

Abstract
Respiratory viral infections are a leading cause of disease and mortality. The severity of these illnesses can vary markedly from mild or asymptomatic upper airway infections to severe wheezing, bronchiolitis or pneumonia. In this article, we review the viral sensing pathways and organizing principles that govern the innate immune response to infection. Then, we reconstruct the molecular networks that differentiate symptomatic from asymptomatic respiratory viral infections, and identify the underlying molecular drivers of these networks. Finally, we discuss unique aspects of the biology and pathogenesis of infections with respiratory syncytial virus, rhinovirus and influenza, drawing on insights from genomics.
Keywords: Respiratory viral infections, Host response, Genomics, Respiratory syncytial virus, Rhinovirus, Influenza, Gene network, Systems biology
Background
Respiratory infections are the leading cause of disease globally [1]. In young children, they are responsible for around one quarter of hospitalizations and up to 60% of visits to the general practitioner [2]. In most cases, respiratory viral infections are self-limiting and confined to the upper airways, where they evoke relatively mild symptoms such as sneezing and a runny nose [3]. However, in susceptible individuals, such as newborns and the elderly, the symptoms can impact on the lower airways, resulting in wheeze, shortness of breath, bronchiolitis or pneumonia [4]. Common respiratory viral pathogens include adenovirus, enterovirus, human coronavirus, human metapneumovirus, rhinovirus (RV), influenza, parainfluenza and respiratory syncytial virus (RSV). Of these viruses, most genomic studies have focused on RSV, RV and influenza. This is because RSV is the most important cause of respiratory tract infections leading to hospitalization among infants [5]. RV causes the majority of colds and around two thirds of asthma exacerbations [6]. Influenza is the most common cause of pneumonia related deaths in developed countries [7]. The mechanisms that determine why some individuals suffer from severe illness whilst others do not are not well understood. In this review we will focus primarily on the contribution of the host response, drawing on insights from genomics.
Gene expression profiling technologies
The advent of microarrays enabled the measurement of gene expression patterns on a genome-wide scale [8]. Microarrays comprise a vast array of oligonucleotide probes of 25 bases or more in length, fixed to a solid surface. The probes bind to labelled target molecules (e.g. cDNA derived from mRNA), and the signal intensity is quantified. Despite this major technological advance, cross-hybridization of related sequences limited the signal-to-noise ratio [9]. Microarrays are slowly being phased out and superseded by deep sequencing technologies for gene expression profiling (RNA-Seq). RNA-Seq entails the generation of a library of cDNA fragments from total RNA or mRNA, followed by ligation of adapters, PCR amplification and high throughput sequencing [10]. This generates millions of short sequencing reads (e.g. 50–200 bases), which are aligned to a reference genome sequence, and summarized as gene- or transcript-level counts. Compared with microarrays, advantages of RNA-Seq include single base resolution, superior sensitivity and dynamic range, and the ability to detect novel transcripts. The data from microarray and RNA-Seq experiments is typically submitted to a public repository at the time of publication (e.g. Gene Expression Omnibus; http://www.ncbi.nlm.nih.gov/geo/; Sequence Read Archive; www.ncbi.nlm.nih.gov/sra), allowing other researchers to freely access the data for reanalyses [11].
Network analysis of genomic data
Most genomic studies focus on identification of differentially expressed genes. However, this approach is limited, because genes do not exist nor function in isolation, they work together [12]. A significant advance in this context was the application of network graph theory to genomic data analysis [13]. The underlying concept is that a functioning biological system can be represented as a network of interconnected nodes and links. The nodes represent genes or their products (mRNA transcripts, proteins) and the links represent functional or statistical relationships between genes. For instance, the links could represent physical binding interactions between proteins, or coexpression relationships between mRNA transcripts. Gene networks have a non-uniform, so-called “scale-free” topology, where most genes are connected to few genes, and a few genes are connected to many [14]. This results in a limited number of highly connected nodes called hubs, which essentially “hold” the network together [14]. An emergent property of scale-free networks is a tolerance to random perturbations and a susceptibility to the disruption of hubs [13]. Another fundamental organizing principle of gene networks is modularity, meaning that genes which function in the same biological process form densely interconnected subgraphs embedded within the network structure [15]. Modules execute biological tasks, and the breakdown of functional modules is thought to underpin disease states [16].
Respiratory viral pathogens and entry mechanisms
RSV is an enveloped, single strand, negative sense RNA virus of the family Paramyxoviridae. There are two antigenic subtypes (RSV-A, RSV-B). The genome encodes seven structural proteins and four non-structural proteins. The envelope is encoded by three glycoproteins, namely the small hydrophobic protein, the attachment protein G and the fusion protein F. Virus entry is mediated by binding of the F protein to host-cell nucleolin [17].
RV is a non-enveloped, single strand, positive sense RNA virus belonging to the Picornaviridae family. It is contained within an icosahedral capsid, encoded by four viral proteins (VP1-VP4). More than 160 strains have been identified, and these are classified into three species (RV-A, −B, −C) on the basis of genome sequence. RV-B and most RV-A strains bind intercellular adhesion molecule (ICAM1) on the cell surface to gain entry. A subgroup of RV-A strains bind to low-density lipoprotein receptor (LDLR) family members for virus entry [18]. The entry mechanism for RV-C remained elusive until recently. Using microarrays, it was demonstrated that expression of cadherin-related family member 3 (CDHR3) was upregulated in cells that were susceptible to RV-C versus cells that were resistant [19]. Moreover, ectopic expression of CDHR3 in resistant cells conferred susceptibility to infection.
Influenza is an enveloped virus from the Orthomyoxoviridae family. The genome is composed of eight segments of single strand, negative sense RNA. It is classified into three subtypes (A, B, C). Influenza A virus (IAV) is the major subtype that circulates in humans, but it can also infect a diverse range of hosts including mammals and birds. The IAV genome encodes 11 proteins; hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), matrix proteins (M1, M2), non-structural proteins (NS1, NS2), polymerase proteins (PB1, PB2, PA), and PB1-F2 and is further classified based on the HA and NA glycoproteins found on the viral envelope. Glycans containing sialic acid mediate IAV attachment. Seasonal IAV binds to ASα2-6Gal, which are found primarily on the surface of epithelial cells in the nasal mucosa, trachea and bronchi. In contrast, highly pathogenic avian H5N1 virus binds to ASα2-3Gal, which is found on alveolar epithelial cells [20].
