

Abstract
Purpose
Implementation of pharmacogenetics into clinical practice has been relatively slow despite substantial scientific progress over the last decade. One barrier that inhibits uptake of pharmacogenetics into routine clinical practice is the lack of knowledge of how to translate a genetic test into a clinical action based on current evidence. The purpose of this paper is to describe the current state of pharmacogenetic evidence and evidence-based resources that facilitate the uptake of pharmacogenetics into clinical practice.
Summary
Controversy exists over the required evidence threshold needed for routine clinical implementation of pharmacogenetics. Large randomized controlled trials are not clinically feasible or necessary for many pharmacogenetic applications. Online resources exist like the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Pharmacogenomics Knowledgebase (PharmGKB) that provide freely available, evidence-based resources that facilitate the translation of genetic laboratory test results into actionable prescribing recommendations for specific drugs.
Conclusion
Resources provided by organizations such as CPIC and PharmGKB that use standardized approaches to evaluate the literature and provide clinical guidance are essential for the implementation of pharmacogenetics into routine clinical practice.
Introduction
The implementation of pharmacogenetic knowledge from the bench to the bedside has been relatively slow despite a growing body of evidence that supports using select pharmacogenetic test results to optimize medication outcomes.1 Controversy over the required level of evidence to use pharmacogenetic test results for medication selection and dosing is one barrier that inhibits the routine application of pharmacogenetics to patient care. Another barrier is the lack of knowledge of how to translate a genetic test into a clinical action.2 Resources are available that provide evidence-based gene-drug recommendations. Here we describe the current state of pharmacogenetic evidence and evidence-based resources that facilitate the uptake of pharmacogenetics into clinical practice.
Current Practice of Evaluating Pharmacogenetic Evidence
Different perspectives exist regarding the required evidence thresholds for applying pharmacogenetic test results to patient care, taking into account clinical utility and cost-effectiveness.3, 4 In general, recommendations for dose adjustments are routinely made with little or no large population-based studies on outcomes of those dose adjustments, often extrapolating from strong mechanistic evidence for inter-patient variability in drug response (i.e. pharmacokinetic studies). Furthermore, drug resources and drug labeling often provide recommendations for dose adjustments based on renal function assessment as evidenced by creatinine clearance calculation; however, very few controlled studies validate those recommendations.5 Since many examples of pharmacogenetic variation ultimately affect pharmacokinetics (e.g., cytochrome P450 2D6 [CYP2D6], CYP2C19, CYP3A5) of certain drugs, adjusting doses using pharmacogenetic information is analogous to using drug doses adjusted according to underlying renal function, adjustments for which randomized clinical trials would neither be practical nor indicated.
When evaluating the pharmacogenetic literature to make implementation decisions, various perspectives should be considered, including clinicians and researchers, and evidence standards must be defined. Although randomized controlled trials (RCTs) are recognized as the ‘gold standard’ by which to evaluate the clinical utility of a new drug, this study design is not ideal to measure the benefit of pharmacogenetic testing, as clinically significant genetic variants are often present in only a small percentage of a given patient population. Furthermore, depending on the current body of evidence linking genotype to drug response, RCTs in patients with specific genetic polymorphisms may be precluded on ethical grounds.6 For example, it would be unethical to randomize patients who are homozygous for non-functional thiopurine S-methyltransferase (TPMT) variants to normal vs. reduced doses of thiopurines, as the mechanism of the TPMT variation is related to drug pharmacokinetics, and it is known that normal doses of the drugs could result in lethal toxicity. Therefore, alternative study designs are often necessary to explore the clinical utility of pharmacogenetic testing.7
One drug for which RCTs have been conducted to assess the clinical utility of pharmacogenetic testing is warfarin.8, 9 Genetic variants in VKORC1, the gene that codes for warfarin's target (vitamin K epoxide reductase), as well as variants in CYP2C9, the gene coding for the enzyme that is principally responsible for S-warfarin metabolism, are associated with increased sensitivity to warfarin.10 The European Pharmacogenetics of Anti-coagulant Therapy (EU-PACT) trial concluded that patients who initially received pharmacogenetic-based warfarin dosing were more likely to be in the therapeutic international normalized ratio (INR) range compared to patients who initially received standard warfarin dosing.8 In contrast, however, the Clarification of Optimal Anticoagulation through Genetics (COAG) trial concluded that genotype-guided warfarin dosing did not improve anticoagulation control compared to a non-genotype based dosing algorithm containing other clinical variables and was associated with less time in the therapeutic INR range among African American patients.9
These RCTs ignited a conversation about the clinical utility of genotype-guided warfarin therapy and prompted a critical evaluation of the methods and generalizability of each.11, 12 For example, one critique of these trials is that in the controlled study environment, patients are closely monitored with frequent INRs, which is not always the case in a real-world setting and could therefore mask the benefit of initial pharmacogenetic dosing. In addition, in the COAG trial, genotype-guided dosing was compared to a complex clinical algorithm in the control group, which is not the standard of care. With respect to the worse outcome for African American patients receiving gene-based dosing in the COAG trial, experts were quick to point out that relevant genetic variants common in the African American population that result in increased warfarin sensitivity were not accounted for and could likely explain that outcome.13 Despite the many strengths of a RCT, these warfarin trials highlight some key limitations that must be considered in the broader context of the current state of pharmacogenetic knowledge.
