

Abstract
Small molecules active in the pathogenic bacterium Staphylococcus aureus are valuable tools for the study of its basic biology and pathogenesis, and many molecules may provide leads for novel therapeutics. We have previously reported a small molecule, 1, which activates endogenous heme biosynthesis in S. aureus, leading to an accumulation of intracellular heme. In addition to this novel activity, 1 also exhibits toxicity towards S. aureus growing under fermentative conditions. To determine if these activities are linked and establish what features of the molecule are required for activity, we synthesized a library of analogs around the structure of 1 and screened them for activation of heme biosynthesis and anaerobic toxicity to investigate structure–activity relationships. The results of this analysis suggest that these activities are not linked. Furthermore, we have identified the structural features that promote each activity and have established two classes of molecules: activators of heme biosynthesis and inhibitors of anaerobic growth. These molecules will serve as useful probes for their respective activities without concern for the off target effects of the parent compound.
Graphical abstract
Staphylococcus aureus is a pathogen of humans, primarily causing skin and soft tissue infections, but can also be responsible for more severe diseases including toxic shock syndrome and necrotizing pneumonias.1 S. aureus has shown a remarkable propensity for developing resistance to antimicrobial therapies, and drug resistant strains have increasingly become a significant threat to public health. Strains continue to demonstrate greater virulence and multidrug resistance in both healthcare and community settings, underscoring the need for the discovery of new antimicrobial compounds and pathways to target.2,3
S. aureus requires iron for normal cellular functions and evasion of the immune system during infection. In addition, the availability of iron controls the expression of many virulence factors.4 In the human host, iron is tightly bound to storage and transport proteins and to heme in hemoproteins. While S. aureus can obtain iron from multiple sources, heme is its preferred iron source during infection,5 and the bacterium utilizes systems to scavenge heme from host hemoproteins, pass it through the cell wall and membrane, and either degrade heme to release free iron or store intact heme for incorporation into its own hemoproteins.6–8
Despite the value of heme as a nutrient iron source, heme is toxic to the bacteria in high concentrations.9 In order to overcome heme toxicity, S. aureus employs a two component system called the heme sensor system (HssRS) to sense heme and activate a response to alleviate heme toxicity. HssS is a transmembrane histidine kinase that senses toxic levels of heme through an undetermined mechanism. Upon activation, HssS autophosphorylates and transfers the phosphate to an aspartate residue of its cognate response regulator, HssR. HssR is a cytoplasmic protein which, upon phosphorylation by HssS, binds the direct repeat of the promoter for the heme regulated transporter (hrtAB), a gene encoding an efflux pump that alleviates heme toxicity (Figure 1).10–12 The mechanism by which HrtAB alleviates heme toxicity is not well understood.
Figure 1.
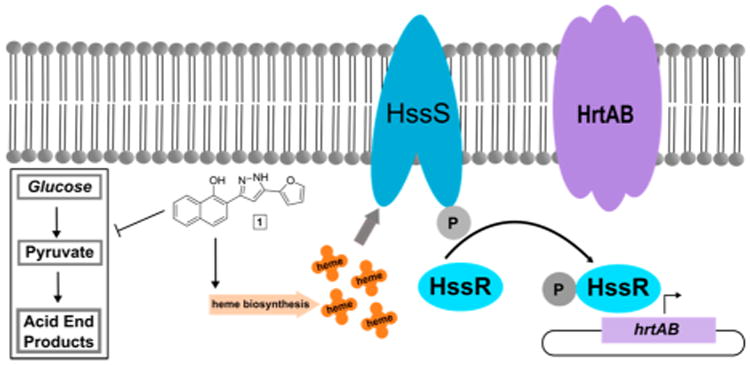
Increase of endogenous heme biosynthesis by compound 1 resulting in the accumulation of heme, which activates the heme sensor system (HssRS) through the sensor histidine kinase HssS. HssS autophosphorylates upon activation and transfers the phosphate to its cognate response regulator, HssR. HssR binds to the promoter of hrtAB, a gene encoding the heme regulated transporter (HrtAB) leading to its expression and resulting in alleviation of heme toxicity. Compound 1 also inhibits the growth of bacteria under anaerobic conditions, potentially through inhibition of a process crucial to fermentation. These two activities may or may not be linked through a single target.