Innate immune sensing of viral infections
The airway epithelium is the primary site for viral infection and replication. Viruses must first penetrate the mucus layer, which provides a first line of defence against invading pathogens. When this mechanism fails, the innate immune system is activated. Innate immunity relies on a series of germ-line encoded receptors that are expressed on epithelial and innate immune cells. These pattern recognition receptors (PRR) are sensors for pathogen-derived, evolutionarily conserved molecular structures, known as pathogen- or microbe-associated molecular patterns (PAMP/MAMP). PRR can also detect endogenous molecules called damage-associated molecular patterns (DAMP). DAMPs are normally sequestered, but released from damaged, dying or infected cells to alert the immune system to the presence of “danger”. There are four major classes of PRR; the Toll-like receptors (TLR), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-1)-like receptors (RLR), and C-type lectin receptors (CLR). Most TLRs are expressed at the cell surface (TLR1, −2, −4, −5, −6, 10), where they interact with bacterial components and viral proteins. For instance, TLR2 is a sensor for bacterial peptidoglycans, lipoproteins, RSV and RV capsid [21, 22]. TLR4 is a sensor for bacterial lipopolysaccharide and RSV F protein, and also responds to DAMPs (e.g. oxidized phospholipids, HMBG1, S100A9) during IAV infection. However, these responses are thought to be detrimental to the host as antagonizing TLR4 signalling during IAV infection can protect mice from lethal disease [23–25]. The remaining TLRs (TLR3, −7, −8, −9) are expressed in endosomes, and RLRs (e.g. RIG-I, MDA5) and NLRs are expressed in the cytoplasm. RNA sensors that elicit responses to RSV include RIG-I, TLR3, TLR7 and Nod2 (NLRC2) [26]. MDA5, TLR3, TLR7, NLRX-1 and PKR mediate responses to RV RNA [27–30]. IAV is detected by RIG-I, TLR3 and TLR7 [31]. The major viral sensing molecules are shown in Fig. 1. Host responses to infection can also be triggered by fusion between viral envelopes and the cell membrane, and virus-induced endoplasmic reticulum stress [32–34].
Fig. 1.
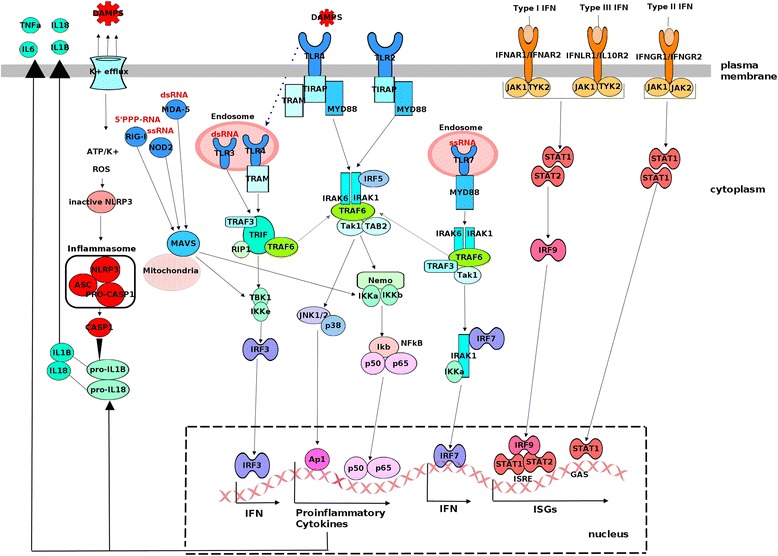
Viral sensing pathways that trigger host immune responses. PRR signalling is triggered by viral proteins and nucleic acids (PAMPs) and host-derived DAMPs. All TLRs except TLR3 signal through the adapter molecule Myd88. TLR3 signals through TRIF, and TLR4 signals via both Myd88- and TRIF-dependant pathways. RIGI, MDA5 and NOD2 signal through the mitochondrial antiviral-signaling protein MAVS (or IPS1). These signalling pathways converge on IRF3/IRF7 and NFkB to activate their respective type I/III interferons and proinflammatory gene programs. Type I (IFN-α, IFN-β) and III interferons (IFN-λ1, IFN-λ2, IFN-λ3 or IL-29, IL-28A, IL-28B) bind to distinct receptor complexes, which activates STAT1 and STAT2 phosphorylation. IRF9 binds to STAT1/STAT2 heterodimers forming the ISGF3 complex, which translocates to the nucleus to induce transcription of ISGs that contain ISRE elements in their promoters. Type II interferon signalling activates STAT1, which translocates to the nucleus to induce transcription of ISGs containing GAS elements in their promoters. Some proinflammatory cytokines (pro-IL-1B, pro-IL-18) are produced in an inactive form. The processing of these cytokines into their bioactive form is mediated by Capase-1, which is activated by the NLRP3 inflammasome pathway
Induction of proinflammatory and antiviral responses
PRR sensing of viruses triggers a series of intracellular signaling cascades that converge on two major families of transcription factors - nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and interferon regulatory factor (IRF) (Fig. 1). These transcription factors translocate to the nucleus where they upregulate their respective proinflammatory and antiviral programs [35, 36]. The major signaling pathways involved are illustrated in Fig. 1.
The interferon system is essential for immunity to most viruses. Innate antiviral responses are primarily mediated by type I (IFN-α, IFN-β) and type III interferons (IFN-λ1, IFN-λ2, IFN-λ3), whereas type II interferon (IFN-γ) promotes macrophage activation and Th1 differentiation [37]. Interferon signaling induces the expression of hundreds of genes known collectively as interferon stimulated genes (ISG, Fig. 1). Almost 1000 genes are upregulated by type I interferon in one or more major immune cell populations, and a core set of 166 genes are upregulated across all cell types [38]. This core set includes archetypal ISGs (e.g. Mx proteins, OAS, PKR, viperin), which induce a robust antiviral state to restrict viral replication and prevent the spread of infection to neighboring cells. Mx proteins trap viral components (e.g. IAV PB2 and nucleocapsid) preventing nuclear import and viral replication. OAS activates RNaseL, which degrades viral and cellular RNA. PKR phosphorylates eukaryotic translation initiation factor eIF2α to shut down the translation of cellular and viral proteins. IFITM proteins block virus-host membrane fusion in the endosome. Although the function of most ISGs is unknown, high throughput functional screens suggest that inhibition of viral translation is a common antiviral strategy [39].
In addition to their classical antiviral function, interferons play diverse roles in infection. For example, interferon signalling drives lymphopenia [40]. Interferons also contribute to tissue injury by upregulating the expression and release of TRAIL, which induces apoptosis in airway epithelial cells [41]. Another emerging function is the promotion of virus-induced sickness behavior and cognitive dysfunction. This is mediated by upregulation of IFNAR1 on brain endothelial and epithelial cells, which in turn produce CXCL10, and this signals via neuronal CXCR3 to inhibit synaptic plasticity [42].
Molecular logic of the innate immune system
The molecular logic underlying innate immune responses to environmental cues is governed by a bow-tie architecture [43]. The bow-tie is an ordered control system, comprised of three crucial elements: (i) input signals or “fan-in”; (ii) the core or “knot”; and (iii) output signals or “fan-out” (Fig. 2). Complex input signals are received by the core, compressed and processed into simpler signalling pathways and converted again to complex functional output signals. Both input and output signals are highly variable, flexible, robust and diverse. If one element fails, another can take over and so the system is resistant to malfunctioning parts. In contrast, the core is specialized, efficient and rigid. This is important, because although the input and output layers are perturbed by genetic variation, which creates a lot of noise and complexity, ultimately a few core pathways are critical for activation of the innate immune response. Indeed, null mutations in core components like IRF7 can result in severe phenotypes [44]. Notably, these mutations are extremely rare, which underscores the importance of this pathway. This principle has been exploited by viruses, which target core molecules to evade the host response. For instance, RSV non-structural proteins suppress the activation and nuclear translocation of IRF3 [45], and inhibit the interaction between RIG-I and MAVS, thus abrogating the production of interferons [46]. IAV NS1 protein also inhibits IRF3 activation and interferon production [47].
