

Abstract
Pharmacogenomics has been lauded as an important innovation in clinical medicine as a result of advances in genomic science. As one of the cornerstones in precision medicine, the vision to determine the right medication in the right dosage for the right treatment with the use of genetic information has not exactly materialised, and few genetic tests have been implemented as the standard of care in health systems worldwide. Here we review the findings from a SWOT analysis to examine the strengths, weaknesses, opportunities and threats around the role of pharmacogenetics in public health and clinical health care, at the micro, meso and macro levels corresponding to the perspectives of the individuals (scientists, patients and physicians), the health-care institutions and the health systems, respectively.
INTRODUCTION
The release of the working draft of the human genome sequence in 20001, 2 promises a new era of genomic medicine where efficacy, dosage or side effects of drugs may be pre-determined with molecular information.3, 4 This is predicated on the knowledge of genetic polymorphisms that alter protein synthesis responsible for drug absorption, distribution, metabolism and elimination, leading to either a piling up of unused drug in the body resulting in drug toxicity, or an overly rapid clearance causing therapeutic failures. Genetically predisposed immune responses can also produce undesirable hypersensitivity reactions, which diminish quality of life and increase morbidity and mortality. Fifteen years since the sequencing of the human genome, genomic medicine has yet to live up to the expectations promised. Despite some initial successes,5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 very few pharmacogenomics tests are actually integrated routinely in clinics, and the translation of pharmacogenomics to standard clinical practices worldwide is often slow or non-existent.
The translation pathway of pharmacogenomics from basic science discovery to clinical implementation can be broadly summarised into seven key steps illustrated in Box 1. The first two steps of discovery and validation establish the clinical relevance and importance of genetic information for a pharmacological response, whereas the subsequent five steps establish the systems-level considerations taken in implementing pharmacogenetics testing in the clinic, by looking at issues around cost-effectiveness, availability of accredited clinical genomics pipelines with a supporting framework for genetic counselling, the training and upgrading of health-care workers to handle molecular pharmacology, and national regulatory and policy infrastructure to guide clinical implementation.
Box 1 Seven steps in establishing a pharmacogenomic indication in the clinic.
Step 1: Clinical discovery of a genetic polymorphism affecting drug response
This is the process of discovering which genetic polymorphisms are associated with a pharmacologic response: drug efficacy, dosage or side effects. This process often takes the form of a retrospective case–control (for dichotomous outcomes on efficacy or side effects) or case-only (for quantitative outcomes such as optimal dosage). Recent investigations typically rely on interrogating the whole genome with a commercial genotyping microarray, although smaller studies may still focus on querying subsets of genetic variants in candidate genes. Genetic polymorphisms here refer to single-nucleotide polymorphisms, insertion–deletions and HLA gene polymorphisms.
Step 2: Clinical validation to ascertain predictive accuracies of genetic testing
Discoveries from Step 1 with clinically useful effect sizes are taken forward in this process, often in the form of a randomized controlled trial, which establishes the empirical sensitivity and specificity of the genetic test. Together with information on the prevalence of 'at-risk' genetic profile contextualised in the local population, this allows the positive and negative predictive values to be estimated in order to determine the number needed to test for one patient to benefit from pharmacogenomic testing.
Step 3: Health technology assessments (HTA) to establish cost-effectiveness
A pharmacogenomic test proven to be clinically relevant (from Step 2) needs to be evaluated for cost-effectiveness for health-care systems to absorb or subsidise the costs of the test. HTA compares the additional costs against the additional benefits obtained from implementing a genetic-informed treatment regimen. This is less important if the expense is borne by the patient as a private consult.
Step 4: Establishing/availability of an accredited clinical genomics pipeline
Clinical genetic testing requires the establishment of an accredited pipeline which addresses the following: (i) what and how much biological material to extract; (ii) the clinical workflow for the extracted biological material to DNA/RNA extraction; (iii) who performs the genotyping or sequencing; (iv) who analyses the data; and (v) who advises the health-care workers on the significance of the results. An example of accreditation is the College of American Pathologists (CAP) Laboratory Accreditation Program, which evaluates the accuracy of the genetic assay, quality of the data analysis and interpretation and the overall turnaround time for the clinical genetic service.
Step 5: Developing the genetic counselling framework
A comprehensive clinical framework needs to be developed to guide health-care workers, patients and their families on what the genetic test results mean and do not mean. In particular, a clinical decision support system is required to direct the physician on genetic-informed treatment protocols.
