

Abstract
A myriad of diseases is caused or characterized by alteration of epigenetic patterns, including changes in DNA methylation, post-translational histone modifications, or chromatin structure. These changes of the epigenome represent a highly interesting layer of information for disease stratification and for personalized medicine. Traditionally, epigenomic profiling required large amounts of cells, which are rarely available with clinical samples. Also, the cellular heterogeneity complicates analysis when profiling clinical samples for unbiased genome-wide biomarker discovery. Recent years saw great progress in miniaturization of genome-wide epigenomic profiling, enabling large-scale epigenetic biomarker screens for disease diagnosis, prognosis, and stratification on patient-derived samples. All main genome-wide profiling technologies have now been scaled down and/or are compatible with single-cell readout, including: (i) Bisulfite sequencing to determine DNA methylation at base-pair resolution, (ii) ChIP-Seq to identify protein binding sites on the genome, (iii) DNaseI-Seq/ATAC-Seq to profile open chromatin, and (iv) 4C-Seq and HiC-Seq to determine the spatial organization of chromosomes. In this review we provide an overview of current genome-wide epigenomic profiling technologies and main technological advances that allowed miniaturization of these assays down to single-cell level. For each of these technologies we evaluate their application for future biomarker discovery. We will focus on (i) compatibility of these technologies with methods used for clinical sample preservation, including methods used by biobanks that store large numbers of patient samples, and (ii) automation of these technologies for robust sample preparation and increased throughput.
Keywords: Genome-wide epigenetic profiling, Biomarker discovery, Miniaturization, Automation, Single cell, DNA methylation, WGBS, ATAC-Seq, Stratification, Precision medicine
Background
Within fundamental and clinical research and in clinical practice, biomarkers play an important role to facilitate disease diagnosis, prognosis, and selection of targeted therapies in patients. As such, biomarkers are critical for personalized medicine to improve disease stratification: the identification of groups of patients with shared (biological) characteristics, such as a favorable response to a particular drug [1, 2]. Biomarkers need to fulfill a number of requirements, the most important of which is to show high predictive value. From a practical perspective, the detection method for a biomarker must be accurate, relatively easy to carry out, and show high reproducibility [3]. Over the last decade, there has been an increasing interest in biomarkers at the hand of rapid developments within high-throughput molecular biology technologies, capable of identifying “molecular biomarkers” [4, 5]. Molecular biomarkers possess a critical advantage over more traditional biomarkers during the exploratory phase of biomarker discovery, as many candidate molecular biomarkers can be assayed in parallel. This particularly involves screening of (epi)genomic features at a genome-wide scale, often making use of powerful next-generation sequencing (NGS)-based technologies. These screens can assess very large numbers of loci for the presence or absence of a certain (epi)genomic feature. Subsequently, these loci can be evaluated as a potential biomarker by determining their correlation between samples with different characteristics, for example, by comparing healthy versus diseased tissue.
To be suitable for biomarker discovery, (epi)genomic profiling assays need to fulfill a number of important requirements. To accommodate sample collection for batch processing, clinical samples are often preserved by freezing or by formaldehyde crosslinking. Therefore, an important requirement for (epi)genomic biomarker screening technologies is that these are compatible with processed samples. Additionally, this allows inclusion of clinical samples that have been processed for biobanking, or to use such samples for replication or validation. Biobanks collect large numbers of samples such as tissues or DNA (deoxyribonucleic acid) and the associated patient information, which is highly valuable for retrospective biomarker studies [6–9]. Exploratory screens for candidate biomarkers mainly rely on the use of patient specimens, which are obtained in small quantities, while also biobanks often contain limited quantities of patient material. Therefore, a second requirement is that assays used for biomarker discovery are compatible with miniaturization to allow processing of low-input samples. Furthermore, robust biomarker discovery is dependent on the screening of large numbers of samples due to the inherent clinical and biological variability between patient samples [10]. Assays used for biomarker discovery therefore benefit from automation and digitalization, facilitating upscaling while reducing the chance of errors due to human handling.
Genomic features that are utilized for molecular biomarker discovery can be separated in two categories: (i) changes in the DNA sequence itself, such as mutations and rearrangements, and (ii) changes in the epigenome, represented by molecules and structures associated with the DNA such as DNA methylation and post-translational histone modifications. This review will focus on the latter category, as recent developments in epigenetic profiling technologies have not only greatly increased our knowledge on epigenetic regulation, but also allow for large-scale discovery of molecular epigenetic biomarkers. The first section of this review provides an overview of epigenetic features and how these can be assayed. We discuss how misregulation of epigenetic processes may lead to disease, providing mechanistic rationale for the use of epigenetic features as biomarkers. The feasibility of applying epigenetic biomarkers in the clinic is demonstrated by examples of DNA methylation biomarkers that have reached clinical stages. In the second part of this review, we will focus on current genome-wide epigenomic profiling technologies, and whether these are already or will likely become compatible with biomarker discovery in the near future. We will evaluate these approaches with three criteria in mind: (i) the possibility to use frozen or chemically fixed material in these assays, (ii) compatibility with miniaturization and single-cell profiling, and (iii) the current level of automation.
Main text
The epigenome
Within a eukaryotic cell, the DNA is packaged to fit into the small volume of the nucleus in a highly organized fashion. The basic unit of chromatin involves the DNA wrapped around nucleosomes consisting of two copies of each of the core histones H2A, H2B, H3, and H4: the so-called beads-on-string structure [11]. Subsequent compaction leads to higher order structures including the formation of very dense arrays of nucleosomes observed in heterochromatin [12, 13]. Despite being tightly packed, the chromatin appears to be highly plastic to allow processes such as transcription, DNA damage repair, DNA remodeling, and DNA replication. This plasticity is facilitated by several factors that influence both local and global chromatin architectures. The most prominent features affecting chromatin structure are reversible covalent modifications of the DNA, e.g., cytosine methylation and hydroxymethylation mainly occurring within the genomic CG context (CpGs), and reversible post-translational modifications of histones, e.g., lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination and sumoylation. These modifications are set by specific classes of enzymes: DNA methyltransferases (DNMTs) in case of cytosine methylation [14] or histone-modifying enzymes [15]. Besides facilitating chromatin compaction, modifications of the DNA and histones are read by adaptor molecules, chromatin-modifying enzymes, and transcription factors (TFs) that contribute to the regulation of transcription and other chromatin-related processes [15, 16]. Next to modifications of DNA and histones, the three-dimensional (3D) conformation of the DNA within the nucleus imposes an additional regulatory layer of gene expression [17].
The chromatin state of a cell, including the genomic localization of modifications of DNA and histones, TF binding sites, and 3D DNA structure, is generally referred to as the epigenome. The epigenome is an important layer that regulates which parts of the genome are accessible and thereby active and which parts are condensed and hence inactive. As such, epigenetic changes are a major driver of development and important to gain and maintain cellular identity. Each of the approximately 200 distinct cell types in the human body has essentially the same genome but has a unique epigenome that serves to instruct specific gene expression programs present within the cells. To gain insight in this variation, the epigenetic features of these cell types (Fig. 1) are comprehensively studied at a genome-wide scale using high-resolution technologies as summarized in Table 1. Most of the approaches are based on NGS, which generally yield higher sensitivity and resolution as compared to alternative readouts such as microarrays, and provide additional information such as allele specificity [18, 19]. The International Human Epigenome Consortium (IHEC; http://www.ihec-epigenomes.org) and associated consortia such as BLUEPRINT and National Institutes of Health (NIH) Roadmap use these technologies to generate human reference data sets for a range of epigenetic features [20–23]. The aim of IHEC is to generate approximately 1000 reference epigenomes which are made publicly available. This data contains a wealth of information on the epigenetic mechanisms acting in healthy cells and serves as a valuable reference for comparisons with malignant cells and tissues [24, 25].
Fig. 1.

Main epigenetic features (indicated by orange arrows) that can be assayed on a genome-wide scale using sequencing-based technologies
Table 1.