Fig. 2.

Innate immunity is governed by a bow-tie architecture. The bow-tie is a multilayered control system, comprising input, core and output signals
Viral challenge studies
Viral challenge studies of adult volunteers with live respiratory viruses provide a powerful model to identify host responses underlying disease pathogenesis in vivo. In these challenge models, around half of the inoculated subjects develop respiratory symptoms and have confirmed viral shedding, whereas the remaining subjects are asymptomatic [3, 48]. These studies have found that symptomatic infections are associated with a heighted immune response in blood [3, 49]. Host response signatures can therefore be leveraged to diagnose viral infections, prior to the onset of peak clinical symptoms, and can also differentiate between RSV, or RV and influenza infections [48, 49].
To characterize the molecular networks underlying symptomatic infections, we downloaded a microarray data set from a viral challenge study in which adult volunteers were inoculated with RSV, RV or IAV [3]. We employed case/control comparisons of symptomatic versus asymptomatic subjects to identify differentially expressed genes for each virus, and we found that the strongest disturbance of the transcriptome was induced by IAV, followed by RV and then RSV. To provide a network-level view of these data, we leveraged experimentally supported molecular interaction data from prior studies to reconstruct the wiring diagram of the underlying gene networks [50]. These networks unveil the innate immune response hubs underlying symptomatic infections (Fig. 3a, c and e). To identify the putative causal pathways that give rise to the observed gene expression patterns, we employing upstream regulator analysis [51]. This analysis suggested that symptomatic responses to RSV were largely driven by IFN-λ, IFN-γ, IFN-α and STAT3 signalling (Fig. 3b). RV responses were also characterized by upregulation of type I, II and III interferon signalling, but lacked a prominent STAT3 signature (Fig. 3d). The hallmark of the IAV response was upregulation of IFN-γ, TNF, IFN-α and IL-1β signalling (Fig. 3f). The proinflammatory component of the IAV response provides a plausible mechanism to explain why this virus elicits a stronger perturbation to the transcriptome.
Fig. 3.

Gene networks and molecular drivers underlying symptomatic host responses to infection. Gene expression patterns were profiled in blood from adult volunteers after experimental infection with RSV, RV or IAV [3]. Differentially expressed were identified between symptomatic versus asymptomatic subjects for each virus, resulting in 934 differentially expressed genes for IAV (adjusted p-value < 0.05), 173 genes for RV (adj p-value < 0.05), and 130 genes for RSV (adj p-value < 0.1). a/c/e; Network wiring diagrams were constructed utilizing experimentally supported findings from prior studies (Ingenuity Systems KnowledgeBase) [50]. Genes coloured red were upregulated and those coloured green were downregulated. b/d/f; Upstream regulator analysis was employed to identify the molecular drivers of the response [51]. The negative log p-value was shaded red to indicate pathway activation, and blue indicates inhibition
Pathogenesis of RSV infections
Most children with RSV develop mild symptoms such as cough, fever, sore throat and runny nose. Up to 40% of infants develop lower airway symptoms (e.g. shortness of breath, wheeze, bronchiolitis) and 0.5-2% of all infected infants are hospitalized [52]. Risk factors for severe disease include premature birth, low birth weight, young age, male gender, chronic lung disease, congenital heart disease, immune deficiency, low socio-economic status and tobacco exposure [52]. The presence of pathogenic bacteria (Streptococcus, Moraxella, Haemophilus) in the nasopharyngeal microbiome increases the risk of the infection spreading from the upper to the lower airways and inducing more severe symptoms [53].
The pathogenesis of RSV infections is thought to be mediated by direct cytopathic effects of the virus on lung epithelial cells in combination with the inflammatory response [54–56]. Airways obstruction results from epithelial damage, including loss of function of motile cilia, epithelial sloughing, apoptosis, mucous hypersecretion, edema of the airway wall, and infiltration of neutrophils and lymphocytes [2]. High viral load correlates with increased disease severity in hospitalized infants, while faster rates of RSV clearance are associated with more rapid disease resolution and shorter hospitalization [57, 58]. In differentiated epithelial cell cultures, RSV induces a relatively mild disturbance to the transcriptome, which is an order of magnitude lower than that of IAV [59]. The RSV response in this system is mainly restricted to an interferon signature, whereas the IAV response comprises type I and III interferons, and a broad range of proinflammatory cytokines and chemokines. However, these models cannot provide insight into the role of innate signalling from inflammatory cells that are recruited to the airways.
A unique and perplexing feature of RSV infections is the disproportionate impact on infants. At birth, the immune and respiratory systems are functionally immature, and susceptibility to lower respiratory illnesses can be explained in part by diminished lung function [60, 61]. On the other hand, the neonatal innate immune system has a reduced capacity to produce cytokines (IL-12, IFN-α, TNF) that promote Th1 responses against intracellular pathogens, but has a similar or augmented capacity to produce cytokines (IL-1b, IL-6, IL-8, IL-10, IL-23) that promote Th2 and Th17 responses [62, 63]. This defect in Th1 function is also apparent within the CD4 T cell compartment [60], and is thought to result in Th2- and Th17-skewed responses to infection [64–66]. Recent evidence from parallel studies in neonatal animal models and in hospitalized infants suggests that RSV-induced IL-33 responses are exaggerated in early life, which in turn activates group 2 innate lymphoid cells (ILC2) that drive Th2-associated pathology (eosinophilic inflammation, mucus hypersecretion, airway hyperresponsiveness) [67]. Th1/Th2 imbalance during RSV bronchiolitis is modified by interactions between TLR4 genotype and environmental LPS exposure levels, thus explaining in part why some individuals are predisposed to severe disease [68].
Gene network patterns in blood have been investigated in a cohort of hospitalized infants with RSV, RV or IAV [69]. RSV response networks were characterized by upregulation of interferon signalling, innate immunity and neutrophil/myeloid signatures, and downregulation of lymphocyte signatures (T cells, B cells, NK cells). Notably, interferon responses continued to rise at one month after hospitalization, whereas expression of the other networks faded. Compared with RV or IAV, RSV was associated with overexpression of neutrophil genes and marked suppression of lymphocyte genes. Younger infants (<6 months) with RSV had impaired innate immune and inflammatory responses compared with older infants. The severity of RSV illness was correlated with heightened expression of networks related to neutrophils, inflammation and erythrocytes, whereas innate immune and interferon responses were similar in moderate and severe infants. Finally, length of hospitalization and total duration of supplemental oxygen was strongly correlated with the extent at which the transcriptome was perturbed in infants with RSV illness relative to healthy baseline controls. These findings implicate excessive activation of neutrophil and inflammatory responses in the pathogenesis of severe RSV disease.
