcriptase polymerase chain reaction (PCR) was performed for the detection of ETV6-RUNX1 fusion transcripts. PCR-based DNA sequencing was performed to assess for mutations in exons 4 to 9 (codons 33 to 331) of TP53.
Acknowledgements
Not applicable.
Funding
No funding was needed for this project.
Availability of data and material
Data available for sharing upon request.
Authors’ contributions
XL designed the study. JG, AR, and XL analyzed and reviewed data. AR collected clinical data and performed SNP array analysis. PL, LF and LJM reviewed the clinical data. KP reviewed the molecular data; CD and KS assisted the literature review. JG, AR, LJM and XL wrote the manuscript and all authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This project has been approved by the institutional review board (IRB) of the University of Texas, MD Anderson Cancer Center. Written informed consent of this report was waived for the publication.
Abbreviations
- ASCT
Allogeneic stem cell transplant
- B-ALL
B cell acute lymphoblastic leukemia
- CNAs
Copy number alterations
- cnLOH
Copy-neutral loss of heterozygosity
- CNS
Central nervous system
- CR
Complete remission
- DGV
Databases of normal genomic variants
- EFS
Event-free survival
- FISH
Fluorescence in situ hybridization
- iAMP21
Intrachromosomal amplification of chromosome 21
- OS
Overall survival
- PCR
Polymerase chain reaction
- SNP array
Single nucleotide polymorphism microarray
- WBC
White blood cell counts
- WHO
World Health Organization
Contributor Information
Jun Gu, Email: jungu@mdanderson.org.
Alexandra Reynolds, Email: AReynolds1@mdanderson.org.
Lianghua Fang, Email: LFang2@mdanderson.org.
Corrie DeGraffenreid, Email: corriedegraffenreid@yahoo.com.
Kenneth Sterns, Email: kcsterns@mdanderson.org.
Keyur P. Patel, Email: kppatel@mdanderson.org
L. Jeffrey Medeiros, Email: ljmedeiros@mdanderson.org.
Xinyan Lu, Email: xinyan.lu@northwestern.edu.
References
- 1.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102. doi: 10.1038/nature13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mardin BR, Drainas AP, Waszak SM, Weischenfeldt J, Isokane M, Stutz AM, et al. A cell-based model system links chromothripsis with hyperploidy. Mol Syst Biol. 2015;11(9):828. doi: 10.15252/msb.20156505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moorman AV, Richards SM, Robinson HM, Strefford JC, Gibson BE, Kinsey SE, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21) Blood. 2007;109(6):2327–30. doi: 10.1182/blood-2006-08-040436. [DOI] [PubMed] [Google Scholar]
- 6.Harrison CJ. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood. 2015;125(9):1383–6. doi: 10.1182/blood-2014-08-569228. [DOI] [PubMed] [Google Scholar]
- 7.Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28(5):1015–21. doi: 10.1038/leu.2013.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichard KK, Kang H, Robinett S. Pediatric B-lymphoblastic leukemia with RUNX1 amplification: clinicopathologic study of eight cases. Mod Pathol. 2011;24(12):1606–11. doi: 10.1038/modpathol.2011.118. [DOI] [PubMed] [Google Scholar]
- 9.Moricke A, Zimmermann M, Reiter A, Gadner H, Odenwald E, Harbott J, et al. Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr. 2005;217(6):310–20. doi: 10.1055/s-2005-872515. [DOI] [PubMed] [Google Scholar]
- 10.Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429–38. doi: 10.1016/S1470-2045(10)70066-8. [DOI] [PubMed] [Google Scholar]
- 11.Johnson RC, Weinberg OK, Cascio MJ, Dahl GV, Mitton BA, Silverman LB, et al. Cytogenetic variation of B-Lymphoblastic leukemia with intrachromosomal amplification of chromosome 21 (iAMP21): a multi-institutional series review. Am J Clin Pathol. 2015;144(1):103–12. doi: 10.1309/AJCPLUYF11HQBYRB. [DOI] [PubMed] [Google Scholar]
- 12.Haltrich I, Csoka M, Kovacs G, Torok D, Alpar D, Ottoffy G, et al. Six cases of rare gene amplifications and multiple copy of fusion gene in childhood acute lymphoblastic leukemia. Pathol Oncol Res. 2013;19(1):123–8. doi: 10.1007/s12253-012-9533-9. [DOI] [PubMed] [Google Scholar]
- 13.Mikhail FM, Serry KA, Hatem N, Mourad ZI, Farawela HM, El Kaffash DM, et al. AML1 gene over-expression in childhood acute lymphoblastic leukemia. Leukemia. 2002;16(4):658–68. doi: 10.1038/sj.leu.2402399. [DOI] [PubMed] [Google Scholar]