Summary of the main epigenetic features and the principles, caveats, and requirements of the main technologies used for their profiling
| DNA methylation. DNA methylation is the process in which a methyl group is added to the 5′ position of cytosines in the DNA, which mainly occurs within the context of CpGs. DNA methylation typically acts to repress gene transcription when located in a gene promoter, while gene-body methylation is positively correlated with expression [153–157]. Distal regulatory regions like enhancers generally contain low DNA methylation levels when active due to binding of TFs [158]. The role or consequence of DNA methylation at other places of the genome is less well understood [14]. Genome-wide profiling of DNA methylation generally relies on (i) affinity purification of methylated DNA fragments or (ii) the use of sodium bisulfite converting unmethylated cytosines into uracil. The technologies referred to by the first method, MBD-Seq/MethylCap-Seq (methyl-CpG binding domain protein-enriched sequencing/methylated DNA capture sequencing) [140, 141, 159] and MeDIP-Seq (methylation DNA immunoprecipitation sequencing) [160, 161], utilize a methyl binding protein domain or an antibody raised against 5-methylcytosine, respectively, to affinity purify methylated DNA fragments from sheared genomic DNA. Although MethylCap-Seq/MeDIP-Seq provides accurate measurements of DNA methylation [162], an important caveat is the aspecific background remaining after the affinity purification. These might cause false positive results (in particular in case of copy number variations) if not properly controlled for. The second method makes use of bisulfite on sheared genomic DNA to convert unmethylated cytosines into uracil, while leaving methylated cytosines unaffected [154]. After subsequent amplification to prepare the DNA for readout, the uracil (representing the unmethylated cytosine) is read as a thymidine, while cytosines represent methylated cytosines in the original sample. The readout of bisulfite-based methods is mainly performed by microarrays (including the Infinium HumanMethylation450 BeadChip array (“450K array”) covering 450,000 of the 28 million genomic CpGs) [163] or by sequencing, referred to as whole-genome bisulfite sequencing (WGBS). In light of the high sequencing costs associated with WGBS, reduced representation bisulfite sequencing (RRBS) selects for CpG-rich fragments before sequencing using methylation-insensitive restriction enzymes such as MspI [164]. An important advantage of bisulfite-based methods (450K array, WGBS, RRBS) over other DNA methylation profiling technologies is that these generate DNA methylation profiles at base-pair resolution. Furthermore, the input requirements for WGBS/RRBS (20 ng of DNA for low-input WGBS/RRBS profiling, equivalent to 3 × 103 cells [121]) are low as compared to the 450K array (500 ng; 7.5 × 104 cells) and MBD-Seq/MethylCap-Seq/MeDIP-Seq (1 μg DNA; 1.5 × 105 cells). Although dependent on sequencing depth, the coverage of WGBS is usually >90% of all CpGs in the genome [165, 166], as compared to 60–90% for MBD-Seq/MethylCap-Seq/MeDIP-Seq and 2% for the 450K array. In view of the superior specifications, WGBS is considered the “golden standard” for determining the DNA methylome. |
| Protein binding sites. Characterization of the genomic locations of post-translational histone modifications, histone variants, TFs, and other chromatin associated proteins is generally performed by chromatin immunoprecipitation (ChIP). ChIP relies on the use of a specific antibody to perform affinity purifications on sheared chromatin to isolate fragments bound by the protein of interest. In most workflows, proteins are crosslinked to the DNA by formaldehyde, after which the chromatin is fragmented by sonication or enzymatic digestion. However, in particular in case of histones, ChIP can also be performed on native (meaning non-crosslinked) chromatin fragmented by micrococcal nuclease (MNase) [167, 168]. After ChIP, the purified DNA fragments are sequenced to determine the protein localization on a genome-wide scale (ChIP-Seq) [169, 170]. Loci in the genome which are enriched for mapped sequencing reads (generally referred to as “peaks” according to their visual appearance in genome browsers) represent protein binding sites. ChIP-Seq heavily relies on the availability of antibodies that are specific for their endogenous target and that are compatible with the ChIP conditions. Since ChIP-Seq relies on an enrichment strategy, it generally requires a relative high number of cells as input to distinguish specific signals from background. The number of input cells for ChIP-Seq is typically 0.5–5 × 106 cells, with profiling of histones requiring less cells than profiling TFs [134]. |
| Chromatin accessibility/footprinting. Transcriptional activation is tightly linked with disruption or eviction of nucleosome organization at control regions such as promoters and enhancers due to binding of TFs. Regulatory DNA thus coincides with open or accessible genomic sites in chromatin [171, 172]. Profiling of these accessible sites is performed using the exonuclease desoxyribonuclease 1 (DNaseI) or using the Tn5 transposase on native chromatin, as both enzymes are able to target accessible genomic regions within chromatin. Selecting and sequencing short fragments (50–150 nt) after treatment with DNaseI (DNAseI-Seq) [173, 174] or transposase (assay for transposase-accessible chromatin (ATAC)-Seq) allows to enrich for TF binding sites, in contrast to larger fragments that might be derived from nucleosomes [175]. Similar to ChIP-Seq, loci in the genome which are enriched for mapped sequencing reads (referred to as “peaks”) represent accessible sites. Within the ATAC-Seq procedure, the Tn5 transposase directly inserts the adapters for sequencing. Therefore, ATAC-Seq has an important advantage in that it requires a relative small number of cells (5 × 104 cells) [175] to start with as compared to DNAseI-Seq (1–10 × 106 cells [172]). Both for ATAC-Seq and DNAseI-Seq, characterization of enriched DNA motifs within the accessible sites can be used to infer the identity of sequence-specific TFs. A complementary approach to infer the identity of TFs that are binding within accessible regions is by the use of so-called “footprints.” Sequence-specific TFs protect the genome from DNAseI and transposase digestion at the exact position where they are binding the DNA. This results in a unique, detectable footprint that can be used for characterization of the factor that is binding [174, 176]. |
| Nucleosome occupancy/positioning. Nucleosomes are the basic core particles of the chromatin, consisting of histones and approximately 147 base pairs of DNA wrapped around it. Although the DNA-protein binding within nucleosomes is very stable, nucleosomes can be remodeled or slide along the DNA, thereby facilitating or inhibiting chromatin-related processes such as transcription. Nucleosome positioning is usually determined with the use of MNase on native chromatin [171, 177]. MNase is an endo-exonuclease that digests and cleaves DNA unless it is protected by proteins. Nucleosome position can be determined by sequencing the DNA fragments (115–195 bp in size) isolated from chromatin treated with MNase (MNase-Seq) [178, 179]. A typical MNAse-Seq profiling experiments requires 1–10 × 106 cells. |
| 3D conformation of the genome. Chromatin loops and further high-order chromatin structures are profiled using chromosome confirmation capture [180]. Chromosome confirmation capture relies on digestion of crosslinked chromatin using restriction enzymes, followed by ligation of the sticky ends. Sequencing of DNA ligation products allows to determine the proximity of the ligated fragments and provides insight into the 3D structure within the nucleus. Chromosomal loci that are far apart on a linear chromosome, but close together in nuclear space, can come into proximity and will hence be ligated [181]. For genome-wide profiling, two different variants of chromosome confirmation capture that are popular include Circular chromosome confirmation capture (4C-Seq) [182] and HiC-Seq [183, 184]. 4C-Seq determines all genomic interaction partners of one specific locus in the genome (referred to as “bait”) at high resolution and sensitivity. In HiC-Seq, all genomic interactions are profiled at low resolution and sensitivity, enabling a global 3D view on the genome. Using HiC-Seq, recent studies in mice and human have revealed that chromosome territories are arranged into large megabase-sized topologically-associating domains (TADs) that are highly conserved and stable across cell types [183, 185]. 4C-Seq experiments typically require 1 × 107 cells [186], while HiC-Seq experiments require 2.5 × 107 cells [187]. |
Comparative analyses of epigenomes are complicated by the epigenetic variability that is present between individuals within a population. Genetic variation such as SNPs (single-nucleotide polymorphisms) or indels in regulatory sequences or mutations in epigenetic enzymes will have a direct effect on the epigenome [26–29]. Furthermore, environmental factors such as lifestyle, stress, and nutrition influence epigenetic patterns [30–33]. Also, epigenetic patterns change during aging. In fact, DNA methylation markers in saliva and blood can be used for accurate estimation of age [34–37]. Thus, epigenetic patterns are plastic and change during development and over time. The variability between individuals has to be accounted for in epigenetic studies including biomarker discovery and hence large cohorts need to be studied to overcome the intra-individual variation. In this respect, it is important to note that the extent of the intra-individual variation is much less as compared to the variation observed between tissues within individuals, at least for DNA methylation [38–40].
It has become increasingly clear that misregulation or mutations of epigenetic enzymes are at the basis of a broad range of syndromes and diseases [41]. Mutations in epigenetic enzymes are frequently observed in cancer [42], intellectual disability [43], neurological disorders such as Alzheimer’s, Parkinson’s, and Huntington’s disease [44], and autoimmune diseases such as rheumatoid arthritis [45–47] and type 1 diabetes [48]. Most studies have been performed in cancer: ~30% of all driver genes characterized in cancer are related to chromatin structure and function [42]. Well-known examples of genes in which mutations can promote or drive tumorigenesis include DNMT3A and TET2, involved in DNA methylation and DNA demethylation, respectively, and EZH2, which is part of the polycomb repressive complex 2 (PRC2) complex that trimethylates lysine 27 on histone 3 (H3K27me3) [49–51]. Apart from mutations in epigenetic enzymes, mistargeting of epigenetic enzymes, such as the silencing of CDKN2A and MLH1 by aberrant promoter DNA methylation, is considered to drive tumor formation [52]. Given their prominent roles in cancer and various other diseases, epigenetics enzymes represent promising targets for therapeutic intervention. For example, small molecules targeting enzymes involved in the post-translational modifications of histones, such as SAHA (suberanilohydroxamic acid; Vorinostat) inhibiting histone deacetylases (HDACs), are effective as therapeutic drugs for a range of tumor types including T cell lymphomas in case of SAHA [53–55]. See Rodriguez and Miller [56], Qureshi and Mehler [57], and various papers within this special issue for excellent recent reviews on the use of small molecules to target epigenetic enzymes and their current status in clinical applications.
Epigenetic biomarkers
Molecular diagnosis and prognosis is traditionally often based on (immuno)histochemistry or immunoassays, for example by assaying prostate-specific antigen (PSA) in case of testing for prostate cancer [58]. Also , changes in RNA (ribonucleic acid) expression, genetic alterations, and chromosomal abnormalities represent powerful biomarkers in various diseases including cancer [59]. Notable examples are mutations in the BRCA1 and BRCA2 genes in breast and ovarian cancer or the presence of the Philadelphia chromosome in leukemia [60–62]. With the growing understanding that changes in the epigenome and chromatin are related with or causative in disease [41], it became clear that epigenetic alterations represent promising features to be used as biomarkers. An important characteristic for their use as biomarker is that epigenetic marks, in particular DNA methylation, are known to survive sample storage conditions reasonably well [63, 64]. Another convenient characteristic is that almost every biological tissue sample or body fluid such as blood or saliva can be used for analysis of DNA methylation and other epigenetic marks [22, 65, 66]. This robustness makes the application of epigenetic biomarkers in a clinical environment attractive.
Over the recent years, it has become clear that epigenetic features contain a high predictive value during various stages of disease. These analyses thus far mainly focused on DNA methylation. DNA methylation has been shown to be informative for disease diagnosis, prognosis, and stratification. Some of the DNA methylation-based epigenetic biomarkers, such as the methylation status of VIM and SEPT9 for colorectal cancer, SHOX2 for lung cancer, and GSTP1 for prostate cancer, are in clinical use and diagnostic kits are commercially available [67–71]. In case of one of the best characterized biomarkers, GSTP1, a meta study (mainly using prostatectomy tissue or prostate sextant biopsies) showed that hypermethylation of the promoter allows to diagnose prostate cancer with a sensitivity of 82% and a specificity of 95% [72]. Importantly, the use of multiple DNA methylation biomarkers (combining hypermethylation of GSTP1, APC, RASSF1, PTGS2, and MDR1) resulted in a sensitivity and specificity of up to 100% [73]. See Heyn and Esteller [74] for a recent comprehensive overview of DNA methylation biomarkers and its potential use in the clinic. In addition to its diagnostic potential, it has been well established that DNA methylation is informative for patient prognosis in terms of tumor recurrence and overall survival. For example, the hypermethylation of four genes, CDKN2A, cadherin 13 (CDH13), RASSF1, and APC, can be used to predict tumor progression of stage 1 non-small cell lung cancer (NSCLC) [75]. In addition to disease prognosis, DNA methylation has been shown to be valuable for patient stratification to predict response to chemotherapeutic treatment. A well-known example is hypermethylation of MGMT in glioblastoma, which render the tumors sensitive to alkylating agents [76, 77] such as carmustine and temozolomide.
