

Summary
Sickle cell disease (SCD) is a significant healthcare burden worldwide, but most affected individuals reside in low-resource areas where access to diagnostic testing may be limited. We developed and validated a rapid, inexpensive, disposable diagnostic test, the HemoTypeSC™, based on novel monoclonal antibodies (MAbs) that differentiate normal adult haemoglobin (Hb A), sickle haemoglobin (Hb S) and haemoglobin C (Hb C). In competitive enzyme-linked immunosorbent assays, each MAb bound only its target with <0.1% cross-reactivity. With the HemoTypeSC™ test procedure, the sensitivity for each variant was <5.0 g/l. The accuracy of HemoTypeSC™ was evaluated on 100 whole blood samples from individuals with common relevant haemoglobin phenotypes, including normal (Hb AA, N=20), carrier or trait (Hb AS, N=22; Hb AC, N=20), SCD (Hb SS, N=22; Hb SC, N=13), and Hb C disease (Hb CC, N=3). The correct haemoglobin phenotype was identified in 100% of these samples. The accuracy of the test was not affected by Hb F (0-94.8% of total Hb) or Hb A2 (0-5.6% of total Hb). HemoTypeSC™ requires <1 μl of whole blood and no instruments or power sources. The total time-to-result is <20 min. HemoTypeSC™ may be a practical solution for point-of-care testing for SCD and carrier status in low-resource settings.
Keywords: sickle cell disease, haemoglobinopathy, point of care, lateral flow immunoassay, monoclonal antibody
Introduction
Sickle cell disease (SCD), a group of related genetic disorders of haemoglobin, is most prevalent in sub-Saharan Africa and in populations with African ancestry. In equatorial Africa, the prevalence of sickle trait is 10-40% (Piel et al, 2010; Mulumba & Wilson, 2015). SCD also occurs elsewhere throughout the world, especially in India, the Americas, Europe and the Middle East. More than 400,000 children are born with SCD and >5,000,000 with sickle trait every year (Piel et al, 2013). Taken together, the worldwide population at risk for SCD or sickle trait is over 2 billion. It has been estimated that 50-90% of newborns with SCD in Africa die in early childhood (Grosse et al, 2011), typically undiagnosed until late in the course of the disease (Chakravorty & Williams, 2015). In many low-resource environments, haemoglobin electrophoresis and neonatal screening programmes for haemoglobinopathies are available only as regional or pilot programmes (Makani & Roberts, 2016; Tshilolo et al, 2008). Therefore, it is likely that few individuals in these areas will know their own carrier status, and most will be unaware of their risk of having children with SCD (World Health Organization, 2010).
An accurate diagnosis is the first step in improving the outcomes of individuals with SCD. In high-resource environments, such as the United States, United Kingdom and many European countries, universal newborn screening for haemoglobinopathies has clearly decreased childhood mortality from SCD (Telfer et al, 2007; Quinn et al, 2010). Diagnostic testing for haemoglobinopathies, whether by screening newborns or later in life, involves a number of laboratory methods, including various electrophoretic techniques (gel- or capillary-based), high-pressure liquid chromatography (HPLC), mass spectrometry and genetic testing. All of these methods require special equipment, oftentimes expensive, and highly trained personnel. However, over 80% of the population at risk for SCD reside in low- and middle-income countries. The high cost and technological barriers of standard diagnostic methods prohibits their implementation.
Recently, several rapid diagnostic methods have been described, including assays based on differential erythrocyte density (Kumar et al, 2014), differential wicking of Hb S and Hb A through filter paper (Yang et al, 2013), and a polyclonal antibody-based capture immunoassay (Kanter et al, 2015). While these represent significant advances in diagnostic capabilities for SCD, each has deficiencies. Each method requires an instrument as either an integral part of the procedure or to achieve maximum sensitivity and specificity (Piety et al, 2016; McGann et al, 2016), which can be prohibitive in low-resource settings. In addition, none of these methods achieve 100% accuracy with or without instrumentation. In the case of the immunoassay device (SickleSCAN®, BioMedomics, Inc., Research Triangle Park, NC, USA), an independent evaluation identified limitations to the specificity and sensitivity of the device in clinical settings, which could be due to the use of polyclonal antibodies (McGann et al, 2016).
We set out to develop monoclonal antibodies specific for each of Hb A, Hb S and Hb C to the exclusion of the other variants. There have been reports of such MAbs (Epstein et al, 1996; Rosenthal et al, 1995; Jensen et al, 1985), but, to our knowledge, none has resulted in field-usable immunoassays. These major haemoglobin variants differ from Hb A by a single amino acid at position 6 of the mature β-globin chain (glutamate to valine for Hb S, glutamate to lysine for Hb C), perhaps explaining the difficulties encountered by previous attempts to make such MAbs. We successfully produced MAbs with the desired specificities, and then produced lateral flow immunoassay test strips in a competitive format – the HemoTypeSC™ test. The procedure for HemoTypeSC™ requires <1 μl of blood, no instrumentation or power sources, and has a total time-to-result of <20 min. We then investigated the performance and accuracy of these simple devices using whole blood from individuals with known haemoglobin phenotypes. The results of this evaluation indicate that the HemoTypeSC™ test is a promising new method for rapid, point-of-care determination of haemoglobin phenotype in low-resource settings.