While heme serves as an iron source during infection, it is also an essential prosthetic group for proteins involved in respiration and detoxification of reactive oxygen species, and S. aureus is capable of de novo heme biosynthesis.13 We previously reported a high throughput screen for activators of the HssRS two component system. The most active hit compound, 1, was determined to activate HssRS by increasing endogenous heme biosynthesis, leading to intracellular accumulation of heme sufficient to activate HssRS but not affect overall growth.14
In the course of studying the mechanism of action of 1, we observed that 1 was toxic to S. aureus growing anaerobically, potentially through the inhibition of a process essential during fermentation. S. aureus is a facultative anaerobe capable of generating energy through respiration or fermentation depending on the availability of terminal electron acceptors. Fermentative growth of S. aureus is significantly inhibited by 1 compared to an untreated control or S. aureus treated with 1 under aerobic conditions.14
To the best of our knowledge, there are no other reports of molecules with either of these activities. Activators of heme biosynthesis are potentially useful chemical tools to study the regulation of heme biosynthesis in bacteria as little is known regarding the regulation of heme import and biosynthesis. Small molecules toxic to fermenting bacteria may serve as the basis for a new class of therapeutics. During certain types of infections, S. aureus relies heavily on fermentation to generate energy. In addition, phenotypic variants of S. aureus known as small colony variants (SCVs) are often obligate fermenters and are generally more resistant to current antimicrobial therapies.15–17
While we initially concluded these two activities were linked through a single target, given the promiscuous nature of molecules identified through high throughput screens, it is possible that these phenotypes are the result of interactions of the same molecule with distinct targets in the bacterium. The need for probes with high target specificity as well as activity is paramount in chemical biology.18
We synthesized a library of compounds around the scaffold of 1 and screened for HssRS activation and anaerobic toxicity to determine how chemical modifications affect the two activities. Using this approach, we have effectively decoupled the two activities of 1 and established that it likely has two or more targets. Furthermore, we have identified the structural features of 1 that promote one activity over the other and present derivatives that maintain each activity while introducing higher specificity.
Results and Discussion
Library Synthesis
Our initial efforts toward varying the structure of 1 focused on four regions of the molecule (Figure 2A); removal of the B ring of the naphthol moiety; modification of the phenol by walking to the m- and p-positions (with B ring removed), O-methylation, and replacement with phenol equivalents (amides and sulfonamide); N-methylation of the pyrazole; and replacement of the furan with aromatic, heteroaromatic, and alkyl groups.
Figure 2.
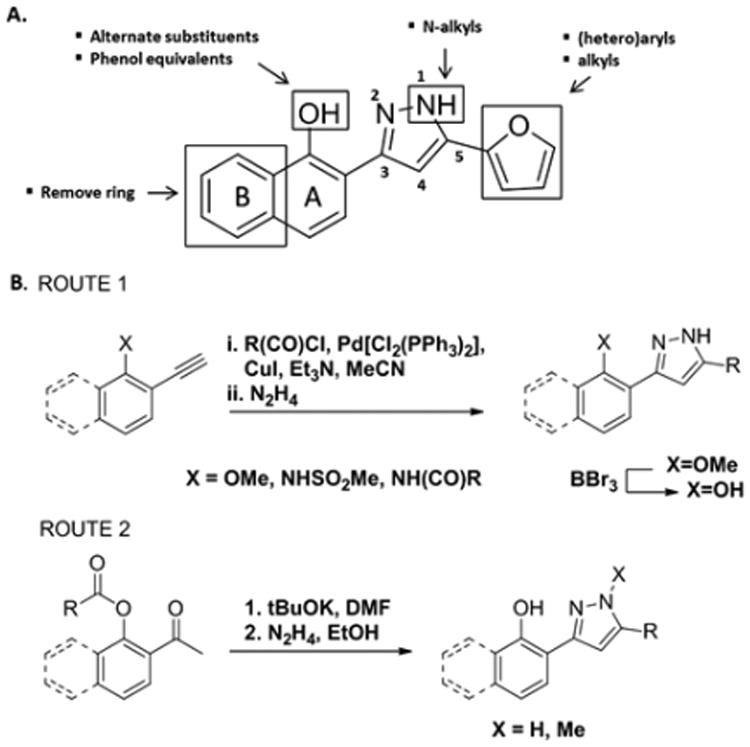
(A) Derivatization strategy to study structure–activity relationships for 1. (B) Synthetic methods utilized to access derivatives for library generation. Et3N = triethyl amine, MeCN = acetonitrile, Me = methyl, tBuOK = potassium tert-butoxide, DMF = N,N-dimethylformamide, EtOH = ethanol.
Many routes to 3,5-substituted pyrazoles have been reported.19 We utilized two routes, (1) cyclocondensation of hydrazine with an alkynone and (2) cyclocondensation of hydrazine with a 1,3-diketone (Figure 2B). The alkynone can be installed by palladium-mediated coupling with the acid chloride containing the corresponding 5-position substituent. This route requires methyl ether protection of the phenol. As such, synthesis of free phenol containing compounds involves subsequent deprotection with boron tribromide. For this reason, this sequence was primarily utilized to synthesize the amide, sulfonamide, and O-methylated derivatives of 1 (Figure 3; 1c, 1d, 1h–1l, 3a, and 3b). The amide derivatives were quite labile in the presence of acid, particularly when substituted with small R groups (Me), and underwent further dehydration to generate 2,5-substituted pyrazolo[1,5-c]quinazolines (Supporting Information, 5a–5c). These compounds will not be discussed due to their divergence from the optimization plan.
Figure 3.
