

Abstract
Studies in vitro have demonstrated that β3-adrenergic receptors (β3-ARs) regulate protein metabolism in skeletal muscle by promoting protein synthesis and inhibiting protein degradation. In this study, we evaluated whether activation of β3-ARs by the selective agonist CL316,243 modifies the functional and structural properties of skeletal muscles of healthy mice. Daily injections of CL316,243 for 15 days resulted in a significant improvement in muscle force production, assessed by grip strength and weight tests, and an increased myofiber cross-sectional area, indicative of muscle hypertrophy. In addition, atomic force microscopy revealed a significant effect of CL316,243 on the transversal stiffness of isolated muscle fibers. Interestingly, the expression level of mammalian target of rapamycin (mTOR) downstream targets and neuronal nitric oxide synthase (NOS) was also found to be enhanced in tibialis anterior and soleus muscles of CL316,243 treated mice, in accordance with previous data linking β3-ARs to mTOR and NOS signaling pathways. In conclusion, our data suggest that CL316,243 systemic administration might be a novel therapeutic strategy worthy of further investigations in conditions of muscle wasting and weakness associated with aging and muscular diseases.
Studies in humans and animal models have revealed that β-adrenergic receptors (β-ARs) stimulation exerts potent anabolic effects on striated muscles1,2. Since activation of β-ARs induces skeletal muscle growth associated, in some cases, with an increase of contractile function3,4, β-AR agonists have been proposed as a therapeutic intervention to counteract muscle wasting correlated with aging or chronic diseases such as muscle dystrophy5,6,7. However, the potential for targeting β-ARs in dystrophies has been diminished because of the mild improvements in skeletal mass/function and adverse cardiac events induced by β1/β2 ARs agonists2. So far, much of our knowledge on the role of β-AR signaling in skeletal muscle is based on studies focused on β2-AR agonists, since β2-AR is considered the predominant subtype in skeletal muscle2. However, β3-ARs have been also identified in human and rodent skeletal muscles8,9. Selective activation of β3-ARs has been established to determine important metabolic responses in skeletal muscle such as glucose uptake, phosphorylation, and oxidation leading to an increase of energy expenditure10. In addition, β3-AR agonists have been shown to affect muscle thermogenesis by increasing the expression of the uncoupling protein-3 (UCP-3), a protein that uncouples mitochondrial respiration from ATP production, thereby dissipating energy in the form of heat11. Even though metabolic effects of β3-AR activation are highly recognized, less is known about the impact of β3-ARs in the regulation of skeletal muscle structure and function. Using a β3-AR selective agonist, CL316,243, we have recently demonstrated in vitro that β3-ARs play a critical role in the regulation of protein metabolism in skeletal muscle12. In particular, we found that CL316,243 induced a significant increase of skeletal muscle constitutive proteins into muscle cell proteins such as myosin heavy chain, myosin light chain, and actin in rat L6 myocytes. Such anabolic effect was associated with the activation of PI3K/Akt/mTOR pathway, via Gi/o protein, resulting in an increase of p70S6 kinase (p70S6K) and protein translation. Another signaling pathway that has been linked to β3-AR is the G protein inhibitory (Gi)–nitric oxide (NO) pathway13. In ventricular muscles, activation of the β3-AR receptors by BRL 37344 is accompanied by decreased contractility via NO production. The β3-AR-induced negative inotropic effect was shown to be inhibited by the NOS inhibitor L-NAME and could be reversed by an excess of the NOS-substrate, L-arginine14.
Based on these lines of evidence, we first examined whether the in vivo administration of the β3-AR agonist CL316,243 affected skeletal muscle strength in adult mice. By using atomic force microscopy (AFM), we next determined whether β3-AR stimulation modifies the mechanical properties of dissociated skeletal muscle fibers. Furthermore, to gain more insight into the molecular mechanism underlying the β3-AR function in skeletal muscle, we investigated whether CL316,243 treatment was associated with an upregulation of the putative β3-AR signaling transduction pathways, involving p70S6K as well as the neuronal nitric oxide synthase (nNOS), which is considered the main source of NO in skeletal muscle15.
Results
CL316,243 treatment induces an increase in skeletal muscle strength in adult healthy mice
Muscular strength was assessed in wild-type healthy mice treated with the selective β3-ARs agonist CL316,243 (CL; 1 mg/kg) or saline once per day for 15 days. As shown in Fig. 1A, CL-treated mice exhibited a significant increase in the strength score on the weight test (23.9 ± 0.1 vs. 17.44 ± 1.23; p < 0.0001). These results were confirmed by the grip strength test, showing that CL316,243 treatment resulted in a 23% increase of peak force with respect to control (0.96 ± 0.04 vs. 0.78 ± 0.03; p = 0.008; Fig. 1B). Furthermore, we found that injections of CL316,243 at this dose and duration did not affect mice body weight (corresponding to 27.81 ± 0.31 grams before CL-treatment vs. 27.93 ± 0.30 grams after CL-treatment; t(17) = 0.275, p = 0.787).
Figure 1. Effects of treatment with CL316,243 on muscular strength in wild type mice.