Together, these examples show the power and feasibility of using epigenetic features, and in particular DNA methylation, as biomarkers. Epigenetic biomarkers are complementary to genetic biomarkers. Whereas genetic mutations can (among others) disrupt protein function due to amino acid changes, epigenetic alterations can de-regulate mechanisms such as transcriptional control, leading to the inappropriate silencing or activation of genes. Notably, epigenetic changes occur early and at high frequencies in a wide range of diseases including cancer [78]. It has been suggested that epigenetic alterations occur at higher percentages of tumors than genetic variations, resulting in a higher sensitivity in the detection of tumors [79].
Genome-wide epigenetic profiling for DNA methylation biomarkers
Thus far, the discovery of the epigenetic biomarkers mostly relied on targeted approaches using individual gene loci known or suspected to be involved in the etiology or progression of the disease or other phenotype under study. Despite the challenges in the identification of biomarkers using such approaches, this yielded a number of important epigenetic biomarkers. However, these approaches require a priori knowledge for the selection of candidate biomarkers.
In order to perform unbiased screens in the exploratory phase of biomarker discovery, genome-wide profiling technologies have spurred molecular biomarker discovery (detailed information on epigenomic profiling assays is presented in Table 1). Using these technologies, the entire (epi)genome can be interrogated for potential biomarkers by comparing healthy versus diseased cells/tissue, malignant versus non-malignant tumors, or drug-sensitive versus drug-resistant tumors. This enables selection of candidate biomarkers that are most informative for disease detection, prognosis, or stratification. The use of genome-wide screens furthermore enables to detect and evaluate combinations of (many) candidate loci, which often results in increased sensitivity and specificity of the biomarker. Importantly, the identification of individual genomic loci or genes as biomarkers from large datasets requires robust statistical testing such as multiple-testing correction (although traditional tests like the Bonferroni correction are over-conservative since there is often correlation between loci, i.e., they are not independent) or stringent false discovery rate (FDR) control (for example, by the Benjamini–Hochberg procedure) [80–82]. To define sets of biomarkers from large dataset, alternative statistical methods (such as sparse principle component analysis (PCA) or sparse canonical correlation analysis (CCA) [83, 84]) are available as well. In light of (i) challenges with the experimental setup when using patient material, (ii) costs, and (iii) the extensive computational analysis associated with the exploratory phase of biomarker discovery, genome-wide screens are often performed on relatively small cohorts. Independent of the (statistical) methods used, it is essential to validate (sets of) candidate biomarkers in follow-up studies on large cohorts using targeted epigenetic approaches before potential application in the clinic [85].
Recent years have seen an increasing number of studies using genome-wide epigenetic profiling to predict disease outcome. For a range of tumors, including childhood acute lymphoblastic leukemia [86], kidney cancer [87], NSCLC [88], rectal cancer [89], cervical cancer [90, 91], breast cancer [92, 93], and glioblastoma [94], DNA methylome analysis has been shown to be of prognostic value. Most of these studies define changes in DNA methylation at single sites or at small subsets of sites that represent potential disease signatures. Although these studies are often restricted to a subset of CpGs within the genome and mostly rely on relatively small sample sizes, they show the power of performing genome-wide biomarker screens.
Currently, the most popular platform used in the exploratory phase of DNA methylation biomarker discovery represents the Infinium HumanMethylation450 BeadChip array (further referred to as “450K array”; see a short explanation of the 450K array within Table 1). The probes on the 450K array mainly represent functional CpG islands and functional elements such as promoters, enhancers, and TF binding sites. Main advantages of the 450K array for the detection of DNA methylation as compared to other DNA methylation platforms include (i) its high reproducibility, (ii) the straightforward analysis methods, (iii) the large number of samples that have been profiled using the 450K array thus far (which can be used for comparative purposes), and (iv) the relatively low costs. A disadvantage, like with all bisulfite-based methods (unless combined with additional chemical procedures), is that the 450K array is unable to distinguish between DNA methylation and DNA hydroxymethylation. Hydroxymethylated cytosines represent an intermediate step during demethylation of methylated cytosines but is relatively stable and is therefore likely to have specific biological functions as well [95]. It should be noted that levels of DNA hydroxymethylation are generally much lower as compared to levels of DNA methylation (for example, DNA hydroxymethylation levels are >95% lower in case of peripheral blood mononuclear cell (PBMC) [96]). A further disadvantage of the 450K array is that genetic differences between samples might result in false positives, in particular since a subset of probes on the 450K array target polymorphic CpGs that overlap SNPs [97, 98]. For association studies using large cohorts, computational methods (based on principle components) have been developed to account for population stratification resulting from differences in allele frequencies [98–100].
To enable robust screening for a (set of) potential biomarker(s), most current studies apply the 450K array on up to several hundred samples. To narrow down and validate candidate biomarkers, more targeted DNA methylation assays are used on the same or a very similar-sized cohort [101]. Subsequently, the remaining candidate biomarkers are further validated on larger cohorts using targeted DNA methylation assays that are compatible with routine clinical use, for example, by amplicon bisulfite sequencing [85]. Using this powerful workflow, tumors for which prognostic biomarkers have been identified include rectal cancer [102], breast cancer [103], hepatocellular carcinoma [104], and chronic lymphocytic leukemia (CLL) [105, 106]. Interestingly, using a similar workflow, sets of DNA methylation biomarkers have recently been identified that are prognostic for the aggressiveness of tumors in prostate cancer [107, 108]. Such studies are very important for improving treatment of prostate cancer by avoiding (radical) prostatectomy in cases where careful monitoring of the tumor over time is preferred.
Biomarkers other than DNA methylation
The majority of epigenetic biomarkers identified thus far involve changes in DNA methylation. However, in light of the various types of epigenetic misregulation associated with diseases, changes in epigenetic features other than DNA methylation are likely to become powerful molecular biomarkers as well. ChIP-Seq profiling has revealed prominent differences in binding sites of post-translational histone modifications and other proteins between healthy and cancer tissue, both in leukemia as well as in solid tumors. For example, localized changes in H3 acetylation have been reported in leukemia (see, for example, Martens et al. [109] and Saeed et al. [110]). For solid tumors, differential estrogen receptor (ER) binding and H3K27me3 as determined by ChIP-Seq has been shown to be associated with clinical outcome in breast cancer [111, 112]. Also, androgen receptor (AR) profiling predicts prostate cancer outcome [113]. A recent study identified tumor-specific enhancer profiles in colorectal, breast, and bladder carcinomas using H3K4me2 ChIP-Seq [114]. Next to ChIP-Seq, DNAseI hypersensitivity assays have identified tumor-specific open chromatin sites for several types of cancer (see, for example, Jin et al. [115]). In terms of chromatin conformation, it has recently been shown that disruption of the 3D conformation of the genome can result in inappropriate enhancer activity causing mis-expression of genes including proto-oncogenes [116, 117]. These examples show that, besides DNA methylation, changes in (i) protein binding sites (including post-translational histone modifications), (ii) accessible (open) chromatin, and (iii) the 3D conformation of the genome represent epigenetic features that are potential effective biomarkers (Fig. 1). The near absence of biomarkers based on these epigenetic features is mainly due to practical reasons. ChIP-Seq as well as other comprehensive epigenetic profiling technologies traditionally require (much) more input material, up to 1 × 106 cells or more, to obtain robust results as compared to DNA methylation profiling (Table 1). This is particularly challenging for (banked) patient samples, which are often available in small quantities that might not be compatible with epigenetic profiling other than DNA methylation profiling. Also, profiling of such epigenetic features often require elaborate and delicate workflows (Table 1). Hence, quantitation and reproducibility of ChIP-Seq and other epigenetic profiling assays besides DNA methylation profiling are challenging. Furthermore, DNA methylation profiling is better compatible with (archived) frozen or fixed samples.
However, the last 2 years have seen a spectacular progress in miniaturization of epigenetic profiling assays. In various instances, this included automation of (part of) the workflow, improving the robustness of the assays and its output. Also, improved workflows for epigenetic profiling of frozen or fixed samples have been reported. Although this involved proof-of-concept studies in basic research settings, these efforts are likely to have significant impetus on genome-wide epigenetic screens for candidate biomarkers. The remainder of this review will provide an overview of the current status of genome-wide epigenetic profiling and the technological advances that facilitate miniaturization, automation, and compatibility with preserved samples.
New developments in epigenetic profiling: compatibility with preservation methods
Most epigenetic profiling assays have been developed using fresh material in order to preserve the native chromatin architecture. However, epigenetic biomarker screens require the use of patient-derived clinical samples that are generally processed to preserve the samples as well as to allow convenient sample handling, for example, for sectioning of biopsies. Also, samples present in biobanks are fixed to allow long-time storage. In particular for retrospective studies, epigenetic profiling technologies that are applied for biomarker screens should therefore be compatible with methodologies that are routinely used for sample preservation: freezing and chemical fixation (in particular FFPE fixation) [118].
Freezing
Freezing of tissue specimens is typically performed by snap-freezing with subsequent storage at −80 °C or in liquid nitrogen [119]. Freezing seems to maintain nuclear integrity and chromatin structure very well (Fig. 2). WGBS [120], ChIP-Seq [121–123], ATAC-Seq [124, 125], and DNAseI-Seq [126, 127] all have been shown to be compatible with frozen cells or tissues.
Fig. 2.
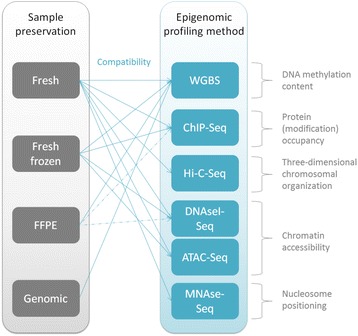
Compatibility of commonly used sample preservation methods with current epigenome profiling assays. A dotted line indicates that these assays would benefit from further optimization
Chemical Fixation (FFPE)
Chemical fixation generally includes overnight crosslinking with formaldehyde at high concentrations (up to 10%), followed by dehydration and paraffin embedding (so-called “FFPE”: formalin-fixed, paraffin-embedded) [128]. Although procedures for FFPE fixation are time-consuming, FFPE fixation has the advantage that samples can be stored at room temperature and that samples can be evaluated by morphology or immunohistochemistry (prior to possible further processing such as epigenetic profiling).
