

Abstract
Nanoformulations have become important tools for modifying drug disposition, be it from the perspective of enabling prolonged drug release, protecting the drug molecule from metabolism, or achieving targeted delivery. When examining the in vivo pharmacokinetic properties of these formulations, most investigations either focus on systemic concentrations of total (encapsulated plus unencapsulated) drug, or concentrations of encapsulated and unencapsulated drug. However, it is rare to find studies that differentiate between protein-bound and unbound (free) forms of the unencapsulated drug. In light of the unique attributes of these formulations, we cannot simply assume it appropriate to rely upon the protein-binding properties of the traditionally formulated or legacy drug when trying to define the pharmacokinetic or pharmacokinetic/pharmacodynamic characteristics of these nanoformulations. Therefore, this commentary explores reasons why it is important to consider not only unencapsulated drug, but also the portion of unencapsulated drug that is not bound to plasma proteins. Specifically, we highlight those situations when it may be necessary to include measurement of unencapsulated, unbound drug concentrations as part of the nanoformulation pharmacokinetic evaluation.
Introduction
Nanomedicine is described as the application of nanotechnology for medical purposes and has shown great promise in the field of drug delivery (Lobatto et al., 2011; Chow and Ho, 2013). Nanomedicines come in a variety of compositions (liposomes, emulsions, micelles, inorganic particles, and solid lipid nanoparticles), sizes (10–1000 nm), shapes, surface chemistries, and charges. These physical and chemical properties of the nanomedicine influence its biologic performance, such as toxicity, stability, and disposition.
These innovative formulations allow for the tailoring of drug absorption and distribution characteristics to address specific therapeutic objectives. In some cases, they may also alter mechanisms of drug clearance. For example, whereas some orally administered nanoformulations provide modified release properties and increased solubility in the gastrointestinal tract, others leave the gut lumen intact by a localization within gut-associated lymphoid tissue, where they act as drug-releasing depots (Jani et al., 1990; Florence and Hussain, 2001). Alternatively, they may be administered s.c. where they can be retained near the site of injection, taken up by dendritic cells, or retained by local lymph nodes where they serve as drug-releasing depots (Cheng et al., 2015). Despite the variety of formulation technologies and routes of administration applied to these products, there are many shared features. Nanomedicines are typically <350 nm, spherical, and have neutral and hydrophilic surfaces. The most common route of administration is i.v. injection, and a push has been made for utilizing this drug delivery strategy for developing creative approaches for cancer therapy (Etheridge et al., 2013).
One of the benefits associated with the use of nanoformulations is enhanced therapeutic efficacy, accomplished by targeted delivery of the active pharmaceutical ingredient (API) to the site of action, thereby minimizing systemic drug exposure and toxicity. Accordingly, most pharmacokinetic (PK) studies of these formulations focus on measuring total (encapsulated plus unencapsulated) drug, or (less frequently) the encapsulated and unencapsulated drug (Ambardekar and Stern, 2015). However, a differentiation of the total versus unbound concentrations of the unencapsulated drug (i.e., plasma, serum, or tissue protein binding) is largely ignored.
Understanding the PK mechanisms by which nanomedicines alter drug disposition is fundamental to the continued advancement and application of nanotechnology as a drug delivery science. Traditional pharmacology maintains that unbound drug is the biologically active form; thus, reliance solely on total drug has the potential to introduce significant errors into the interpretation of drug delivery mechanisms and PK/pharmacodynamic (PD) relationships. Recently, national and international regulatory bodies have introduced generic nanomedicine guidance focusing on the evaluation of encapsulated and unencapsulated PK profiles, within the context of establishing the bioequivalence (PK equivalence) (Ambardekar and Stern, 2015). For generic versions of the popular nanomedicine Abraxane, Food and Drug Administration guidance has also included the evaluation of unencapsulated, unbound drug profiles. A commentary on the need to examine the active unbound drug fraction of nanomedicines is long overdue.
For reasons discussed in this commentary, we cannot simply assume that in vitro protein-binding information on the API itself (Otagiri, 2005) can be translated directly into free versus bound concentrations associated with the drug delivery platform, a concern that is only further magnified by the potential impact of disease on plasma protein-binding characteristics (Tesseromatis and Alevizou, 2008). Thus, in light of the unique attributes of these formulations and potential changes in protein binding that can occur as a function of disease, a simple reliance on the in vitro protein-binding characteristics of the API may not be appropriate for defining the PK or PK/PD relationships of nanoformulations.
With these considerations in mind, this commentary focuses on when it may be necessary to measure not only encapsulated versus unencapsulated drug, but also the concentrations of unencapsulated, unbound drug as part of the PK assessment of nanoformulations. Although plasma protein binding has been explored relative to the development of a protein corona surrounding some nanomedicines (Caracciolo, 2015), we view this as part of the PK of the encapsulated drug fraction that is carrier-dependent and, therefore, will only be considering protein binding of the unencapsulated drug in this manuscript. In addition, this article does not provide a general overview of the PK attributes of the various nanoformulations because such information has already been published in some outstanding reviews (Mukherjee et al., 2014; Onoue et al., 2014; Lucas et al., 2015).