Pathogenesis of RV infection
RV is the most important trigger of asthma exacerbations, however, it is frequently detected in children with asthma in the absence of significant symptoms [4]. This suggests that specific environmental conditions are necessary for RV to provoke exacerbations [70]. Natural allergen exposure in sensitized subjects markedly increases risk for experiencing a severe exacerbation with RV [71]. Pathogenic bacteria can also increase the severity of RV infections [72]. Another contributing factor is the virus itself. RV-C is the most common virus found in children presenting to hospital with acute asthma [73]. RV-C-induced exacerbations are more severe, and associated with an increased risk of recurrence, especially in atopics [73, 74]. A variant in CDHR3 (Cys529 → Tyr, rs6967330) that enhances RV-C binding and replication by 10-fold is associated with recurrent severe exacerbations [19, 75]. RV-B species on the other hand have reduced replication rates, elicit lower levels of cytokine production, and induce less severe respiratory symptoms compared with RV-A and RV-C [76, 77]. Antibody responses to RV-C are much lower than RV-A or RV-B, providing further evidence that RV species elicit differential immune responses [78].
There is evidence suggesting that genetic variation modulates host responses to RV and the expression of respiratory symptoms [79, 80]. Variants in the 17q21 locus are associated with RV-induced wheezing but not RSV-induced wheezing [80]. Although the underlying mechanisms are unknown, expression levels of only two genes (ORMDL3, GSDMB) in this region are induced by RV and are correlated with the 17q variants [80]. Transgenic overexpression of human ORMDL3 in mice results in increased levels of airways remodelling, a hallmark of asthma [81]. Of note, a unique aspect of the airway epithelial cell response to RV is the induction of a broad range of genes involved in epithelial repair and remodelling [82, 83]. In vitro studies have shown that the capacity of the airway epithelium to repair is impaired in subjects with asthma, and this capacity diminishes even further in the presence of RV [84]. These data highlight a potential role for dysregulated repair and remodelling in the pathogenesis of RV infections.
Numerous studies have investigated the role of innate immunity in asthma. Ex vivo studies in airway epithelial cells found that RV-induced IFN-β and IFN-λ responses were impaired in subjects with asthma, leading to increased viral replication and shedding and exaggerated secondary responses [85, 86]. However, this finding was replicated by some groups but not others (reviewed [87]). A simple explanation for these inconsistent findings is that asthma is a highly heterogeneous disease that can be divided into Th2High versus Th2Low subgroups [88]. Given that type I and III interferon and Th2 responses are mutually antagonistic, variations in airway epithelial cell responses to RV ex vivo may reflect the nature of the inflammatory microenvironment that was present during specimen collection rather than a defect in the capacity to respond to viruses [89–93]. Indeed, genomic studies in peripheral blood mononuclear cells (PBMC) have found that asthma exacerbations are heterogeneous conditions and comprise at least three molecular subphenotypes [94]. The first phenotype was characterized by activation of innate immunity genes. The second was enriched with antigen driven pathways of adaptive immunity and the third was not associated with any dominant biological pathways. An alternative explanation for impaired interferon responses in subjects with asthma is decreased expression of pathways that promote these responses (e.g. TLR7) or increased expression of pathways that inhibit these responses (e.g. SOCS1) [29, 95].
We have characterized the inflammatory mechanisms that underpin virus-induced asthma exacerbations in vivo. We found that interferon gene networks were upregulated in the upper (nasal wash) and lower (sputum) airways of children during acute asthma exacerbations compared to 7–14 days later [50, 96]. However, expression of Th1 and type I interferon gene networks was reduced in those asthmatic children with evidence of chronic airflow limitation, suggesting a protective role for interferons [96]. Other groups have reported that expression of interferons is increased and correlated with expression of respiratory symptoms during naturally acquired RV-induced exacerbations [97, 98]. Taken together, these studies highlight the potential for interferons to play a dual role in asthma, in both the regulation of antiviral immunity and expression of respiratory symptoms [99]. Variable expression of interferon production in asthma may also be related to viral susceptibility or loads, or variations in the timing from infection onset to peak symptom expression.
Genomic studies from our group found that childhood asthma exacerbation responses in PBMC were associated with upregulation of innate immune and Th2-associated signatures, suggesting that these two pathways interact to drive disease pathogenesis [100]. The mechanisms that link innate antiviral responses with Th2-associated effector mechanisms are not well understood, but recent studies suggest a role for IL-25 and IL-33. Expression of both of these factors is increased in the airways of asthmatic subjects after experimental RV infection in vivo [101, 102]. In allergic mice, blockade of IL-25 signalling markedly suppressed RV-induced production of mucus and airways inflammation [101]. Moreover, supernatants from RV-infected bronchial epithelial cells stimulate Th2 cytokine production in Th2 and ILC2 cells in an IL-33 dependant manner [102]. The mechanisms that regulate the production of IL-25 and IL-33 during RV infections are not well understood. Employing siRNA-mediated gene silencing, we found that IRF7 promotes innate antiviral responses to RV in airway epithelial cells, and limits IL-33 responses [103]. These data are consistent with the mutually antagonistic relationship between the interferon and Th2 response system described above.
Pathogenesis of IAV infections
IAV is ranked amongst the top 10 causes of death in the USA [7]. Mortality rates can be even higher during pandemics such as the 1918 “Spanish Flu”, which resulted in an estimated 50 million deaths [104]. Symptoms of influenza include fever, headache, sore throat, sneezing, nausea, body aches and fatigue. Risk factors for severe disease include old age, very young age, pregnancy, immune deficiency and chronic diseases like asthma, COPD and obesity [105]. The pathogenesis of IAV infections is thought to be determined by cell tropism for human airway and alveolar epithelial cells, viral replication rate and the intensity and dynamics of the inflammatory response [105]. Large swathes of epithelium are denuded or damaged by IAV infection, resulting in exudation of fluid into the airways and alveolar spaces, and in severe cases this can lead to hypoxaemia and respiratory failure [105]. A robust repair response is crucial to restore the epithelial barrier and lung microarchitecture following viral clearance [106]. However, the mechanisms that act to resolve inflammation and restore tissue homeostasis also impair immunity, and this increases susceptibility to secondary bacterial pneumonia, especially during pandemics [107].
Host responses to IAV infection in the lung are highly dynamic and comprise multiple sequential waves of gene expression, and similar findings have been reported in blood [108, 109]. The first wave peaks around 2–5 days post infection, and is characterized by upregulation of innate immune networks, interferon and NK cell responses, and proinflammatory cytokines and chemokines. The next wave spikes on day 8 post infection, and this is defined by upregulation of T cell activation and induction of apoptosis. This is followed by upregulation of B cell activation and proliferation, which is maximal around day 14. On day 30, differentiation and tissue repair processes are upregulated, and these responses persist out to 60 days post infection, suggesting long term or permanent alterations to the lung [108]. Notably, restoration of epithelial barrier integrity and lung function after IAV infection depends on an IL-33-ILC2-amphiregulin axis [110]. This contrasts with the known pathogenic role of IL-33/ILC2 in the context of RSV and RV illness.