Due to the limitations of performing pharmacogenetic RCTs (i.e., number of participants needed, ethical issues, cost, etc.), knowledge must be derived from non-RCT sources, including observational studies (e.g, case reports, cross-sectional studies, case-control studies), and pharmacokinetic/pharmacodynamic studies, including in vivo and in vitro studies, linking drug effect to genetic variation. Although observational studies are more susceptible to bias, these studies offer advantages over RCTs for studying pharmacogenetic associations including the ability to compare larger numbers of subjects at a lower cost with few ethical concerns.
At this time, implementing pharmacogenetics and genomic medicine often requires a strategic commitment from the organization, and pharmacists are well-positioned to lead the implementation.14-16 In this context, pharmacogenetic testing can be viewed as a patient safety strategy, and the evidence for the use of pharmacogenetic testing can be compared with the evidence threshold needed for other safety strategies an organization pursues. Other medication safety technologies are becoming widely used without RCT evidence, and persuasive arguments have been made for implementing patient safety interventions without waiting for RCTs.17
Evidence-based resources for pharmacogenetics implementation: CPIC and PharmGKB
Clinical Pharmacogenetics Implementation Consortium (CPIC)
CPIC™ was established as a shared project between PharmGKB18 and the Pharmacogenomics Research Network (PGRN)19 to address the need for clinical practice guidelines that facilitate the translation of genetic laboratory test results into actionable prescribing recommendations for specific drugs. PharmGKB is a comprehensive online resource established in 2000 whose scientific team at Stanford University collects, curates, and disseminates knowledge about the impact of human genetic variation on drug responses.18 The PGRN, also founded in 2000, is a National Institutes of Health (NIH) -funded group of investigators that lead research in the discovery of how genomic variation impacts therapeutic and adverse drug effects.20 CPIC membership has grown to over 160 pharmacogenetics experts (clinicians and scientists) from 86 institutions and 16 countries with multiple observers from the NIH and the U.S. Food and Drug Administration (FDA). To date, CPIC has published 19 gene-drug guidelines, six of which have recently been updated16, 18-39 (Table 1).
Table 1.
Current CPIC guidelines and associated PharmGKB clinical annotation levels and FDA label information
| Genea,b | Drug | PharmGKB clinical associationsc | Pharmacogenetics on FDA Labeld | CPIC guideline reference |
|---|---|---|---|---|
| CFTR | ivacaftor | 1A | Genetic testing required | 46 |
| CYP2C19 | amitriptyline | 1A | 27 | |
| CYP2C19 | clopidogrel | 1A | Genetic testing recommended | 30 |
| CYP2C19 | doxepin | 3 | Actionable pharmacogenetics | 27 |
| CYP2C19 | imipramine | 2A | 27 | |
| CYP2C19 | trimipramine | 2A | 27 | |
| CYP2C19 | voriconazole | 2A | Actionable pharmacogenetics | In progress |
| CYP2C19 | citalopram | 1A | Actionable pharmacogenetics | 38 |
| CYP2C19 | escitalopram | 1A | 38 | |
| CYP2C9 | phenytoin | 1B | 31 | |
| CYP2C9 | warfarin | 1A | Actionable pharmacogenetics | 21 |
| CYP2D6 | amitriptyline | 1A | Actionable pharmacogenetics | 27 |
| CYP2D6 | codeine | 1A | Actionable pharmacogenetics | 23 |
| CYP2D6 | desipramine | 1A | Actionable pharmacogenetics | 27 |
| CYP2D6 | doxepin | 1A | Actionable pharmacogenetics | 27 |
| CYP2D6 | fluvoxamine | 1A | Informative pharmacogenetics | 38 |
| CYP2D6 | imipramine | 1A | Actionable pharmacogenetics | 27 |
| CYP2D6 | nortriptyline | 1A | Actionable pharmacogenetics | 27 |
| CYP2D6 | paroxetine | 1A | Informative pharmacogenetics | 38 |
| CYP2D6 | tamoxifen | 2A | In progress | |