FFPE conditions do not affect DNA methylation, and also formaldehyde and paraffin do not interfere with the WGBS profiling procedure [129]. However, epigenetic assays other than bisulfite-based DNA methylation profiling are cumbersome with FFPE samples (Fig. 2). In case of ChIP-Seq, crosslinking generally occurs in much milder conditions (1% formaldehyde for 10 min) as compared to the harsh conditions used for FFPE fixation [120], which can complicate shearing and epitope accessibility. Pathology tissue (PAT)-ChIP has been reported to prepare FFPE samples for ChIP-Seq by the use of deparaffinization, rehydration, and MNase treatment followed by sonication at high power [130, 131]. However, PAT-ChIP comes with various limitations including the long running time of the protocol (up to 4 days) and the fact that it is not compatible with all ChIP-grade antibodies. Interestingly, some of these issues have been resolved in the very recently developed fixed-tissue (FiT)-Seq procedure, which might open up new avenues for ChIP-Seq profiling of FFPE samples [114]. DNaseI-Seq on FFPE samples has been reported at the expense of a drop in signal-to-noise ratios of around 50% as compared to the use of fresh material [115].
Despite new developments for ChIP-Seq and DNaseI-Seq, this overview shows that DNA methylation is still the most robust of all epigenetic marks for profiling of samples that are processed by freezing or chemical fixation. Although most other epigenetic profiling assays are compatible with frozen samples (at the expense of signal-to-noise ratios for some of the assays), they are generally not or poorly compatible with FFPE specimens (Fig. 2). This also implies that for these assays, it is much more challenging to make use of laser microdissection to select specific regions of interest from specimens for epigenetic analysis, for example, to separate tumor cells from stromal cells [132, 133]. An additional advantage of using DNA methylation for biomarker screening is that, in contrast to the other epigenetic profiling assays discussed, the profiling can be performed on isolated genomic DNA. This enables the use of genomic DNA from clinical DNA banks to be included in DNA methylation biomarker screens.
It should be noted that in contrast to retrospective studies, it might be feasible to use fresh or fresh-frozen patient material for screening in prospective biomarker studies. However, the use of fresh(-frozen) material in these studies could interfere with further development of potential biomarkers if it turns out that these biomarkers are incompatible with (FFPE-)fixed patient material present in the clinic. In all cases, when collecting patient samples for profiling of epigenetic marks, it is important to keep the time between surgical removal and fixation or freezing as short as possible to avoid epitope destruction and/or breakdown of the chromatin. It would therefore be helpful if the procedure time up to fixation would be documented for banked samples, so as to evaluate whether such banked samples are suitable for the epigenetic profiling technology of choice.
New developments in epigenetic profiling: miniaturization and automation
The recent years saw great progress in low-input epigenetic profiling without significantly affecting signal-to-noise ratios (Fig. 3). Also, all main genome-wide epigenetic profiling assays are now compatible with single-cell readouts. An overview of the main technological advances that allowed miniaturization and single-cell readout is described in Table 2. Besides miniaturization, various epigenetic profiling assays, in particular ChIP-Seq, have been (partly) automated to improve reproducibility and to allow higher throughput. In this section, we briefly evaluate these new technological developments with respect to biomarker discovery.
Fig. 3.

Level of comprehensiveness of epigenetic data from global epigenetic profiling assays using an increasing number of cells as input
Table 2.
Overview of the main technological advances that allowed miniaturization and single-cell readout of genome-wide epigenetic profiling assays
| WGBS Conventional WGBS profiling is compatible with a relatively low number of cells (Table 1). Recently, WGBS was adapted to enable single-cell profiling (scBS-Seq; single-cell bisulfite sequencing) [188]. Single cells were captured by fluorescence-activated cell sorting (FACS). To cope with the extensive DNA damage caused by the bisulfite treatment, Smallwood et al. [188] performed tagging of the DNA fragments with sequencing adaptors after bisulfite conversion as developed by Miura et al. [189], instead of before conversion as performed in traditional WGBS. scBS-Seq allows to get coverage of up to 48.4% over all CpGs. A subsequent study by Farlik et al. [190] used a similar approach for scBS-Seq but adapted it such that the whole process of library preparation following bisulfite treatment and cleanup is performed in a single tube, minimizing DNA loss and reducing contamination risk [190]. |
| ChIP-Seq. Traditionally, ChIP-Seq requires a large number of cells (at least several hundred thousands). However, improvements in the sample preparation procedure to prepare the ChIPped DNA for sequencing allowed to perform ChIP-Seq profiling on 1 × 104 cells for H3K4me3 and H3K27me3 [191–194] and recently even 200 cells for H3K4me3 [195]. Also, the use of MNase for chromatin digestion has been shown to facilitate low-input ChIP-Seq [196, 197]. An alternative approach for downscaling the number of cells for ChIP-Seq is to use carrier material, such as inert proteins and/or mRNA, which do not interfere with the ChIP-Seq procedure but increase efficiency and sensitivity [198]. This strategy allowed to perform ChIP-Seq on the TF Estrogen Receptor (ER) on 1 × 104 cells. Similarly, bacterial DNA has been used as carrier, although this comes at the cost of increased sequencing depth as the bacterial DNA remains included in the sequencing procedure [199]. In more recent studies aiming to obtain ChIP-Seq information from low numbers of cells, barcodes or adaptors for sequencing are ligated or transposed before or during the ChIP procedure instead of after the ChIP. ChIPmentation, the use of transposase to add adaptors to DNA during the ChIP, was shown to be highly efficient and compatible with as low as 1 × 103 cells [200]. An alternative recent strategy for low-input ChIP-Seq relies on the addition of histone octamers during ChIP to outcompete unspecific binding [201]. Ligation of adaptors before the ChIP (indexing-first ChIP (iChIP)) allows to pool multiple samples during the ChIP-Seq procedure, after which the sequence tags can be mapped back to the original sample [202, 203]. Bernstein and his coworkers developed this further using direct adaptor ligation on MNAse treated chromatin in an automated droplet-based microfluidic device to obtain single-cell resolution for H3K4me3 and H3K4me2 ChIP-Seq [152]. Efficient immunoprecipitations were performed by pooling 100 single cells with the addition of carrier material that is not amplified during preparation of the ChIPped DNA for sequencing. This workflow enables the profiling of thousands of individual cells in parallel, mainly due to the continuous flow of droplets that is being generated to capture the individual cells (Fig. 4). Inherent to single-cell enrichment techniques, the coverage per single cell is sparse (~1000 unique reads per cell) and does not allow comprehensive analysis of protein binding sites in individual cells. However, the single-cell ChIP-Seq was shown to be very powerful in identifying functionally-relevant subpopulations within embryonic stem cells [152]. |
| ATAC-Seq/DNAseI-Seq. ATAC-Seq has been downscaled to less than 200 cells [135, 175]. Next to this, Buenrostro et al. [151] reported ATAC-Seq to be compatible with single-cell profiling by performing transposition on single cells captured on a commercial microfluidics platform (Fluidigm C1; Fig. 4). This allows capturing of 96 single cells in parallel and subsequent processing steps toward a full library ready for sequencing. Together, this automated epigenetic platform represents the first of its kind in which a single-cell suspension is loaded on a platform that subsequently generates a full library for sequencing without any further manual intervention. An alternative approach for single-cell ATAC-Seq has been developed by Cusanovich et al. [204]. They performed the transposase reaction in intact nuclei on small pools, while simultaneously performing indexing of the tagged sides. Pooling followed by redistribution of the small cell numbers combined with the introduction of a second barcode for each cell allowed to map back the tags obtained after sequencing to individual cells. The advantage of this strategy is that it allows for a higher throughput as shown by the 15,000 individual cells profiled by Cusanovich et al. [204]. Recently, also DNaseI-Seq has been further developed to facilitate low-input profiling (between 1 × 102 and 1 × 104 cells) as well as single-cell profiling [115]. Critically, after FACS sorting of single cells followed by lysis and DNaseI digestion, large amounts of circular plasmid DNA were added during further sample preparation for sequencing. The genomic coverage of both DNaseI-Seq and ATAC-Seq in single cells is inherently low due to the fact that each cell only contains two copies of the genome. The average number of sequence reads per cell was about 317,000 reads for DNaseI-Seq [115] and 73,000 [151] or 35,000 [204] reads for ATAC-Seq after deep sequencing of the libraries. Clearly, these numbers of sequencing reads do not allow to investigate individual genomic loci within single cells. Rather, the computational analysis in both studies makes use of DNaseI hypersensitive sites (DHSs) determined in pools of cells in order to call DHSs in single cells. Despite this limitation, the single-cell chromatin accessibility assays were shown to be powerful in identifying cell-type specific transcription factors, and their variation on genomic binding within individual cells on a global scale [115, 151]. |
| 4C-Seq and HiC-Seq. 4C-Seq and HiC-Seq are relatively new techniques [182–184], for which optimization to low cell numbers have not been extensively reported yet. However, it has been shown that HiC-Seq is compatible with single-cell profiling by performing in-nuclei DNA digestion and ligation and subsequent manual picking of individual nuclei. Using single-cell HiC-Seq it was shown that the large megabase-sized TADs that have been identified in large populations of cells are also present in single cells [205, 206]. Furthermore, single-cell HiC-Seq was shown to be very powerful to reconstruct chromosome folding. Although providing information at single-cell resolution, the single-cell HiC-Seq protocol requires 1 × 107 cells as starting material to facilitate the early steps of the protocol. Inherent to the HiC-Seq protocol, the resolution obtained in individual cells is low. Currently, between 10,000 and 30,000 ligation events are profiled per cell [205]. |
Miniaturization of epigenetic profiling
As summarized in Table 2, Fig. 3, and Table 3, the amount of cells required for three of the main epigenetic profiling assays is currently well compatible with amounts present in patient-derived specimens or amounts present in banked patient samples. For bisulfite-based DNA methylation profiling, a starting amount of 7.5 × 104 cells for the 450K array or 3 × 103 cells for WGBS/RRBS is sufficient to obtain high-quality genome-wide profiles. For ChIP-Seq, the minimum amount of starting material is highly dependent on the protein to be profiled and the antibody that is used [134]. Although both histone modification and TF binding sites (such as ER [111, 112]) are potentially powerful as biomarker, the minimum number of cells required for histone modification profiling (~1–5 × 104 cells) is much more compatible with patient samples than the number of cells required for TF profiling (generally 1 × 105 cells or more; Tables 1 and 2). ATAC-Seq and DNAseI-Seq are compatible with as low as 200 cells and 1 × 103 cells, respectively (Table 2) [115, 135]. Together, this shows that the input requirements for bisulfite-based DNA methylation profiling, ChIP-Seq (in particular for histone modifications), and ATAC-Seq/DNAseI-Seq are well compatible with most clinical samples. The minimum number of cells currently required for 4C-Seq and HiC-Seq, at least 1 × 107 cells, is currently too high for clinical use.