The biology of asymptomatic IAV infections is poorly understood. Viral challenge studies are particularly informative in this context, because only a subset of the participants develops symptoms and has evidence of viral shedding [3, 111]. Of the remaining asymptomatic subjects, a subset of them has detectable viral shedding, albeit levels are lower and delayed in onset [111]. The temporal dynamics of the host response to IAV in blood is strikingly different in symptomatic versus asymptomatic subjects [111]. Expression of clinical symptoms is strongly associated with upregulation of viral sensing pathways and interferon-stimulated genes, as well as neutrophil activation, proinflammatory responses and inflammasome signaling (NOD2, NALP3, CASP5, IL-1b) [111]. In contrast, a hallmark of the asymptomatic response was upregulation of pathways that restrain cytokine signaling (SOCS2, SOCS5) and oxidative stress (SOD1, STK25), together with suppression of inflammasome signaling pathways. Of note, myeloid derived suppressor cells (MDSC) accumulate in the lung during IAV infections, and may contribute to the immunosuppressive environment via production of IL-10 [112, 113]. These findings suggest that a unique host response is mobilized during asymptomatic infections, and this could potentially be leveraged to develop novel therapeutics.
Genomic studies of host responses to highly pathogenic avian H5N1 virus and the 1918 (H1N1) pandemic virus have provided unique insights into disease pathogenesis. The 1918 virus was recreated from the genomic sequence using reverse genetics [114]. In mice and cynomolgus macaques, 1918 infection results in a lethal disease phenotype, which is characterized by high viral titers, severe lung pathology and marked and sustained activation of interferon-stimulated genes and pro-inflammatory cytokines and chemokines until death [115, 116]. Notably, expression of multiple type I interferon subtypes was suppressed despite high levels of ISGs, suggesting that the 1918 virus elicits an aberrant interferon response, and/or has reduced sensitivity to the interferon system. Highly pathogenic avian H5N1 infections are also characterized by severe lung pathology together with early and sustained activation of proinflammatory and interferon responses [117, 118]. One interpretation of these data is that although a robust innate immune response becomes activated, it is not able to control the virus, resulting in death due to direct viral damage to the airways. An alternative view is that an unconstrained “cytokine storm” drives lethal disease [119]. With regards to the former hypothesis, the virulence of the 1918 viral proteins has been demonstrated by introducing them into less pathogenic strains. These experiments found that the 1918 HA protein confers high viral loads and aberrant host responses resulting in mortality, whereas the polymerase and NP proteins confers high viral loads, but does not induce lethal disease [120]. The NS1 protein blocks interferon signaling and lipid metabolism, the PB2 protein enhances inflammation and suppresses pathways involved in lung repair [121, 122] and a mutation in the 1918 NP protein confers resistance to Mx1 [123]. These data shed light on the exceptional virulence of the 1918 virus. With regards to the cytokine storm hypothesis, attempts to curtail the 1918-induced host response using gene deficient mice can prolong (Nos2−/−, TNFR−/−) or shorten (IL1R1−/−, IFNAR1−/−) survival, but does not prevent mortality [118, 124, 125]. In contrast, therapy with the ROS scavenger EUK-207 increased survival in mice with 1918 [126].
It is important to acknowledge that the findings from gene deficient mice are oversimplified, because immune mechanisms that are important in viral control may also damage the host, and this dual role of the immune system may be dependent on the timing and magnitude of gene expression. In this context it is noteworthy that lethal influenza in mice is associated with excessive activation of neutrophils. Increased survival in this model was achieved by attenuating rather than ablating the neutrophil response, without compromising viral clearance [127].
Conclusions
The pathogenesis of respiratory viral infections involves the complex interplay between viral virulence factors, environmental conditions, the magnitude and temporal dynamics of the host response, and host susceptibility factors. Symptomatic infections are associated with increased viral shedding and a heightened host immune response. The disproportionate impact of respiratory viral infections in early life and in individuals with chronic respiratory diseases like asthma can be explained by an aberrant host response. It is also apparent that the immune response whilst essential for viral control also promotes the expression of respiratory symptoms and causes collateral damage to the tissues, which in some cases can lead to mortality. This dual role of the immune system can be difficult to dissect using conventional knockout mouse models, thus more systems-levels analyses of multi-omic data sets will be essential to unlock the underlying mechanisms.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
NT and AB drafted and edited the manuscript. NT and AB approved the final version of the manuscript.
Competing interests
The authors declare that that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Contributor Information
Niamh M. Troy, Email: niamh.troy@telethonkids.org.au
Anthony Bosco, Email: anthony.bosco@telethonkids.org.au.
References
- 1.Mizgerd JP. Lung infection--a public health priority. PLoS Med. 2006;3:e76. doi: 10.1371/journal.pmed.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tregoning JS, Schwarze J. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev. 2010;23:74–98. doi: 10.1128/CMR.00032-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaas AK, Chen M, Varkey J, Veldman T, Hero AO, 3rd, Lucas J, Huang Y, Turner R, Gilbert A, Lambkin-Williams R, et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009;6:207–217. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olenec JP, Kim WK, Lee WM, Vang F, Pappas TE, Salazar LE, Evans MD, Bork J, Roberg K, Lemanske RF, Jr, Gern JE. Weekly monitoring of children with asthma for infections and illness during common cold seasons. J Allergy Clin Immunol. 2010;125:1001–1006. doi: 10.1016/j.jaci.2010.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kusel MM, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: a birth cohort study. Pediatr Infect Dis J. 2006;25:680–686. doi: 10.1097/01.inf.0000226912.88900.a3. [DOI] [PubMed] [Google Scholar]
- 6.Gern JE. How rhinovirus infections cause exacerbations of asthma. Clin Exp Allergy. 2015;45:32–42. doi: 10.1111/cea.12428. [DOI] [PubMed] [Google Scholar]
- 7.Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol. 2016;33(4):471–482. doi: 10.1007/s00281-016-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proud D, Turner RB, Winther B, Wiehler S, Tiesman JP, Reichling TD, Juhlin KD, Fulmer AW, Ho BY, Walanski AA, et al. Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am J Respir Crit Care Med. 2008;178:962–968. doi: 10.1164/rccm.200805-670OC. [DOI] [PubMed] [Google Scholar]