For clarity, the following terms will be used throughout this commentary:
Encapsulated drug: drug molecule that is physically associated with the nanoformulation.
Unencapsulated drug: drug molecule that has been released from the nanoformulation.
Total drug concentrations: encapsulated plus unencapsulated drug.
Bound drug: unencapsulated drug that is bound to plasma or tissue proteins.
Unbound drug: unencapsulated drug that is not bound to plasma or tissue proteins.
Traditional Protein-Binding PK Paradigms
The critical PK variables for small molecules are typically the intrinsic clearance and the volume of distribution associated with the unbound (free) drug fraction (fu). When measuring drug concentrations, substantial error can potentially be introduced into the PK and PK/PD data interpretation if the relationship between total (bound + unbound) and unbound drug concentrations is not considered (Zeitlinger et al., 2011).
In the majority of cases, conditions such as disease-induced changes in protein binding, drug-drug interactions, or nonlinear protein binding will not affect the extent of unbound drug exposure [expressed as area under the unbound concentration versus time profile (AUCu)], even though total drug exposure may change (exception is high extraction ratio drugs administered via i.v. injection). This point was extensively discussed by Benet and Hoener (2002). Nevertheless, changes in the relationship between total versus unbound drug concentrations can still be important in disease-induced alterations in protein binding (e.g., liver and renal diseases, dehydration, infection and inflammation, and diseases such as Crohn’s and Celiac) (Smith et al., 2010), as it can result in clinically relevant changes in the shape of the tissue and/or plasma profiles of the unbound drug concentrations. Furthermore, failure to consider the relationship between bound and unbound drug concentrations can lead to substantial bias in conclusions derived from therapeutic drug monitoring when only total drug concentrations are measured. For example, the relationship between total drug exposure and therapeutic response for narrow therapeutic window drugs (e.g., intensive care patients with hypoalbuminemia or increased levels of α1-acid glycoprotein receiving sedatives and pain medications) can differ from those without similar changes in plasma protein binding (Smith et al., 2012).
Incorporating traditional PK paradigms with protein-binding considerations (e.g., due to disease) results in the following relationships, displayed in Tables 1 and 2 [based upon information in Schmidt et al. (2010)], between the extent of exposure [expressed as concentration steady state (Css)], shape of the profile [expressed as concentration maximum (Cmax)] or clearance (expressed as CL), and fraction unbound in plasma (fu = unbound/total drug concentrations in plasma). To allow for a qualitative graphic presentation of these relationships, the effect of altered fu is described in terms “increase,” “no change,” and “decrease.”
TABLE 1.
Impact of increasing fraction unbound in plasma
Administration route and drug characteristics.
| Parameter | Par Low E Low V | Par Low E High V | Par High E Low V | Par High E High V | Oral Low E Low V | Oral Low E High V | Oral High E Low V | Oral High E High V |
|---|---|---|---|---|---|---|---|---|
| CL total | ↑ | ↑ | — | — | ↑ | ↑ | — | — |
| Cmax total | ↓ | ↓ | — | ↓ | ↓ | ↓ | ↓ | ↓ |
| Css total | ↓ | ↓ | — | — | ↓ | ↓ | ↓ | ↓ |
| Cmax unbound | ↑ | — | ↑ | ↑ | ↑ | — | — | ↓ |
| Css unbound | — | — | ↑ | ↑ | — | — | — | — |
CL, total drug clearance; high E, high extraction ratio; high V, high volume of distribution (distributes extensively to peripheral tissues); low E, low extraction ratio; low V, low volume of distribution (confined primarily to central compartment); oral, oral administration; par, parenteral administration.
TABLE 2.
Impact of decreasing fraction unbound in plasma
Administration route and drug characteristics.
| Parameter | Par Low E Low V | Par Low E High V | Par High E Low V | Par High E High V | Oral Low E Low V | Oral Low E High V | Oral High E Low V | Oral High E High V |
|---|---|---|---|---|---|---|---|---|
| CL total | ↓ | ↓ | — | — | ↓ | ↓ | — | — |
| Cmax total | ↑ | ↑ | — | ↑ | ↑ | ↑ | ↑ | ↑ |
| Css total | ↑ | ↑ | — | — | ↑ | ↑ | ↑ | ↑ |
| Cmax unbound | ↓ | — | ↓ | ↓ | ↓ | — | — | ↑ |
| Css unbound | — | — | ↓ | ↓ | — | — | — | — |
CL, total drug clearance; high E, high extraction ratio; high V, high volume of distribution (distributes extensively to peripheral tissues); low E, low extraction ratio; low V, low volume of distribution (confined primarily to central compartment); oral, oral administration; par, parenteral administration.