Step 6: Continuing medical education (CME) and training future health-care workers
The vast majority of the currently practicing physicians are not trained in genomic medicine, highlighting the need for CME to plug existing and new knowledge gaps in the ever-expanding role and validity of genetic-guided pharmacology. Curricula in medical schools globally need to expand and include pharmacogenomics in the training of future physicians.
Step 7: Laying the national regulatory legislation and policies in pharmacogenomics
National health-care agencies need to establish legislative guidelines that conform to international standards around pharmacogenomics regulatory communications and nomenclature. State policies around ownership and residency of genomic data need to protect individuals against unwarranted, unethical or commercial abuse of the information for purposes unrelated to meeting health-care needs, including on the management of incidental findings.
The advent of genome-wide association studies (GWAS) facilitated the discoveries of new pharmacogenomics markers, as the majority of the human genome can be efficiently queried within a single experiment allowing multiple genetic markers to be identified concurrently. For example, a recent GWAS looking at response to human immunodeficiency virus (HIV)-1 infection treatment simultaneously identified 27 genes to be associated with response to abacavir-containing treatment, and 35 genes with efavirenz-containing treatments.17 The advent of next-generation sequencing also means there is an increasing amount of research on the impact of rare and private mutations on drug response variation.18, 19 As of 1 February 2016, there were 157 studies listed in the NHGRI-EBI GWAS Catalog20 reporting a total of 1425 genetic markers associated with drug outcomes for a spectrum of disorders including cancers, diabetes and HIV. And yet, genetic testing is recommended or required by the United States Food and Drug Administration (US FDA) for only 39 drugs, many of the genetic markers predate the discoveries made by GWAS.21
Here we perform a SWOT analysis examining the strengths, weaknesses, opportunities and threats around the role of pharmacogenomics in public health and clinical medicine. The outcome of our analysis explains the dichotomy between the accelerated pace of discovery and the sluggish uptake of pharmacogenomics in standard clinical care. The SWOT analysis also identifies existing gaps in the current situation impeding the process of clinical translation, and provides a guide to facilitate health systems research and developments geared at enabling regulatory policies around pharmacogenomic testing to be formulated.
Strengths of pharmacogenomics in clinical medicine
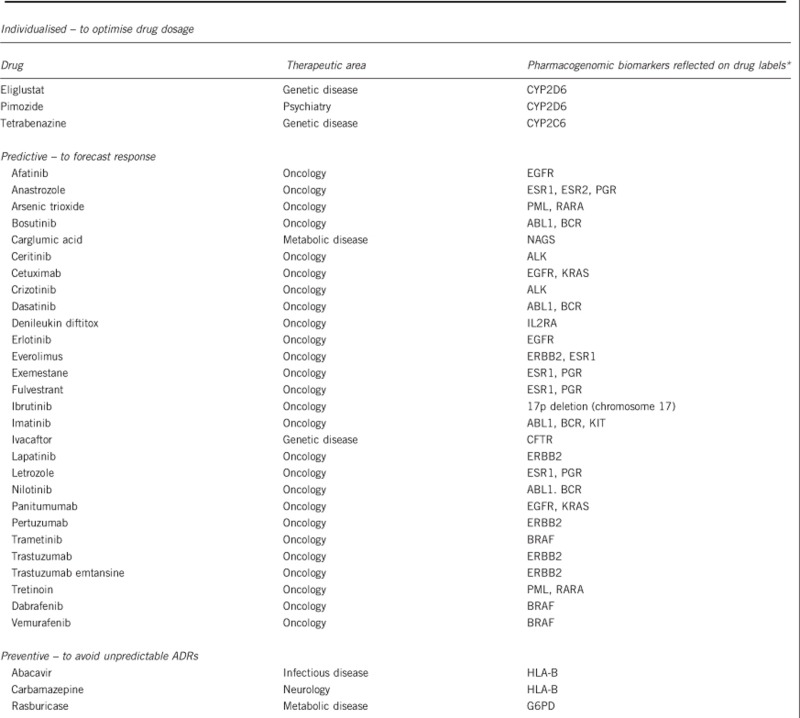
The benefits of pharmacogenomic testing over conventional practice of clinical medicine lie in the intention to stratify patients according to the expected pharmacologic requirements or outcomes, broadly in the three categories of: (i) predicting the optimal drug dosage; (ii) identifying patients at risk of drug-induced toxicity or adverse side effects; or (iii) whether an indicated drug will be efficacious. Box 2 summarises the list of FDA drug product labels, which state mandatory genetic testing prior to prescription. Targeted therapies that genuinely improve treatment efficacy and minimise unintended mortality and morbidity can also (iv) generate considerable cost savings to the health systems, even beyond the additional expenses incurred in performing the genetic tests.