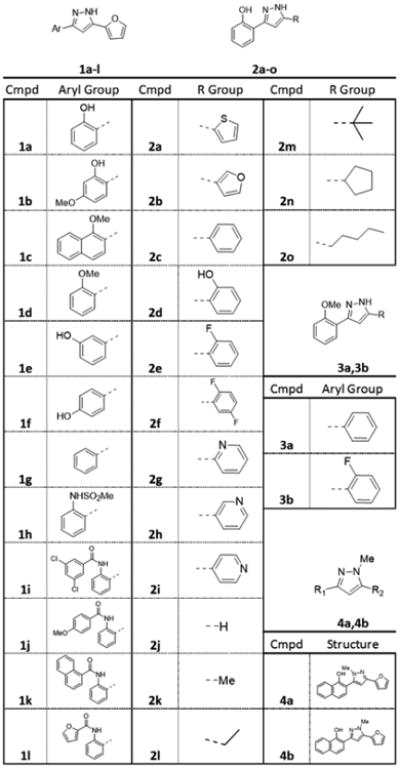
Analogs of 1 synthesized in this work.
Using route 2, the 5-position was diversified by acylating the 2′-phenol of the corresponding acetophenone to generate the ester substrate for an intramolecular Claisen condensation to provide the 1,3-diketone. Compounds 2a–2c, 2e–2i, and 2k–2o were afforded by cyclocondensation with hydrazine. Compound 2d was prepared by acylating 2′-hydroxyacetophone with 2-methoxybenzoyl chloride, carrying the ester through this reaction sequence, and subsequently removing the methyl group using boron tribromide. Compound 2j was synthesized by reacting hydrazine with the corresponding chromone under the same conditions as the cyclocondensation. Using an analogous reaction sequence but acylating 3′- and 4′-hydroxyacetophenone allowed synthesis of 1e and 1f. Compound 1g was prepared by intermolecular Claisen condensation of the lithium enolate of acetophenone, prepared by reaction with LHMDS in toluene, with furoyl chloride to provide the 1,3-diketone. Methylation of the pyrazole nitrogen was achieved by reaction of the intermediate 1,3-diketone with methylhydrazine followed by HPLC separation of the resulting isomeric pyrazoles 4a and 4b.
HssRS Activation
The library was screened for activation of the heme stress response as an indicator for activation of heme biosynthesis in S. aureus. We began by screening at the single point concentration of 50 μM using a previously described reporter assay (Figure 4).11 In this assay, Newman harboring a plasmid with the hrt promoter fused to xylE, a gene encoding a catechol oxidase, is treated with compound for 6 h, the bacteria lysed, and the oxidation of catechol by XylE quantified by spectrophotometry. An increase in absorbance due to the oxidation of catechol indicates elevated levels of xylE transcription and hrt promoter activity. Results are expressed as the fraction of activation of HssRS by the compound compared to 1. An arbitrary cutoff of 0.05 was chosen to define active versus inactive compounds.
Figure 4.
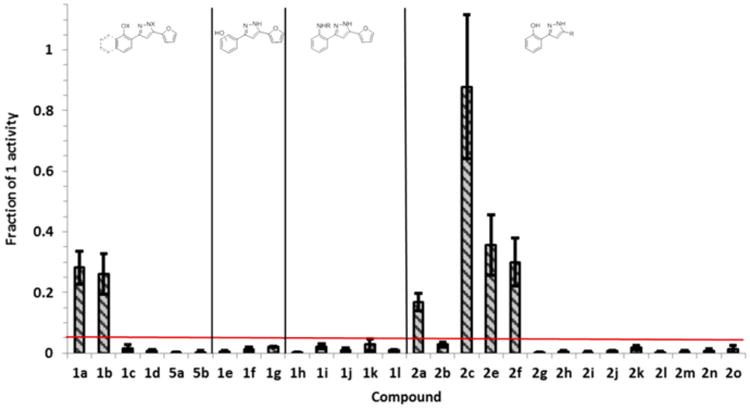
Identification of HssRS activators by XylE assay. Activation of HssRS is presented as the fraction of activation by the indicated derivative relative to 1 at 50 μM. Data are the average of three replicates. Error bars represent one standard deviation from the mean. The red bar indicates the cutoff value for activity (above = active; below = inactive).
Replacement of the naphthol substituent with phenol (1a) or 4-methoxyphenol (1b) resulted in a modest loss of activity compared to 1 but still retained significant ability to activate HssRS. O- and N-methylation of 1 (1c, 4a, and 4b) resulted in complete loss of activity. These data suggest that disruption of the hydrogen bonding properties of 1 has a significant effect on its ability to activate heme biosynthesis. To test this further, several modifications to the phenol of 1a were made. O-Methylation (1d), movement to m- or p-positions (1e and 1f), removal (1g), and replacement with a methyl sulfonamide (1h) or various aryl or heteroaryl amides (1i—1l) resulted in a loss of HssRS activation compared to 1 and 1a. This suggests that the ortho-OH is required for activity.