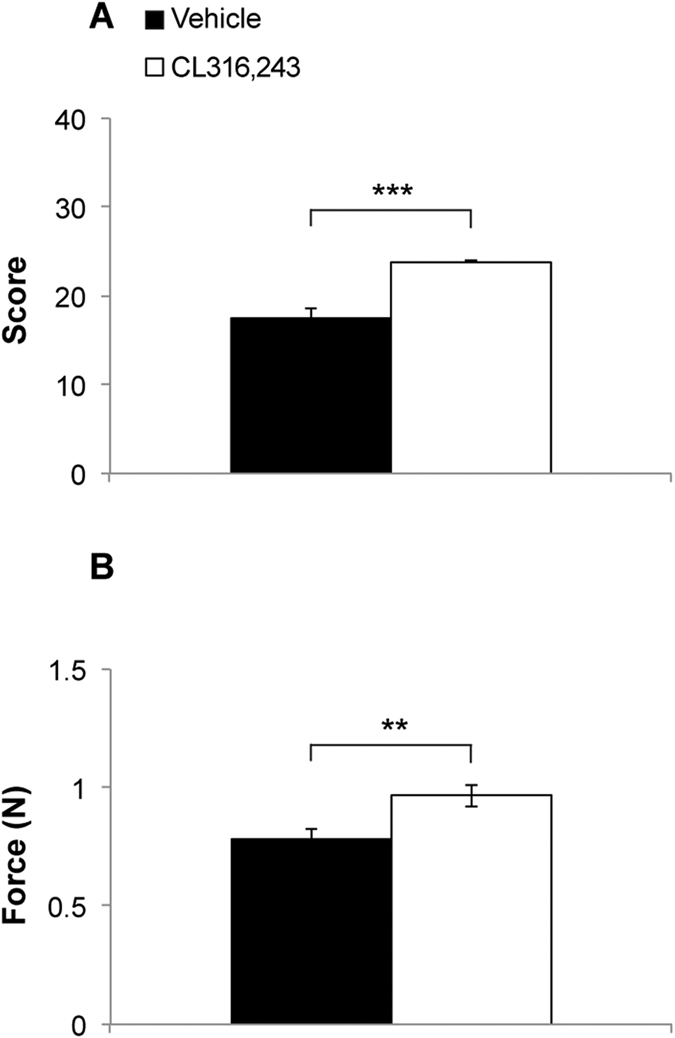
(A) Mice treated with the β3-AR agonist CL316,243 show an increase in the strength score. The score was calculated as the product of the number of links in the heaviest chain held for the full 3 sec, multiplied by the time (sec) it was held (n = 10 CL316,243-treated mice vs n = 9 vehicle-treated mice, unpaired t-test: t(17) = 5.495, p < 0.0001). (B) Grip test shows an increase in peak force in CL316,243-treated mice compared to vehicle (n = 10/9; unpaired t-test: t(17) = 2.978, p = 0.008). ***p < 0.0001, **p < 0.01.
CL316,243 regulates the mechanical properties of skeletal muscle fibers
To determine whether β3-AR activation can also affect the mechanical properties of the cytoskeleton, we measured the transversal stiffness of CL316,243-treated flexor digitorum brevis (FDB) fibers in the relaxed state by means of AFM-based nanoindentation technique. AFM is a useful tool for studying cell mechanics since it allows to apply controlled loads in the nanoNewtown range to living cells and measure the corresponding cell deformation with nanometer resolution16,17,18 (Fig. 2A and B). AFM measurements were performed on single dissociated muscle fibers incubated with either control solution or 1 μM CL316,243 for 3 or 12 hours. As shown in Fig. 2C, treatment with CL316,243 induced a significant reduction of transversal stiffness in the sub-sarcolemma region, at an indentation depth of 200 nm, when compared to untreated fibers at both time points (p < 0.0001).
Figure 2. Effects of CL316,243 treatment on muscle stiffness.

(A) Immunofluorescence image of a dissociated FDB muscle fiber to visualize Z-bands (red), F-actin (green) and nuclei (blue). (B) Bright field image of a single FDB fiber and the AFM rectangular cantilever during nanoindentation measurements. (C) Normalized stiffness indicated as Young’s modulus, was obtained by AFM nanoindentation measurements from single dissociated muscle fibers, at 200 nm penetration depth. Fibers were incubated with CL316,243 or vehicle and stiffness was measured after 3 or 12 h treatment (n = 4–5 fibers/treatment, two-way ANOVA for treatment F(1,14) = 81.022, p < 0.0001, time F(1,14) = 0.087; p = 0.772, and treatment x time interaction F(1,14) = 1.218; p = 0.288). ***p < 0.0001. Calibration bar = 100 μm.
CL316,243 treatment induces an increase in muscle fiber cross-sectional area in adult healthy mice
To assess whether the increase in muscle strength induced by CL316,243 was associated with muscle hypertrophy, we measured the cross-sectional area (CSA) of hindlimb of CL-treated mice. For this purpose, we selected the soleus and the tibialis anterior (TA), as representative of slow-twitch and fast-twitch muscles, respectively. Morphometric analysis of muscle fibers revealed that CL316,243 induced a significant increase by 83% in TA muscle fiber CSA (p < 0.0001), whereas in soleus CSA was slightly increased by 11% (p = 0.13) compared to control (Fig. 3A and B). The examined muscles did not show any sign of degeneration such as intracytoplasmic vacuoles and centralized nuclei.
Figure 3. Effects of CL316,243 treatment on fiber cross-sectional area.