| CYP2D6 | tramadol | 1B | Actionable pharmacogenetics | 34 |
| CYP2D6 | trimipramine | 1A | Actionable pharmacogenetics | 27 |
| CYP3A5 | tacrolimus | 1A | 39 | |
| DPYD | capecitabine | 1A | Actionable pharmacogenetics | 25 |
| DPYD | fluorouracil | 1A | Actionable pharmacogenetics | 25 |
| DPYD | tegafur | 1A | 25 | |
| G6PD | rasburicase | 1A | Genetic testing required | 20 |
| HLA-B | abacavir | 1A | Genetic testing required | 18, 47 |
| HLA-B | allopurinol | 1A | 16, 26 | |
| HLA-B | carbamazepine | 1A | Genetic testing required | 28 |
| HLA-B | phenytoin | 1A | Actionable pharmacogenetics | 31 |
| IFNL3 | peg interferon alfa-2b | 1A | Actionable pharmacogenetics | 36 |
| SLCO1B1 | simvastatin | 1A | 24 | |
| TPMT | azathioprine | 1A | Genetic testing recommended | 19 |
| TPMT | mercaptopurine | 1A | Genetic testing recommended | 19 |
| TPMT | thioguanine | 1A | Actionable pharmacogenetics | 19 |
| UGT1A1 | atazanavir | 1A | 37 | |
| UGT1A1 | irinotecan | 2A | Actionable pharmacogenetics | planned |
| VKORC1 | warfarin | 1A | Actionable pharmacogenetics | 21 |
Assigning CPIC levels to genes and drugs and grouping together genes and drugs for planned CPIC guidelines is a dynamic process that is continually updated. CPIC levels are ultimately decided by the guideline writing committees, who may modify dosing recommendations only after a detailed review of the evidence for genes and drugs. This list was current as of November 2015. The list posted on PharmGKB is the most current list http://www.pharmgkb.org/cpic/pairs.
CFTR = cystic fibrosis transmembrane conductance regulator; CYP2C19 = cytochrome P450 family 2 subfamily C member 19; CYP2C9 = cytochrome P450 family 2 subfamily C member 9; CYP2D6 = cytochrome P450 family 2 subfamily D member 6; CYP3A5 = cytochrome P450 family 3 subfamily A member 5; DPYD = dihydropyrimidine dehydrogenase; G6PD = glucose-6-phosphate dehydrogenase; HLA-B = major histocompatibility complex, class I, B; IFNL3 = interferon, lambda 3; SLCO1B1 = solute carrier organic anion transporter family member 1B1; TPMT = thiopurine S-methyltransferase; UGT1A1 = UDP glucuronosyltransferase family 1 member A1; VKORC1 = vitamin K epoxide reductase complex subunit 1
PharmGKB Clinical Annotation Levels of Evidence as defined at https://www.pharmgkb.org/page/clinAnnLevels.
FDA Label categories created and assigned by PharmGKB, defined at https://www.pharmgkb.org/page/drugLabelLegend#PGxLevel.
CPIC guidelines are designed to help clinicians understand how available genetic test results should be used to optimize drug therapy and not whether to order a genetic test. This is an important distinction that separates CPIC guidelines from other disease-specific guidelines that may address pharmacogenetic testing. For example, CPIC's guideline for CYP2C19/clopidogrel offers genotype-based clopidogrel prescribing recommendations for patients with a known CYP2C19 genotype.30 In contrast, the American College of Cardiology Foundation, American Heart Association Task Force on Practice Guidelines, and the Society for Cardiovascular Angiography and Interventions recommended against routine CYP2C19 genotyping in their 2011 guideline for percutaneous coronary intervention, as did the American Heart Association and the American College of Cardiology in their 2014 guideline for the management of patients with non-ST-elevation acute coronary syndromes. 40, 41 The underlying assumption governing CPIC guidelines is that genomic testing results will increasingly be available and clinicians will be faced with having a patient's relevant pharmacogenetic genotype available, even if they did not order a test with a specific gene or drug in mind. Therefore, the question will become not whether to test, but how to effectively use the pharmacogenetic information that is becoming increasingly available.