Methods
Monoclonal Antibodies
Female 6- to 10-week-old Balb/C or BDF-1 mice were immunised with immunogen constructs containing the N-terminal sequences of human Hb A (VHLTPEEKSAVTAL), human Hb S (VHLTPVEKSAVTAL) and human Hb C (VHLTPKEKSAVTAL). Splenocyte fusions were performed as described (Shirahata et al, 1998; Greenfield, 2013) and the resulting clones were screened by direct and competitive enzyme-linked immunosorbent assays (ELISAs) as described below. Hybridoma clones were selected if they 1) bound to the correct Hb variant; 2) did not bind to the other two variants; 3) could be competed by diluted, lysed blood samples from homozygous donors of the correct phenotype but not of the other two phenotypes; and 4) were of the IgG isotype, as determined by a commercial kit (SBA Clonotyping System – AP, Southern Biotechnology Associates, Birmingham, AL, USA; Cat. # 5300-04). Clones were stabilised by multiple rounds of limiting dilution subcloning and cryopreserved. The monoclonal antibodies described here are commercially available from Silver Lake Research Corporation, Azusa, CA, USA (http://www.silverlakeresearch.com). Monoclonal antibodies for subsequent ELISAs and lateral flow test strip construction were purified by Protein G chromatography (Sigma-Aldrich, St. Louis, MO, USA; Cat. # P4691) or other methods.
ELISA
The methodology of indirect competitive ELISA has been described previously (Kemeny & Challacombe, 1988). Briefly, ferrous stabilised human Hb A (Sigma-Aldrich, St. Louis, MO, USA; Cat. # H0267) and Hb S (Sigma-Aldrich, St. Louis, MO, USA; Cat. # H0392), were used as capture antigens. As Hb C is not commercially available, an analogue, BSA-C, was produced by conjugating bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA; Cat. # A2934), to a synthetic peptide containing the N-terminal 14 amino acids of Hb C (VHLTPKEKSAVTAL) through the heterobifunctional linker ε-maleimidocaproic acid N-hydroxysuccinimide ester (EMCS; Prochem, Rockford, IL, USA, Cat. # CL-222) (Chen et al, 2003). Ninety-six-well enzyme immunoassay plates were coated with 50 μl of 1 μg /ml of each capture antigen for 1 h and washed three times with PBS containing 0.05% Tween 20 (PBST). For direct ELISA, antibodies were added to the wells at 50 μl/well. For competitive ELISAs, 25 μl of lysed blood samples, diluted in H2O, were added as competitors to the wells, followed by addition of 25 μl of anti-haemoglobin antibodies (1029-31, 1029-13, and 1029-8) to a final concentration of 1 μg/ ml and incubated for 45 min. After three additional washes, 50 μl of alkaline phosphatase (AP)-conjugated goat anti-mouse immunoglobulin G (American Qualex, San Clemente, CA, USA) was added at 1 μg/ ml in PBST and incubated for 45 min. The plates were again washed three times with PBST and 50 μl of 1 mg/ml p-nitrophenyl phosphate substrate (Thermo Fisher Scientific, Waltham, MA, USA) was added to the wells. The plates were read at 405 nm after allowing the substrate to develop for 30 min. Data points were performed in triplicate. Data are presented as % of maximal reading (in the absence of competitor) after subtraction of background optical density (OD) (in the absence of primary antibody), mean of triplicates.
HemoTypeSC™ Lateral Flow Test Strips
The methods for construction of lateral flow immunochromatographic test strips have been well described (Wong & Tse, 2008). Briefly, for the HemoTypeSC™ test, Hb A, Hb S and BSA-C (described above), as well as an irrelevant mouse immunoglobulin, were conjugated to colloidal gold nanoparticles and dried onto the surface of polypropylene test vials. Capture-purified monoclonal antibodies 1029-31, 1029-13 and 1029-8, as well as the control capture antibody (goat anti-mouse-Ig polyclonal serum) were deposited in discrete lines onto the surface of plastic-backed nitrocellulose membranes. Test strips were comprised of laminated fiberglass sample pads, antibody-impregnated nitrocellulose and a cellulosic wick, so that a liquid sample can flow sequentially through the three materials in order. An assay buffer, containing detergents and non-specific blocking reagents, was used in the procedure to rehydrate the dried gold conjugate and dilute the lysed blood sample. The test procedure for the HemoTypeSC™ test entailed: (1) lysis of the blood sample by adding 1 μl whole blood to 1 ml of distilled H2O; (2) rehydration of the dried gold conjugate in the test vial with 150 μl of assay buffer; (3) adding 15 μl of lysed blood to the fiberglass portion of the test strip; (4) placing the test strip into the test vial; (5) allowing the liquid to migrate through each portion of the test strip for 10 min; (6) taking the test strip out of the vial and visually determining the presence or absence of a red-coloured line at each of four locations (representing, in order, Hb C, Hb S, Hb A and Control). Following the competitive principle, the absence of a line indicates the presence of the specific haemoglobin variant.