- 9.Piccolo SR, Sun Y, Campbell JD, Lenburg ME, Bild AH, Johnson WE. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics. 2012;100:337–344. doi: 10.1016/j.ygeno.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouillard AD, Wang Z, Ma'ayan A. Reprint of “Abstraction for data integration: Fusing mammalian molecular, cellular and phenotype big datasets for better knowledge extraction”. Comput Biol Chem. 2015;59:123–138. doi: 10.1016/j.compbiolchem.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 12.de la Fuente A. From ‘differential expression’ to ‘differential networking’ - identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26:326–333. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 14.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 15.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med. 2011;17:1132–1135. doi: 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 18.Bochkov YA, Gern JE. Rhinoviruses and their receptors: implications for allergic disease. Curr Allergy Asthma Rep. 2016;16:30. doi: 10.1007/s11882-016-0608-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, Palmenberg AC, Gern JE. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A. 2015;112(17):5485–5490. doi: 10.1073/pnas.1421178112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 21.Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol. 2009;83:1492–1500. doi: 10.1128/JVI.00671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Triantafilou K, Vakakis E, Richer EA, Evans GL, Villiers JP, Triantafilou M. Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence. 2011;2:22–29. doi: 10.4161/viru.2.1.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirey KA, Lai W, Patel MC, Pletneva LM, Pang C, Kurt-Jones E, Lipsky M, Roger T, Calandra T, Tracey KJ, et al. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 2016;9(5):1173–1182. doi: 10.1038/mi.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, Tardif MR, Cesaro A, Bose S. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 2014;10:e1003848. doi: 10.1371/journal.ppat.1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TH, Lee HK. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. 2014;47:184–191. doi: 10.5483/BMBRep.2014.47.4.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards MR, Hewson CA, Laza-Stanca V, Lau HT, Mukaida N, Hershenson MB, Johnston SL. Protein kinase R, IkappaB kinase-beta and NF-kappaB are required for human rhinovirus induced pro-inflammatory cytokine production in bronchial epithelial cells. Mol Immunol. 2007;44:1587–1597. doi: 10.1016/j.molimm.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Miller DJ, Bowman ER, Nagarkar DR, Schneider D, Zhao Y, Linn MJ, Goldsmith AM, Bentley JK, Sajjan US, Hershenson MB. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011;7:e1002070. doi: 10.1371/journal.ppat.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatchwell L, Collison A, Girkin J, Parsons K, Li J, Zhang J, Phipps S, Knight D, Bartlett NW, Johnston SL, et al. Toll-like receptor 7 governs interferon and inflammatory responses to rhinovirus and is suppressed by IL-5-induced lung eosinophilia. Thorax. 2015;70:854–861. doi: 10.1136/thoraxjnl-2014-205465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, Zhao Y, McHenry CL, Burgens RV, Miller DJ, et al. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J Immunol. 2009;183:6989–6997. doi: 10.4049/jimmunol.0901386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holm CK, Rahbek SH, Gad HH, Bak RO, Jakobsen MR, Jiang Z, Hansen AL, Jensen SK, Sun C, Thomsen MK, et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun. 2016;7:10680. doi: 10.1038/ncomms10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hrincius ER, Liedmann S, Finkelstein D, Vogel P, Gansebom S, Samarasinghe AE, You D, Cormier SA, McCullers JA. Acute lung injury results from innate sensing of viruses by an ER stress pathway. Cell Rep. 2015;11:1591–1603. doi: 10.1016/j.celrep.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147:853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dussurget O, Bierne H, Cossart P. The bacterial pathogen Listeria monocytogenes and the interferon family: type I, type II and type III interferons. Front Cell Infect Microbiol. 2014;4:50. doi: 10.3389/fcimb.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, et al. Parsing the interferon transcriptional network and its disease associations. Cell. 2016;164:564–578. doi: 10.1016/j.cell.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- 41.Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, et al. Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 2013;9:e1003188. doi: 10.1371/journal.ppat.1003188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blank T, Detje CN, Spiess A, Hagemeyer N, Brendecke SM, Wolfart J, Staszewski O, Zoller T, Papageorgiou I, Schneider J, et al. Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity. 2016;44:901–912. doi: 10.1016/j.immuni.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Oda K, Kitano H. A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol. 2006;2:2006.0015. doi: 10.1038/msb4100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, Lafaille FG, Trouillet C, Schmolke M, Albrecht RA, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348:448–453. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spann KM, Tran KC, Collins PL. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol. 2005;79:5353–5362. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyapalle S, Wong T, Garay J, Teng M, San Juan-Vergara H, Mohapatra S, Mohapatra S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS One. 2012;7:e29386. doi: 10.1371/journal.pone.0029386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol. 2000;74:7989–7996. doi: 10.1128/JVI.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, Veldman T, Kingsmore SF, Huang Y, Lambkin-Williams R, et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS One. 2013;8:e52198. doi: 10.1371/journal.pone.0052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andres-Terre M, McGuire HM, Pouliot Y, Bongen E, Sweeney TE, Tato CM, Khatri P. Integrated, multi-cohort analysis identifies conserved transcriptional signatures across multiple respiratory viruses. Immunity. 2015;43:1199–1211. doi: 10.1016/j.immuni.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bosco A, Ehteshami S, Panyala S, Martinez FD. Interferon regulatory factor 7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J Allergy Clin Immunol. 2012;129:88–94. doi: 10.1016/j.jaci.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Troy NM, Hollams EM, Holt PG, Bosco A. Differential gene network analysis for the identification of asthma-associated therapeutic targets in allergen-specific T-helper memory responses. BMC Med Genomics. 2016;9:9. doi: 10.1186/s12920-016-0171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McNamara PS, Smyth RL. The pathogenesis of respiratory syncytial virus disease in childhood. Br Med Bull. 2002;61:13–28. doi: 10.1093/bmb/61.1.13. [DOI] [PubMed] [Google Scholar]
- 53.Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt BJ, Hales BJ, Walker ML, Hollams E, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17:704–715. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang P, Zheng J, Wang S, Liu P, Xie M, Zhao D. Respiratory syncytial virus nonstructural proteins 1 and 2 are crucial pathogenic factors that modulate interferon signaling and Treg cell distribution in mice. Virology. 2015;485:223–232. doi: 10.1016/j.virol.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 55.Fonceca AM, Flanagan BF, Trinick R, Smyth RL, McNamara PS. Primary airway epithelial cultures from children are highly permissive to respiratory syncytial virus infection. Thorax. 2012;67:42–48. doi: 10.1136/thoraxjnl-2011-200131. [DOI] [PubMed] [Google Scholar]
- 56.Liesman RM, Buchholz UJ, Luongo CL, Yang L, Proia AD, DeVincenzo JP, Collins PL, Pickles RJ. RSV-encoded NS2 promotes epithelial cell shedding and distal airway obstruction. J Clin Invest. 2014;124:2219–2233. doi: 10.1172/JCI72948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeVincenzo JP, El Saleeby CM, Bush AJ. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J Infect Dis. 2005;191:1861–1868. doi: 10.1086/430008. [DOI] [PubMed] [Google Scholar]