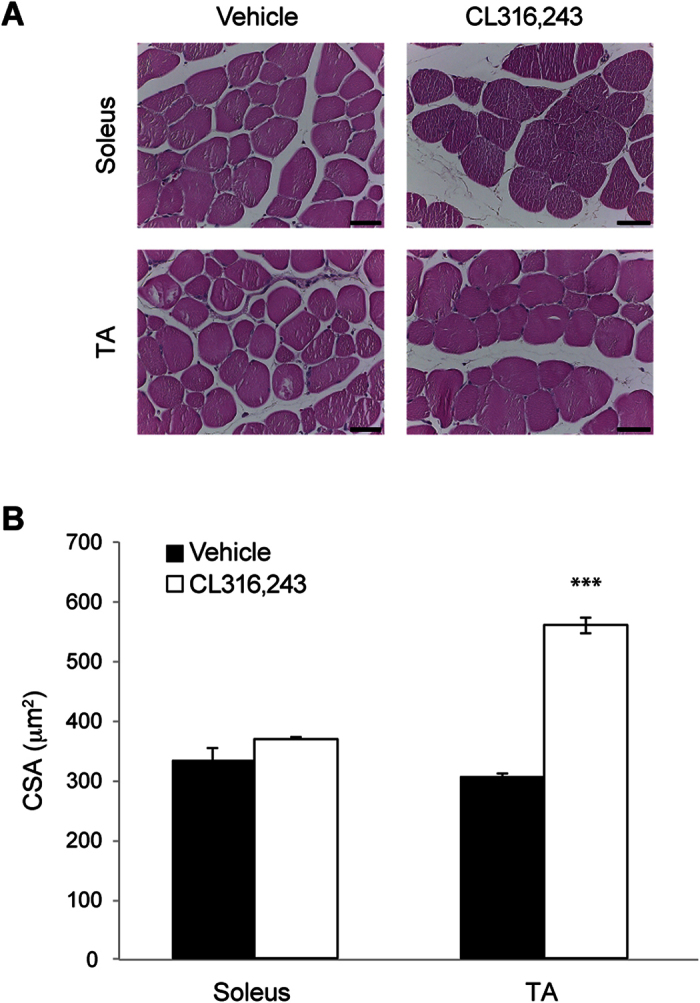
(A) Representative images of hematoxylin and eosin staining of soleus (upper panels) and TA (bottom panels) muscles from mice treated with CL316,243 or vehicle. The stained muscle sections were analyzed for CSA. (B) Bar Graph showing the increase in TA CSA after treatment with CL316,243 compared to vehicle (TA CSA, n = 3–4 samples/treatment; unpaired t-test: t(5) = 15.08, p < 0.0001; soleus CSA n = 3 soleus muscle samples/treatment; unpaired t-test: t(4) = 1.86; p = 0.13). ***p < 0.0001. Calibration bar = 20 μm.
CL316,243 treatment increases the skeletal muscle expression level of p70S6K and rpS6
According to our previous studies in vitro, β3-ARs stimulation up-regulates protein synthesis in myocyte cultures and this effect is likely mediated by the PI3K– mTOR- p70S6K signaling pathway activation. Indeed, the CL316,243-induced increase of p70S6K was markedly inhibited by wortmannin, a PI3K inhibitor, and rapamycin, a specific inhibitor of mTOR12. Based on these observations, we examined whether the in vivo administration of CL316,243 modulates the expression of p70S6K and its downstream target, rpS6, in skeletal muscles obtained from mice treated with CL316,243 or vehicle. As shown in Fig. 4A and B, western blot analysis revealed that the expression level of phospho-p70S6K was significantly higher in both TA and soleus muscles of CL316,243-treated mice with respect to vehicle-treated mice (p < 0.0001). This up-regulation of p70S6K was associated with an increased expression of phospho-rpS6 in TA (p < 0.0001) and soleus (p < 0.05) when compared to control conditions (Fig. 4A and C).
Figure 4. Effects of CL316,243 treatment on phospho-p70S6K and phospho-rpS6 expression levels in skeletal muscles.

(A) Representative western blot showing the p-p70S6K and p-rpS6 protein expression in TA and soleus skeletal muscles of mice treated with CL316,243 (CL) or vehicle (Veh). α-Tubulin was used as internal loading control. (B) Densitometric quantification of protein shows significant increase of of p-p70S6K in both soleus and TA after treatment with CL316,243 (n = 5/5; for soleus unpaired t-test: t(8) = 8.51, p < 0.0001; for TA unpaired t-test: t(8) = 6.04, p < 0.0001). (C) Densitometric analysis shows significant increase of p-rpS6 p70S6K in both soleus and TA after treatment with CL316,243 (n = 5/5; for soleus unpaired t-test: t(8) = 2.93, p = 0.019; for TA unpaired t-test: t(8) = 5.91, p < 0.0001). *p < 0.05, ***p < 0.0001.
CL316,243 treatment increases the skeletal muscle expression level of neuronal-NOS
Studies in murine myocardium have demonstrated that application of the β3-AR agonist, BRL 37344, modulates NOS activity and increases NO formation19. Additional evidence for nNOS coupling to β3-AR comes from studies showing that the β3-AR-induced negative inotropic effect is absent in cardiomyocytes of nNOS-deficient (NOS1−/−) mice as well as in control cardiomyocytes with acute nNOS inhibition20. We, therefore, examined whether CL316,243 treatment in vivo affected the expression level of nNOS, which is the most abundant NOS isoform in skeletal muscle. As shown in Fig. 5A and B, nNOS appeared to be regulated in both TA (p < 0.01) and soleus (p < 0.0001) of CL316,243-treated mice with respect to vehicle-treated mice. These data suggest that nNOS may be involved in the β3-AR effects on skeletal muscle.
Figure 5. Effects of CL316,243 treatment on nNOS expression levels in skeletal muscles.