Note that with the exception of parenteral high extraction compounds, changes in protein binding do not influence Css unbound, although changes in Cmax unbound may occur (e.g., in the case of oral drug with altered drug partitioning into peripheral tissues).
The important messages conveyed through Tables 1 and 2 are twofold:
Traditionally, changes in protein binding will only influence the extent of unbound drug exposure (reflected as Css unbound) when the drug is characterized as having a high extraction ratio, low V, and is administered parenterally. In all other cases, changes in fu will not influence Css unbound. However, Cmax unbound values can differ as a consequence of altered protein binding when the drug has a low volume of distribution (oral or parenteral) or when it is administered parenterally and has a high extraction ratio. Therefore, even in situations when total exposure may not change, if Cmax unbound can influence therapeutic response or toxicity, altered protein binding may need to be considered.
Evaluations based upon total drug concentrations will be misleading when trying to predict PK/PD relationships as it can suggest changes in drug exposure that are without clinical relevance (based upon the absence of a corresponding change in unbound drug concentrations).
Nanoparticle PK
Our current understanding is that encapsulation of the drug within a nanoparticle can dramatically alter the drug’s PK by altering tissue distribution and clearance. The distribution and clearance of i.v. administered nanomedicines are primarily dependent on the enhanced permeability and retention (EPR) effect and upon the mononuclear phagocyte system (MPS), respectively (Zamboni et al., 2012) (Fig. 1). The MPS also appears to be responsible for saturable clearance of some formulations (e.g., liposomes) at higher doses, and for a higher intersubject variability in the extent of drug exposure as compared with that seen with conventional formulations (Song et al., 2012). This variability can influence the clinical response (safety and effectiveness) (Schell et al., 2014).
Fig. 1.

General schematic of nanomedicine PK. CDrug, concentration of unencapsulated drug in central compartment; CNM, concentration of nanomedicine (NM) encapsulated drug in the central compartment; CLD, distributional clearance for unencapsulated drug; CLEL, grouped elimination clearance pathways for unencapsulated drug; CLEPR, EPR clearance of NM encapsulated drug; CLMPS, MPS clearance of NM encapsulated drug; KRS, NM encapsulated drug release rate; VC, volume of central compartment for unencapsulated drug; VNM, volume central compartment for NM encapsulated drug; VP, volume of peripheral compartment.
Nanoparticles not cleared by MPS demonstrate longer circulatory half-lives and better tumor accumulation by EPR (Briley-Saebo et al., 2008; Maeda, 2012). The EPR effect is a well-known phenomenon that describes the accumulation of nanoscale drug formulations at sites of inflammation and solid tumors due to the leaky tumor vasculature and suppressed lymphatic system (Maeda, 2012). Such leaky vessels are not typically found in normal healthy tissues, thereby allowing for a more targeted drug delivery (Nakamura et al., 2015). Heterogeneity of solid tumors among patients and interpatient variability of nanoparticle PK create additional challenges in predicting the safety and efficacy of a nanomedicine formulation (Zamboni et al., 2009; Sidone et al., 2007; Caron et al., 2011; Prabhakar et al., 2013).
Although a drug is encapsulated in a nanomedicine (e.g., liposome), its PK is dependent upon the physiochemical characteristics of the carrier until the drug is released from the carrier. Conversely, the inherent PK of the drug itself dictates the PK for the unencapsulated drug fraction (Ambardekar and Stern, 2015). Therefore, the PK profiles of both the encapsulated and unencapsulated drug fractions for a given nanomedicine formulation are critical for understanding how the PK properties of the nanomedicine influence product PD and toxicodynamics. However, whereas for many kinds of nanoformulations the unencapsulated concentration/effect relationships can be derived from unbound/total drug ratio of the legacy (traditionally) formulated API, there are situations when this is not the case.
Nonlinear Changes in Protein Binding in Which Formulation Has the Potential to Alter This Binding Behavior.
An example of where the PK of the traditionally formulated API should not have been relied upon to predict behavior of a nanoformulation is exemplified by liposomal amphotericin B (AmBisome). Bekersky et al. (2002) evaluated and compared the PK of a traditional i.v. formulation (Amphotericin B Deoxycholate) to the liposomal formulation. They used a nonlinear relationship between drug concentration and protein binding to differentiate encapsulated and unencapsulated liposomal fractions. Adapting the ultrafiltration method for fractionation of amphotericin B nanoliposome in plasma samples, the protein-bound drug fraction was interpolated from an established correlation between free (ultrafilterable) drug concentrations and protein binding. The encapsulated amphotericin B fraction was estimated by subtracting the ultrafilterable and protein-bound amphotericin B concentrations from the total amphotericin B concentrations. Thus, their estimation technique was based upon an assumption that the nanoformulation does not influence amphotericin protein-binding behavior. The error in utilizing this approach, with the assumption that protein binding was formulation-independent, has since been demonstrated using stable isotope tracers to account for formulation-mediated changes in binding behavior (Skoczen et al., 2015).