Table 3.
Overview of the number of cells required for the various epigenetic profiling assays
| Epigenomic profiling method | Cell input using traditional profiling on bulk cells to obtain optimal data quality | Cell input using miniaturized profiling | Compatible with single-cell readout | Compatible with single cell as input |
|---|---|---|---|---|
| WGBS | 3 × 103 | 3 × 103 | ✓ | ✓ |
| ChIP-Seq | 0.5–5 × 106* | 1 × 104 or more | ✓ | |
| DNAseI-Seq | 1–10 × 106 | 1 × 103 | ✓ | ✓ |
| ATAC-Seq | 5 × 104 | 2 × 102 | ✓ | ✓ |
| Hi-C-Seq | 2.5 × 107 | 1 × 107 | ✓ | |
| MNase-Seq | 1 × 106 | – |
*Depending on histone modification/TF
Interestingly, all main epigenetic profiling assays can now provide single-cell readouts (Table 2, Table 3). The possibility to assay individual cells within populations allows interrogation of heterogeneity which in “bulk” profiling would be averaged. This is very informative for clinical samples which can be highly heterogeneous [136]. Single-cell profiling has been shown to be powerful in obtaining molecular signatures of heterogeneous populations that shift in cell type composition [137]. As such, an important clinical application of single-cell profiling is to screen for resistant versus non-resistant cells after drug treatment [138] or to monitor disease progression [139]. In terms of biomarker discovery, the use of single-cell assays will allow to screen for cell types that are most informative for disease stratification. Also, the level of heterogeneity as measured by single-cell studies might possibly by itself be informative for disease stratification. From a practical perspective, epigenetic profiling of single cells is challenging. Since one cell only contains two copies for each genomic locus to be assayed, any loss of material during washing or enrichments steps such as immunoprecipitations will significantly impact the outcome of the assay. Similarly, background signals are hard to distinguish from true signal. One of the main strategies to account for false negative signals as well as for aspecific background is to include a large number of cells in single-cell epigenetic assays to enable proper statistics. However, this results in (very) large datasets, for which computational and statistical analysis are generally challenging. For single-cell epigenetic profiling of clinical samples, there are two additional issues to consider: (i) generation of single-cell suspensions from patient samples might be challenging, and (ii) the number of cells required as input for single-cell epigenetic profiling is generally higher than for miniaturized epigenetic profiling in order to enable capturing of single cells (Fig. 4), which might affect compatibility with patient samples. Since single-cell technologies emerged very recently, further developments in technology (to increase sensitivity and specificity) and in computational analysis (for more robust statistical testing and model development) are to be expected. Once single-cell epigenetic profiling has fully matured, it will be very powerful for biomarker discovery in heterogeneous cell populations such as human blood samples and biopsies.
Fig. 4.

State-of-the-art microfluidic systems capable of performing single-cell epigenomic profiling. Simplified representation of a Fluidigm C1 integrated fluidic circuit design capable of capturing 96 single cell for ATAC-Seq [151] (a). Droplet microfluidic workflow applying barcoding of single-cell chromatin to enable pooling for subsequent ChIP experiments [152] (b). Alternatively, single cells can be captured by FACS (not shown)
Automation of epigenetic profiling
The use of genome-wide epigenetic profiling for biomarker discovery strongly benefits from automated procedures that are compatible with upscaling to facilitate large-scale screens. Main advantages of automation include (i) a reduction in variability and batch effects, both of which are frequently observed in epigenetic profiling, (ii) increased throughput, (iii) reduced procedure and/or hands-on time, and (iv) lower error rates. In light of the limited number of cells within clinical samples, a combination of automation and miniaturization is likely to be beneficial in most cases. This comes with the additional advantage of reduced reagent cost, which can be substantial considering the high costs associated with epigenetic profiling. It should be noted that epigenetic profiling thus far is mainly being performed within basic research settings on relatively small sample sizes, which are well compatible with manual handling. Therefore, most automated platforms have been developed recently to cope with the increasing sample sizes and the profiling of more challenging (clinical) samples. In this section, we focus on automation of bulk and miniaturized epigenetic profiling; information on automation of single-cell technologies is included in Table 2.
Efforts to design automated workflows for epigenetic profiling have mainly been focused on ChIP-Seq and to a lesser extent on DNA methylation profiling. This can be explained by the fact that DNA methylation profiling, and chromatin profiling (ATAC-Seq/DNAseI-Seq) as well, is relatively straightforward and therefore well compatible with manual handling. Considering 4C-Seq and HiC-Seq, these are relatively new technologies for which automated workflows have not been reported yet. For DNA methylation profiling, (parts of) the workflow for MBD-Seq, MethylCap-Seq, and MeDIP-Seq have been designed on custom-programmed robotic liquid handling systems [140–142]. For ChIP-Seq, immunoprecipitations and subsequent sample preparation for sequencing have been designed on the same or similar robotic systems [143–146]. However, these robotic workflows require large amounts of starting material in the range of 1 × 106 cells or more. Clearly, with such input requirements, these platforms are not readily compatible with biomarker discovery.
More recently, miniaturized automated platforms have been described for ChIP-Seq using PDMS (polydimethylsiloxane)-based microfluidic devices that have been designed to perform automated immunoprecipitations. These platforms allow to perform ChIP-Seq using as low as 1 × 103 cells [147] or 100 cells [148] due to very small reaction volumes, providing proof-of-principle that automated low-input ChIP-Seq profiling is feasible. However, to facilitate high-throughput profiling, it would be important to increase the number of parallel samples to be profiled, as currently these platforms contain a maximum of assaying four samples in parallel [147, 148]. Furthermore, integration with the labor-intensive DNA library preparation procedure would be desirable; stand-alone library preparation platforms on microfluidic devices have been reported [149, 150]. For DNA methylation profiling, various commercial low-input bisulfite conversion kits have been shown to be compatible with automation. However, a fully automated miniaturized DNA methylation profiling platform has not been reported yet.
Conclusions
Biomarkers are highly valuable and desirable in a wide range of clinical settings, ranging from pharmacodynamics to monitoring treatment. Here, we have provided an overview of recent developments within genome-wide profiling technologies that may enable future large-scale screens for candidate epigenetic biomarkers. When comparing compatibility with miniaturization, automation and tissue preservation methods, bisulfite-based DNA methylation profiling is currently by far superior to other epigenetic profiling technologies for large-scale biomarker discovery. DNA methylation assays are technically less challenging than most other profiling assays, as it is not dependent on delicate enzymatic reactions or on immunoprecipitation, but on chemical conversion. A critical advantage of DNA methylation profiling over other assays is that is not affected by freezing or chemical fixation, and therefore very well compatible with (archived) clinical samples. DNA methylation profiling has the additional advantage that it requires a relatively low number of cells as input. In line with these advantages, most of the epigenetic biomarkers that have been identified thus far involve changes in DNA methylation.
Despite the advantages of DNA methylation, various other epigenetic marks are promising biomarkers. Histone-modifying enzymes are frequently mutated in a range of diseases, often directly affecting epigenetic patterns of post-translational histone modifications. The main methodology to profile these post-translational histone modifications is ChIP-Seq. ChIP-Seq is challenging on samples containing low numbers of cells as well as on archived samples, often resulting in variability in signal-to-noise ratios. However, in view of the continuous improvements in ChIP-Seq procedures for (ultra-)low input samples and for fixed samples, large scale ChIP-Seq-based screens for candidate biomarkers is likely to become feasible in the near future. These screens might benefit from the automated ChIP(-Seq) platforms that are currently being developed. The development of such automated platforms will also facilitate robust integration of ChIP assays as a diagnostic tool in clinical practice.
Of the remaining technologies discussed in this paper, ATAC-Seq and DNAseI-Seq seem most compatible with profiling of clinical samples, requiring as low as several hundred cells as input. Both ATAC-Seq and DNAseI-Seq are compatible with frozen patient samples [125–128], while DNAseI-Seq was recently successfully applied on FFPE samples [115]. However, as compared to DNAseI-Seq, the workflow of ATAC-Seq is much more straightforward as the adaptors for sequencing are inserted as part of the transposition. Also, at least for single-cell ATAC-Seq, a fully automated platform has been developed [151]. For biomarker discovery, compatibility of ATAC-Seq with FFPE samples would be highly desirable, as this would enable to include clinical samples from biobanks in large-scale ATAC-Seq profiling studies. This might be achieved by incorporating critical steps from the FFPE-compatible DNAseI-Seq. Although the use of open chromatin as an epigenetic biomarker has been rare thus far, the flexibility and ease of the recently developed ATAC-Seq (and possibly DNAseI-Seq) will undoubtedly boost the use of open chromatin in clinical research and clinical practice.
Together, this review shows that genome-wide epigenetic profiling technologies have very rapidly matured over the past decade. While originally these technologies were only compatible with large numbers of (in vitro cultured) cells, most of these can now be applied on samples containing very low numbers of primary cells down to single cells. Combined with an increasing number of sophisticated workflows and (automated) platforms, this will pave the way for large-scale epigenetic screens on clinical patient material. Such screens are essential to fill the need for new biomarkers for disease diagnosis, prognosis, and selection of targeted therapies, necessary for personalized medicine.