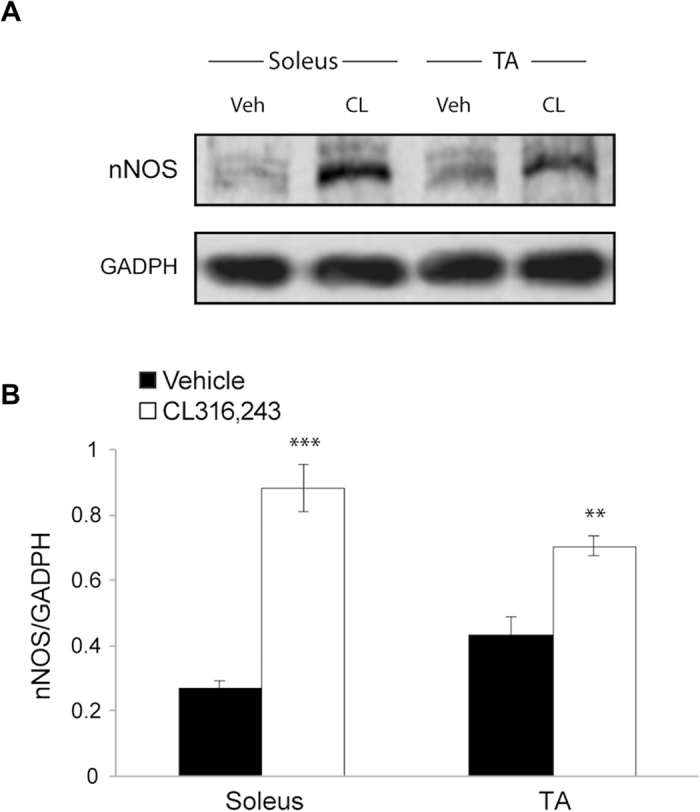
(A) Representative western blot showing the nNOS protein expression in TA and soleus skeletal muscles of mice treated with CL316,243 (CL) or vehicle (Veh). GADPH was used as internal loading control. (B) Densitometric quantification of protein shows significant increase of nNOS in both soleus and TA after treatment with CL316,243 (n = 5/5; for soleus unpaired t-test: t(8) = 8.41, p < 0.0001; for TA unpaired t-test: t(8) = 4.57, p = 0.002). **p < 0.01, ***p < 0.0001.
Discussion
This study provides the first demonstration that β3-AR activation has important anabolic effects on skeletal muscle. In particular, we show that CL316,243 treatment induces a significant increase of muscle CSA and strength in adult healthy mice compared to controls. The increase in CSA, indicative of muscle hypertrophy, was particularly evident in TA compared to soleus muscle, suggesting a difference in response to β3-agonists between fast- and slow-twitch skeletal muscles. The increase of CSA in TA, a muscle containing a high quantity of fast-contracting fibers, is in agreement with several studies reporting a hypertrophic effect of β2-AR agonist mainly in fast-twitch fibers21,22. Conversely, there is conflicting evidence concerning whether the slow-contracting fibers are affected by β2-AR agonist treatment in intact animals23. In addition, when mice were challenged with specific behavioral tests to evaluate skeletal muscle function, we found an increase of muscle strength in CL316,243-treated mice compared to vehicle-treated animals. Such positive effect was confirmed by the grip strength test, which allowed us to evaluate the peak resistance force, and by the weights test, evaluating the maximal isometric strength.
Several agents have been shown to increase skeletal muscle mass and force24 by regulating protein synthesis, including anabolic steroids, growth hormones, IGF and β2-AR agonists4,25,26,27,28. Most of these factors control the rate of protein turnover at the level of transcription, translation, degradation or a combination of these29. According to our previous studies in vitro, β3-ARs stimulation up-regulates protein synthesis and this effect is likely due to the activation of components of the translational machinery, including the ribosomal protein S612. Here, the use of an in vivo model confirmed our previous in vitro data, thus providing a further demonstration that CL316,243 has the potential to regulate protein metabolism in skeletal muscle by increasing the expression of mTOR targets in the long term. The importance of mTOR in muscle size regulation has been demonstrated by both pharmacological and genetic studies30,31,32. For example, inhibition of mTOR by rapamycin prevented the hypertrophy of myotubes induced in vitro by IGF as well as the skeletal muscle hypertrophy in vivo induced by overload or clenbuterol25,33,34,35. A decrease in muscle mass and fiber CSA has also been revealed in mTOR and p70S6K knockout mice36,37. According to Navegantes and collaborators38, the anabolic effects of CL316,243 in skeletal muscles are in part due to an inhibition of muscle proteolysis. Such anti-proteolytic effect was particularly evident in rat soleus, but not in extensor digitorum longus, suggesting a different response to β3-ARs agonist of slow-twitch and fast-twitch muscle. However, the effects of β3-AR activation in slow-twitch muscle fibers are quite complex since they can also involve the regulation of mitochondrial uncoupling proteins. In particular, immunohistochemical studies have revealed an increase of UCP-3 signals in slow-twitch muscles of obese mice following a chronic administration of CL316,243, which may contribute to the thermogenic effect of β3-AR agonists11,39. In this study, we have also shown that CL316,243 treatment was associated with an increased expression of the nNOS protein in both fast- and slow-twitch muscles. In skeletal muscle, NO has been identified as a physiological intracellular messenger modulating the contractile activity of skeletal muscle, blood flow, exercise-induced skeletal muscle hypertrophy and glucose homeostasis40,41,42,43,44. The precise role of the nNOS isoform in skeletal muscle is still a matter of debate, although several lines of evidence suggested that NO plays a key role in the hypertrophic response of skeletal muscle to mechanical and metabolic stimulations45,46. NOS activity has been shown to promote transcription of contractile proteins, such as skeletal α-actin and type I myosin heavy chain mRNA, during chronic skeletal muscle overload44. According to our previous studies in vitro47, the anabolic effect of NOS might be mediated by the mTOR/p70S6K signaling pathway since NOS inhibition by L-NAME prevented the activation of p70S6K in response to glucose deprivation. The importance of nNOS in skeletal muscle has been clearly revealed by studies carried out in nNOS-deficient (NOS1−/−) mice48. These mice presented a significant reduction of muscle mass and CSA of tibialis anterior accompanied by a decrease of muscle force and decreased resistance to fatigue, compared with control mice. Interestingly, the TA of NOS1−/− mice showed also lower levels of phosphorylated rpS6, 4E-BP1, and Akt than controls, suggesting that the AKT/mTOR pathway activation was reduced in absence of nNOS. In addition, recent studies have demonstrated that restoration of NO signaling by nNOS overexpression can reduce muscle pathology in mouse models of muscular dystrophy (named mdx mice) by preventing muscle membrane injury and promoting regeneration49. A novel important finding of this study is that the acute treatment with CL316,243 induced a significant decrease of the transversal stiffness of single muscle fibers within the first 1000 nm of the fiber surface. This suggests that β3-AR activation may modulate the elastic properties of near-membrane components, such as the external basal membrane, sarcolemma, cytoskeletal network, and cytoplasm50. Further studies are still necessary to clarify the mechanisms by which CL316,243 lowers muscle stiffness and its potential role under pathological conditions associated with high muscle stiffness, including muscle dystrophies. Interestingly, β-adrenergic stimulation has been demonstrated to have also an acute effect on the myocardial stiffness of rabbits51. In particular, it has been found that exposure to isoprenaline, a non-selective β-AR agonist, induced a concentration-dependent reduction of myocardial stiffness in papillary muscles isolated from the rabbit’s ventricle. In this case, titin phosphorylation was hypothesized as the molecular mechanism responsible for the observed change in stiffness.