To explore modification at the 5-position, we chose the o-hydroxyphenyl substituent at the 3-position instead of the o-hydroxynaphthyl of the parent molecule for convenience. Replacement of the furan with hydrogen (2j), alkyl (2k–2o), or pyridyl (2g–2i) substituents eliminates activity compared to 1 and 1a. The furan can be replaced with several aromatic or heteroaromatic groups and retain considerable activity. In particular, replacement of the furan with an unsubstituted phenyl group, 2c, seems to restore activity comparable to 1 without the presence of the naphthol B-ring. However, fluorination of 2c decreases activity though 2e and 2f are comparable to 1a in activity. Substitution with hydroxyl at the ortho position of the phenyl ring of 2c to provide 2d renders the molecule toxic under the single point assay conditions and is therefore not included in Figure 4. However, 2d activates HssRS at lower concentrations, and its activity was explored further.
Concentration response curves for the top six activators from the single point screen (1a, 1b, 2a, 2c, 2e, and 2f) and 2d were generated to determine EC50 values as a measure of compound potency (Table 1, supplemental Figure 2). Compound efficacy is presented as percent activation compared to 1 as displayed in Figure 4 since most compounds reach their Emax at or below 50 μM. Exceptions to this are 2a, which did not reach a plateau below its solubility limit (∼80 μM), 1b which plateaus around 100 μM, and 2d which is toxic in the XylE assay at 50 μM. For the most part, compound potency does not significantly deviate from that of 1. In addition, 2e and 2f maintain comparable potency to 1 while exhibiting a ∼70% drop in efficacy indicating potency and efficacy do not correlate well. These data suggest that efficacy is a more important quantitative descriptor of compound activity.
Table 1. EC50, pEC50, and Efficacy Values for 1 and the Top Six HssRS Activators from the Single Point Screena.
| Cmpd | Structure | EC50(μM) | pEC50 | Efficacy (%) |
|---|---|---|---|---|
| 1 |
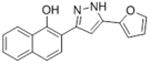
|
11.6 | 4.90 ± 0.37 | 100 |
| la |
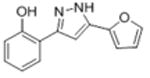
|
30.4 | 4.49 ± 0.083 | 28.2 ± 5.4 |
| lb |
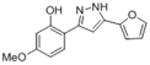
|
50.2 | 4.30 ± 0.20 | 26.2 ± 6.7 |
| 2a |
|
ND | ND | 17.0 ± 2.9 |
| 2c |
|
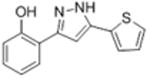
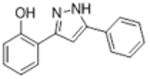
14.6 | 4.66 ± 0.016 | 87.8 ± 23.7 |
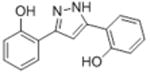
| 2d |
|
5.81 | 5.24 ± 0.076 | ND |
| 2e |
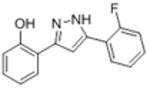
|
10.7 | 4.97 ± 0.033 | 35.7 ± 10.1 |
| 2f |
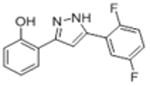
|
13.6 | 4.90 ± 1.1 | 30.1 ± 8.0 |
EC50 and pEC50 were calculated from concentration response curves after 6 h of growth using the XylE reporter assay.
An EC50 for 2a could not be determined because the concentration response curve did not plateau below its solubility limit. Efficacy is the percent activation of HssRS compared to 1 at 50 μM (all compounds reach Emax at or below 50 μM except 2a). All data were collected in triplicate, and error values are one standard deviation from the mean.
Finally, the top activators were assayed for their ability to preadapt S. aureus to heme as an indication that they can activate HssRS outside the context of a reporter assay. Pretreatment of cultures with a compound that induces HrtAB expression through activation of HssRS at subtoxic concentrations allows the bacteria to survive and grow when subcultured into a toxic concentration of heme. We compared pretreatment with 4 μM heme, 40 μM 1, and vehicle with 40 μM of each of the top activators (excluding 2d because of toxicity). All derivatives were able to preadapt S. aureus to a toxic concentration of heme (20 μM) as well as 4 μM heme and 40 μM 1 (Figure 5). The discrepancy between XylE assay and heme adaptation activity is likely due to the length of treatment with compound (6 h vs 15 h). Pretreatment with 2d at the nontoxic concentration of 20 μM induced excellent preadaptation to heme toxicity (supplemental Figure 3A). Four inactive derivatives from the single point screen, 1i, 2i, 2o, and 3a, were also assayed for preadaptation to heme toxicity. Growth of bacteria subcultured into 20 μM heme after pretreatment with 40 μM compound was indistinguishable from the vehicle, confirming that these compounds do not activate HssRS (supplemental Figure 3B).
Figure 5.