Acknowledgements
We thank members of the Marks’ and Stunnenberg laboratories for discussion and insight. We thank Dr. Arjen Brinkman, Dr. Richard Bartfai, and Dr. Joost Martens for their input on the manuscript. We thank COST action “CM1406- Epigenetic Chemical Biology” for their financial support regarding the publication fee.
Funding
Research in the group of HGS is supported by the European Union grant BLUEPRINT (FP7/2011: 282510) and ERC-2013-ADG-339431 “SysStemCell.” Research in the group of HM is supported by a grant from the Netherlands Organization for Scientific Research (NWO-VIDI 864.12.007).
Availability of data and materials
Not applicable.
Authors’ contributions
HM prepared the main text and tables with help of RD and HGS. RD prepared the figures. All authors contributed to the content. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 3D
Three-dimensional
- 450K array
Infinium HumanMethylation450 BeadChip array
- 4C
Circular chromosome conformation capture
- AR
Androgen receptor
- ATAC
Assay for transposase-accessible chromatin
- CCA
Canonical correlation analysis
- ChIP
Chromatin immunoprecipitation
- CLL
Chronic lymphocytic leukemia
- CpG
CG dinucleotide
- DHS
DNAseI hypersensitive site
- DNA
Deoxyribonucleic acid
- DNAseI
Desoxyribonuclease 1
- ER
Estrogen receptor
- FACS
Fluorescence-activated cell sorting
- FDR
False discovery rate
- FFPE
Formalin-fixed paraffin-embedded sample
- FiT-Seq
Fixed-tissue ChIP-Seq
- HDAC
Histone deacetylase
- IHEC
International Human Epigenome Consortium
- MBD
Methyl-CpG binding domain protein-enriched
- MeDIP
Methylation DNA immunoprecipitation
- MethylCap
Methylated DNA capture
- MNAse
Micrococcal nuclease
- NGS
Next-generation sequencing
- NIH
National Institutes of Health
- NSCLC
Non-small cell lung cancer
- PAT-ChIP
Pathology tissue chromatin immunoprecipitation
- PBMC
Peripheral blood mononuclear cell
- PCA
Principle component analysis
- PDMS
Polydimethylsiloxane
- PRC
Polycomb repressive complex
- PSA
Prostate-specific antigen
- RNA
Ribonucleic acid
- RRBS
Reduced representation bisulfite sequencing
- SAHA
Suberanilohydroxamic acid (Vorinostat)
- scBS
single-cell bisulfite sequencing
- -Seq
followed by sequencing
- SNP
Single-nucleotide polymorphism
- TAD
Topologically associating domain
- TF
Transcription factor
- WGBS
Whole-genome bisulfite sequencing
Contributor Information
René A. M. Dirks, Email: R.Dirks@ncmls.ru.nl
Hendrik G. Stunnenberg, Email: H.Stunnenberg@ncmls.ru.nl
Hendrik Marks, Email: H.Marks@ncmls.ru.nl.
References
- 1.Brower V. Biomarkers: portents of malignancy. Nature. 2011;471(7339):S19–21. doi: 10.1038/471S19a. [DOI] [PubMed] [Google Scholar]
- 2.Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–52. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 3.Biomarkers Definitions Working Group Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 4.Hu ZZ, Huang H, Wu CH, Jung M, Dritschilo A, Riegel AT, et al. Omics-based molecular target and biomarker identification. Methods Mol Biol (Clifton, NJ) 2011;719:547–71. doi: 10.1007/978-1-61779-027-0_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poste G. Bring on the biomarkers. Nature. 2011;469(7329):156–7. doi: 10.1038/469156a. [DOI] [PubMed] [Google Scholar]
- 6.Wichmann HE, Kuhn KA, Waldenberger M, Schmelcher D, Schuffenhauer S, Meitinger T, et al. Comprehensive catalog of European biobanks. Nat Biotechnol. 2011;29(9):795–7. doi: 10.1038/nbt.1958. [DOI] [PubMed] [Google Scholar]
- 7.Baker M. Biorepositories: building better biobanks. Nature. 2012;486(7401):141–6. doi: 10.1038/486141a. [DOI] [PubMed] [Google Scholar]
- 8.Knoppers BM, Chisholm RL, Kaye J, Cox D, Thorogood A, Burton P, et al. A P3G generic access agreement for population genomic studies. Nat Biotechnol. 2013;31(5):384–5. doi: 10.1038/nbt.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement B, Yuille M, Zaltoukal K, Wichmann HE, Anton G, Parodi B, et al. Public biobanks: calculation and recovery of costs. Sci Transl Med. 2014;6(261):261fs45. doi: 10.1126/scitranslmed.3010444. [DOI] [PubMed] [Google Scholar]
- 10.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 11.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 12.Song F, Chen P, Sun D, Wang M, Dong L, Liang D, et al. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science (New York, NY) 2014;344(6182):376–80. doi: 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- 13.Finch JT, Klug A. Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci U S A. 1976;73(6):1897–901. doi: 10.1073/pnas.73.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 15.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Jenuwein T, Allis CD. Translating the histone code. Science (New York, NY) 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 17.Dekker J. Gene regulation in the third dimension. Science (New York, NY) 2008;319(5871):1793–4. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marks H, Chow JC, Denissov S, Francoijs KJ, Brockdorff N, Heard E, et al. High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 2009;19(8):1361–73. doi: 10.1101/gr.092643.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks H, Kerstens HH, Barakat TS, Splinter E, Dirks RA, van Mierlo G, et al. Dynamics of gene silencing during X inactivation using allele-specific RNA-Seq. Genome Biol. 2015;16:149. doi: 10.1186/s13059-015-0698-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martens JH, Stunnenberg HG. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 2013;98(10):1487–9. doi: 10.3324/haematol.2013.094243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck S, Bernstein BE, Campbell RM, Costello JF, Dhanak D, Ecker JR, et al. A blueprint for an international cancer epigenome consortium. A report from the AACR Cancer Epigenome Task Force. Cancer Res. 2012;72(24):6319–24. doi: 10.1158/0008-5472.CAN-12-3658. [DOI] [PubMed] [Google Scholar]
- 22.Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30(3):224–6. doi: 10.1038/nbt.2153. [DOI] [PubMed] [Google Scholar]
- 23.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agirre X, Castellano G, Pascual M, Heath S, Kulis M, Segura V, et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015;25(4):478–87. doi: 10.1101/gr.180240.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kretzmer H, Bernhart SH, Wang W, Haake A, Weniger MA, Bergmann AK, et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat Genet. 2015;47(11):1316–25. doi: 10.1038/ng.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol. 2012;13(2):R8. doi: 10.1186/gb-2012-13-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12(1):R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, et al. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86(3):411–9. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6(5):e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duee PH, et al. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25(7):1227–36. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 31.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63(22):7563–70. [PubMed] [Google Scholar]
- 32.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13(2):97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 33.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232(2):227–34. [PMC free article] [PubMed] [Google Scholar]
- 34.Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24. doi: 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, et al. Epigenetic predictor of age. PLoS One. 2011;6(6):e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet. 2009;18(24):4808–17. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38(12):1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13(6):R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 42.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science (New York, NY) 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25(10):1473–81. doi: 10.1101/gr.190629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8(11):1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 45.Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17(5):690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 46.Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60(12):3613–22. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–7. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 2008;57(12):3189–98. doi: 10.2337/db08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 50.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beggs AD, Jones A, El-Bahrawy M, Abulafi M, Hodgson SV, Tomlinson IP. Whole-genome methylation analysis of benign and malignant colorectal tumours. J Pathol. 2013;229(5):697–704. doi: 10.1002/path.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–6. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 55.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–91. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez R, Miller KM. Unravelling the genomic targets of small molecules using high-throughput sequencing. Nat Rev Genet. 2014;15(12):783–96. doi: 10.1038/nrg3796. [DOI] [PubMed] [Google Scholar]
- 57.Qureshi IA, Mehler MF. Developing epigenetic diagnostics and therapeutics for brain disorders. Trends Mol Med. 2013;19(12):732–41. doi: 10.1016/j.molmed.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pellicciari C, Malatesta M. Identifying pathological biomarkers: histochemistry still ranks high in the omics era. Eur J Histochem. 2011;55(4):e42. doi: 10.4081/ejh.2011.e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mishra A, Verma M. Cancer biomarkers: are we ready for the prime time? Cancers. 2010;2(1):190–208. doi: 10.3390/cancers2010190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (New York, NY) 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 61.Moorman AV, Chilton L, Wilkinson J, Ensor HM, Bown N, Proctor SJ. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood. 2010;115(2):206–14. doi: 10.1182/blood-2009-07-232124. [DOI] [PubMed] [Google Scholar]
- 62.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science (New York, NY) 1994;265(5181):2088–90. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 63.Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, et al. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods (San Diego, Calif) 2010;52(3):248–54. doi: 10.1016/j.ymeth.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 64.Wong NC, Ashley D, Chatterton Z, Parkinson-Bates M, Ng HK, Halemba MS, et al. A distinct DNA methylation signature defines pediatric pre-B cell acute lymphoblastic leukemia. Epigenetics. 2012;7(6):535–41. doi: 10.4161/epi.20193. [DOI] [PubMed] [Google Scholar]
- 65.Souren NY, Tierling S, Fryns JP, Derom C, Walter J, Zeegers MP. DNA methylation variability at growth-related imprints does not contribute to overweight in monozygotic twins discordant for BMI. Obesity (Silver Spring, Md) 2011;19(7):1519–22. doi: 10.1038/oby.2010.353. [DOI] [PubMed] [Google Scholar]
- 66.Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62(8):2370–7. [PubMed] [Google Scholar]
- 67.Li YW, Kong FM, Zhou JP, Dong M. Aberrant promoter methylation of the vimentin gene may contribute to colorectal carcinogenesis: a meta-analysis. Tumour Biol. 2014;35(7):6783–90. doi: 10.1007/s13277-014-1905-1. [DOI] [PubMed] [Google Scholar]
- 68.Payne SR. From discovery to the clinic: the novel DNA methylation biomarker (m)SEPT9 for the detection of colorectal cancer in blood. Epigenomics. 2010;2(4):575–85. doi: 10.2217/epi.10.35. [DOI] [PubMed] [Google Scholar]
- 69.Dietrich D, Hasinger O, Liebenberg V, Field JK, Kristiansen G, Soltermann A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn Mol Pathol. 2012;21(2):93–104. doi: 10.1097/PDM.0b013e318240503b. [DOI] [PubMed] [Google Scholar]