Together, these results suggest that targeting β3-AR may be an effective therapeutic strategy for enhancing muscle growth and strength in a variety of disorders associated with muscle loss and degeneration.
Methods
Animals and treatment with CL316,243
C57Bl/6 J wild type male mice aged 3 months were obtained from a breeding colony kept at the University of Naples and University of Catania. Mice were maintained at a controlled temperature (21 °C ± 1 °C) and humidity (50%) on a 12 h light/dark cycle (light from 06:00 to 18:00), with ad libitum food and water. All animal experimentation was conducted in accordance with the guidelines laid down by the European Community Council (2010/63/EU). The experimental protocols have been approved by the University Institutional Animal Care and Use Committee from the University of Naples (#0016945, 02/16/2012). Experiments were performed in parallel using 2 groups of mice treated with vehicle (n = 9) or CL316,243 (n = 10) at a concentration of 1 mg/kg in saline by subcutaneous injections for 15 days. Two hours after the last injection, mice underwent behavioral assessment of muscular strength by weights test52 and grip strength test53. Mice were then sacrificed by cervical dislocation and muscles were removed and frozen for western blot or processed for CSA evaluation.
Behavioral assessment of muscle strength
The weights test was performed as previously described52. We used a series of chain links of different weight (from 14 to 74 gr) attached to a ball of fine wire mesh. Each mouse was held by the tail and was allowed to grasp a series of increasing weight steel chain links placed on the laboratory bench. Based on the number of chain links that the mouse was able to grasp and hold for at least 3 seconds, a specific score was assigned. If the mouse dropped the weight in less than 3 sec, the trial was repeated for 3 times and the maximum time/weight achieved was considered for the final scoring. After a rest period of about 20 sec, the next heaviest weight was tested until the mouse failed for 3 consecutive trials. A final total score was calculated as the product of the number of links in the heaviest chain held for the full 3 sec, multiplied by the time (sec) it was held. If the heaviest weight was dropped before 3 sec an appropriate intermediate value was calculated. For example, a mouse holding a 5-link weight for 3 seconds, but unable to lift a 6-link weight, was assigned a score of (5 × 3) = 15. If it held the 6-link weight for 1 second, the score was (5 × 3) + (1) = 16.
Grip strength test was performed as previously described53 by using an apparatus equipped with a mouse horizontal forelimb bar (Bioseb, Model GT3). Mice were held by the tail and were allowed to grasp the horizontal bar with the forelimb paws. The mice were then gently pulled backward until they released the grid. The peak force applied by the forelimbs of the mouse was recorded in Newton (N). Each mouse received 3 test trials (with a rest of 2 minutes) for two consecutive sessions (1 hour apart).
Preparation and Culture of Muscle Fibers
Untreated adult mice (2–3 months old) were sacrificed by cervical dislocation and FDB muscles were quickly dissected and placed in a small petri dish filled with Tyrode Solution (in mM: 140 NaCl, 2 KCl, 2 CaCl2, 10 HEPES, and 5 glucose). FDB muscles were exposed to enzymatic digestion by using 0.2–0.3% Collagenase type I in Tyrode solution for 1 hour at 4 °C and then incubated in 5% CO2 for 1 hour at 37 °C. After three washes in Tyrode solution containing 10% FBS to block the collagenase effect and stabilize the fibers, FDB muscles were gently triturated to dissociate individual muscle fibers. The fibers were finally plated on laminin-coated culture dishes in serum-containing Tyrode solution and incubated in 5% CO2 at 37 °C until use.
Immunofluorescence
For double immunofluorescence microscopy, the skeletal fibers in culture were fixed in 4% formaldehyde in PBS and permeabilized with PBS-Triton-X100 for 10 min at room temperature. Samples were then incubated for 4 hours with primary antibody against sarcomeric α-actinin (Sigma A-7811) in order to visualize Z-band on myofilaments. After washing in PBS, fibers were incubated for 1 hour in PBS 1% BSA with TRITC-conjugated goat anti-mouse (AlexaFluor 546). We used FITC-conjugated phalloidin (Sigma-Aldrich; St. Louis) to stain filamentous actin (F-actin) and 4′-6-diamidino-2-phenylindol (DAPI) for nuclei labeling. The specimens were visualized, with an Olympus IX-70 epifluorescence microscope equipped with a Hamamatzu-Orca ER II camera. Image ProPlus was used for the image acquisition.