Blood Samples for Clinical Validation
Blood samples for clinical validation of the HemoTypeSC™ were obtained from the Erythrocyte Diagnostic Laboratory at Cincinnati Children’s Hospital Medical Center (CCHMC). Standard clinical diagnostic methods were followed for identification and quantitation of haemoglobins. Briefly, all samples were first tested by capillary zone electrophoresis (CZE; CAPILLARYS™ 2 FLEX Piercing [Sebia, Inc., Norcross, GA, USA]) or HPLC (Alliance Separation Module 2690/2695, dual wavelength absorbance detector 2487 [Waters, Inc., Milford, MA, USA]). Detected Hb variants that were consistent with HbS or HbC in the heterozygous state by CZE or HPLC were confirmed by acid agarose gel electrophoresis. Other variants and HbS and HbC in apparent homozygous or compound heterozygous states were confirmed by both acid agarose gel electrophoresis and isoelectric focusing. Total haemoglobin concentration of blood samples and spiked solutions of Hb A and Hb S were determined by the pyridine-haemochromagen method (Barr & Guo, 2015) and confirmed by direct spectroscopy (Prahl, 2012).
A convenience sample set of approximately 100 blood samples was obtained to represent normal individuals, the trait state for Hb S and Hb C, the two major forms of SCD (Hb SS and Hb SC), and haemoglobin C disease (Hb CC). All blood samples were left-over, de-identified specimens that were originally obtained for clinically-indicated diagnostic testing (haemoglobin analysis) that would otherwise have been discarded. Therefore, the requirement for informed consent was waived, and the study was exempt from full-board review by the local Institutional Review Board. Each sample was phenotyped using standard clinical methods (see above) and used in the HemoTypeSC™ procedure as described. Results were read visually by two operators and photographed; a third individual confirmed the interpretation of results from electronic images. All three individuals’ interpretations of each HemoTypeSC™ result were concordant in all samples.
Results
Monoclonal antibodies specific for Hb A, Hb S, and Hb C
MAbs were produced by screening hybridoma clones for exclusive binding to purified Hb A and Hb S, and to BSA conjugated to synthetic peptides with the N-terminal sequence of Hb C (purified Hb C is not commercially available). MAbs were further selected by competing the binding to the specific antigen by lysed blood from Hb AA, Hb SS and Hb CC donors. In this format, specificity of antibodies is determined by testing for binding of free antibody with free antigen in solution, which decreases the binding of the antibody to a capture antigen immobilised on a surface. In the indirect competitive ELISA format, the selected MAbs (1029-31 for Hb A, 1029-13 for Hb S and 1029-8 for Hb C) each bound exclusively to their respective haemoglobin variant and that binding was competed by lysed blood samples containing the respective haemoglobin variant (Figure 1). This set of three competitive ELISAs correctly identified each relevant homozygous and heterozygous phenotype, including normal (Hb AA), carrier (Hb AS and Hb AC), sickle cell disease (Hb SS and Hb SC) and Hb C disease (Hb CC). No competition by fetal haemoglobin (Hb F, α2γ2) or by Hb A2 (α2δ2) was seen with any of these three MAbs (data not shown). Taken together, these results are consistent with the binding of each MAb being dependent on the presence of a variant-specific residue in position 6 of the Hb β chain. For at least the anti-Hb A MAb 1029-31, the binding is also dependent on the presence of β-chain-specific residues in positions 9 and 12 of the Hb β chain, as these positions are different in the δ chain of Hb A2.
Figure 1. Binding of anti-haemoglobin MAbs to SCD-associated haemoglobin variants.



ELISA plates were coated with Hb A (Panel A), Hb S (Panel B) and BSA conjugated to the N-terminal peptide of Hb C (Panel C). Monoclonal antibodies (MAbs) 1029-31, 1029-13 and 1029-8 were applied in the presence of competitor haemoglobins - whole blood of known Hb phenotype, lysed and diluted 1:400 in H2O – or no competitor (H2O only). After washing, binding of MAb to the plate surface was determined by addition of secondary antibody (goat anti-mouse-Ig conjugated to alkaline phosphatase), followed by addition of the colourimetric phosphatase substrate p-nitrophenyl phosphate. Results were determined by optical density at 405nm (OD405). For each antigen / MAb / competitor combination, data are presented as percentage of maximum MAb binding to that antigen (defined as OD405 of highest-binding MAb with no competitor).
In dilution experiments in the same competitive ELISAs, whole blood of the targeted phenotype competed the binding of each antibody at dilutions up to 1:105 - 1:106, while little or no competition was seen with blood of the non-targeted phenotypes even at dilutions of 1:100 (Figure 2). The IC50 (half maximal inhibitory concentration) of each targeted Hb variant was at least 1000-fold lower than the IC50 of the other two variants–in fact, the non-targeted variants did not reach 50% inhibition of binding at any measured concentration. These results indicate that these MAbs are capable of specifically binding each haemoglobin variant in solution, with < 0.1% cross-reactivity with non-targeted variants.
Figure 2. Cross-reactivity of anti-haemoglobin MAbs with HbA, HbS and HbC.



ELISA plates were coated with Hb A for monoclonal antibody (MAb) 1029-31 (Panel A), with Hb S for MAb 1029-13 (Panel B) and with BSA-C for MAb 1029-8 (Panel C). Competitive ELISAs were performed as in Figure 1, using lysed whole blood from phenotyped homozygous donors (Hb AA, Hb SS, and Hb CC) as competitors. Blood samples were serially diluted in H2O. Data are presented as percentage of maximum MAb binding (with no competitor).