Box 2 List of medications where genetic information is included in the US FDA drug product inserts, and where genetic testing is mandatory prior to prescription.
Faster achievement of optimal drug dosages
Conventional practice of clinical medicine relies on the judgement of the physician and routine monitoring of the patient to establish a working dosage of drug regimen. For the majority of the drugs,
commercially prepared dosage forms are available and these typically contain the amount of active drug components that are necessary and suitable for most patients, or which can be easily adjusted according to patient-specific biometric factors such as weight and age. However, a handful of pharmaceutical drugs demand careful titration of the dose in order to achieve the desired clinical effect within a narrow therapeutic index. Before the discovery of the pharmacogenomic link between genetic profile and optimal drug dosage, this process of continual adjustment was in part a trial-and-error approach accompanied by strict and repeated monitoring of patient response. Warfarin,5, 6, 22, 23 irinotecan15, 24, 25, 26 and atomoxetine27, 28 are examples of medications where dosage determination with clinical factors, such as age and weight, performed poorly in relation to the carriage of particular combinations of genetic variants, which strongly correlate with different dose–response curves. Here we specifically emphasise that genetic test results do not displace the need for clinical monitoring of patient response. Instead, knowing the genetic profile of the individual patient guides the initial dose, thereby accelerating the process of arriving at the optimal drug dosage with minimal toxicity.
Minimising toxicity and adverse side effects
Adverse events from medications constituted one of the leading causes of mortality in the United States of America,25 with a similar trend increasing throughout the rest of the world.26, 29 Recent studies have uncovered numerous genetic linkages with drug-induced toxicity and/or side effects, where knowledge of the individual genetic profile can indicate the likelihood of an undesirable outcome and direct the physician towards alternative medications if necessary and available. Abacavir and carbamazepine are two prime examples, especially as patients who do not carry the HLA-B*57:01 and HLA-B*15:02 alleles, respectively, for the two drugs will almost never experience abacavir-induced hypersensitivity or carbamazepine-induced Stevens–Johnson syndrome. Conversely, patients who do carry these alleles present a perceptible risk of unpredictable and potentially fatal reactions and physicians almost always recommend alternatives as a matter of good clinical practice. Prospective trials of HLA-B*57:01 screening before abacavir prescription have reported striking successes at reducing incidents of abacavir hypersensitivity among HIV patients by substituting treatment regimen with non-abacavir-containing medications for HLA-B*57:01 carriers.8, 30
Identifying efficacious drugs
Genetic profiles of individual patients can be used to identify which patients will respond to medication. This forms the cornerstone for personalising the treatment of diseases such as cancers, metabolic disorders and infectious diseases. Using a prospective approach in investigating the role of pharmacogenetics tests in clinical medicine, the Pharmacogenomic Resource for Enhanced Decisions in Care and Treatment (PREDICT) program from Vanderbilt University has already illustrated how genetic test results have improved patient safety by simplifying the prescription of the appropriate drug and dosage of compatible statins to heart patients who, otherwise, have been coping with ineffective treatments.31 Trastuzumab, commonly known as Herceptin, is also an effective therapy for breast or gastric cancer patients who are specifically HER2-positive, improving survival and reducing 3-year risk of cancer relapse by almost 10%32 conversely, the same medication has no beneficial effect to patients who are HER2-negative. The ability to accurately pinpoint an efficacious treatment, particularly for a debilitating and psychologically traumatic condition such as cancer, vastly increases patient confidence in both the treatment regimen and genetic tests, who otherwise is faced with the double whammy of a rapid decline in quality of life and the price tag of potentially harmful treatment with dubious effectiveness.
Reduce overall costs to the health-care system
One aspect of pharmacogenomic testing that is often overlooked is the potential of cost savings to the health-care system – an aspect often overshadowed by the higher upfront costs to the individuals due to the additional expense of genetic tests. Medications such as abacavir and simvastatin can be considerably cheaper than their respective alternatives (tenofovir, alirocumab) except usage may be accompanied by undesirable side effects. Assigning all the patients to costlier alternatives incur unnecessary expenses to the health-care system and risk the presentation of alternative forms of adverse responses, whereas unguided use of the cheaper alternatives can result in increased morbidity and a corresponding loss in quality of life due to the side effects. Knowledge of the individual genetic profiles of the patients means costlier alternatives can be assigned to only those who cannot tolerate the cheaper medications, and this can produce significant cost savings to the health-care system even after including the price of pre-treatment genetic screening. This was exactly the case for HLA-B*15:02 screening before carbamazepine prescription in East Asians,33, 34, 35 delivering overall savings to the health-care system despite having to screen a greater number of patients to deliver the benefit to one patient. However, it should be reminded that the findings of cost savings are context-specific and not directly transferable across different health systems, such as the discovery of HLA-B*57:01 testing being not cost-effective for East and Southeast Asian populations,36, 37 despite being ascertained to be so in European populations.38, 39, 40 Disparities in local allele prevalence and health-care cost structure can produce opposing conclusions to the economic modelling.