Atomic force microscopy
A commercial atomic force microscope (Keysight Technologies AFM model 5500) mounted on an inverted optical microscope (Olympus IX70) was used to assess the transversal stiffness of single dissociated skeletal muscle fibers17,54. Silicon cantilevers with a nominal spring constant k = 0.03 N/m and conical tips (CSC21, MikroMasch, Germany) were used. The spring constant of each cantilever was determined by a thermal noise based method, which ensures a level of accuracy of 10%–15%55. To probe the mechanical stiffness onto and below the sarcolemma, we performed force versus distance measurements and evaluated the contact region of the obtained curves. The transversal stiffness was measured as the Young’s modulus calculated by considering an approximate purely elastic response of the indented fiber, as proposed by Oliver and collaborators56. The stiffness was calculated at different penetration depths by applying controlled forces in the 0.5–2 nN range. Each measurement consisted of 256 force versus distance curves taken on the same 3 × 3 μm2 region. At least three measurements were performed on a single fiber for each time point (3 and 12 hours) and at least three different fibers were probed for each treated or control condition.
Histology
Vehicle and CL316,243-treated mice were sacrificed by cervical dislocation and the limb muscles, TA and soleus, were harvested. Muscles were cross-cut, fixed in 10% buffered formalin, embedded in paraffin, then cut into 6 μm-thick serial sections and mounted on polylysine coated slides. Sections were stained with Hematoxylin and Eosin staining kit (Bio-optica, Milan, Italy), according to manufacturer protocol. Microscopic observation was performed by Leica DM2000LED (Leica Microsystems, Wetzlar, Germany) light microscope equipped with Leica ICC50HD digital camera for photodocumentation. Digital images acquired were then analyzed with SigmaScan Pro 5.0 software (SYSTAT, San Jose, CA, USA) to measure CSA. Measurements were performed by three independent observers and expressed as mean surface area (μm2). A total of 300–350 muscle fibers were analyzed for each muscle.
Western blot analysis
TA and soleus of both vehicle and CL316,243-treated mice (n = 5 for each condition) were homogenized in lysis buffer (1:2, w/v) solution containing 0.5 M β-glycerophosphate, 20 mM MgCl2, 10 mM ethylene glycol tetraacetic acid, and supplemented with 100 mM dithiothreitol and protease/phosphatase inhibitors (100 mM dimethylsulphonyl fluoride, 2 mg/ml apronitin, 2 mM leupeptin, and 10 mM Na3VO4). Protein concentration was determined by the Bio-Rad protein assay (Bio-Rad, Milan, Italy). Samples containing 100 μg of proteins were denatured, separated on a 10% (for p70S6K and rpS6) or 8% (for nNOS) SDS-polyacrylamide gel, and electro-transferred onto a nitrocellulose membrane using a Bio-Rad Trans-Blot (Bio-Rad, Italy). Western blotting detection reagents were obtained from Amersham Biosciences (UK); the nitrocellulose membrane was from Hybond ECL (GE Healthcare, UK). Proteins were visualized by reversible staining with Ponceau S solution and destained in PBS57. Membranes were blocked at room temperature in milk buffer (1Χ PBS, 5–10% v/v non-fat dry milk, 0.2% v/v Tween-20) and then incubated at 4 °C overnight with the following primary antibodies: anti-phospho-p70S6K (1:1000; Cell Signaling Technology, Massachusetts, USA), anti-phospho-rpS6 (1:1000; Cell Signaling Technology, Massachusetts, USA), anti-nNOS (1:300; Santa Cruz, California, USA); anti-α-tubulin antibody (1:1000; Cell Signaling Technology, Massachusetts, USA) or anti-GAPDH (1:8000; Sigma Aldrich, Milan, Italy). The membranes were then incubated for 90 min at room temperature with 1:5000 horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse antibodies. The resulting complexes were visualized using chemiluminescence Western blotting detection reagents. The western blot images were scanned using GS-800 imaging densitometer (Bio-Rad, Italy) and analyzed using Quantity One software (Biorad, Italy). The background-subtracted density of the bands in all blots was measured and normalized using α-tubulin or GAPDH.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed by using Systat software (Chicago, IL, USA). To compare the experimental conditions, we used unpaired Student’s t-test. A single TA and soleus muscle were examined for each treated animal. Two-way ANOVA was used for the AFM data. The level of significance was set at p < 0.05.
Additional Information
How to cite this article: Puzzo, D. et al. CL316,243, a β3-adrenergic receptor agonist, induces muscle hypertrophy and increased strength. Sci. Rep. 6, 37504; doi: 10.1038/srep37504 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
We thank Angelo Russo and Giovanni Esposito for their valuable technical assistance. The present research was supported by Bank of Italy grant #744460/13 to Maria Concetta Miniaci. The funding sources had no involvement in the study design, collection, analysis or interpretation of data, in the writing of the report, or in the decision to submit for publication.
Footnotes
The authors declare no competing financial interests.
Author Contributions D.P. and W.G. performed behavioral experiments; C.C. performed cross-sectional area experiments; E.P., R.C. and P.L. performed western blot experiments; M.T. performed isolation, culturing and immunocytochemistry of muscle fibers; R.R. and L.D. performed atomic force microscopy; D.P., R.R., A.P., and P.S. contributed through numerous discussions; M.C.M. and D.P. wrote the paper; P.S. and M.C.M supervised the project.