HemoTypeSC™ Test Strip
The HemoTypeSC™ test was designed as a competitive lateral flow immunochromatographic assay (LFA) (Ngom et al, 2010) in order to translate the performance of the three competitive ELISAs into a rapid, inexpensive test kit that requires no instrumentation and few procedural steps. The HemoTypeSC™ test strip incorporates each of three independent competitive immunoassays for Hb A, Hb S and Hb C. Each immunoassay comprises red-coloured colloidal gold particles conjugated to variant-specific antigens Hb A, Hb S and BSA-C, and the corresponding capture MAbs (1029-31 for Hb A, 1029-13 for Hb S and 1029-8 for Hb C) immobilised at a specific location on the LFA test strip (Figure 3A). The flow of the sample through the strip carries the particles through capture MAb-impregnated membrane lines. In the absence of the specific haemoglobin variant, the three MAb-conjugated colour particles bind to their specific immobilised antigens, producing red-coloured lines on the test strip. The presence of each haemoglobin variant in a sample is indicated by the absence of a colour signal at a specific line location on the test strip (Figures 3B, 3C). The test strip also has a control line to indicate that sample flow through the strip proceeded correctly.
Figure 3. HemoTypeSC™ test strip.
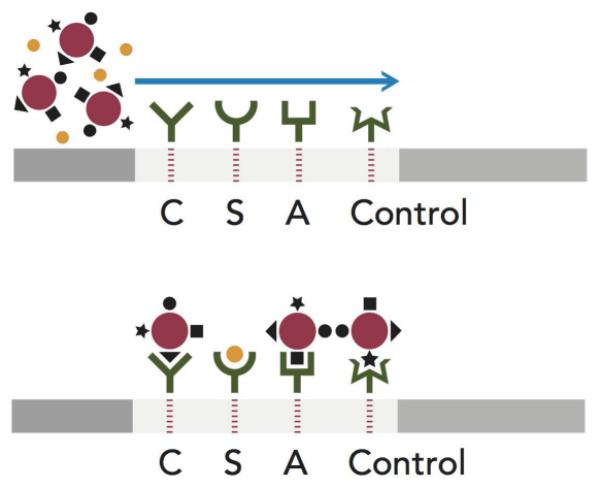


(A): Design of competitive lateral flow immunoassay. The top panel shows a schematic of the lateral flow test strip with a sample containing Hb S ( ) and colloidal gold particles (
) and colloidal gold particles ( ) conjugated to Hb A (■), Hb S (●), BSA-C(▲)and an irrelevant mouse immunoglobulin (◆). The test strip has monoclonal antibodies (MAbs) 1029-8 (anti-Hb C), 1029-13 (anti-Hb S), 1029-31 (anti-Hb A) and goat anti-mouse-immunoglobulin deposited at lines C, S, A, and Control, respectively. Flow of this sample through the test strip (bottom panel) results in the capture of red gold particles at the C, A, and Control lines of the strip, but not at the S line, where the Hb S from the sample competes for the MAb binding sites. (B): Appearance of test strip results for Hb SS phenotype; (C): Schematic of appearance of test results for each relevant phenotype.
) conjugated to Hb A (■), Hb S (●), BSA-C(▲)and an irrelevant mouse immunoglobulin (◆). The test strip has monoclonal antibodies (MAbs) 1029-8 (anti-Hb C), 1029-13 (anti-Hb S), 1029-31 (anti-Hb A) and goat anti-mouse-immunoglobulin deposited at lines C, S, A, and Control, respectively. Flow of this sample through the test strip (bottom panel) results in the capture of red gold particles at the C, A, and Control lines of the strip, but not at the S line, where the Hb S from the sample competes for the MAb binding sites. (B): Appearance of test strip results for Hb SS phenotype; (C): Schematic of appearance of test results for each relevant phenotype.
It was found that the optimal visual result was obtained using a 15-μl sample of 1:1000 diluted whole blood. One μl of whole blood was diluted in 1 ml H2O to lyse erythrocytes immediately. The testing procedure entails adding the 15-μl sample to the strip, then placing the strip into a sample vial containing rehydrated colloidal gold particles. The presence of each line on the test strip is determined visually. The entire procedure is completed in about 20 min.
In the competitive LFA format, the limit of detection of the assay is determined by the minimal concentration of analyte that completely eliminates the colour signal at the capture antigen location. The sensitivity of the HemoTypeSC™ test for each Hb variant was determined using samples of decreasing concentrations of Hb A, Hb S and Hb C, created by spiking blood from Hb SS donors with Hb A, Hb AA blood with Hb S and Hb AA blood with Hb CC blood, respectively (Figure 4). Using the measured total haemoglobin concentration and the fraction of total Hb represented by the variant Hb, it was determined that the limit of detection (LOD) of HemoTypeSC™ was <4.0 g/l for Hb A, <5.0 g/l for Hb S, and <2.0 g/l for Hb C. In a typical heterozygous sample with a total haemoglobin concentration of 150 g/l, these sensitivities correspond to <2.7% for Hb A, <3.3% for Hb S and <1.3% for Hb C.
Figure 4. Limits of detection of HemoTypeSC™ for HbA, HbS, and HbC.