Weaknesses of pharmacogenomics in clinical medicine
There are several elements of pharmacogenomics that invariably makes genetic-guided therapies unfavourable when compared with conventional health care.
Costs of pharmacogenomic tests to the individuals
The additional expense of the genetic test has to be borne as an out-of-pocket expenditure by the patient, or by health-care coverage for the individual provided by either the state or insurance. We emphasise the difference between the costs of the genetic tests to the individuals, versus what we described earlier as cost savings to the entire health-care system. Producing genetic information for an individual incurs an additional expense on top of standard clinical care. In the case of patient-requested genetic tests, the cost is usually borne by the patient as part of enhanced or private medical consultation. However, tests that have undergone health technology assessments and are found to be cost-effective for the health-care system satisfy the criteria for reimbursement in part or entirely by universal health care (UHC). Regrettably, most of the health-care systems currently pass on the expense of genetic tests to the individuals, and even for countries with comprehensive UHC, such as the United Kingdom, Singapore and Thailand,41 the spectrum of reimbursable pharmacogenomic tests is still typically limited to only a handful of cancer-related and HLA-targeted34 pharmacogenetics tests (eg HLA-B*15:02 testing in Singapore and Thailand).
Speed of testing to the physicians
For pharmacogenomics testing to have a respected and useful part in health care, the speedy generation of the test results is just as important as the accuracy on the clinical impact of genetic information. Presently, it takes between 3–7 working days for the test results to reach the physician, which in the specific case of warfarin is already sufficient to adjust the international normalised ratio (INR) to therapeutic range through rigorous monitoring. Testing for HLA alleles to inform prescription of medications such as abacavir, carbamazepine or allopurinol can take even longer, although prescription immediacy is often relatively less urgent for patients requiring these treatments. The undesirably long turnaround time for genetic tests is often related to logistical, infrastructural and cost-related impediments, and seldom an inherent difficulty with the actual genetic test. We also stress the important distinction between performing the test and deriving the actual benefits from the test, which includes accessing and interpreting the test results to the physician. Therein lies an opportunity for developing companion diagnostics to facilitate the implementation of point-of-care genetic tests, except this increases the complexity of accrediting both the genetic test and the accompanying diagnostic kit.
Imperfect understanding of genetic determinants of drug response
Except for a handful of gene–drug associations mostly to do with HLA allele-induced ADR or cancer treatments, there is imperfect understanding of the genetic determinants for the majority of pharmacogenomically linked drug treatments. The promising scenario where being genotype-negative confers almost zero risk of adverse drug reactions (or equivalently, a specificity of almost 100% for genetic testing) really only extends to HLA-B*15:02 and HLA-B*57:01 for carbamazepine33 and abacavir,8 respectively, whereas most of the genetic markers hardly achieve such dichotomy for clinical decisions. For example, the loss-of-function alleles in CYP2C19 accounts for only 12% of clopidogrel response variability in spite of the fact that the heritable variability is estimated to be 72%,42, 43 and this means there are other genes involved on top of CYP2C19 or other yet unknown markers in CYP2C19 that contribute to explain the variation in clopidogrel response. It is, especially, unclear how gain-of-function alleles compensate against loss-of-function alleles in the same gene to affect outcomes.44 Even the alleles in VKORC1 and CYP2C9 commonly used for predicting warfarin dosing account for <40% of the INR dosing variation,45, 46 and rare and private mutations in known pharmacogenomic genes have recently been reported to account for unexplained genetic variability.47 This incomplete understanding of the genetic impact on drug outcomes is likely to affect the confidence of physicians and patients to genetic testing, as it reduces the process of clinical decision-making to an interpretation of probabilities and likelihoods.
Opportunities of pharmacogenomics in clinical medicine
Digitising the health records of an individual facilitates the practice of health-care analytics and precision medicine. The personalised genetic make-up comprises another valuable data source that can inform clinical health care, and we discuss here the factors and opportunities for the growth of pharmacogenomics.