Equilibrium Formulations.
The potential for unbound drug to associate with certain nanomedicine platforms (e.g., micelles, liposomes), thereby becoming transiently bound to the formulation in a manner similar to that of plasma protein binding, can potentially influence the rate and extent of unbound drug exposure in tissues and plasma (Sparreboom et al., 1999; van Tellingen et al., 1999; van Zuylen et al., 2000). For example, the released drug in plasma may recombine with some nanoformulations [e.g., cremophor (CRE) nanomicelle], thereby reforming into carrier-associated nanoparticles.
Equilibrium Formulations
From a drug release standpoint, nanomedicines can be broadly classified as solubilizing formulations, modified release formulations, and equilibrium formulations. As the name implies, solubilizing formulations are unstable, releasing the entire drug immediately upon injection. Modified release nanomedicines function as circulating depots, liposomes being an example that is capable of releasing active drug in a controlled manner. Equilibrium formulations are a unique example of a kind of formulation that provides the shared characteristics of solubilizing agents and modified release formulations. Similar to conventional modified release products, equilibrium formulation platforms are stable, remaining intact systemically. However, unlike conventional modified release formulations, the unencapsulated, unbound drug is in a dynamic equilibrium with the formulation (Brouwer et al., 2000) (Fig. 2). As a result, although the drug is immediately 100% bioavailable (similar to a simple solubilizing agent), an alteration in unbound drug fraction (due to drug binding to the equilibrium formulation) could have therapeutic implications that vary from the classic small-molecule paradigms.
Fig. 2.

Simplified schematic of nanomedicine PK incorporating equilibrium binding to unbound drug.
The most representative example of unbound drug binding to a nanomedicine in equilibrium is the case of Taxol. Taxol is a CRE nanomicelle formulation of paclitaxel with indications for the treatment of solid tumors (de Weger et al., 2014). There are several examples in which the PK of Taxol have been described as dose dependent (i.e., nonlinear), presenting with both saturable distribution and saturable elimination (Sonnichsen et al., 1994; Gianni et al., 1995). It is important to note that this nonlinearity was identified in reference to total drug in plasma (i.e., encapsulated, protein bound, and unbound). The inclusion of unbound drug binding to CRE nanomicelles as part of various paclitaxel PK models explained not only the nonlinearity of total plasma drug, but also the linearity of whole blood and unbound plasma drug (Henningsson et al., 2001; Bulitta et al., 2009). In these models, the nonlinearity of total plasma drug concentrations is a consequence of CRE nanomicelle binding and a decrease in the unbound drug fraction at higher Taxol doses and at decreased infusion times. Plasma unbound fraction studies in vitro, as well as blood:plasma partitioning studies ex vivo, support paclitaxel binding to CRE micelles at clinically relevant drug concentrations (Brouwer et al., 2000; van Zuylen et al., 2001). The ability of CRE micelle to bind drug in vivo is further supported by studies showing Taxol-drug interactions leading to increased total area under the total drug concentration versus time curve (AUC) for coadministered doxorubicin and irinotecan, thought to be due in part to micelle binding of drug (Vigano et al., 2001; Kasai et al., 2002).
The PK equations provided below for small-molecule binding to protein mathematically describe how decreased unbound drug fraction can result in changes to total drug clearance and volume of distribution (Benet and Hoener, 2002):
| (1) |
| (2) |
where V = volume of distribution, VP = volume of the central compartment (plasma), VT = volume of the peripheral compartment (tissue), fuT = the unbound drug fraction in tissue, CL = total systemic clearance, fuP = the unbound drug fraction in plasma, and CLint = intrinsic clearance.
Despite these changes in total drug volume and clearance, one would not anticipate that these changes would have therapeutic consequences for a low extraction drug such as paclitaxel, because the clinically relevant unbound drug exposure (AUCu) is unaffected by changes in unbound fraction (Benet and Hoener, 2002):
| (3) |
These small-molecule paradigms, however, may not be appropriate for equilibrium binding of drugs to nanomedicine formulations. In fact, there is evidence that equilibrium binding of paclitaxel to the Taxol CRE micelle may actually decrease tissue distribution, negatively affecting Taxol therapeutic utility in comparison with Abraxane, a novel albumin-based nanoparticle formulation of paclitaxel. Abraxane is currently approved for treatment of pancreatic cancer, whereas Taxol is not (Goldstein et al., 2015).