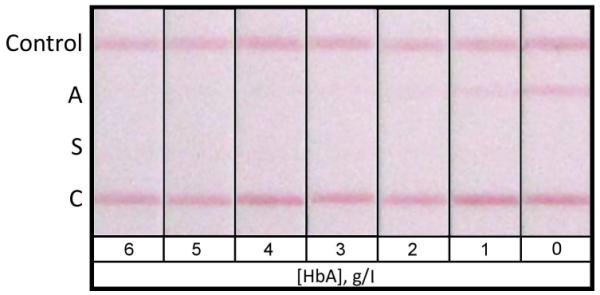
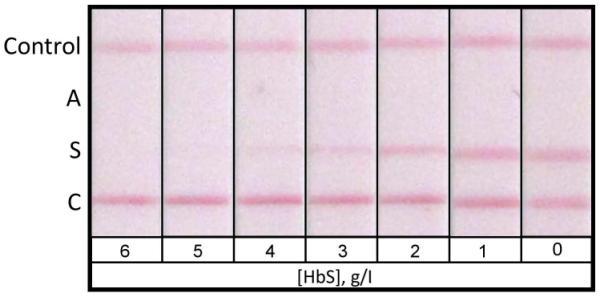

For determination of the limit of detection of HemoTypeSC™ for each haemoglobin variant, tests were performed on samples of Hb SS blood spiked with Hb A to known concentrations (0-6.0 g/l, Panel A); samples of Hb AA blood spiked with Hb S (Panel B); and samples of Hb AA blood spiked with Hb CC blood of known total Hb and known percentage of Hb C (Panel C). The result windows of HemoTypeSC™ test strips are shown for each sample. The limit of detection of the HemoTypeSC™ test for each Hb variant is the lowest concentration of that variant that results in a total absence of the respective result line in the test results.
Clinical Validation of HemoTypeSC™ Using Whole Blood Samples
We tested whole blood samples from 100 unique individuals with established haemoglobin phenotypes (Table I). The test procedure was performed at CCHMC by personnel with no prior training, and the results were interpreted by two observers (who reported no discrepant results). The correct haemoglobin phenotype was identified in 100% of these samples by the HemoTypeSC™ test strip, including normal haemoglobin type (Hb AA), sickle trait (Hb AS), Hb C trait (Hb AC), homozygous sickle cell anaemia (Hb SS), sickle-haemoglobin C disease (Hb SC), and haemoglobin C disease (Hb CC). In this sample set, sensitivity and specificity were 100% for each Hb variant that the test was designed to identify (HbA, HbS and HbC). The test had 100% accuracy across a broad range of clinically relevant concentrations of these Hb types (Hb A: 6 - 98.2% of total Hb; Hb S: 4.3 - 86.9%; Hb C: 4.5 - 99.4%). As expected, the test was “blind” to both Hb F and Hb A2 because they are not recognized by the anti-Hb MAbs in HemoTypeSC™, and the presence or absence of Hb F (0 - 94.8% of total Hb in this sample set) or HbA2 (0 - 5.7% of total Hb in this sample set) does not affect the results of haemoglobin phenotyping. Similarly, the test is “blind” to Hb Bart’s.
Table 1.
Results of HemoTypeSC™ for the diagnosis of individuals with normal haemoglobin type, sickle cell trait, Hb C trait, sickle cell anaemia, sickle-Hb C disease, and Hb C disease.
| Patient Category | N | Range of Hb levels in clinical Hb analysis (%) |
Results of clinical Hb analysis 2 |
Results of HemoTypeSC™ 3 |
|---|---|---|---|---|
| Normal (HbAA)1 | 20 | A: 93.3 - 98.2 F: 0 - 2 A2: 1.8 - 4.7 |
A (19) A F (1) |
A |
| Sickle cell trait (HbAS) | 22 | A: 6 - 66.5 S: 4.3 - 40.8 F: 0 - 89.7 A2: 0 - 3.3 |
A S (10) A S F (4) F A S (8) |
A S |
| Hb C trait (HbAC) | 20 | A: 8.1 - 66 C: 4.5 - 35.4 F: 0 - 87.4 A2: 0 - 4.1 |
A C (5) A C F (10) F A C (5) |
A C |
| Sickle cell anaemia (HbSS) |
22 | S: 11.5 - 86.9 F: 0 - 88.5 A2: 0 - 5.7 |
S S (6) S F (14) F S (2) |
S |
| Sickle-haemoglobin C disease (HbSC) |
13 | S: 19.5 - 51 C: 16.7 - 45 F: 1 - 62 A2: 1.8 - 4.7 |
S C (6) S C F (6) F S C (1) |
S C |
| Hb C disease (HbCC) | 3 | C: 5.2 - 99.4 F: 0.2 - 94.8 A2: 0 |
C C (2) F C (1) |
C |
Includes 2 individuals with increased Hb A2 (>3.3%).
All haemoglobin types detected are listed in order of decreasing abundance (e.g., “AF” indicates Hb A > Hb F). The numbers in parentheses indicate the number of individuals with the specified haemoglobin pattern.
All haemoglobin types detected by the HemoTypeSC™ test strip. The order in which the haemoglobins are listed is arbitrary.