Technological innovations and falling prices of genetic tests
The price tag of sequencing the human genome has fallen exponentially from US $3 billion in 2001 to less than US $1000 in 2016. Unprecedented knowledge of genome diversity gleaned from sequencing the entire genomes of thousands of people have enabled the design of commercial genotyping microarrays that are able to query almost a million genetic variants, including those with the ability to inform pharmacogenomic therapy, at a cost of less than US $50. At such price points, genetic information can be synthesised at a more attainable cost and augmented with the health records of an individual even before the need for medication arises. This circumvents a weakness described above regarding the speed, or lack thereof, of producing the test results in a timely fashion to be of practical use. Technological innovations have also fashioned a market for direct-to-consumer genetic tests, where the decision to genotype is taken by the individual instead of the health-care provider. Rapid point-of-care test kits also mean that general practitioners and specialists alike can order a genetic test, without necessarily involving a laborious and specialised process chain ranging from DNA extraction to bioinformatics interpretations. These developments greatly enhance the accessibility of genetic tests to both physician and patient, and lay the foundation for pharmacogenomics to play a bigger role in health care.
Innovation in health care that impacts drug development and utility
Pharmacogenomics is a significant innovation in health care that possesses the potential to change the paradigm in the practice of medicine, not solely in the way drugs are prescribed but also in the way drugs are discovered and developed.48, 49 The principle of pharmacogenomics-guided clinical trials is to improve and accelerate drug development by correlating genetic profiles of patients with treatment outcomes (chiefly around safety and efficacy) in early phases of the clinical trials, and subsequently extending Phase III trials only to individuals possessing the genetic predispositions linked to the safe and effective use of the developmental drugs. This transforms the current hit-and-miss approach in drug development to one that is significantly more precise, producing a lower attrition of drug candidates and lowering the price tag of developing a new drug. In fact, it is anticipated that pharmaceutical companies are likely to be the main drivers in pushing pharmacogenomics as part of standard of care, when drug product labels carry contraindication warnings and dosing guidance on the basis of individual genetic subtypes, thereby compelling the need to genotype the patient before prescription.
Public pharmacogenomics network on clinical implementation
The Clinical Pharmacogenomics Implementation Consortium (CPIC, https://cpicpgx.org/) is an online resource managed jointly by PharmGKB and the Pharmacogenomics Research Network to provide rigorously curated evidence and guidance on the role of genetic tests in augmenting and optimising drug therapy.50, 51, 52 The CPIC guidelines focus on how the test results should be interpreted to guide treatment, instead of counselling on whether genetic tests should be ordered in the first place. Two other consortiums, IGNITE (Implementing Genomics in Practice, a NIH-funded initiative) and the Dutch Pharmacogenetics Working Group (DPWG) in Europe,53 have also been established with the explicit mandate to focus on the implementation and interpretation of genetic tests to guide clinical decision-making. These consortiums are important enablers in implementing pharmacogenomic services in the clinics, as physicians can be confident that the respective one-stop portal will be able to supplement and update their knowledge of new pharmacogenomic guidelines. This is increasingly necessary with the rising popularity of direct-to-consumer genetic tests, where an individual can order a test independently of medical needs and present the results to the physician with the intention to guide treatment.
Threats of pharmacogenomics in clinical medicine
There are numerous factors that can hinder or derail the implementation, translation and uptake of pharmacogenomics. Here we provide a review of these factors and why they threaten the role of genetic tests to guide prescriptions.
Availability of non-genetics alternatives to physicians
Perhaps the greatest weakness of pharmacogenomics is the perception that molecular information is not essential and can be easily substituted by other clinical measurements or aggressive monitoring, which is already part of routine care with medications known to cause serious toxicity or adverse reactions. Several recent clinical trials have challenged the overall benefits of genetics-based warfarin dosing by illustrating near-similar results of dosage optimisation using other clinical factors and INR monitoring.54, 55 A reconciliatory note here is that the availability of non-genetic alternatives, particularly that of aggressive monitoring, is predicated on having an efficient and rigorous health-care system that is able to diagnose and circumvent adverse effects or re-adjust dosages in a timely manner. A less rigorous (or more negligent) health-care system may see greater occurrences of missed diagnosis or delays in acknowledging adverse events or drug failures, which genetic tests act as early preventive measures to minimise.