As described by Caudle et al.31, CPIC guidelines closely follow the Institute of Medicine's Standards for Developing Trustworthy Clinical Practice Guidelines and have established methods for guideline development, including a rigorous literature review and grading of the scientific literature, input of a writing committee of clinicians and researchers with expertise in the guideline subject, a standard format, and extensive pre- and post-submission peer review approval process. CPIC guidelines also meet the strict criteria for inclusion into the National Guideline Clearinghouse or guidelines.gov. The American Society of Health-System Pharmacists (ASHP) has endorsed seven CPIC guidelines to date42 and is in the process of endorsing more. The American Society of Clinical Pharmacology and Therapeutics has also endorsed the CPIC guideline development process and endorsement of individual guidelines is underway. CPIC guidelines are freely available at the CPIC and PharmGKB websites and at PubMed Central.
CPIC guidelines provide the information a clinician would need to translate patient-specific diplotypes for each gene into clinical phenotypes (e.g., CYP2C19 poor metabolizer) or drug prescribing groups (e.g., HLA-B*15:02 positive) and provide therapeutic recommendations based on these predicted phenotypes or groupings. Phenotype and allele function assignment has been variable in the literature, but CPIC has recently led an effort to establish standardized terminology for these assignments.31 Each guideline contains tables that assign likely function to relevant alleles and phenotypes, and a comprehensive table is posted on PharmGKB that defines all phenotypes for all possible diplotypes (e.g., 6,668 diplotype combinations for CYP2D6). The guideline text also includes 1) background information for both the gene and drug, 2) information regarding the interpretation of the genetic test, 3) incidental findings (i.e., diseases or conditions that have or have not been linked to variation of the gene, unrelated to medication use), 4) other considerations for critical issues about the gene or drug, and 5) description of the evidence linking genetic variability to variability in drug-related phenotypes, and 6) potential benefits and harm for the patient (i.e., the toxicities or adverse reactions that may be avoided by pharmacogenetic-based dosing) as well as any potential risks from incidental findings or use of alternative drugs or dosing (e.g., differences in efficacy). Of course, the “heart” of the guideline is the therapeutic recommendation which is based on the current level of evidence for the gene/drug pair, as well as evidence for alternative therapies.
CPIC's therapeutic recommendations are based on assessing the evidence from a combination of preclinical functional and clinical data, as well as on any existing consensus guidelines.31 Examples of types of evidence reviewed include, but are not limited to, “randomized clinical trials with pharmacogenetic-based prescribing versus dosing not based on genetics, pre-clinical and clinical studies demonstrating that drug effects or concentration are linked to functional pharmacogenetic loci, case studies associating rare variants with drug effects, in vivo pharmacokinetic/pharmacodynamics studies for drug or reference drug plus variant type, and in vitro metabolic and/or transport capacity for the drug plus variant type”.31 Where available, evidence evaluating the outcomes when prescribing has been altered based on genetic testing is included. As stated previously, for most genes/drugs, RCTs comparing clinical outcomes with genotype-guided dosing versus conventional dosing are not available. Furthermore, evidence related to the appropriateness of alternative medications or dosing that may be used based on genetics must be weighed in assigning the strength of the recommendation. Overall, the therapeutic recommendations are simplified to allow rapid interpretation by clinicians and are presented in a guideline table and occasionally in an algorithm. To assign strength to a recommendation, CPIC uses a transparent three category system for rating recommendations.17, 31 Therapeutic recommendations are graded as strong where “the evidence is high quality and the desirable effects clearly outweigh the undesirable effects”; moderate, in which “there is a close or uncertain balance” as to whether the evidence is high quality and the desirable clearly outweigh the undesirable effects; and optional, in which the desirable effects of pharmacogenetic-based dosing are closely balanced with undesirable effects and there is room for differences in opinion as to the need for the recommended course of action. Each recommendation also includes an assessment of its usefulness in pediatric patients.31
With community and member feedback, CPIC has learned that there is a critical need to provide classification of gene/drug groupings into those that are likely actionable versus not, and to develop gene/drug guidelines beyond those with strong prescribing recommendations. CPIC now classifies genes/drugs as CPIC level A, B, C, or D (Figure 1; Table 2). As gene results are placed into patients' medical records for those primary drugs with strong or moderate recommendations (termed CPIC level A guidelines), clinicians are faced with deciding how the same gene test results should (or should not) be used for secondary drugs for which there may be substantial literature references or even clinical laboratory interpretations, but drugs which CPIC's experts have deemed actionability as “optional” (termed CPIC level B guidelines) or not actionable (termed CPIC level C) (Figure 1, Table 2). Definitive recommendations on lack of actionability (CPIC level C) can be just as useful to the clinician as actionable recommendations. There are a few examples of other “non-CPIC” genes/drugs that are marketed by companies or advocated for testing in the literature, yet CPIC does not deem as actionable (CPIC level C). In these cases, clinicians also need an unbiased and well-referenced guidance, based on standardized criteria, to assist in decision making and provide the basis for not changing prescribing based on the test results. Level D gene/drug associations are considered in the CPIC prioritization process, and are generally annotated on the PharmGKB as clinical annotations, but CPIC deems that guidelines are not currently warranted for these genes/drugs. It should be noted that the posted list of genes and drugs and their assignment to CPIC guideline levels is based on current evidence; the process is dynamic and genes and drugs may be added or change CPIC levels in response to new evidence or available testing options. The full list of gene-drug pairs is available on the CPIC website.34
Figure 1. Initial prioritization considerations for new gene/drug groups.