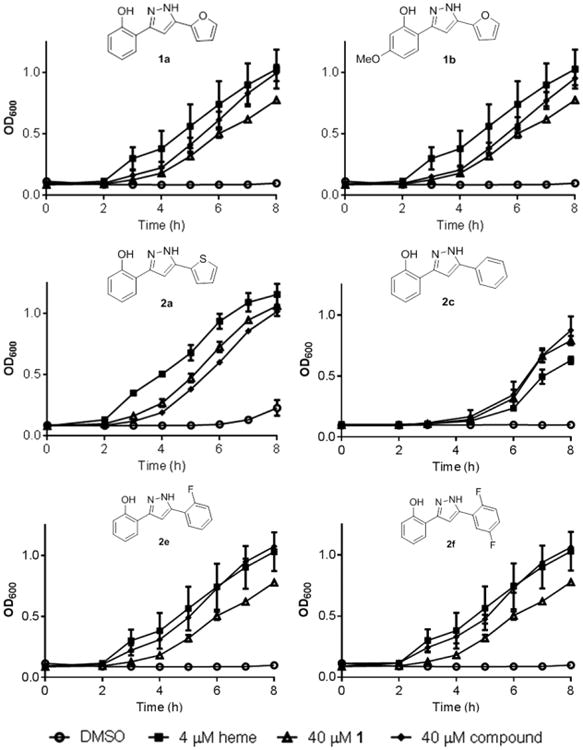
Adaptation for heme toxicity by the indicated compound compared to vehicle (DMSO), 4 μM heme, and 40 μM 1. Overnight cultures were grown in medium with the indicated additive and then subcultured into medium containing a toxic concentration of heme (20 μM). Growth was monitored by reading OD600 at indicated time points. Data are the average of three replicates, and error bars represent one standard deviation from the mean.
Compound 1 Does Not Strongly Bind Iron
The results of the HssRS activation screen suggest that any disruption to the hydrogen bonding ability of the molecule eliminates this activity. While this may be important for binding to a protein target, it may also interfere with the potential for the molecule to bind to metals of biological significance. The hydrogen bond donor–acceptor orientation is potentially capable of binding iron.20 Since its primary activity is associated with heme-iron metabolism, the ability of 1 to bind iron was determined using the Chromazural-S (CAS) assay.21 When compared to the known iron chelator deferasirox, 1 does not strongly bind iron (supplemental Figure 4). Therefore, it is unlikely that iron chelation is involved in the activity of these molecules.
Anaerobic Toxicity
Next, the compound library was screened for anaerobic toxicity by generating concentration response curves for 9 h of growth and determining IC50 values (Table 2). Wild type S. aureus strain Newman was grown in an anaerobic chamber in appropriate media to ensure exclusion of terminal electron acceptors sufficient to force the bacteria to ferment. In addition, an isogenic menB mutant was used as a positive control as this strain lacks the electron carrier menaquinone, rendering the bacterium incapable of generating energy through respiration in the presence of any terminal electron acceptor.16 In parallel, the IC50 values were determined in S. aureus grown aerobically to identify derivatives that exhibit toxicity independent of respiration. The arbitrary cutoff point for toxicity of 60 μM was chosen as many of the compounds were not entirely soluble above this concentration.
Table 2. IC50 Values (μM) for All Compounds Derived from Concentration Response Curves of the Fraction of Growth Compared to Untreated Control after 9 h of Growth of the Indicated Strain and Conditiona.
| Cmpd | Newman aerobic | Newman anaerobic | ΔmenB |
|---|---|---|---|
| 1 | >60 | 13.5 (8.42–21.7) | 1.66 (0.491–5.63) |
| 1a | >60 | >60 | 35.6 (31.0–41.5) |
| 1b | >60 | >60 | >60 |
| 1c | >60 | >60 | >60 |
| 1d | >60 | >60 | >60 |
| 4a | >60 | 26.5 (21.9–32.1) | 15.7 (11.5–21.5) |
| 4b | >60 | 4.27 (3.39–4.65) | 3.3 (1.28–8.55) |
| 1e | >60 | >60 | >60 |
| 1f | >60 | >60 | >60 |
| 1g | >60 | >60 | >60 |
| 1h | >60 | >60 | >60 |
| 1i | >60 | 19.6 (16.9–22.8) | 8.69 (7.65–9.88) |
| 1j | >60 | 11.6 (10.2–13.0) | 6.63 (5.02–8.76) |
| 1k | >60 | 13.4 (12.3–14.7) | 5.89 |
| 1l | >60* | 28.8 (22.0–37.7) | 21.4 (13.9–32.8) |
| 2a | >60 | >60 | >60 |
| 2b | >60 | >60 | >60 |
| 2c | >60 | 24.0 (20.0–28.7) | 25.2 (7.84–81.2) |
| 2d | 42.5 (40.2–44.9) | 15.8 (15.3–16.4) | 9.33 (4.89–20.1) |
| 2e | >60 | >60 | >60 |
| 2f | >60 | >60 | >60 |
| 2g | >60 | >60 | >60 |
| 2h | >60 | >60 | >60 |
| 2i | >60 | >60 | >60 |
| 2j | >60 | >60 | >60 |
| 2k | >60 | >60 | >60 |
| 2l | >60 | >60 | >60 |
| 2m | >60 | >60 | >60 |
| 2n | >60 | 25.3 (23.4–27.2) | 29.7 (24.6–35.9) |
| 2o | 48.6 (37.9–62.3) | 11.6 (10.3–13.1) | 13.2 (10.4–16.6) |
| 3a | >60 | 20.8 (17.9–24.2) | 24.9 (10.2–60.4) |
| 3b | >60 | >60 | >60 |
Data were collected in triplicate.