Misaligned incentives for clinical discovery versus clinical implementation activities
The majority of efforts and funds in pharmacogenomic initiatives are currently being disproportionately invested in clinical discovery and validation (Box 1, steps 1–2), instead of clinical translation (Box 2, steps 3–7). It is not difficult to see why, as the first two steps are more straightforward to conduct and are often within the sole jurisdiction and control of the principal investigator responsible for the research. Such micro establishments contain a relatively low degree of uncertainty, and the main impediments to success are usually the availability of funding and access to the right patients as study subjects. This is in contrast to the stages in clinical implementation, which involve lobbying for systems-level changes at both the meso (implementation in the clinics) and macro (national policies and international guidelines) levels – something that clinician scientists and geneticists regularly do not engage in or are in fact averse to. For example, it is profoundly more challenging to establish an accredited pipeline that operationalise pharmacogenomics from blood drawing to the production of a certified report post-bioinformatic analysis in the clinics; to additionally devote manpower resources and training to support post-testing genetic counselling; to alter current syllabi in medical schools to impart knowledge of pharmacogenomics relevance; and to produce the necessary evidence to guide the development of national and international regulatory legislations. These activities require structural transformations to the foundation of health care, and involve the buy-in of governments, universities and hospital executives. Even the carrot-and-stick system of scientific research via academic promotions, publications and grant funding is often more favourable of clinical discovery research than translational and implementation sciences, thus driving the majority of actual research activities and funding requests around clinical discovery. The status quo of disconnect cannot remain, however, especially when the public and funding agencies expect or even demand the translation of research outcomes. This is especially pertinent as the artificial and strictly regulated nature of clinical trials often fail to represent the reality in health-care establishments, overlooking operational issues such as accessibility to health-care facilities and genetic-informed physicians, acceptance of genetic testing by patients and their family members and availability of infrastructural support of genetic tests.
Lack of nationwide genetics-compatible data grid for health care
The implementation of genomic medicine requires the seamless integration of genetic information with medical records. Ideally, this genetic-compatible health-care database has to be accessible throughout the different health-care sectors from primary to tertiary care, in order to avoid the imbalance of care provision that may arise when one care sector (usually the specialist or tertiary care) is privileged to additional information over another care sector (such as the general practitioners offering primary care), when drug prescriptions are made by physicians throughout the spectrum of care sectors. However, few health systems in the world currently possess a data grid that is simultaneously able to accommodate and display genetic information, and yet accessible by all care sectors.
Mistrust over management and use of genetic information
Genetics does not solely offer insights into the biology and disease risks of the individual, but also to other members in the family. Confidentiality of genetic records must be safeguarded by the overall health-care system, just as standard operating procedures must be in place to guide the management of incidental findings that emerged from pharmacogenomic tests, especially those around paternity and disease risks. The fear and mistrust over how genetic information as a whole will be used by the society to discriminate can erode the reputation of genetic screens.
Lack of buy-in by health-care financiers
The dilemma with genetic testing is the double whammy faced by health-care financiers to bear the costs of the genetic tests and also the higher expenses from pricier alternatives for genotype-positive patients, as these people carry the predisposition for drug failures or adverse reactions with standard treatment regimes. Given the additional knowledge of possible risks, the physician is obliged to prescribe alternative drugs despite the fact that only a fraction of the genotype-positive individuals will experience a suboptimal outcome, and this is presently already arrested with routine monitoring. For chronic conditions requiring lifelong medication, there is, thus, a compelling case to attempt standard treatment before switching to alternatives. The financiers are ultimately motivated to offer a reasonable level of care balancing costs to the health system and benefits to the patients, whereas the individuals are motivated to seek the best possible care with maximum efficacy and minimum side effects. This disconnect between universal health-care coverage and patient-centric health care can manifest in the unwillingness for health-care financiers to absorb what may be perceived as an unnecessary cost.
Widening inequality in health-care accessibility
One of the unintended consequences with the availability of genetic testing is to widen the inequality in health-care accessibility between the wealthy and the less well-off. It is likely that the majority of genetic tests will not pass the assessment of cost-effectiveness, which means health-care financiers are unlikely to underwrite the additional costs, rendering these tests as an out-of-pocket expense. This disparity exists not only in the form of affordability between individuals, but also in the form of accessibility between health systems of developing and developed countries, where the former may deem the advanced genomics technology as secondary to the provision of basic essential health care, whereas the latter has the latitude to offer new technologies aimed at improving quality of care and empowering patients' preferences.
What the health-care community desperately needs is not just more clinical discovery research, but also greater emphasis and efforts in transforming existing discoveries to feasible solutions in the clinics and to address implementation barriers in the health system. Convincing the health systems to make the necessary changes must stem from an evidence-based approach that rigorously interrogates whether a genetic test genuinely improves the quality of care in a cost-effective manner, and this must happen before regulatory agencies can be convinced on which genetic tests ought to be implemented in the health system.