- 70.Wu T, Giovannucci E, Welge J, Mallick P, Tang WY, Ho SM. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: a meta-analysis. Br J Cancer. 2011;105(1):65–73. doi: 10.1038/bjc.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes. 2014;5(3):821–64. doi: 10.3390/genes5030821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Neste L, Herman JG, Otto G, Bigley JW, Epstein JI, Van Criekinge W. The epigenetic promise for prostate cancer diagnosis. Prostate. 2012;72(11):1248–61. doi: 10.1002/pros.22459. [DOI] [PubMed] [Google Scholar]
- 73.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64(6):1975–86. doi: 10.1158/0008-5472.CAN-03-3972. [DOI] [PubMed] [Google Scholar]
- 74.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13(10):679–92. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 75.Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358(11):1118–28. doi: 10.1056/NEJMoa0706550. [DOI] [PubMed] [Google Scholar]
- 76.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 77.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 78.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7(1):21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 79.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225–9. [PubMed] [Google Scholar]
- 80.Simmer F, Brinkman AB, Assenov Y, Matarese F, Kaan A, Sabatino L, et al. Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics. 2012;7(12):1355–67. doi: 10.4161/epi.22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gosho M, Nagashima K, Sato Y. Study designs and statistical analyses for biomarker research. Sensors (Basel, Switzerland) 2012;12(7):8966–86. doi: 10.3390/s120708966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bock C. Epigenetic biomarker development. Epigenomics. 2009;1(1):99–110. doi: 10.2217/epi.09.6. [DOI] [PubMed] [Google Scholar]
- 83.Rousu J, Agranoff DD, Sodeinde O, Shawe-Taylor J, Fernandez-Reyes D. Biomarker discovery by sparse canonical correlation analysis of complex clinical phenotypes of tuberculosis and malaria. PLoS Comput Biol. 2013;9(4):e1003018. doi: 10.1371/journal.pcbi.1003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taguchi YH, Murakami Y. Principal component analysis based feature extraction approach to identify circulating microRNA biomarkers. PLoS One. 2013;8(6):e66714. doi: 10.1371/journal.pone.0066714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.BLUEPRINT-consortium Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34(7):726–37. doi: 10.1038/nbt.3605. [DOI] [PubMed] [Google Scholar]
- 86.Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood. 2010;115(6):1214–25. doi: 10.1182/blood-2009-04-214668. [DOI] [PubMed] [Google Scholar]
- 87.Lasseigne BN, Burwell TC, Patil MA, Absher DM, Brooks JD, Myers RM. DNA methylation profiling reveals novel diagnostic biomarkers in renal cell carcinoma. BMC Med. 2014;12:235. doi: 10.1186/s12916-014-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo S, Yan F, Xu J, Bao Y, Zhu J, Wang X, et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC) Clin Epigenetics. 2015;7(1):3. doi: 10.1186/s13148-014-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Exner R, Pulverer W, Diem M, Spaller L, Woltering L, Schreiber M, et al. Potential of DNA methylation in rectal cancer as diagnostic and prognostic biomarkers. Br J Cancer. 2015;113(7):1035–45. doi: 10.1038/bjc.2015.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boers A, Wang R, van Leeuwen RW, Klip HG, de Bock GH, Hollema H, et al. Discovery of new methylation markers to improve screening for cervical intraepithelial neoplasia grade 2/3. Clin Epigenetics. 2016;8:29. doi: 10.1186/s13148-016-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lendvai A, Johannes F, Grimm C, Eijsink JJ, Wardenaar R, Volders HH, et al. Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics. 2012;7(11):1268–78. doi: 10.4161/epi.22301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Legendre C, Gooden GC, Johnson K, Martinez RA, Liang WS, Salhia B. Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clinical epigenetics. 2015;7(1):100. doi: 10.1186/s13148-015-0135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stirzaker C, Zotenko E, Song JZ, Qu W, Nair SS, Locke WJ, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun. 2015;6:5899. doi: 10.1038/ncomms6899. [DOI] [PubMed] [Google Scholar]
- 94.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–59. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 96.Udali S, Guarini P, Moruzzi S, Ruzzenente A, Tammen SA, Guglielmi A, et al. Global DNA methylation and hydroxymethylation differ in hepatocellular carcinoma and cholangiocarcinoma and relate to survival rate. Hepatology. 2015;62(2):496–504. doi: 10.1002/hep.27823. [DOI] [PubMed] [Google Scholar]
- 97.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Houtepen LC, Vinkers CH, Carrillo-Roa T, Hiemstra M, van Lier PA, Meeus W, et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat Commun. 2016;7:10967. doi: 10.1038/ncomms10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol. 2014;38(3):231–41. doi: 10.1002/gepi.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Daca-Roszak P, Pfeifer A, Zebracka-Gala J, Rusinek D, Szybinska A, Jarzab B, et al. Impact of SNPs on methylation readouts by Illumina Infinium HumanMethylation450 BeadChip Array: implications for comparative population studies. BMC Genomics. 2015;16:1003. doi: 10.1186/s12864-015-2202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10(10):949–55. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 102.Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 2013;8(9):921–34. doi: 10.4161/epi.25577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Veldhoven K, Polidoro S, Baglietto L, Severi G, Sacerdote C, Panico S, et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clinical epigenetics. 2015;7(1):67. doi: 10.1186/s13148-015-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology (Baltimore, Md) 2015;61(6):1945–56. doi: 10.1002/hep.27732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 106.Queiros AC, Villamor N, Clot G, Martinez-Trillos A, Kulis M, Navarro A, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2015;29(3):598–605. doi: 10.1038/leu.2014.252. [DOI] [PubMed] [Google Scholar]
- 107.Wu Y, Davison J, Qu X, Morrissey C, Storer B, Brown L, et al. Methylation profiling identified novel differentially methylated markers including OPCML and FLRT2 in prostate cancer. Epigenetics. 2016;11(4):247–58. doi: 10.1080/15592294.2016.1148867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao S, Geybels MS, Leonardson A, Rubicz R, Kolb S, Yan Q, et al. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men with clinically localized prostate cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, et al. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell. 2010;17(2):173–85. doi: 10.1016/j.ccr.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 110.Saeed S, Logie C, Francoijs KJ, Frige G, Romanenghi M, Nielsen FG, et al. Chromatin accessibility, p300, and histone acetylation define PML-RARalpha and AML1-ETO binding sites in acute myeloid leukemia. Blood. 2012;120(15):3058–68. doi: 10.1182/blood-2011-10-386086. [DOI] [PubMed] [Google Scholar]
- 111.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481(7381):389–93. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jansen MP, Knijnenburg T, Reijm EA, Simon I, Kerkhoven R, Droog M, et al. Hallmarks of aromatase inhibitor drug resistance revealed by epigenetic profiling in breast cancer. Cancer Res. 2013;73(22):6632–41. doi: 10.1158/0008-5472.CAN-13-0704. [DOI] [PubMed] [Google Scholar]
- 113.Stelloo S, Nevedomskaya E, van der Poel HG, de Jong J, van Leenders GJ, Jenster G, et al. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol Med. 2015;7(11):1450–64. doi: 10.15252/emmm.201505424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cejas P, Li L, O’Neill NK, Duarte M, Rao P, Bowden M, et al. Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat Med. 2016;22(6):685–91. doi: 10.1038/nm.4085. [DOI] [PubMed] [Google Scholar]
- 115.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528(7580):142–6. doi: 10.1038/nature15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science (New York, NY) 2016;351(6280):1454–8. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161(5):1012–25. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lou JJ, Mirsadraei L, Sanchez DE, Wilson RW, Shabihkhani M, Lucey GM, et al. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin Biochem. 2014;47(4-5):267–73. doi: 10.1016/j.clinbiochem.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shabihkhani M, Lucey GM, Wei B, Mareninov S, Lou JJ, Vinters HV, et al. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin Biochem. 2014;47(4-5):258–66. doi: 10.1016/j.clinbiochem.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Q, Gu L, Adey A, Radlwimmer B, Wang W, Hovestadt V, et al. Tagmentation-based whole-genome bisulfite sequencing. Nat Protoc. 2013;8(10):2022–32. doi: 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- 121.Dahl JA, Collas P. A rapid micro chromatin immunoprecipitation assay (microChIP) Nat Protoc. 2008;3(6):1032–45. doi: 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- 122.Lei H, Liu J, Fukushige T, Fire A, Krause M. Caudal-like PAL-1 directly activates the bodywall muscle module regulator hlh-1 in C. elegans to initiate the embryonic muscle gene regulatory network. Development. 2009;136(8):1241–9. doi: 10.1242/dev.030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Savic D, Gertz J, Jain P, Cooper GM, Myers RM. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenetics Chromatin. 2013;6(1):30. doi: 10.1186/1756-8935-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Milani P, Escalante-Chong R, Shelley BC, Patel-Murray NL, Xin X, Adam M, et al. Cell freezing protocol suitable for ATAC-Seq on motor neurons derived from human induced pluripotent stem cells. Sci Rep. 2016;6:25474. doi: 10.1038/srep25474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scharer CD, Blalock EL, Barwick BG, Haines RR, Wei C, Sanz I, et al. ATAC-Seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Scientific reports. 2016;6:27030. doi: 10.1038/srep27030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cumbie JS, Filichkin SA, Megraw M. Improved DNase-Seq protocol facilitates high resolution mapping of DNase I hypersensitive sites in roots in Arabidopsis thaliana. Plant Methods. 2015;11:42. doi: 10.1186/s13007-015-0087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ling G, Waxman DJ. Isolation of nuclei for use in genome-wide DNase hypersensitivity assays to probe chromatin structure. Methods Mol Biol. 2013;977:13–9. doi: 10.1007/978-1-62703-284-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002;161(6):1961–71. doi: 10.1016/S0002-9440(10)64472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013;23(9):1522–40. doi: 10.1101/gr.156539.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fanelli M, Amatori S, Barozzi I, Minucci S. Chromatin immunoprecipitation and high-throughput sequencing from paraffin-embedded pathology tissue. Nat Protoc. 2011;6(12):1905–19. doi: 10.1038/nprot.2011.406. [DOI] [PubMed] [Google Scholar]