- 58.El Saleeby CM, Bush AJ, Harrison LM, Aitken JA, Devincenzo JP. Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J Infect Dis. 2011;204:996–1002. doi: 10.1093/infdis/jir494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, Flano E. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol. 2012;86:5422–5436. doi: 10.1128/JVI.06757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holt PG, Upham JW, Sly PD. Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J Allergy Clin Immunol. 2005;116:16–24. doi: 10.1016/j.jaci.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 61.Martinez FD, Morgan WJ, Wright AL, Holberg CJ, Taussig LM. Diminished lung function as a predisposing factor for wheezing respiratory illness in infants. N Engl J Med. 1988;319:1112–1117. doi: 10.1056/NEJM198810273191702. [DOI] [PubMed] [Google Scholar]
- 62.Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, Lavoie PM, Furlong J, Fortuno ES, 3rd, Hajjar AM, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol. 2009;183:7150–7160. doi: 10.4049/jimmunol.0901481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol. 2007;7:379–390. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- 64.Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. 2003;168:633–639. doi: 10.1164/rccm.200210-1148OC. [DOI] [PubMed] [Google Scholar]
- 65.Kristjansson S, Bjarnarson SP, Wennergren G, Palsdottir AH, Arnadottir T, Haraldsson A, Jonsdottir I. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. J Allergy Clin Immunol. 2005;116:805–811. doi: 10.1016/j.jaci.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 66.Stoppelenburg AJ, de Roock S, Hennus MP, Bont L, Boes M. Elevated Th17 response in infants undergoing respiratory viral infection. Am J Pathol. 2014;184:1274–1279. doi: 10.1016/j.ajpath.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 67.Saravia J, You D, Shrestha B, Jaligama S, Siefker D, Lee GI, Harding JN, Jones TL, Rovnaghi C, Bagga B, et al. Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog. 2015;11:e1005217. doi: 10.1371/journal.ppat.1005217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caballero MT, Serra ME, Acosta PL, Marzec J, Gibbons L, Salim M, Rodriguez A, Reynaldi A, Garcia A, Bado D, et al. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J Clin Invest. 2015;125:571–582. doi: 10.1172/JCI75183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mejias A, Dimo B, Suarez NM, Garcia C, Suarez-Arrabal MC, Jartti T, Blankenship D, Jordan-Villegas A, Ardura MI, Xu Z, et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013;10:e1001549. doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martinez FD. Managing childhood asthma: challenge of preventing exacerbations. Pediatrics. 2009;123(Suppl 3):S146–S150. doi: 10.1542/peds.2008-2233D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, Custovic A. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61:376–382. doi: 10.1136/thx.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kloepfer KM, Lee WM, Pappas TE, Kang TJ, Vrtis RF, Evans MD, Gangnon RE, Bochkov YA, Jackson DJ, Lemanske RF, Jr, Gern JE. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J Allergy Clin Immunol. 2014;133:1301–1307. doi: 10.1016/j.jaci.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, Martin AC, Khoo SK, Cox DW, Geelhoed GC, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J. 2011;37:1037–1042. doi: 10.1183/09031936.00092410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cox DW, Bizzintino J, Ferrari G, Khoo SK, Zhang G, Whelan S, Lee WM, Bochkov YA, Geelhoed GC, Goldblatt J, et al. Human rhinovirus species C infection in young children with acute wheeze is associated with increased acute respiratory hospital admissions. Am J Respir Crit Care Med. 2013;188:1358–1364. doi: 10.1164/rccm.201303-0498OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, den Dekker HT, Husby A, Sevelsted A, Faura-Tellez G, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46:51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 76.Lee WM, Lemanske RF, Jr, Evans MD, Vang F, Pappas T, Gangnon R, Jackson DJ, Gern JE. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med. 2012;186:886–891. doi: 10.1164/rccm.201202-0330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakagome K, Bochkov YA, Ashraf S, Brockman-Schneider RA, Evans MD, Pasic TR, Gern JE. Effects of rhinovirus species on viral replication and cytokine production. J Allergy Clin Immunol. 2014;134:332–341. doi: 10.1016/j.jaci.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iwasaki J, Smith WA, Khoo SK, Bizzintino J, Zhang G, Cox DW, Laing IA, Le Souef PN, Thomas WR, Hales BJ. Comparison of rhinovirus antibody titers in children with asthma exacerbations and species-specific rhinovirus infection. J Allergy Clin Immunol. 2014;134:25–32. doi: 10.1016/j.jaci.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 79.Caliskan M, Baker SW, Gilad Y, Ober C. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet. 2015;11:e1005111. doi: 10.1371/journal.pgen.1005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caliskan M, Bochkov YA, Kreiner-Moller E, Bonnelykke K, Stein MM, Du G, Bisgaard H, Jackson DJ, Gern JE, Lemanske RF, Jr, et al. Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N Engl J Med. 2013;368:1398–1407. doi: 10.1056/NEJMoa1211592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, Doherty TA, McGeough MD, Pena CA, Suzukawa M, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. 2014;192:3475–3487. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim TK, Bheda-Malge A, Lin Y, Sreekrishna K, Adams R, Robinson MK, Bascom CC, Tiesman JP, Isfort RJ, Gelinas R. A systems approach to understanding human rhinovirus and influenza virus infection. Virology. 2015;486:146–157. doi: 10.1016/j.virol.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kicic A, Stevens PT, Sutanto EN, Kicic-Starcevich E, Ling KM, Looi K, Martinovich KM, Garratt LW, Iosifidis T, Shaw NC, et al. Impaired airway epithelial cell responses from children with asthma to rhinoviral infection. Clin Exp Allergy. 2016;46(11):1441–1455. doi: 10.1111/cea.12767. [DOI] [PubMed] [Google Scholar]
- 85.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, Holgate ST, Davies DE. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 87.Ritchie AI, Jackson DJ, Edwards MR, Johnston SL. Airway epithelial orchestration of innate immune function in response to virus infection. A focus on asthma. Ann Am Thorac Soc. 2016;13(Suppl 1):S55–S63. doi: 10.1513/AnnalsATS.201507-421MG. [DOI] [PubMed] [Google Scholar]
- 88.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baraldo S, Contoli M, Bazzan E, Turato G, Padovani A, Marku B, Calabrese F, Caramori G, Ballarin A, Snijders D, et al. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J Allergy Clin Immunol. 2012;130:1307–1314. doi: 10.1016/j.jaci.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 90.Patel DA, You Y, Huang G, Byers DE, Kim HJ, Agapov E, Moore ML, Peebles RS, Jr, Castro M, Sumino K, et al. Interferon response and respiratory virus control are preserved in bronchial epithelial cells in asthma. J Allergy Clin Immunol. 2014;134:1402–1412. doi: 10.1016/j.jaci.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Contoli M, Ito K, Padovani A, Poletti D, Marku B, Edwards MR, Stanciu LA, Gnesini G, Pastore A, Spanevello A, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. 2015;70:910–920. doi: 10.1111/all.12627. [DOI] [PubMed] [Google Scholar]
- 92.Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, Eva MM, Gauchat JF, Qureshi ST, Mazer BD, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2015;17(1):65–75. doi: 10.1038/ni.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pritchard AL, Carroll ML, Burel JG, White OJ, Phipps S, Upham JW. Innate IFNs and plasmacytoid dendritic cells constrain Th2 cytokine responses to rhinovirus: a regulatory mechanism with relevance to asthma. J Immunol. 2012;188:5898–5905. doi: 10.4049/jimmunol.1103507. [DOI] [PubMed] [Google Scholar]