References
- Kim Y. S. & Sainz R. D. Beta-adrenergic agonists and hypertrophy of skeletal muscles. Life sciences 50, 397–407 (1992). [DOI] [PubMed] [Google Scholar]
- Lynch G. S. & Ryall J. G. Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiological reviews 88, 729–767, doi: 10.1152/physrev.00028.2007 (2008). [DOI] [PubMed] [Google Scholar]
- Ryall J. G., Gregorevic P., Plant D. R., Sillence M. N. & Lynch G. S. Beta 2-agonist fenoterol has greater effects on contractile function of rat skeletal muscles than clenbuterol. Am J Physiol Regul Integr Comp Physiol 283, R1386–R1394, doi: 10.1152/ajpregu.00324.2002 (2002). [DOI] [PubMed] [Google Scholar]
- Ryall J. G., Sillence M. N. & Lynch G. S. Systemic administration of beta2-adrenoceptor agonists, formoterol and salmeterol, elicit skeletal muscle hypertrophy in rats at micromolar doses. British journal of pharmacology 147, 587–595, doi: 10.1038/sj.bjp.0706669 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltin C. A., Hay S. M., Delday M. I., Lobley G. E. & Reeds P. J. The action of the beta-agonist clenbuterol on protein metabolism in innervated and denervated phasic muscles. The Biochemical journal 261, 965–971, doi: 10.1042/bj2610965 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman R. J., Peng H., Danon M. J. & Etlinger J. D. Clenbuterol reduces degeneration of exercised or aged dystrophic (mdx) muscle. Muscle & nerve 23, 521–528, doi: 10.1002/(SICI)1097-4598(200004)23:4<521::AID-MUS10>3.0.CO;2-8 (2000). [DOI] [PubMed] [Google Scholar]
- Beitzel F. et al. Beta2-adrenoceptor agonist fenoterol enhances functional repair of regenerating rat skeletal muscle after injury. J Appl Physiol (1985) 96, 1385–1392, doi: 10.1152/japplphysiol.01081.2003 (2004). [DOI] [PubMed] [Google Scholar]
- Evans B. A., Papaioannou M., Bonazzi V. R. & Summers R. J. Expression of beta 3-adrenoceptor mRNA in rat tissues. British journal of pharmacology 117, 210–216 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P. D. et al. The tissue distribution of the human beta3-adrenoceptor studied using a monoclonal antibody: direct evidence of the beta3-adrenoceptor in human adipose tissue, atrium and skeletal muscle. Int J Obes Relat Metab Disord 23, 1057–1065, doi: 10.1038/sj.ijo.0801039 (1999). [DOI] [PubMed] [Google Scholar]
- Board M., Doyle P. & Cawthorne M. A. BRL37344, but not CGP12177, stimulates fuel oxidation by soleus muscle in vitro. European journal of pharmacology 406, 33–34, doi: 10.1016/S0014-2999(00)00671-3 (2000). [DOI] [PubMed] [Google Scholar]
- Nakamura Y. et al. Beta 3-adrenergic agonist up-regulates uncoupling proteins 2 and 3 in skeletal muscle of the mouse. The Journal of veterinary medical science/the Japanese Society of Veterinary Science 63, 309–314, doi: 10.1292/jvms.63.309 (2001). [DOI] [PubMed] [Google Scholar]
- Miniaci M. C. et al. CL316,243, a selective beta3-adrenoceptor agonist, activates protein translation through mTOR/p70S6K signaling pathway in rat skeletal muscle cells. Pflugers Archiv European journal of physiology 465, 509–516, doi: 10.1007/s00424-012-1213-9 (2013). [DOI] [PubMed] [Google Scholar]
- Gauthier C. et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. The Journal of clinical investigation 102, 1377–1384, doi: 10.1172/JCI2191 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniotte S. et al. Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 103, 1649–1655, doi: 10.1161/01.CIR.103.12.1649 (2001). [DOI] [PubMed] [Google Scholar]
- Stamler J. S. & Meissner G. Physiology of nitric oxide in skeletal muscle. Physiological reviews 81, 209–237 (2001). [DOI] [PubMed] [Google Scholar]
- Thomas C. R., Stenson J. D. & Zhang Z. Measuring the mechanical properties of single microbial cells. Adv Biochem Eng Biotechnol 124, 83–98, doi: 10.1007/10_2010_84 (2011). [DOI] [PubMed] [Google Scholar]
- Khairallah R. J. et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal 5, ra56, doi: 10.1126/scisignal.2002829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase K. & Pelling A. E. Investigating cell mechanics with atomic force microscopy. J R Soc Interface 12, 20140970, doi: 10.1098/rsif.2014.0970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brixius K. et al. Beta3-adrenergic eNOS stimulation in left ventricular murine myocardium. Can J Physiol Pharmacol 84, 1051–1060, doi: 10.1139/y06-033 (2006). [DOI] [PubMed] [Google Scholar]
- Idigo W., Zhang M. H., Zhang Y. H. & Casadei B. The negative inotropic effect of [beta]3-adrenergic receptor stimulation in nNOS-/- mice is restored by oxypurinol. Heart 92, e1–008 (2006). [Google Scholar]
- Zeman R. J., Ludemann R., Easton T. G. & Etlinger J. D. Slow to fast alterations in skeletal muscle fibers caused by clenbuterol, a beta 2-receptor agonist. The American journal of physiology 254, E726–E732 (1988). [DOI] [PubMed] [Google Scholar]
- Mounier R., Cavalie H., Lac G. & Clottes E. Molecular impact of clenbuterol and isometric strength training on rat EDL muscles. Pflugers Archiv: European journal of physiology 453, 497–507, doi: 10.1007/s00424-006-0122-1 (2007). [DOI] [PubMed] [Google Scholar]
- Sato S., Shirato K., Tachiyashiki K. & Imaizumi K. Muscle plasticity and beta(2)-adrenergic receptors: adaptive responses of beta(2)-adrenergic receptor expression to muscle hypertrophy and atrophy. Journal of biomedicine & biotechnology 2011, 729598, doi: 10.1155/2011/729598 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaauw B. et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 23, 3896–3905, doi: 10.1096/fj.09-131870 (2009). [DOI] [PubMed] [Google Scholar]