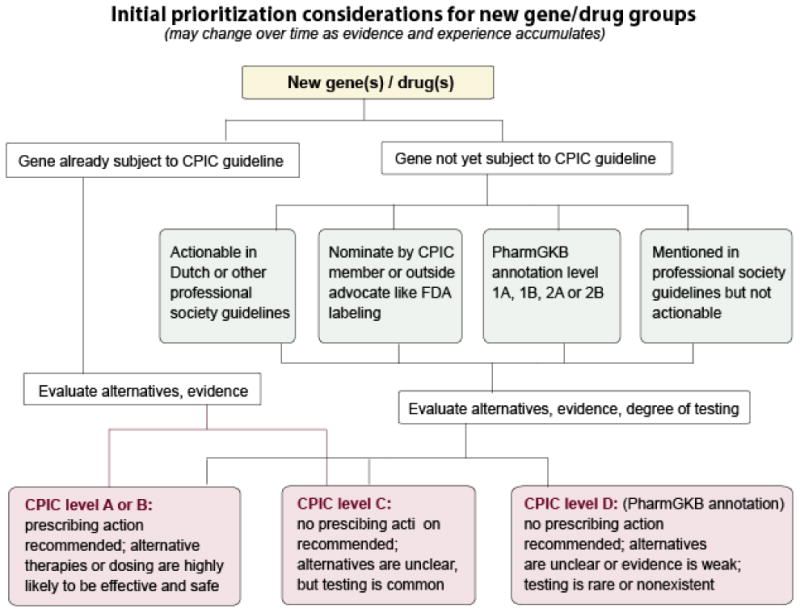
Used with permission from PharmGKB. PharmGKB® is a registered trademark of the U.S. Department of Health and Human Services and is financially supported by NIH/NIGMS. It is managed at Stanford University.
Table 2. CPIC Level Definitions for Genes and Drugs.
| CPIC Level | Clinical Context | Level of evidence | Strength of Recommendation |
|---|---|---|---|
| A | Genetic information should be used to change prescribing of affected drug | Preponderance of evidence is high or moderate in favor of changing prescribing | At least one moderate or strong action (change in prescribing) recommended. |
| B | Genetic information could be used to change prescribing of the affected drug because alternative therapies/dosing are extremely likely to be as effective and as safe as non-genetically based dosing | Preponderance of evidence is weak with little conflicting data | At least one optional action (change in prescribing) is recommended. |
| C | There are published studies at varying levels of evidence, some with mechanistic rationale, but no prescribing actions are recommended because (a) dosing based on genetics convincingly makes no difference or (b) alternatives are unclear, possibly less effective, more toxic, or otherwise impractical. Most important for genes that are subject of other CPIC guidelines or genes that are commonly included in clinical or DTC tests. | Evidence levels can vary | No prescribing actions are recommended. |
| D | There are few published studies, clinical actions are unclear, little mechanistic basis, mostly weak evidence, or substantial conflicting data. If the genes are not widely tested for clinically, evaluations are not needed. | Evidence levels can vary | No prescribing actions are recommended. |
Used with permission from PharmGKB. PharmGKB® is a registered trademark of the U.S. Department of Health and Human Services and is financially supported by NIH/NIGMS. It is managed at Stanford University.
The Pharmacogenomics Knowledgebase (PharmGKB)
Like CPIC, PharmGKB uses an evidence-based, tiered system of grading pharmacogenetic associations.43 An annotated pharmacogenetic summary of FDA-approved labeling, European Medicines Association (EMA), the Pharmaceuticals and Medical Devices Agency, Japan (PMDA), and Health Canada (Santé Canada) (HCSC) can also be found on the PharmGKB website.