We tested whole blood samples from 11 additional unique individuals with established haemoglobin phenotypes that the HemoTypeSC™ test strip was not designed to identify and differentiate (Table II). All non-Hb S and non-Hb C structural variants were not identified by the test strip. Heterozygotes for these variants appeared to have only Hb A. One compound heterozygote for Hb S and Hb D (Hb SD disease) appeared to have Hb S and Hb A, indicating that anti-Hb A MAb cross-reacts with Hb D. An individual with sickle-β0-thalassaemia was correctly identified to have only Hb S present. An individual with sickle-β+-thalassaemia was correctly identified to have both HbS and HbA present, but this qualitative determination does not differentiate it from HbS trait.
Table II.
Results of HemoTypeSC™ for other structural haemoglobin variants and thalassaemia.
| Patient Category | Type | Results of clinical Hb analysis 1 |
Results of HemoTypeSC™ 2 |
|---|---|---|---|
| Hb I trait | α-globin variant | A: 70.8% I: 27.2% A2: 2% |
A |
| Hb G-Philadelphia trait |
α-globin variant | A: 69.6% F: 5.1% G-Phil: 22.2% A2: 1.4% G2-Phil: 1.7% |
A |
| α-thalassaemia trait | α-globin gene deletion |
A: 69.6% F: 28.8% A2: 1.3% Barts: 0.3% |
A |
| Hb D-Punjab trait | β-globin variant | A: 57.4% D: 39.7% A2: 2.9% |
A |
| Hb E trait | β-globin variant | A: 71.1% E: 25.8% A2: 3.1% |
A |
| Hb O-Arab trait | β-globin variant | A: 58.5% O-Arab + A2: 41.55 |
A |
| Hb Hope trait | β-globin variant | A: 51.9% Hope: 43.7% A2: 3.8% |
A |
| Hb Lepore trait | δβ fusion product | A: 81.1% Lepore: 16.7% A2: 2.2% |
A |
| Sickle-haemoglobin D disease | Compound heterozygous SCD | S+D: 72.6% F: 24.8% A2: 2.6% |
A S |
| Sickle-β0-thalassaemia | Compound heterozygous SCD | S: 80.7% F: 13.4% A2: 5.9% |
S |
| Sickle-β+-thalassaemia | Compound heterozygous SCD | S: 80.7% A: 4.4% F: 12.1% A2: 2.8% |
A S |
When Hb fractions could not be resolved by the electrophoretic method, the sum of the two is indicated.
All haemoglobin types detected by the HemoTypeSC™ test strip. The order in which the haemoglobins are listed is arbitrary.
Discussion
The HemoTypeSC™ test is a promising new method for rapid, point-of-care determination of haemoglobin phenotypes, specifically for the major subtypes of SCD and related carrier states, in low-resource settings. HemoTypeSC™ requires <1 μl of blood and no instrumentation or power sources. The interpretation of test results is visual and qualitative, so extensive training is not required. The total time-to-result is <20 min. The test is designed for use at the site of blood collection, without the need to prepare a dried blood spot or transport it elsewhere for testing. This timeframe gives healthcare providers the opportunity for parent education and immediate intervention, without the disadvantage of locating the affected individuals after a long delay of obtaining test results. The simple test-strip design minimises the costs of production and packaging for low-resource settings. The estimated cost of materials for this version of HemoTypeSC™ is less than U.S. $0.25. Similar tests using the same format (competitive LFA), but for other analytes, have a shelf life of >2 years at ambient temperatures of up to 40°C, which is a key advantage of this method.
One advantage of the competitive format is the absence of a “prozone” effect. In sandwich-type assays, high concentrations of analyte can give a low signal or a false negative result. (Gillet et al, 2009) This is caused by an excess of antigen relative to the binding sites on the sandwich antibodies, so that all sites are saturated by antigen without a “sandwich” being formed. Assay developers must use a variety of procedural adjustments to counteract potential prozone effects. This is a particular problem in detecting haemoglobin variants, which can be present at > 150 mg/ml in whole blood samples. In the competitive format used in HemoTypeSC™, any concentration of Hb above the limit of detection results in a total elimination of signal, thus the prozone effect does not exist.
HemoTypeSC™ was designed with highly-specific MAbs to Hb A, Hb S and Hb C to differentiate the following conditions: normal Hb type (Hb AA), sickle cell trait (Hb AS), Hb C trait (Hb AC), homozygous sickle cell anaemia (Hb SS), sickle-Hb C disease (Hb SC) and Hb C disease (Hb CC). The sensitivity and specificity of HemoTypeSC™ was 100% when tested against a panel of whole blood specimens from 100 unique individuals with these combinations of haemoglobins. By comparison, no other rapid, non-instrument test for haemoglobinopathies has reported specificities over 98.5% (Yang et al, 2013; Kumar et al, 2014; Kanter et al, 2015; Piety et al, 2016; McGann et al, 2016). This difference may be significant because of the low prevalence of SCD, even in areas of relatively high penetration of the genetic trait. For example, in Uganda, with one of the world’s highest reported frequencies of Hb S trait carriers, the percentage of children born with SCD is about 2% (Okwi et al, 2010). A newborn screening test with a specificity of 98.5% could potentially identify 1.5% of non-SCD infants as SCD-positive, in addition to correctly identifying the vast majority of the 2% of true SCD infants as SCD-positive. In this case, the predictive value of the positive result would be only 57%, which may result in unnecessary treatment for a large number of individuals. In areas with <0.5% incidence of SCD births, such as regions of Brazil (Lervolino et al, 2011), the predictive value of the positive result of a test with 98.5% specificity would decrease to <25%. The specificity, sensitivity, and the predictive value of both positive and negative results of the HemoTypeSC™ test in this study were 100%; further studies are needed to ascertain whether this accuracy can be approached when the test method is used in low-resource settings.