- 131.Fanelli M, Amatori S, Barozzi I, Soncini M, Dal Zuffo R, Bucci G, et al. Pathology tissue-chromatin immunoprecipitation, coupled with high-throughput sequencing, allows the epigenetic profiling of patient samples. Proc Natl Acad Sci U S A. 2010;107(50):21535–40. doi: 10.1073/pnas.1007647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Amatori S, Ballarini M, Faversani A, Belloni E, Fusar F, Bosari S, et al. PAT-ChIP coupled with laser microdissection allows the study of chromatin in selected cell populations from paraffin-embedded patient samples. Methods. 2014;7:18. doi: 10.1186/1756-8935-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schillebeeckx M, Schrade A, Lobs AK, Pihlajoki M, Wilson DB, Mitra RD. Laser capture microdissection-reduced representation bisulfite sequencing (LCM-RRBS) maps changes in DNA methylation associated with gonadectomy-induced adrenocortical neoplasia in the mouse. Nucleic Acids Res. 2013;41(11):e116. doi: 10.1093/nar/gkt230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Furey TS. ChIP-Seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13(12):840–52. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534(7609):652–7. doi: 10.1038/nature18606. [DOI] [PubMed] [Google Scholar]
- 136.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 137.Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25(10):1491–8. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, Chung W, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015;16:127. doi: 10.1186/s13059-015-0692-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–5. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Aberg KA, McClay JL, Nerella S, Xie LY, Clark SL, Hudson AD, et al. MBD-Seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case–control samples. Epigenomics. 2012;4(6):605–21. doi: 10.2217/epi.12.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-Seq. Methods (San Diego, Calif) 2010;52(3):232–6. doi: 10.1016/j.ymeth.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 142.Butcher LM, Beck S. AutoMeDIP-Seq: a high-throughput, whole genome, DNA methylation assay. Methods (San Diego, Calif) 2010;52(3):223–31. doi: 10.1016/j.ymeth.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aldridge S, Watt S, Quail MA, Rayner T, Lukk M, Bimson MF, et al. AHT-ChIP-Seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 2013;14(11):R124. doi: 10.1186/gb-2013-14-11-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Berguet G, Hendrickx J, Sabatel C, Laczik M, Squazzo S, Mazon Pelaez I, et al. Automating ChIP-Seq experiments to generate epigenetic profiles on 10,000 HeLa cells. J Vis Exp. 2014:(94). doi:10.3791/52150. [DOI] [PMC free article] [PubMed]
- 145.Gasper WC, Marinov GK, Pauli-Behn F, Scott MT, Newberry K, DeSalvo G, et al. Fully automated high-throughput chromatin immunoprecipitation for ChIP-Seq: identifying ChIP-quality p300 monoclonal antibodies. Scientific reports. 2014;4:5152. doi: 10.1038/srep05152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wallerman O, Nord H, Bysani M, Borghini L, Wadelius C. lobChIP: from cells to sequencing ready ChIP libraries in a single day. Epigenetics & chromatin. 2015;8:25. doi: 10.1186/s13072-015-0017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shen J, Jiang D, Fu Y, Wu X, Guo H, Feng B, et al. H3K4me3 epigenomic landscape derived from ChIP-Seq of 1,000 mouse early embryonic cells. Cell Res. 2015;25(1):143–7. doi: 10.1038/cr.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cao Z, Chen C, He B, Tan K, Lu C. A microfluidic device for epigenomic profiling using 100 cells. Nat Methods. 2015;12(10):959–62. doi: 10.1038/nmeth.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim H, Jebrail MJ, Sinha A, Bent ZW, Solberg OD, Williams KP, et al. A microfluidic DNA library preparation platform for next-generation sequencing. PLoS One. 2013;8(7):e68988. doi: 10.1371/journal.pone.0068988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tan SJ, Phan H, Gerry BM, Kuhn A, Hong LZ, Min Ong Y, et al. A microfluidic device for preparing next generation DNA sequencing libraries and for automating other laboratory protocols that require one or more column chromatography steps. PLoS One. 2013;8(7):e64084. doi: 10.1371/journal.pone.0064084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, et al. Single-cell ChIP-Seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33(11):1165–72. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105(35):12979–84. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39(2):237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 157.Wolf SF, Jolly DJ, Lunnen KD, Friedmann T, Migeon BR. Methylation of the hypoxanthine phosphoribosyltransferase locus on the human X chromosome: implications for X-chromosome inactivation. Proc Natl Acad Sci U S A. 1984;81(9):2806–10. doi: 10.1073/pnas.81.9.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–5. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 159.Li N, Ye M, Li Y, Yan Z, Butcher LM, Sun J, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods (San Diego, Calif) 2010;52(3):203–12. doi: 10.1016/j.ymeth.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 160.Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, et al. Methylome analysis using MeDIP-Seq with low DNA concentrations. Nat Protoc. 2012;7(4):617–36. doi: 10.1038/nprot.2012.012. [DOI] [PubMed] [Google Scholar]
- 161.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37(8):853–62. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 162.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28(10):1106–14. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6(6):692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 164.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33(18):5868–77. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;15(10):647–61. doi: 10.1038/nrg3772. [DOI] [PubMed] [Google Scholar]
- 166.Sun Z, Wu Y, Ordog T, Baheti S, Nie J, Duan X, et al. Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics. 2014;9(8):1184–93. doi: 10.4161/epi.29676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cuddapah S, Barski A, Cui K, Schones DE, Wang Z, Wei G, et al. Native chromatin preparation and Illumina/Solexa library construction. Cold Spring Harb Protoc. 2009;2009(6):pdb.prot5237. doi: 10.1101/pdb.prot5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kasinathan S, Orsi GA, Zentner GE, Ahmad K, Henikoff S. High-resolution mapping of transcription factor binding sites on native chromatin. Nat Methods. 2014;11(2):203–9. doi: 10.1038/nmeth.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 170.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science (New York, NY) 2007;316(5830):1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 171.Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet. 2014;15(11):709–21. doi: 10.1038/nrg3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tsompana M, Buck MJ. Chromatin accessibility: a window into the genome. Epigenetics & chromatin. 2014;7(1):33. doi: 10.1186/1756-8935-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6(4):283–9. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489(7414):83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, et al. Refined DNase-Seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;11(1):73–8. doi: 10.1038/nmeth.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods in molecular biology (Clifton, NJ) 2012;833:413–9. doi: 10.1007/978-1-61779-477-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42(4):343–7. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science (New York, NY) 2002;295(5558):1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 181.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26(1):11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJ, Zhu Y, et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011;25(13):1371–83. doi: 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, NY) 2009;326(5950):289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–5. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.van de Werken HJ, de Vree PJ, Splinter E, Holwerda SJ, Klous P, de Wit E, et al. 4C technology: protocols and data analysis. Methods Enzymol. 2012;513:89–112. doi: 10.1016/B978-0-12-391938-0.00004-5. [DOI] [PubMed] [Google Scholar]
- 187.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods (San Diego, Calif) 2012;58(3):268–76. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817–20. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40(17):e136. doi: 10.1093/nar/gks454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schonegger A, Klughammer J, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10(8):1386–97. doi: 10.1016/j.celrep.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Adli M, Zhu J, Bernstein BE. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat Methods. 2010;7(8):615–8. doi: 10.1038/nmeth.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ng JH, Kumar V, Muratani M, Kraus P, Yeo JC, Yaw LP, et al. In vivo epigenomic profiling of germ cells reveals germ cell molecular signatures. Dev Cell. 2013;24(3):324–33. doi: 10.1016/j.devcel.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 193.Sachs M, Onodera C, Blaschke K, Ebata KT, Song JS, Ramalho-Santos M. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 2013;3(6):1777–84. doi: 10.1016/j.celrep.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shankaranarayanan P, Mendoza-Parra MA, Walia M, Wang L, Li N, Trindade LM, et al. Single-tube linear DNA amplification (LinDA) for robust ChIP-Seq. Nat Methods. 2011;8(7):565–7. doi: 10.1038/nmeth.1626. [DOI] [PubMed] [Google Scholar]
- 195.Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature. 2016;537(7621):553–7. doi: 10.1038/nature19361. [DOI] [PubMed] [Google Scholar]
- 196.Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, Lorincz MC. An ultra-low-input native ChIP-Seq protocol for genome-wide profiling of rare cell populations. Nat Commun. 2015;6:6033. doi: 10.1038/ncomms7033. [DOI] [PubMed] [Google Scholar]
- 197.Liu X, Wang C, Liu W, Li J, Li C, Kou X, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016;537(7621):558–62. doi: 10.1038/nature19362. [DOI] [PubMed] [Google Scholar]
- 198.Zwart W, Koornstra R, Wesseling J, Rutgers E, Linn S, Carroll JS. A carrier-assisted ChIP-Seq method for estrogen receptor-chromatin interactions from breast cancer core needle biopsy samples. BMC Genomics. 2013;14:232. doi: 10.1186/1471-2164-14-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jakobsen JS, Bagger FO, Hasemann MS, Schuster MB, Frank AK, Waage J, et al. Amplification of pico-scale DNA mediated by bacterial carrier DNA for small-cell-number transcription factor ChIP-Seq. BMC Genomics. 2015;16:46. doi: 10.1186/s12864-014-1195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schmidl C, Rendeiro AF, Sheffield NC, Bock C. ChIPmentation: fast, robust, low-input ChIP-Seq for histones and transcription factors. Nat Methods. 2015;12(10):963–5. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537(7621):548–52. doi: 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science (New York, NY) 2014;345(6199):943–9. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.van Galen P, Viny AD, Ram O, Ryan RJ, Cotton MJ, Donohue L, et al. A multiplexed system for quantitative comparisons of chromatin landscapes. Mol Cell. 2016;61(1):170–80. doi: 10.1016/j.molcel.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, et al. Epigenetics. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science (New York, NY) 2015;348(6237):910–4. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nagano T, Lubling Y, Yaffe E, Wingett SW, Dean W, Tanay A, et al. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat Protoc. 2015;10(12):1986–2003. doi: 10.1038/nprot.2015.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.