Scientific curators annotate published literature with variant-drug phenotypes, capturing information such as statistical significance, population size and association type (e.g., drug efficacy, toxicity, etc.). Curators group together like associations across publications and summarize the impact of each genotype on the drug phenotype in what PharmGKB calls “clinical associations”. Clinical associations are assigned a level of evidence to indicate the strength of the literature support, and therefore the confidence in the association as determined by PharmGKB curators.
PharmGKB assigns a level from 1A (highest strength) for annotations for variant-drug combinations in a CPIC or medical society-endorsed PGx guidelines, or implemented at a PGRN site or in another major health system, to level 4 (lowest strength) for annotations based on a case report, non-significant study or in vitro, molecular or functional assay evidence only. The assignment of evidence level is based on several criteria, including replication of the association, statistical parameters and population size. The evidence level for a clinical annotation can change over time as new studies are published. Association evidence may accumulate and as new literature is annotated by PharmGKB curators, the corresponding clinical annotations are re-evaluated. If the clinical annotation meets the criteria for a higher level of evidence with the new literature, the curator adjusts the level. Conversely, a preliminary association may not be replicated by future studies, and so the level of evidence would be adjusted down. Therefore, the level of evidence assignment is dynamic.
Organizations such as CPIC and PharmGKB that use standardized approaches to evaluate the literature are becoming important to other areas of genomic and precision medicine such as ClinGen44 and ClinVar23, 45 which are central resources that define the clinical relevance of gene and variants. These types of relationships unite a relatively new field in developing gold standards for clinical pharmacogenetic evidence review and the description of gene and variant functionality, all requirements for implementation into clinical practice.
Conclusion
Controversy exists over the required evidence threshold needed for routine clinical implementation of pharmacogenetics, and the level of evidence required to mandate ordering genetic tests vs. the level required to act on preemptively available evidence may differ. Resources provided by organizations such as CPIC and PharmGKB that use standardized approaches to evaluate the literature and provide clinical guidance are essential for the implementation of pharmacogenetics into routine clinical practice.
Key Points.
Controversy exists over the required evidence threshold needed for routine clinical implementation of pharmacogenetics.
One barrier that inhibits uptake of pharmacogenetics into routine clinical practice is the lack of knowledge of how to translate a genetic test into a clinical action based on current evidence.
Resources provided by organizations such as CPIC and PharmGKB that use standardized approaches to evaluate the literature and provide clinical guidance are essential for the implementation of pharmacogenetics into routine clinical practice.
Acknowledgments
This work was funded by the National Institutes of Health (NIH) CPIC (R24GM115264) and PharmGKB (R24 GM61374).
Conflict of Interest: TEK and MWC are scientific advisors to Rxight Pharmacogenetics and are funded by NIH grant R24 GM61374 for PharmGKB. MVR, KEC, TEK, JMH and MWC are funded by NIH grant R24GM115264 for CPIC.
References
- 1.Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–50. doi: 10.1038/nature15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Relling MV, Klein TE. Cpic: Clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clinical pharmacology and therapeutics. 2011;89(3):464–7. doi: 10.1038/clpt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gillis NK, Innocenti F. Evidence required to demonstrate clinical utility of pharmacogenetic testing: The debate continues. Clinical pharmacology and therapeutics. 2014;96(6):655–7. doi: 10.1038/clpt.2014.185. [DOI] [PubMed] [Google Scholar]
- 4.Ratain MJ, Johnson JA. Meaningful use of pharmacogenetics. Clinical pharmacology and therapeutics. 2014;96(6):650–2. doi: 10.1038/clpt.2014.188. [DOI] [PubMed] [Google Scholar]
- 5.Khanal A, Castelino RL, Peterson GM, et al. Dose adjustment guidelines for medications in patients with renal impairment: How consistent are drug information sources? Internal medicine journal. 2014;44(1):77–85. doi: 10.1111/imj.12291. [DOI] [PubMed] [Google Scholar]
- 6.Young B, Squires K, Patel P, et al. First large, multicenter, open-label study utilizing hla-b*5701 screening for abacavir hypersensitivity in north america. AIDS. 2008;22(13):1673–5. doi: 10.1097/QAD.0b013e32830719aa. [DOI] [PubMed] [Google Scholar]