- 94.Bjornsdottir US, Holgate ST, Reddy PS, Hill AA, McKee CM, Csimma CI, Weaver AA, Legault HM, Small CG, Ramsey RC, et al. Pathways activated during human asthma exacerbation as revealed by gene expression patterns in blood. PLoS One. 2011;6:e21902. doi: 10.1371/journal.pone.0021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gielen V, Sykes A, Zhu J, Chan B, Macintyre J, Regamey N, Kieninger E, Gupta A, Shoemark A, Bossley C, et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol. 2015;136:177–188. doi: 10.1016/j.jaci.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bosco A, Ehteshami S, Stern DA, Martinez FD. Decreased activation of inflammatory networks during acute asthma exacerbations is associated with chronic airflow obstruction. Mucosal Immunol. 2010;3:399–409. doi: 10.1038/mi.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miller EK, Hernandez JZ, Wimmenauer V, Shepherd BE, Hijano D, Libster R, Serra ME, Bhat N, Batalle JP, Mohamed Y, et al. A mechanistic role for type III IFN-lambda1 in asthma exacerbations mediated by human rhinoviruses. Am J Respir Crit Care Med. 2012;185:508–516. doi: 10.1164/rccm.201108-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schwantes EA, Manthei DM, Denlinger LC, Evans MD, Gern JE, Jarjour NN, Mathur SK. Interferon gene expression in sputum cells correlates with the asthma index score during virus-induced exacerbations. Clin Exp Allergy. 2014;44(6):813–821. doi: 10.1111/cea.12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coleman L, Laing IA, Bosco A. Rhinovirus-induced asthma exacerbations and risk populations. Curr Opin Allergy Clin Immunol. 2016;16(2):179–185. doi: 10.1097/ACI.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 100.Subrata LS, Bizzintino J, Mamessier E, Bosco A, McKenna KL, Wikstrom ME, Goldblatt J, Sly PD, Hales BJ, Thomas WR, et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J Immunol. 2009;183:2793–2800. doi: 10.4049/jimmunol.0900695. [DOI] [PubMed] [Google Scholar]
- 101.Beale J, Jayaraman A, Jackson DJ, Macintyre JD, Edwards MR, Walton RP, Zhu J, Ching YM, Shamji B, Edwards M, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. 2014;6:256ra134. doi: 10.1126/scitranslmed.3009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, Jerico D-R, Telcian AG, Nikonova A, Zhu J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in Vivo. Am J Respir Crit Care Med. 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bosco A, Wiehler S, Proud D. Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genomics. 2016;17:76. doi: 10.1186/s12864-016-2405-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taubenberger JK, Morens DM. Influenza: the mother of all pandemics. Emerg Infect Dis. 1918;2006(12):15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J. 2015;45:1463–1478. doi: 10.1183/09031936.00186214. [DOI] [PubMed] [Google Scholar]
- 106.Gorski SA, Hufford MM, Braciale TJ. Recent insights into pulmonary repair following virus-induced inflammation of the respiratory tract. Curr Opin Virol. 2012;2:233–241. doi: 10.1016/j.coviro.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van der Sluijs KF, van der Poll T, Lutter R, Juffermans NP, Schultz MJ. Bench-to-bedside review: bacterial pneumonia with influenza - pathogenesis and clinical implications. Crit Care. 2010;14:219. doi: 10.1186/cc8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pommerenke C, Wilk E, Srivastava B, Schulze A, Novoselova N, Geffers R, Schughart K. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS One. 2012;7:e41169. doi: 10.1371/journal.pone.0041169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhai Y, Franco LM, Atmar RL, Quarles JM, Arden N, Bucasas KL, Wells JM, Nino D, Wang X, Zapata GE, et al. Host transcriptional response to influenza and other acute respiratory viral infections--a prospective cohort study. PLoS Pathog. 2015;11:e1004869. doi: 10.1371/journal.ppat.1004869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1038/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang Y, Zaas AK, Rao A, Dobigeon N, Woolf PJ, Veldman T, Oien NC, McClain MT, Varkey JB, Nicholson B, et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011;7:e1002234. doi: 10.1371/journal.pgen.1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jeisy-Scott V, Davis WG, Patel JR, Bowzard JB, Shieh WJ, Zaki SR, Katz JM, Sambhara S. Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS One. 2011;6:e25242. doi: 10.1371/journal.pone.0025242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, Porubsky S, Booth S, Veerapen N, Besra GS, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. 2008;118:4036–4048. doi: 10.1172/JCI36264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 115.Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 117.Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, Garcia-Sastre A, Tolnay AE, Albrecht R, Pyles JA, Olson PH, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci U S A. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cilloniz C, Pantin-Jackwood MJ, Ni C, Goodman AG, Peng X, Proll SC, Carter VS, Rosenzweig ER, Szretter KJ, Katz JM, et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J Virol. 2010;84:7613–7624. doi: 10.1128/JVI.00553-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Korth MJ, Tchitchek N, Benecke AG, Katze MG. Systems approaches to influenza-virus host interactions and the pathogenesis of highly virulent and pandemic viruses. Semin Immunol. 2013;25:228–239. doi: 10.1016/j.smim.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watanabe T, Tisoncik-Go J, Tchitchek N, Watanabe S, Benecke AG, Katze MG, Kawaoka Y. Influenza virus hemagglutinin (HA) and the viral RNA polymerase complex enhance viral pathogenicity, but only HA induces aberrant host responses in mice. J Virol. 1918;2013(87):5239–5254. doi: 10.1128/JVI.02753-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Billharz R, Zeng H, Proll SC, Korth MJ, Lederer S, Albrecht R, Goodman AG, Rosenzweig E, Tumpey TM, Garcia-Sastre A, Katze MG. The NS1 protein of the 1918 pandemic influenza virus blocks host interferon and lipid metabolism pathways. J Virol. 2009;83:10557–10570. doi: 10.1128/JVI.00330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Forero A, Tisoncik-Go J, Watanabe T, Zhong G, Hatta M, Tchitchek N, Selinger C, Chang J, Barker K, Morrison J, et al. The 1918 influenza virus PB2 protein enhances virulence through the disruption of inflammatory and Wnt-mediated signaling in Mice. J Virol. 2016;90:2240–2253. doi: 10.1128/JVI.02974-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Manz B, Dornfeld D, Gotz V, Zell R, Zimmermann P, Haller O, Kochs G, Schwemmle M. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 2013;9:e1003279. doi: 10.1371/journal.ppat.1003279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Perrone LA, Belser JA, Wadford DA, Katz JM, Tumpey TM. Inducible nitric oxide contributes to viral pathogenesis following highly pathogenic influenza virus infection in mice. J Infect Dis. 2013;207:1576–1584. doi: 10.1093/infdis/jit062. [DOI] [PubMed] [Google Scholar]
- 125.Belisle SE, Tisoncik JR, Korth MJ, Carter VS, Proll SC, Swayne DE, Pantin-Jackwood M, Tumpey TM, Katze MG. Genomic profiling of tumor necrosis factor alpha (TNF-alpha) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-alpha signaling and increased severity of 1918 pandemic influenza virus infection. J Virol. 2010;84:12576–12588. doi: 10.1128/JVI.01310-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kash JC, Xiao Y, Davis AS, Walters KA, Chertow DS, Easterbrook JD, Dunfee RL, Sandouk A, Jagger BW, Schwartzman LM, et al. Treatment with the reactive oxygen species scavenger EUK-207 reduces lung damage and increases survival during 1918 influenza virus infection in mice. Free Radic Biol Med. 2014;67:235–247. doi: 10.1016/j.freeradbiomed.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.