In the case of Taxol, formulation components contribute substantially to maximum tolerated dose (MTD), as CRE is immunotoxic and potentially neurotoxic; Abraxane has a 1.7-fold higher MTD than Taxol due to the absence of CRE (Sparreboom et al., 2005). At comparable clinical MTD (i.e., at a higher dose of Abraxane on the basis of dose calculated as mg/m2), Abraxane has a 50% greater total drug clearance and volume of distribution than Taxol (Sparreboom et al., 2005). This difference in Abraxane and Taxol PK is also observed in animal models (Sparreboom et al., 2005). As discussed above, Taxol in vivo and in vitro PK studies support the stability of the Taxol CRE micelle in vivo, and its ability to bind paclitaxel in equilibrium with the unbound form. By contrast, the Abraxane formulation is unstable, with the nanoparticles rapidly dissociating into their component albumin molecules upon systemic administration (Svenson, 2014). The rapid dissociation of the Abraxane nanoparticles is consistent with the fact that, despite a higher administered dose of Abraxane (260 mg/m2) versus Taxol (175 mg/m2), the two formulations resulted in comparable total exposure estimates (expressed as the AUC of the total drug concentrations in plasma). However, at an equivalent total drug exposure as Taxol, Abraxane was associated with a greater unbound drug fraction, and at a greater unbound drug exposure, Abraxane’s higher unbound drug fraction potentially explains its greater total drug clearance (eq. 2) and volume of distribution (eq. 1). In the case of greater unbound drug fraction, terminal half-life does not change because clearance and volume of distribution increase proportionately for a moderate-high volume of distribution drug like paclitaxel, according to eq. 4 (Schmidt et al., 2010):
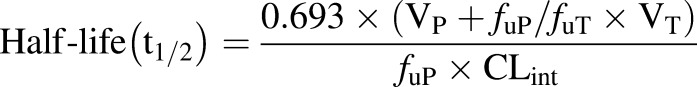 |
(4) |
This expected similarity in terminal half-life is consistent with clinical observations for Abraxane and Taxol, further supporting changes in unbound drug fraction as being the underlying mechanism responsible for differences in their PK (Sparreboom et al., 2005; Gardner et al., 2008).
In support of the higher volume of distribution observed preclinically and clinically, greater paclitaxel tissue AUC has also been observed for the Abraxane formulation in comparison with Taxol for organs such as prostate, pancreas, and xenograft tumor in preclinical models at equal doses (Sparreboom et al., 2005; Desai et al., 2006). Apart from the increase in systemic unbound drug exposure resulting from the higher MTD, these differences in Abraxane versus Taxol tissue distribution could be clinically meaningful and a reason for the increased efficacy of Abraxane over Taxol in pancreatic cancer.
Redefining Biologically Active Drug Fraction and Its Impact on the PK/PD of Nanoformulations
In addition to our prior PK discussions, the biologically active drug fraction may also need to be redefined from a PK/PD perspective. This is particularly true with regard to nanoformulations in which delivery of the active agent bypasses the unbound form as a result of direct transfer to the target cells. In cases where the carrier delivers the active drug directly to the target cell (e.g., tumor), the significance of the unbound drug fraction with regard to efficacy is greatly diminished, although still having importance for off-target exposure-toxicity relationships. An example of such targeted delivery systems includes nanomicelles and nanoliposomes that bind to amphipathic peptide and small-molecule drugs, thereby protecting these compounds from rapid metabolic clearance, although paradoxically also increasing drug potency (Kirchherr et al., 2009; Banerjee and Onyuksel, 2012b; Nie et al., 2012). To illustrate, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (poly-[ethylene glycol])-2000] micelles of pancreatic polypeptide and vasoactive intestinal polypeptide show resistance to proteases in vitro, an increase in total drug exposure (i.e., total drug AUC), and a marked improvement in in vivo potency (Banerjee and Onyuksel, 2012a, 2013; Sethi et al., 2013). In the case of the vasoactive intestinal peptide, its systemic clearance decreased 147-fold and its volume of distribution decreased fivefold in mice (Sethi et al., 2013). In the case of the antidiabetic pancreatic polypeptide, nanomicellar formulation increased glycogen storage and insulin sensitivity 1.7- and 1.8-fold, respectively, over equivalent doses of unformulated peptide in a rat model of diabetes (Banerjee and Onyuksel, 2013). Although these are high clearance drugs, and unbound drug concentrations decreased upon formulation in both of these examples, drug therapeutic potency actually increased (which would seem to contradict the traditional small-molecule paradigm of activity being a function of unbound drug concentrations). In these cases, the enhanced efficacy appears to be a result of direct distribution of active drug to the target organ by the nanoformulation, bypassing the unbound form (Yano et al., 2010; Wang et al., 2012). Thus, to accurately describe the PK/PD relationships for some nanoformulations, we need to understand how target tissue exposure to encapsulated and unencapsulated, unbound forms of the drug relates to drug concentrations at the intracellular site of action. Indeed, for some nanomedicines, the biologically active form of the drug may be the encapsulated drug, not the unencapsulated, unbound drug, diminishing the relevance of the unbound drug fraction from a PK/PD perspective.