The authors declare no conflict of interest.
References
- Lander ES, Linton LM, Birren B et al: Initial sequencing and analysis of the human genome. Nature 2001; 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW et al: The sequence of the human genome. Science 2001; 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Collins FS, McKusick VA: Implications of the Human Genome Project for medical science. JAMA 2001; 285: 540–544. [DOI] [PubMed] [Google Scholar]
- Ojha RP, Thertulien R: Health care policy issues as a result of the genetic revolution: implications for public health. Am J Public Health 2005; 95: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TE, Altman RB, Eriksson N et al: Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360: 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz UI, Ritchie MD, Bradford Y et al: Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallal S, Nolan D, Witt C et al: Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002; 359: 727–732. [DOI] [PubMed] [Google Scholar]
- Mallal S, Phillips E, Carosi G et al: HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358: 568–579. [DOI] [PubMed] [Google Scholar]
- Chung WH, Hung SI, Hong HS et al: Medical genetics: a marker for Stevens-Johnson syndrome. Nature 2004; 428: 486. [DOI] [PubMed] [Google Scholar]
- Hung SI, Chung WH, Liu ZS et al: Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 2010; 11: 349–356. [DOI] [PubMed] [Google Scholar]
- Hung SI, Chung WH, Liou LB et al: HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA 2005; 102: 4134–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somkrua R, Eickman EE, Saokaew S et al: Association of HLA-B*5801 allele and allopurinol-induced Stevens Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. BMC Med Genet 2011; 12: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Y, Saka H, Ando M et al: Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 2000; 60: 6921–6926. [PubMed] [Google Scholar]
- Innocenti F, Undevia SD, Iyer L et al: Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol 2004; 22: 1382–1388. [DOI] [PubMed] [Google Scholar]
- Rouits E, Boisdron-Celle M, Dumont A et al: Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin Cancer Res 2004; 10: 5151–5159. [DOI] [PubMed] [Google Scholar]
- Koren G, Cairns J, Chitayat D et al: Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368: 704. [DOI] [PubMed] [Google Scholar]
- Lehmann DS, Ribaudo HJ, Daar ES et al: Genome-wide association study of virologic response with efavirenz-containing or abacavir-containing regimens in AIDS clinical trials group protocols. Pharmacogenet Genomics 2015; 25: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Butte AJ, Wheeler MT et al: Clinical assessment incorporating a personal genome. Lancet 2010; 375: 1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani B, Nakaoka H, Akhondzadeh S et al: Next generation sequencing: implications in personalized medicine and pharmacogenomics. Mol Biosyst 2016; 12: 1818–1830. [DOI] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J et al: The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 2014; 42: D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirl-Carrillo M, McDonagh EM, Hebert JM et al: Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Therap 2012; 92: 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Suo C, Lee SC et al: Translational aspects of genetic factors in the prediction of drug response variability: a case study of warfarin pharmacogenomics in a multi-ethnic cohort from Asia. Pharmacogenomics J 2012; 12: 312–318. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Johnson JA, Langaee TY et al: A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008; 112: 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh S, Hoskins JM: Irinotecan pharmacogenomics. Pharmacogenomics 2010; 11: 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L, Das S, Janisch L et al: UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002; 2: 43–47. [DOI] [PubMed] [Google Scholar]
- Hoskins JM, Goldberg RM, Qu P et al: UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 2007; 99: 1290–1295. [DOI] [PubMed] [Google Scholar]
- Brown JT, Bishop JR: Atomoxetine pharmacogenetics: associations with pharmacokinetics, treatment response and tolerability. Pharmacogenomics 2015; 16: 1513–1520. [DOI] [PubMed] [Google Scholar]
- Michelson D, Read HA, Ruff DD et al: CYP2D6 and clinical response to atomoxetine in children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry 2007; 46: 242–251. [DOI] [PubMed] [Google Scholar]
- Sultana J, Cutroneo P, Trifiro G: Clinical and economic burden of adverse drug reactions. J Pharmacol Pharmacother 2013; 4: S73–S77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Nolan D, Martin A et al: Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study. Clin Infect Dis 2006; 43: 99–102. [DOI] [PubMed] [Google Scholar]
- Whitney K: PREDICT helps pinpoint right statin for patient: Reporter: Vanderbilt University Medical Center: Nashville, TN, USA, 2012.