Error values are represented as the 95% confidence interval. IC50 values greater than 60 μM could not be reliably determined because of solubility problems at high concentrations, and compounds with an IC50 > 60 μM are considered nontoxic.
IC50 close to 60 μM, but solubility issues prevented accurate determination.
The majority of compounds were essentially nontoxic to aerobically growing Newman with the exceptions of 2d and 2o with IC50's of 42.2 and 48.6 μM, respectively. These data indicate that the majority of derivatives do not exhibit general toxicity.
Compounds 1a and 1b were relatively nontoxic to fermenting S. aureus, suggesting that the B-ring of 1 is important for toxicity. O-methylation (1c) also eliminates toxicity. However, N-methylation does not decrease the toxicity of the molecules. In addition, the regiochemistry of N-methylation appears to exert a significant effect on toxicity. Compound 4a is approximately five times less toxic to fermenting wildtype S. aureus and ΔmenB than 4b, while the toxicity of 1 is intermediate between the two.
Substitution of the o-hydroxyl of 1a with aromatic amides (1i–1l) seems to impart toxicity to fermenting S. aureus comparable to 1. However, substitution with a sulfonamide renders the compound nontoxic under fermentative conditions. This may be a consequence of the differing properties of amides and sulfonamides (pKa, hydrogen bonding, etc.) or may be related to the added aromatic bulk of the amides while the methyl group of 1h is innocuous. We were unable to test this due to the propensity of amides with smaller R-groups to dehydrate to pyrazolo[1,5-c]quinazolines such as 5a (Supporting Information).
Replacement of the furan with most aromatic or heteroaromatic groups resulted in nontoxic molecules under most conditions, the notable exceptions being 2c and 2d, with the furan replaced by phenyl and o-hydroxyphenyl, respectively. These compounds exhibit anaerobic toxicity similar to 1. Substitution of the phenyl ring with fluorine(s) (2e, 2f) eliminates this toxicity. Replacement of the furan with large (>4C) alkyl groups also produced molecules with anaerobic toxicity. 2n and 2o were comparably toxic to 1 while 2j, 2k, 2l, and 2m, with hydrogen or smaller alkyl groups were nontoxic under all conditions.
While O-methylation of 1 eliminates toxicity, O-methylation of 2c (4a), maintains toxicity comparable to the parent molecule. This suggests that O-alkylation is not a major contributor to the toxic character of these molecules.
Compound 2e was previously reported to exhibit anaerobic toxicity and efficacy in a mouse model of S. aureus infection.14 The difference in these toxicity results is likely due to the method of IC50 determination. The previous method relied on a significant back-dilution of overnight cultures prior to compound addition to account for the time needed for the apparatus to become anaerobic. Bacteria grown at this low cell density could experience greater toxicity from the compound than the higher cell density used in this work. Although, 2e is not bacteriostatic under the conditions tested here, the ability of the compound to activate HssRS was corroborated (Figure 4). This suggests that derivatives that either activate HssRS or inhibit growth under anaerobiosis may both be valid antibacterial approaches. This is supported by previous studies that have reported that HssRS activation affects virulence during infection.11
Relationship between HssRS Activation and Anaerobic Toxicity
Any disruption to the hydrogen bond donating ability of the molecule through O- or N-methylation or replacement with alternate hydrogen bond donor groups removes its ability to activate heme biosynthesis and is absolutely required for this activity. In contrast, the hydrogen bonding character seems to be less important for toxicity as several O- and N-methylated derivatives maintain toxicity levels comparable to 1. Comparing the methylated derivatives of 1; 1c, 4a, and 4b, O-methylation eliminates toxicity while the regiochemistry of N-methylation has a significant impact on the magnitude of toxicity.
Modification at the 5-position significantly affects which activity is favored. Aromatic or heteroaromatic groups are required to activate heme biosynthesis while large alkyl groups favor toxicity. Despite this, some overlap between HssRS activation and toxicity is evident. Replacement of the furan with a phenyl ring (1a to 2c) restores toxicity comparable to 1 and maintains HssRS activity. O-methylation of this derivative (3a) removes HssRS activity as expected while maintaining toxicity. Fluorination of 2c to 2e maintains HssRS activity but removes toxicity. In addition, fluorination of 3a to 3b eliminates toxicity. Interestingly, 2d, which is highly symmetric, exhibits both activities.
In summary, using a medicinal chemistry approach, we have decoupled the two activities of 1 and established two classes of molecules: activators of heme biosynthesis and inhibitors of anaerobic growth (Figure 6). These molecules will serve as probes for further study of their respective activities and mechanisms of action without concern for the off target effects of the parent molecule. Future directions will focus on optimizing the activity of both classes of molecules in S. aureus as well as evaluation of activity in other Gram positive and Gram negative bacteria.