When blood was tested from individuals with haemoglobinopathies for which HemoTypeSC™ was not designed to differentiate (i.e., not Hb S or Hb C), these structural variants gave results consistent with Hb A. For sickle-β0-thalassaemia, HemoTypeSC™ gave the correct Hb phenotype (presence of Hb S only). Homozygous sickle cell anaemia (Hb SS) and sickle-β0-thalassaemia have the same Hb phenotype, and their clinical management is identical, so the conflation of these genotypes with HemoTypeSC™ would have no direct clinical consequences. For sickle-β+-thalassaemia, HemoTypeSC™ again gave the correct Hb phenotype (presence of Hb A and Hb S). However, this non-quantitative result does not differentiate it from sickle cell trait, where Hb A > Hb S, while in sickle-β+-thalassaemia Hb S > Hb A. Sickle-haemoglobin D disease was misidentified as having Hb A and Hb S–as expected and explained by the sequence identity in the N-terminal region between β chains of Hb A and Hb D. The SickleSCAN® assay was shown to have the same limitations, and any antibody-based test specific for N-terminal differences between the β chains of Hb A, S, and C can be expected to perform in the same manner. The inability to discriminate sickle-β+-thalassaemia and sickle-haemoglobin D disease from sickle cell trait does have direct clinical consequences.
For all forms of protein-based testing, the differentiation of sickle cell trait from sickle-β+-thalassaemia requires a quantitative analysis. In both conditions, Hb A and Hb S will be present but differ in proportion (Hb S > Hb A for sickle-β+-thalassaemia). If applied to newborn screening, the HemoTypeSC™ would properly identify three of the most common forms of SCD without additional testing: homozygous sickle cell anaemia, sickle-haemoglobin C disease, and sickle-β0-thalassaemia. In general, these are the most severe forms of SCD. However, sickle-β+-thalassaemia, the third most common form of SCD, and other rare compound heterozygous disease forms, would not be properly identified with one-stage testing.
The HemoTypeSC™ test would be best suited for low-resource settings in which Hb S and Hb C are the predominant β-haemoglobinopathies in the population, especially sub-Saharan Africa. In this environment, the positive and negative predictive values of HemoTypeSC™ for the most common forms of SCD and their carrier states results will be extremely high. Users of this test system for populations in which Hb D, Hb E, or Hb O are common, such as in the Middle East, South Asia or Southeast Asia, would need to take into account that clinically significant, compound heterozygous forms of SCD would probably be misidentified as Hb S trait.
In summary, HemoTypeSC™ test is a promising new method for rapid, point-of-care determination of haemoglobin phenotypes in low-resource settings, such as sub-Saharan Africa, where Hb S and Hb C are the predominant β-haemoglobinopathies in the population. Only a small amount of whole blood is needed, and no instrumentation or power sources are required. It is inexpensive compared to standard diagnostic methods, and does not require extensive operator training. The HemoTypeSC™ test has the potential to improve the outcomes of individuals with SCD in low-resource settings by providing the necessary first step of an accurate diagnosis. In addition, an accurate and inexpensive method of determining carrier status can inform parental choices and possibly reduce the incidence of SCD births.
Acknowledgments
CTQ, MP, RKD, AP and MG performed the research. CTQ, RKD, AP and MG analysed the data. CTQ and MG wrote the paper.
Conflict of Interest Statement
RKD, AP and MG are employees of Silver Lake Research Corporation. CTQ is a consultant for Silver Lake Research Corporation. CTQ and MP receive research funding from Silver Lake Research Corporation through a grant from NIH-NHLBI to Silver Lake Research Corporation (R43HL123670).
This study is supported by Grant R43HL123670 from NIH-NHLBI to Silver Lake Research Corporation.