- 7.Filipski KK, Mechanic LE, Long R, et al. Pharmacogenomics in oncology care. Frontiers in genetics. 2014;5:73. doi: 10.3389/fgene.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pirmohamed M, Burnside G, Eriksson N, et al. A randomized trial of genotype-guided dosing of warfarin. The New England journal of medicine. 2013;369(24):2294–303. doi: 10.1056/NEJMoa1311386. [DOI] [PubMed] [Google Scholar]
- 9.Kimmel SE, French B, Kasner SE, et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. The New England journal of medicine. 2013;369(24):2283–93. doi: 10.1056/NEJMoa1310669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker WL, Johnson SG. Pharmacogenetics and oral antithrombotic drugs. Curr Opin Pharmacol. 2016;27:38–42. doi: 10.1016/j.coph.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Zineh I, Pacanowski M, Woodcock J. Pharmacogenetics and coumarin dosing--recalibrating expectations. The New England journal of medicine. 2013;369(24):2273–5. doi: 10.1056/NEJMp1314529. [DOI] [PubMed] [Google Scholar]
- 12.Furie B. Do pharmacogenetics have a role in the dosing of vitamin k antagonists? The New England journal of medicine. 2013;369(24):2345–6. doi: 10.1056/NEJMe1313682. [DOI] [PubMed] [Google Scholar]
- 13.Drozda K, Wong S, Patel SR, et al. Poor warfarin dose prediction with pharmacogenetic algorithms that exclude genotypes important for african americans. Pharmacogenet Genomics. 2015;25(2):73–81. doi: 10.1097/FPC.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans WE, Crews KR, Pui CH. A health-care system perspective on implementing genomic medicine: Pediatric acute lymphoblastic leukemia as a paradigm. Clinical pharmacology and therapeutics. 2013;94(2):224–9. doi: 10.1038/clpt.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson SG. Leading clinical pharmacogenomics implementation: Advancing pharmacy practice. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists. 2015;72(15):1324–8. doi: 10.2146/ajhp140613. [DOI] [PubMed] [Google Scholar]
- 16.Saito Y, Stamp LK, Caudle KE, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for human leukocyte antigen b (hla-b) genotype and allopurinol dosing: 2015 update. Clinical pharmacology and therapeutics. 2015 doi: 10.1002/cpt.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leape LL, Berwick DM, Bates DW. What practices will most improve safety? Evidence-based medicine meets patient safety. Jama. 2002;288(4):501–7. doi: 10.1001/jama.288.4.501. [DOI] [PubMed] [Google Scholar]
- 18.Martin MA, Klein TE, Dong BJ, et al. Clinical pharmacogenetics implementation consortium guidelines for hla-b genotype and abacavir dosing. Clinical pharmacology and therapeutics. 2012;91(4):734–8. doi: 10.1038/clpt.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clinical pharmacology and therapeutics. 2011;89(3):387–91. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Relling MV, McDonagh EM, Chang T, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for rasburicase therapy in the context of g6pd deficiency genotype. Clinical pharmacology and therapeutics. 2014;96(2):169–74. doi: 10.1038/clpt.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson JA, Gong L, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for cyp2c9 and vkorc1 genotypes and warfarin dosing. Clinical pharmacology and therapeutics. 2011;90(4):625–9. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott SA, Sangkuhl K, Gardner EE, et al. Clinical pharmacogenetics implementation consortium guidelines for cytochrome p450-2c19 (cyp2c19) genotype and clopidogrel therapy. Clinical pharmacology and therapeutics. 2011;90(2):328–32. doi: 10.1038/clpt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for codeine therapy in the context of cytochrome p450 2d6 (cyp2d6) genotype. Clinical pharmacology and therapeutics. 2012;91(2):321–6. doi: 10.1038/clpt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilke RA, Ramsey LB, Johnson SG, et al. The clinical pharmacogenomics implementation consortium: Cpic guideline for slco1b1 and simvastatin-induced myopathy. Clinical pharmacology and therapeutics. 2012;92(1):112–7. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caudle KE, Thorn CF, Klein TE, et al. Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clinical pharmacology and therapeutics. 2013;94(6):640–5. doi: 10.1038/clpt.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershfield MS, Callaghan JT, Tassaneeyakul W, et al. Clinical pharmacogenetics implementation consortium guidelines for human leukocyte antigen-b genotype and allopurinol dosing. Clinical pharmacology and therapeutics. 2013;93(2):153–8. doi: 10.1038/clpt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hicks JK, Swen JJ, Thorn CF, et al. Clinical pharmacogenetics implementation consortium guideline for cyp2d6 and cyp2c19 genotypes and dosing of tricyclic antidepressants. Clinical pharmacology and therapeutics. 2013;93(5):402–8. doi: 10.1038/clpt.