Figure 6.
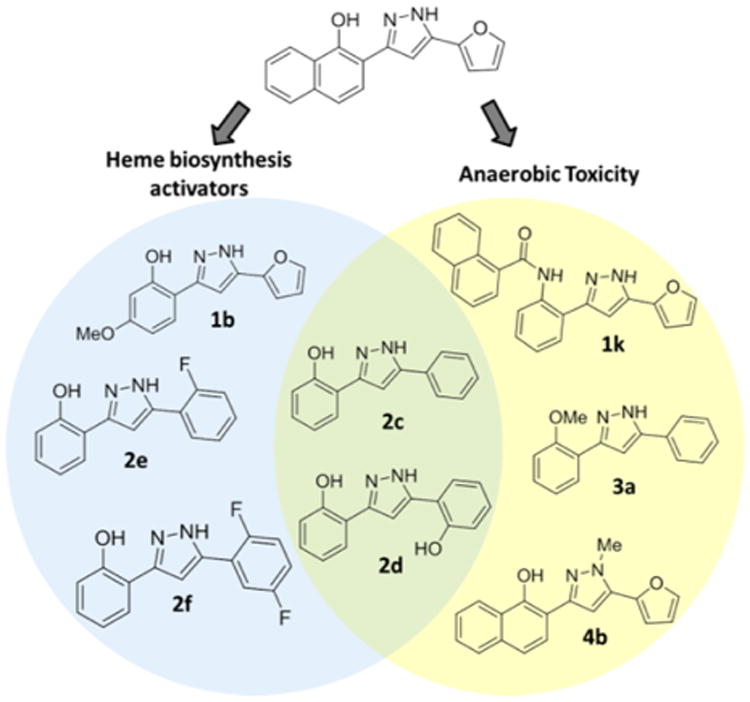
Molecules produced in this work divided into three categories: activators of heme biosynthesis, inhibitors of anaerobic growth, and hybrids exhibiting both activities.
Methods
Chemical Synthesis
Detailed experimental procedures for all reactions and compound characterization data can be found in the Supporting Information.
Pyrazole Synthesis
Route 1
To a stirred solution of terminal alkyne (1 equiv) in THF (0.2 M) was added triethylamine (3 equiv), bistriphenylphosphine palladium dichloride (0.05 equiv), copper(I) iodide (0.1 equiv), and acid chloride (1.5 equiv) at RT. The reaction was stirred until conversion of starting material was observed by TLC. The reaction was diluted 1:1 with acetonitrile followed by the addition of hydrazine hydrate (4 equiv). The reaction was stirred until complete as determined by TLC. The reaction was filtered through Celite, concentrated, and purified by preparative scale reverse phase HPLC.
Route 2
To a solution of crude diketone in ethanol (0.25 M) in a microwave vial was added hydrazine hydrate (2 equiv). The vial was sealed and heated to 150 °C in a microwave reactor for 5 min. The reaction was concentrated, and the product was purified by preparative scale reverse phase HPLC.
XylE Assay
Previously reported strains were used.11 Cultures grown overnight in 5 mL of TSB with 10 μg mL−1 chloramphenicol for 15–18 h were subcultured 1:100 into 0.5 mL TSB with 10 μg mL−1 chloramphenicol containing compound and incubated at 37 °C, at 180 rpm for 6 h. Cells were washed and lysed as previously described. A total of 200 μL of a 200 μM catechol solution in 100 mM potassium phosphate (pH 8.0) was added to 20 μL of lysate, and the oxidation of catechol was followed by monitoring absorbance at 375 nm for 10 min. Samples were normalized to protein concentration as determined by BCA assay (Pierce).
HssRS Activation Dose Response Curves and EC50 Determination
The above XylE procedure was followed using different concentrations of compound. The data were then entered into Graphpad Prism 6 and fit to a curve to determine EC50 values.
IC50 Determination
Cultures of wild type S. aureus strain Newman and ΔmenB14 were grown in aeration tubes aerobically at 37 °C with shaking for 15–18 h. Anaerobic cultures were prepared by growing bacteria at 37 °C without shaking in an anaerobic chamber for 15–18 h. Bacteria from each condition were subcultured 1:100 into TSB containing various concentrations of compound in a 96 well plate. Aerobic wild type and ΔmenB plates were incubated aerobically at 37 °C with shaking while anaerobic plates were grown in an anaerobic chamber (Coy) at 37 °C without shaking. The absorbance at 600 nm (OD600) was determined after 9 h of growth and the fraction of growth at each compound concentration is determined by dividing the OD600 by the vehicle control (DMSO) value. IC50's were calculated using Graphpad Prism 6, and errors are reported as 95% confidence intervals.