- 14.Ma SK, Wan TS, Cheuk AT, Fung LF, Chan GC, Chan SY, et al. Characterization of additional genetic events in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion: a molecular cytogenetics study. Leukemia. 2001;15(9):1442–7. doi: 10.1038/sj.leu.2402202. [DOI] [PubMed] [Google Scholar]
- 15.Knez VM, Carstens BJ, Swisshelm KL, McGranahan AN, Liang X. Heterogeneity of Abnormal RUNX1 Leading to Clinicopathologic Variations in Childhood B-Lymphoblastic Leukemia. Am J Clin Pathol. 2015;144(2):305–14. doi: 10.1309/AJCPVY5E5OMMYBFJ. [DOI] [PubMed] [Google Scholar]
- 16.Soulier J, Trakhtenbrot L, Najfeld V, Lipton JM, Mathew S, Avet-Loiseau H, et al. Amplification of band q22 of chromosome 21, including AML1, in older children with acute lymphoblastic leukemia: an emerging molecular cytogenetic subgroup. Leukemia. 2003;17(8):1679–82. doi: 10.1038/sj.leu.2403000. [DOI] [PubMed] [Google Scholar]
- 17.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muhlbacher V, Zenger M, Schnittger S, Weissmann S, Kunze F, Kohlmann A, et al. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93% Genes Chromosomes Cancer. 2014;53(6):524–36. doi: 10.1002/gcc.22163. [DOI] [PubMed] [Google Scholar]
- 19.Hof J, Krentz S, van Schewick C, Korner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185–93. doi: 10.1200/JCO.2011.34.8144. [DOI] [PubMed] [Google Scholar]
- 20.Hong M, Hao S, Patel KP, Kantarjian HM, Garcia-Manero G, Yin CC, et al. Whole-arm translocation of der(5;17)(p10;q10) with concurrent TP53 mutations in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): A unique molecular-cytogenetic subgroup. Cancer Genet. 2016;209(5):205–14. doi: 10.1016/j.cancergen.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1-2):59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vijayakrishnan J, Henrion M, Moorman AV, Fiege B, Kumar R, da Silva Filho MI, et al. The 9p21.3 risk of childhood acute lymphoblastic leukaemia is explained by a rare high-impact variant in CDKN2A. Sci Rep. 2015;5:15065. doi: 10.1038/srep15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leibowitz ML, Zhang CZ, Pellman D. Chromothripsis: a new mechanism for rapid karyotype evolution. Annu Rev Genet. 2015;49:183–211. doi: 10.1146/annurev-genet-120213-092228. [DOI] [PubMed] [Google Scholar]
- 25.Storchova Z, Kloosterman WP. The genomic characteristics and cellular origin of chromothripsis. Curr Opin Cell Biol. 2016;40:106–13. doi: 10.1016/j.ceb.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Robinson HM, Harrison CJ, Moorman AV, Chudoba I, Strefford JC. Intrachromosomal amplification of chromosome 21 (iAMP21) may arise from a breakage-fusion-bridge cycle. Genes Chromosomes Cancer. 2007;46(4):318–26. doi: 10.1002/gcc.20412. [DOI] [PubMed] [Google Scholar]
- 27.Baughn LB, Biegel JA, South ST, Smolarek TA, Volkert S, Carroll AJ, et al. Integration of cytogenomic data for furthering the characterization of pediatric B-cell acute lymphoblastic leukemia: a multi-institution, multi-platform microarray study. Cancer Genet. 2015;208(1-2):1–18. doi: 10.1016/j.cancergen.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duployez N, Boudry-Labis E, Decool G, Grzych G, Grardel N, Abou Chahla W, et al. Diagnosis of intrachromosomal amplification of chromosome 21 (iAMP21) by molecular cytogenetics in pediatric acute lymphoblastic leukemia. Clin Case Rep. 2015;3(10):814–6. doi: 10.1002/ccr3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117(25):6848–55. doi: 10.1182/blood-2011-01-329961. [DOI] [PubMed] [Google Scholar]
- 30.Sinclair PB, Parker H, An Q, Rand V, Ensor H, Harrison CJ, et al. Analysis of a breakpoint cluster reveals insight into the mechanism of intrachromosomal amplification in a lymphoid malignancy. Hum Mol Genet. 2011;20(13):2591–602. doi: 10.1093/hmg/ddr159. [DOI] [PubMed] [Google Scholar]
- 31.Strefford JC, van Delft FW, Robinson HM, Worley H, Yiannikouris O, Selzer R, et al. Complex genomic alterations and gene expression in acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Proc Natl Acad Sci U S A. 2006;103(21):8167–72. doi: 10.1073/pnas.0602360103. [DOI] [PMC free article] [PubMed] [Google Scholar]