- Moja L, Tagliabue L, Balduzzi S et al: Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst Rev 2012; 4: CD006243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Sung C, Finkelstein EA: Cost-effectiveness of HLA-B*1502 genotyping in adult patients with newly diagnosed epilepsy in Singapore. Neurology 2012; 79: 1259–1267. [DOI] [PubMed] [Google Scholar]
- Rattanavipapong W, Koopitakkajorn T, Praditsitthikorn N et al: Economic evaluation of HLA-B*15:02 screening for carbamazepine-induced severe adverse drug reactions in Thailand. Epilepsia 2013; 54: 1628–1638. [DOI] [PubMed] [Google Scholar]
- Tiamkao S, Jitpimolmard J, Sawanyawisuth K et al: Cost minimization of HLA-B*1502 screening before prescribing carbamazepine in Thailand. Int J Clin Pharm 2013; 35: 608–612. [DOI] [PubMed] [Google Scholar]
- Kapoor R, Martinez-Vega R, Dong D et al: Reducing hypersensitivity reactions with HLA-B*5701 genotyping before abacavir prescription: clinically useful but is it cost-effective in Singapore? Pharmacogenet Genomics 2015; 25: 60–72. [DOI] [PubMed] [Google Scholar]
- Park WB, Choe PG, Song KH et al: Should HLA-B*5701 screening be performed in every ethnic group before starting abacavir? Clin Infect Dis 2009; 48: 365–367. [DOI] [PubMed] [Google Scholar]
- Schackman BR, Scott CA, Walensky RP et al: The cost-effectiveness of HLA-B*5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS 2008; 22: 2025–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves Calatrava D, Calle-Martin Ode L, Iribarren-Loyarte JA et al: Cost-effectiveness analysis of HLA-B*5701 typing in the prevention of hypersensitivity to abacavir in HIV+ patients in Spain. Enferm Infecc Microbiol Clin 2010; 28: 590–595. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Vilar FJ, Ward CC et al: Cost-effectiveness analysis of HLA B*5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenetics 2004; 14: 335–342. [DOI] [PubMed] [Google Scholar]
- Shotelersuk V, Limwongse C, Mahasirimongkol S: Genetics and genomics in Thailand: challenges and opportunities. Mol Genet Genomic Med 2014; 2: 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuldiner AR, O'Connell JR, Bliden KP et al: Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302: 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare G, Eikelboom JW, Sibbing D et al: Testing should not be done in all patients treated with clopidogrel who are undergoing percutaneous coronary intervention. Circ Cardiovasc Interv 2011; 4: 514–521, discussion 521. [DOI] [PubMed] [Google Scholar]
- Li Y, Tang HL, Hu YF et al: The gain-of-function variant allele CYP2C19*17: a double-edged sword between thrombosis and bleeding in clopidogrel-treated patients. J Thromb Haemost 2012; 10: 199–206. [DOI] [PubMed] [Google Scholar]
- Chan SL, Suo C, Chia KS et al: The population attributable fraction as a measure of the impact of warfarin pharmacogenetic testing. Pharmacogenomics 2012; 13: 1247–1256. [DOI] [PubMed] [Google Scholar]
- Yin T, Miyata T: Warfarin dose and the pharmacogenomics of CYP2C9 and VKORC1 - rationale and perspectives. Thromb Res 2007; 120: 1–10. [DOI] [PubMed] [Google Scholar]
- Ramsey LB, Bruun GH, Yang W et al: Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res 2012; 22: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesko LJ, Woodcock J: Pharmacogenomic-guided drug development: regulatory perspective. Pharmacogenomics J 2002; 2: 20–24. [DOI] [PubMed] [Google Scholar]
- Surendiran A, Pradhan SC, Adithan C: Role of pharmacogenomics in drug discovery and development. Indian J Pharmacol 2008; 40: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relling MV, McDonagh EM, Chang T et al: Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for rasburicase therapy in the context of G6PD deficiency genotype. Clin Pharmacol Ther 2014; 96: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle KE, Klein TE, Hoffman JM et al: Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab 2014; 15: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relling MV, Klein TE: CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther 2011; 89: 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swen JJ, Nijenhuis M, de Boer A et al: Pharmacogenetics: from bench to byte—an update of guidelines. Clin Pharmacol Ther 2011; 89: 662–673. [DOI] [PubMed] [Google Scholar]
- Kimmel SE, French B, Kasner SE et al: A pharmacogenetic versus a clinical algorithm for Warfarin Dosing. N Engl J Med 2013; 369: 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker WL, Chamberlin KW: New oral anticoagulants vs. warfarin treatment: no need for pharmacogenomics? Clin Pharmacol Ther 2014; 96: 17–19. [DOI] [PubMed] [Google Scholar]