Heme Adaptation Assays
A total of 500 μL of TSB were inoculated with a colony of S. aureus and incubated at 37 °C for 3 – 4 h. Bacteria were then subcultured 1:100 into 500 μL of TSB containing a compound in 1.5 mL tubes and incubated at 37 °C with shaking for 15 h. Bacteria from the compound treated cultures were then subcultured 1:100 into 100 μL of TSB containing heme and incubated at 37 °C with shaking for 8 h. Growth was monitored by reading the OD600 on a Biotek microplate reader at the defined time intervals.
Iron Chelation Assay
Iron chelation by 1 was characterized using the CAS assay. Solutions were prepared as described.21 The clinical iron chelator deferasirox (AK Scientific) was used as a control. Samples were incubated in 1 mL cuvettes at RT for 30 min after addition of compound. The maximum concentration of compound used was 30 μM, which is a 4:1 stoichiometry of 1 to Chromeazural-S. Absorbance at 630 nm was measured on a Varian UV/vis spectrophotometer.
Supplementary Material
General methods, materials, instrumentation, synthetic procedures and compound characterization data, and supplemental Figures 1–4 (PDF)
Acknowledgments
We thank G. M. Kurkis for compound preparation. We thank J. E. Hempel and L. J. Lojek for critical reading of the manuscript. We thank C. D. Weaver for important discussions. B.F.D. was supported by National Institutes of Health Grant T32 GM065086. L.A.M. was supported by National Institutes of Health Grant T32 HL069765. This work was funded by National Institutes of Health R01 AI069233 and National Institutes of Health R01 AI073843.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.5b00934.
Notes: The authors declare no competing financial interest.
References
- 1.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto T, Nishiyama A, Takano T, Yabe S, Higuchi W, Razvina O, Shi D. Community-acquired methicillin-resistant Staphylococcus aureus: community transmission, pathogenesis, and drug resistance. J Infect Chemother. 2010;16:225–254. doi: 10.1007/s10156-010-0045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan C. Fresh approaches to anti-infective therapies. Sci Transl Med. 2012;4:140sr2. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Somerville GA, Proctor RA. At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol Mol Biol Rev. 2009;73:233–248. doi: 10.1128/MMBR.00005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. Iron-source preference of Staphylococcus aureus infections. Science. 2004;305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- 6.Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Passage of heme-iron across the envelope of Staphylococcus aureus. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- 7.Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol. 2006;188:8421–8429. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pishchany G, Dickey SE, Skaar EP. Subcellular localization of the Staphylococcus aureus heme iron transport components IsdA and IsdB. Infect Immun. 2009;77:2624–2634. doi: 10.1128/IAI.01531-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anzaldi LL, Skaar EP. Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect Immun. 2010;78:4977–4989. doi: 10.1128/IAI.00613-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP. Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathog. 2006;2:0777–0789. doi: 10.1371/journal.ppat.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, Anderson KL, Dunman PM, Joyce S, Skaar EP. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe. 2007;1:109–119. doi: 10.1016/j.chom.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stauff DL, Torres VJ, Skaar EP. Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. J Biol Chem. 2007;282:26111–26121. doi: 10.1074/jbc.M703797200. [DOI] [PubMed] [Google Scholar]
- 13.Johansson P, Hederstedt L. Organization of genes for tetrapyrrole biosynthesis in Gram-positive bacteria. Microbiology. 1999;145:529–538. doi: 10.1099/13500872-145-3-529. [DOI] [PubMed] [Google Scholar]
- 14.Mike LA, Dutter BF, Stauff DL, Moore JL, Vitko NP, Aranmolate O, Kehl-Fie TE, Sullivan S, Reid PR, DuBois JL, Richardson AR, Caprioli RM, Sulikowski GA, Skaar EP. Activation of heme biosynthesis by a small molecule that is toxic to fermenting Staphylococcus aureus. Proc Natl Acad Sci U S A. 2013;110:8206–8211. doi: 10.1073/pnas.1303674110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohler C, von Eiff C, Liebeke M, McNamara PJ, Lalk M, Proctor RA, Hecker M, Engelmann S. A defect in menadione biosynthesis induces global changes in gene expression in Staphylococcus aureus. J Bacteriol. 2008;190:6351–6364. doi: 10.1128/JB.00505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol. 2006;4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 18.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Muller S, O'Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundstrom M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ, Frederiksen M. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fustero S, Sánchez-Roselló M, Barrio P, Simón-Fuentes A. From 2000 to mid-2010: a fruitful decade for the synthesis of pyrazoles. Chem Rev. 2011;111:6984–7034. doi: 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- 20.Richardson DR, Bernhardt PV. Crystal and molecular structure of 2-hydroxy-1-naphthaldehyde isonicotinoyl hydrazone (NIH) and its iron(III) complex: an iron chelator with anti-tumour activity. JBIC, J Biol Inorg Chem. 1999;4:266–273. doi: 10.1007/s007750050312. [DOI] [PubMed] [Google Scholar]
- 21.Schwyn B, Neilands JB. Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 1987;160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General methods, materials, instrumentation, synthetic procedures and compound characterization data, and supplemental Figures 1–4 (PDF)