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leckband SG, Kelsoe JR, Dunnenberger HM, et al. Clinical pharmacogenetics implementation consortium guidelines for hla-b genotype and carbamazepine dosing. Clin Pharmacol Ther. 2013;94(3):324–8. doi: 10.1038/clpt.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clinical pharmacology and therapeutics. 2013;93(4):324–5. doi: 10.1038/clpt.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott SA, Sangkuhl K, Stein CM, et al. Clinical pharmacogenetics implementation consortium guidelines for cyp2c19 genotype and clopidogrel therapy: 2013 update. Clinical pharmacology and therapeutics. 2013;94(3):317–23. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: The clinical pharmacogenetics implementation consortium (cpic) guideline development process. Current drug metabolism. 2014;15(2):209–17. doi: 10.2174/1389200215666140130124910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsey LB, Johnson SG, Caudle KE, et al. The clinical pharmacogenetics implementation consortium guideline for slco1b1 and simvastatin-induced myopathy: 2014 update. Clinical pharmacology and therapeutics. 2014 doi: 10.1038/clpt.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clancy JP, Johnson SG, Yee SW, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for ivacaftor therapy in the context of cftr genotype. Clinical pharmacology and therapeutics. 2014 doi: 10.1038/clpt.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical pharmacogenetics implementation consortium guidelines for cytochrome p450 2d6 genotype and codeine therapy: 2014 update. Clinical pharmacology and therapeutics. 2014;95(4):376–82. doi: 10.1038/clpt.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin MA, Hoffman JM, Freimuth RR, et al. Clinical pharmacogenetics implementation consortium guidelines for hla-b genotype and abacavir dosing: 2014 update. Clinical pharmacology and therapeutics. 2014 doi: 10.1038/clpt.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muir AJ, Gong L, Johnson SG, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for ifnl3 (il28b) genotype and peg interferon-alpha-based regimens. Clinical pharmacology and therapeutics. 2014;95(2):141–6. doi: 10.1038/clpt.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gammal RS, Court MH, Haidar CE, et al. Clinical pharmacogenetics implementation consortium (cpic) guideline for ugt1a1 and atazanavir prescribing. Clinical pharmacology and therapeutics. 2015 doi: 10.1002/cpt.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hicks JK, Bishop JR, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium (cpic) guideline for cyp2d6 and cyp2c19 genotypes and dosing of selective serotonin reuptake inhibitors. Clinical pharmacology and therapeutics. 2015;98(2):127–34. doi: 10.1002/cpt.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birdwell KA, Decker B, Barbarino JM, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for cyp3a5 genotype and tacrolimus dosing. Clinical pharmacology and therapeutics. 2015;98(1):19–24. doi: 10.1002/cpt.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine GN, Bates ER, Blankenship JC, et al. 2011 accf/aha/scai guideline for percutaneous coronary intervention. A report of the american college of cardiology foundation/american heart association task force on practice guidelines and the society for cardiovascular angiography and interventions. J Am Coll Cardiol. 2011;58(24):e44–122. doi: 10.1016/j.jacc.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 aha/acc guideline for the management of patients with non-st-elevation acute coronary syndromes: A report of the american college of cardiology/american heart association task force on practice guidelines. J Am Coll Cardiol. 2014;64(24):e139–228. doi: 10.1016/j.jacc.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 42.American Society of Health-System Pharmacists. Endorsed documents. [accessed 2016 March 16]; http://www.ashp.org/menu/PracticePolicy/PolicyPositionsGuidelinesBestPractices/BrowsebyDocumentType/EndorsedDocuments.aspx.
- 43.Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clinical pharmacology and therapeutics. 2012;92(4):414–7. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clinical genome resource. [accessed 2016 March 16]; https://www.clinicalgenome.org/
- 45.Gram LF. Plasma level monitoring of tricyclic antidepressant therapy. Clinical pharmacokinetics. 1977;2(4):237–51. doi: 10.2165/00003088-197702040-00001. [DOI] [PubMed] [Google Scholar]
- 46.Clancy JP, Johnson SG, Yee SW, et al. Clinical pharmacogenetics implementation consortium (cpic) guidelines for ivacaftor therapy in the context of cftr genotype. Clinical pharmacology and therapeutics. 2014;95(6):592–7. doi: 10.1038/clpt.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin MA, Hoffman JM, Freimuth RR, et al. Clinical pharmacogenetics implementation consortium guidelines for hla-b genotype and abacavir dosing: 2014 update. Clinical pharmacology and therapeutics. 2014;95(5):499–500. doi: 10.1038/clpt.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]