Debating the Question: When Unbound Fractions May Need to be Considered
Clearly, knowing the protein-binding characteristics of a drug is an essential component to translating total drug (bound plus unbound) concentration data to its clinical effects (safety or toxicity). For a nanoformulation, the authors also express the opinion that in addition to characterizing unencapsulated and encapsulated drug, differentiating unbound and total (bound plus unbound) concentrations of the unencapsulated drug is necessary. In some situations, the protein-binding characteristics for the unencapsulated drug can be extrapolated from what is known about the drug molecule itself. However, in other situations, direct measurement of the bound and total unencapsulated drug is prudent.
On the basis of the discussions contained within this commentary, we conclude that the following are situations when extrapolation of the unbound/total drug concentration relationship of the legacy or traditionally formulated drug may not adequately describe the PK or PK/PD relationships of their nanoformulations, and direct measurement of unbound versus total unencapsulated drug concentration is necessary:
Equilibrium formulations (where unbound drug fraction may change as a function of formulation).
When there are known nonlinear relationships between total versus unbound drug concentration that could be influenced by the nanoformulation.
Oral nanomedicine formulations of high extraction drugs that are absorbed intact systemically and bypass first pass effect, thereby acting as i.v. formulations with regard to small-molecule protein-binding paradigms.
When the unbound drug is the same as the unencapsulated fraction, such as for protein nanoparticle formulations of small molecules, like Abraxane, and peptide drug nanomedicine formulations.
In the absence of an existing well-characterized relationship between total and unbound drug concentration that can be extrapolated to the nanomedicine formulation.
Ultimately, it is only when the unbound/total drug concentration relationships are understood that we can rely upon unencapsulated drug concentrations to predict changes in PK or PK/PD (safety and/or effectiveness) for a nanoformulation. Without knowledge of the unbound drug concentrations resulting from the administration of nanoformulations, we cannot appreciate the dose-exposure-response relationships that are integral to the safe and effective use of these formulations across a diverse patient population.
Efforts to describe the PK and PK/PD of nanoparticles are generally based on the evaluation of total (encapsulated plus unencapsulated), or encapsulated and unencapsulated drug. We have expressed our opinion that in certain instances such studies need to further define unencapsulated drug from the perspective of total versus unbound drug concentrations. Our hope is that this commentary will stimulate additional discussion and research on this important topic.
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Comments made in this review reflect the views of the authors and are not intended to represent those of the Food and Drug Administration.
Abbreviations
- API
active pharmaceutical ingredient
- AUC
area under the total drug concentration versus time curve
- AUCu
area under the unbound drug concentration versus time curve
- Cmax
concentration maximum
- CRE
cremophor
- Css
clearance steady state
- EPR
enhanced permeability and retention effect
- fu
unbound drug fraction in plasma
- MPS
mononuclear phagocyte system
- MTD
maximum tolerated dose
- PD
pharmacodynamics
- PK
pharmacokinetics
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Stern, Martinez, Stevens.
Footnotes
This work was supported in whole or in part with federal funds from the National Institutes of Health National Cancer Institute [Grant HHSN261200800e001E].
Leidos Biomedical Research is a subcontractor of National Institutes of Health.
References
- Ambardekar VV, Stern ST. (2015) NBCD pharmacokinetics and drug release methods, in Non-Biological Complex Drugs; the Science and the Regulatory Landscape (Crommelin DJA, de Vlieger JSB. eds) pp 261–287, Springer International Publishing, Cham, Switzerland. [Google Scholar]
- Banerjee A, Onyuksel H. (2012a) Human pancreatic polypeptide in a phospholipid-based micellar formulation. Pharm Res 29:1698–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Onyuksel H. (2012b) Peptide delivery using phospholipid micelles. Wiley Interdiscip Rev Nanomed Nanobiotechnol 4:562–574. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Onyuksel H. (2013) A novel peptide nanomedicine for treatment of pancreatogenic diabetes. Nanomedicine 9:722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. (2002) Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob Agents Chemother 46:834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet LZ, Hoener BA. (2002) Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther 71:115–121. [DOI] [PubMed] [Google Scholar]