References
- Barr I, Guo F. Pyridine hemochromagen assay for determining the concentration of heme in purified protein solutions. Bio-protocol. 2015;5:e1594. doi: 10.21769/bioprotoc.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Archives of Disease in Childhood. 2015;100:48–53. doi: 10.1136/archdischild-2013-303773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Stokes DL, Rice WJ, Jones LR. Spatial and dynamic interactions between phospholamban and the canine cardiac Ca2+ pump revealed with use of heterobifunctional cross-linking agents. The Journal of Biological Chemistry. 2003;278:48348–48356. doi: 10.1074/jbc.M309545200. [DOI] [PubMed] [Google Scholar]
- Epstein N, Epstein M, Boulet A, Fibach E, Rodgers GP. Monoclonal antibody-based methods for quantitation of hemoglobins: application to evaluating patients with sickle cell anemia treated with hydroxyurea. European Journal of Haematology. 1996;57:17–24. doi: 10.1111/j.1600-0609.1996.tb00484.x. [DOI] [PubMed] [Google Scholar]
- Gillet P, Mori M, Van Esbroeck M, Van den Ende J, Jacobs J. Assessment of the prozone effect in malaria rapid diagnostic tests. Malaria Journal. 2009;8:271. doi: 10.1186/1475-2875-8-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield EA, editor. Antibodies: A Laboratory Manual. 2nd ed Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2013. [Google Scholar]
- Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. American Journal of Preventive Medicine. 2011;41:S398–405. doi: 10.1016/j.amepre.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RH, Vanderlaan M, Grabske RJ, Branscomb EW, Bigbee WL, Stanker LH. Monoclonal antibodies specific for sickle cell hemoglobin. Hemoglobin. 1985;9:349–362. doi: 10.3109/03630268508997010. [DOI] [PubMed] [Google Scholar]
- Kanter J, Telen MJ, Hoppe C, Roberts CL, Kim JS, Yang X. Validation of a novel point of care testing device for sickle cell disease. BMC Medicine. 2015;13:225. doi: 10.1186/s12916-015-0473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemeny DM, Challacombe SJ, editors. ELISA and Other Solid Phase Immunoassays. John Wiley & Sons; New York, NY: 1988. [DOI] [PubMed] [Google Scholar]
- Kumar AA, Patton MR, Hennek JW, Lee SYR, D'Alesio-Spina G, Yang X, Kanter J, Shevkoplyas SS, Brugnara C, Whitesides GM. Density-based separation in multiphase systems provides a simple method to identify sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:14864–14869. doi: 10.1073/pnas.1414739111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lervolino LG, Baldin PEA, Picado SM, Calil KB, Viel AA, Campos LAF. Prevalence of sickle cell disease and sickle cell trait in national neonatal screening studies. Revista Brasileira de Hematologia e Hemoterapia. 2011;33:49–54. doi: 10.5581/1516-8484.20110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makani J, Roberts DJ. Hematology in Africa. Hematology/Oncology Clinics of North America. 2016;30:457–475. doi: 10.1016/j.hoc.2015.12.002. [DOI] [PubMed] [Google Scholar]
- McGann PT, Schaefer BA, Paniagua M, Howard TA, Ware RE. Characteristics of a rapid, point-of-care lateral flow immunoassay for the diagnosis of sickle cell disease. American Journal of Hematology. 2016;91:205–210. doi: 10.1002/ajh.24232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulumba LL, Wilson L. Sickle cell disease among children in Africa: An integrative literature review and global recommendations. International Journal of Africa Nursing Sciences. 2015;3:56–64. [Google Scholar]
- Ngom B, Guo Y, Wang X, Bi D. Development and application of lateral flow test strip technology for detection of infectious agents and chemical contaminants: a review. Analytical and Bioanalytical Chemistry. 2010;397:1113–1135. doi: 10.1007/s00216-010-3661-4. [DOI] [PubMed] [Google Scholar]
- Okwi AL, Byarugaba W, Ndugwa CM, Parkes A, Ocaido M, Tumwine JK. An up-date on the prevalence of sickle cell trait in Eastern and Western Uganda. BMC Blood Disorders. 2010;10:5. doi: 10.1186/1471-2326-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, Weatherall DJ, Hay SI. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nature Communications. 2010;1:104. doi: 10.1038/ncomms1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piety NZ, Yang X, Kanter J, Vignes SM, George A, Shevkoplyas SS. Validation of a Low-Cost Paper-Based Screening Test for Sickle Cell Anemia. PLOS ONE. 2016;11:e0144901. doi: 10.1371/journal.pone.0144901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahl S. [Accessed June 21, 2016];Tabulated molar extinction coefficient for hemoglobin in water. 2012 Available at: http://omlc.org/spectra/hemoglobin/summary.html.
- Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447–3452. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal MA, Campbell TA, Epstein N. Binding specificity of a monoclonal antibody to human Hb A. Hemoglobin. 1995;19:191–196. doi: 10.3109/03630269509036939. [DOI] [PubMed] [Google Scholar]
- Shirahata S, Katakura Y, Teruya K. Cell hybridization, hybridomas, and human hybridomas. Methods in Cell Biology. 1998;57:111–145. doi: 10.1016/s0091-679x(08)61575-7. [DOI] [PubMed] [Google Scholar]
- Telfer P, Coen P, Chakravorty S, Wilkey O, Evans J, Newell H, Smalling B, Amos R, Stephens A, Rogers D, Kirkham F. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92:905–912. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]
- Tshilolo L, Kafando E, Sawadogo M, Cotton F, Vertongen F, Ferster A, Gulbis B. Neonatal screening and clinical care programmes for sickle cell disorders in sub-Saharan Africa: lessons from pilot studies. Public health. 2008;122:933–941. doi: 10.1016/j.puhe.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Wong R, Tse H, editors. Lateral Flow Immunoassay. Springer Science & Business Media; Totowa, NJ: 2008. [Google Scholar]
- World Health Organization [Accessed June 21, 2016];Sickle cell disease: A strategy for the WHO African Region. 2010 Available at http://www.afro.who.int/en/clusters-a-programmes/dpc/non-communicable-diseases-managementndm/npc-publications.html.
- Yang X, Kanter J, Piety NZ, Benton MS, Vignes SM, Shevkoplyas SS. A simple, rapid, low-cost diagnostic test for sickle cell disease. Lab on a Chip. 2013;13:1464–1467. doi: 10.1039/c3lc41302k. [DOI] [PubMed] [Google Scholar]