- Briley-Saebo KC, Mani V, Hyafil F, Cornily JC, Fayad ZA. (2008) Fractionated Feridex and positive contrast: in vivo MR imaging of atherosclerosis. Magn Reson Med 59:721–730. [DOI] [PubMed] [Google Scholar]
- Brouwer E, Verweij J, De Bruijn P, Loos WJ, Pillay M, Buijs D, Sparreboom A. (2000) Measurement of fraction unbound paclitaxel in human plasma. Drug Metab Dispos 28:1141–1145. [PubMed] [Google Scholar]
- Bulitta JB, Zhao P, Arnold RD, Kessler DR, Daifuku R, Pratt J, Luciano G, Hanauske AR, Gelderblom H, Awada A, et al. (2009) Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients. Cancer Chemother Pharmacol 63:1049–1063. [DOI] [PubMed] [Google Scholar]
- Caracciolo G. (2015) Liposome-protein corona in a physiological environment: challenges and opportunities for targeted delivery of nanomedicines. Nanomedicine 11:543–557. [DOI] [PubMed] [Google Scholar]
- Caron WP, Clewell H, Dedrick R, Ramanathan RK, Davis WL, Yu N, Tonda M, Schellens JH, Beijnen JH, Zamboni WC. (2011) Allometric scaling of pegylated liposomal anticancer drugs. J Pharmacokinet Pharmacodyn 38:653–669. [DOI] [PubMed] [Google Scholar]
- Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. (2015) A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov 14:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EKH, Ho D. (2013) Cancer nanomedicine: from drug delivery to imaging. Sci Transl Med 5:216rv4. [DOI] [PubMed] [Google Scholar]
- de Weger VA, Beijnen JH, Schellens JH. (2014) Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel: a review. Anticancer Drugs 25:488–494. [DOI] [PubMed] [Google Scholar]
- Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, et al. (2006) Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res 12:1317–1324. [DOI] [PubMed] [Google Scholar]
- Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. (2013) The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine 9:1–14. Erratum in: Clin Cancer Res (2006) 12:3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence AT, Hussain N. (2001) Transcytosis of nanoparticle and dendrimer delivery systems: evolving vistas. Adv Drug Deliv Rev 50 (Suppl 1):S69–S89. [DOI] [PubMed] [Google Scholar]
- Gardner ER, Dahut WL, Scripture CD, Jones J, Aragon-Ching JB, Desai N, Hawkins MJ, Sparreboom A, Figg WD. (2008) Randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel. Clin Cancer Res 14:4200–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni L, Kearns CM, Giani A, Capri G, Viganó L, Lacatelli A, Bonadonna G, Egorin MJ. (1995) Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol 13:180–190. [DOI] [PubMed] [Google Scholar]
- Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, Scheithauer W, Siena S, Tabernero J, Teixeira L, et al. (2015) nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst 107 DOI: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- Henningsson A, Karlsson MO, Viganò L, Gianni L, Verweij J, Sparreboom A. (2001) Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol 19:4065–4073. [DOI] [PubMed] [Google Scholar]
- Jani P, Halbert GW, Langridge J, Florence AT. (1990) Nanoparticle uptake by the rat gastrointestinal mucosa: quantitation and particle size dependency. J Pharm Pharmacol 42:821–826. [DOI] [PubMed] [Google Scholar]
- Kasai T, Oka M, Soda H, Tsurutani J, Fukuda M, Nakamura Y, Kawabata S, Nakatomi K, Nagashima S, Takatani H, et al. (2002) Phase I and pharmacokinetic study of paclitaxel and irinotecan for patients with advanced non-small cell lung cancer. Eur J Cancer 38:1871–1878. [DOI] [PubMed] [Google Scholar]
- Kirchherr AK, Briel A, Mäder K. (2009) Stabilization of indocyanine green by encapsulation within micellar systems. Mol Pharm 6:480–491. [DOI] [PubMed] [Google Scholar]
- Lobatto ME, Fuster V, Fayad ZA, Mulder WJ. (2011) Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov 10:835–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas AT, Madden AJ, Zamboni WC. (2015) Formulation and physiologic factors affecting the pharmacology of carrier-mediated anticancer agents. Expert Opin Drug Metab Toxicol 11:1419–1433. [DOI] [PubMed] [Google Scholar]
- Maeda H. (2012) Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release 164:138–144. [DOI] [PubMed] [Google Scholar]
- Mukherjee B, Das S, Chakraborty S, Satapathy BS, Das PJ, Mondal L, Hossain CM, Dey NS, Chaudhury A. (2014) Potentials of polymeric nanoparticle as drug carrier for cancer therapy: with a special reference to pharmacokinetic parameters. Curr Drug Metab 15:565–580. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Fang J, Maeda H. (2015) Development of next-generation macromolecular drugs based on the EPR effect: challenges and pitfalls. Expert Opin Drug Deliv 12:53–64. [DOI] [PubMed] [Google Scholar]
- Nie T, Wong CC, Alston N, Aro P, Constantinides PP, Rigas B. (2012) Phospho-ibuprofen (MDC-917) incorporated in nanocarriers: anti-cancer activity in vitro and in vivo. Br J Pharmacol 166:991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue S, Yamada S, Chan HK. (2014) Nanodrugs: pharmacokinetics and safety. Int J Nanomedicine 9:1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otagiri M. (2005) A molecular functional study on the interactions of drugs with plasma proteins. Drug Metab Pharmacokinet 20:309–323. [DOI] [PubMed] [Google Scholar]
- Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. (2013) Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res 73:2412–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell RF, Sidone BJ, Caron WP, Walsh MD, White TF, Zamboni BA, Ramanathan RK, Zamboni WC. (2014) Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine 10:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Gonzalez D, Derendorf H. (2010) Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci 99:1107–1122. [DOI] [PubMed] [Google Scholar]
- Sethi V, Rubinstein I, Kuzmis A, Kastrissios H, Artwohl J, Onyuksel H. (2013) Novel, biocompatible, and disease modifying VIP nanomedicine for rheumatoid arthritis. Mol Pharm 10:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidone B, Edwards RP, Zamboni B, Strychor S, Maruca L, Zamboni WC. (2007) Evaluation of body surface area (BSA) based dosing, age, and body composition as factors affecting the pharmacokinetic (PK) variability of STEALTH liposomal doxorubicin (Doxil). Mol Cancer Ther 6:3561s(abstract C107) [Google Scholar]
- Skoczen S, McNeil SE, Stern ST. (2015) Stable isotope method to measure drug release from nanomedicines. J Control Release 220:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BS, Yogaratnam D, Levasseur-Franklin KE, Forni A, Fong J. (2012) Introduction to drug pharmacokinetics in the critically ill patient. Chest 141:1327–1336. [DOI] [PubMed] [Google Scholar]
- Smith DA, Di L, Kerns EH. (2010) The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9:929–939. [DOI] [PubMed] [Google Scholar]
- Song G, Wu H, Yoshino K, Zamboni WC. (2012) Factors affecting the pharmacokinetics and pharmacodynamics of liposomal drugs. J Liposome Res 22:177–192. [DOI] [PubMed] [Google Scholar]
- Sonnichsen DS, Hurwitz CA, Pratt CB, Shuster JJ, Relling MV. (1994) Saturable pharmacokinetics and paclitaxel pharmacodynamics in children with solid tumors. J Clin Oncol 12:532–538. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N. (2005) Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res 11:4136–4143. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, Pillay M, Nooter K, Stoter G, Verweij J. (1999) Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res 59:1454–1457. [PubMed] [Google Scholar]
- Svenson S. (2014) What nanomedicine in the clinic right now really forms nanoparticles? Wiley Interdiscip Rev Nanomed Nanobiotechnol 6:125–135. [DOI] [PubMed] [Google Scholar]
- Tesseromatis C, Alevizou A. (2008) The role of the protein-binding on the mode of drug action as well the interactions with other drugs. Eur J Drug Metab Pharmacokinet 33:225–230. [DOI] [PubMed] [Google Scholar]
- van Tellingen O, Huizing MT, Panday VR, Schellens JH, Nooijen WJ, Beijnen JH. (1999) Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br J Cancer 81:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zuylen L, Gianni L, Verweij J, Mross K, Brouwer E, Loos WJ, Sparreboom A. (2000) Inter-relationships of paclitaxel disposition, infusion duration and cremophor EL kinetics in cancer patients. Anticancer Drugs 11:331–337. [DOI] [PubMed] [Google Scholar]
- van Zuylen L, Karlsson MO, Verweij J, Brouwer E, de Bruijn P, Nooter K, Stoter G, Sparreboom A. (2001) Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother Pharmacol 47:309–318. [DOI] [PubMed] [Google Scholar]
- Vigano L, Locatelli A, Grasselli G, Gianni L. (2001) Drug interactions of paclitaxel and docetaxel and their relevance for the design of combination therapy. Invest New Drugs 19:179–196. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang Y, Liang W. (2012) Delivery of drugs to cell membranes by encapsulation in PEG-PE micelles. J Control Release 160:637–651. [DOI] [PubMed] [Google Scholar]
- Yano K, Masaoka Y, Kataoka M, Sakuma S, Yamashita S. (2010) Mechanisms of membrane transport of poorly soluble drugs: role of micelles in oral absorption processes. J Pharm Sci 99:1336–1345. [DOI] [PubMed] [Google Scholar]
- Zamboni WC, Ramalingam S, Friedland DM, Edwards RP, Stoller RG, Strychor S, Maruca L, Zamboni BA, Belani CP, Ramanathan RK. (2009) Phase I and pharmacokinetic study of pegylated liposomal CKD-602 in patients with advanced malignancies. Clin Cancer Res 15:1466-1472 Erratum in: Clin Cancer Res (2009) 15:2949–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni WC, Torchilin V, Patri AK, Hrkach J, Stern S, Lee R, Nel A, Panaro NJ, Grodzinski P. (2012) Best practices in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res 18:3229–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger MA, Derendorf H, Mouton JW, Cars O, Craig WA, Andes D, Theuretzbacher U. (2011) Protein binding: do we ever learn? Antimicrob Agents Chemother 55:3067–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]