- Bodine S. C. et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature cell biology 3, 1014–1019, doi: 10.1038/ncb1101-1014 (2001). [DOI] [PubMed] [Google Scholar]
- Monda M. et al. Inhibition of prostaglandin synthesis reduces the induction of MyoD expression in rat soleus muscle. J Muscle Res Cell Motil 30, 139–144, doi: 10.1007/s10974-009-9182-0 (2009). [DOI] [PubMed] [Google Scholar]
- Kimball S. R. & Jefferson L. S. Control of translation initiation through integration of signals generated by hormones, nutrients, and exercise. The Journal of biological chemistry 285, 29027–29032, doi: 10.1074/jbc.R110.137208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman R. et al. Cellular mechanisms underlying temporal changes in skeletal muscle protein synthesis and breakdown during chronic {beta}-adrenoceptor stimulation in mice. The Journal of physiology 588, 4811–4823, doi: 10.1113/jphysiol.2010.196725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S., Dyar K. A., Ciciliot S., Blaauw B. & Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. The FEBS journal 280, 4294–4314, doi: 10.1111/febs.12253 (2013). [DOI] [PubMed] [Google Scholar]
- Goodman C. A. et al. A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Molecular biology of the cell 21, 3258–3268, doi: 10.1091/mbc.E10-05-0454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. A. et al. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. The Journal of physiology 589, 5485–5501, doi: 10.1113/jphysiol.2011.218255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier R. et al. Antagonistic control of muscle cell size by AMPK and mTORC1. Cell Cycle 10, 2640–2646, doi: 10.4161/cc.10.16.17102 (2011). [DOI] [PubMed] [Google Scholar]
- Kline W. O., Panaro F. J., Yang H. & Bodine S. C. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol 102, 740–747, doi: 10.1152/japplphysiol.00873.2006 (2007). [DOI] [PubMed] [Google Scholar]
- Rommel C. et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nature cell biology 3, 1009–1013, doi: 10.1038/ncb1101-1009 (2001). [DOI] [PubMed] [Google Scholar]
- Bentzinger C. F. et al. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet Muscle 3, 6, doi: 10.1186/2044-5040-3-6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risson V. et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. The Journal of cell biology 187, 859–874, doi: 10.1083/jcb.200903131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I. et al. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PloS one 4, e5618, doi: 10.1371/journal.pone.0005618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navegantes L. C., Resano N. M., Baviera A. M., Migliorini R. H. & Kettelhut I. C. CL 316,243, a selective beta3-adrenergic agonist, inhibits protein breakdown in rat skeletal muscle. Pflugers Archiv: European journal of physiology 451, 617–624, doi: 10.1007/s00424-005-1496-1 (2006). [DOI] [PubMed] [Google Scholar]
- Yoshida T. et al. Beta 3-Adrenergic agonist induces a functionally active uncoupling protein in fat and slow-twitch muscle fibers. The American journal of physiology 274, E469–E475 (1998). [DOI] [PubMed] [Google Scholar]
- Kobzik L., Reid M. B., Bredt D. S. & Stamler J. S. Nitric oxide in skeletal muscle. Nature 372, 546–548, doi: 10.1038/372546a0 (1994). [DOI] [PubMed] [Google Scholar]
- Reid M. B. Role of nitric oxide in skeletal muscle: synthesis, distribution and functional importance. Acta Physiol Scand 162, 401–409, doi: 10.1046/j.1365-201X.1998.0303f.x (1998). [DOI] [PubMed] [Google Scholar]
- Bradley S. J., Kingwell B. A. & McConell G. K. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes 48, 1815–1821, doi: 10.2337/diabetes.48.9.1815 (1999). [DOI] [PubMed] [Google Scholar]
- Anderson J. E. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Molecular biology of the cell 11, 1859–1874 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr F., Gehlert S., Grau M. & Bloch W. Skeletal muscle function during exercise-fine-tuning of diverse subsystems by nitric oxide. International journal of molecular sciences 14, 7109–7139; doi: 10.3390/ijms14047109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. S., Kraus W. E. & Truskey G. A. Stretch-induced nitric oxide modulates mechanical properties of skeletal muscle cells. American journal of physiology. Cell physiology 287, C292–C299, doi: 10.1152/ajpcell.00018.2004 (2004). [DOI] [PubMed] [Google Scholar]
- Ito N., Ruegg U. T., Kudo A., Miyagoe-Suzuki Y. & Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nature medicine 19, 101–106, doi: 10.1038/nm.3019 (2013). [DOI] [PubMed] [Google Scholar]
- Miniaci M. C. et al. Glucose deprivation promotes activation of mTOR signaling pathway and protein synthesis in rat skeletal muscle cells. Pflugers Archiv: European journal of physiology 467, 1357–1366, doi: 10.1007/s00424-014-1583-2 (2015). [DOI] [PubMed] [Google Scholar]
- De Palma C. et al. Deficient nitric oxide signalling impairs skeletal muscle growth and performance: involvement of mitochondrial dysregulation. Skelet Muscle 4, 22, doi: 10.1186/s13395-014-0022-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M., Spencer M. J. & Tidball J. G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. The Journal of cell biology 155, 123–131, doi: 10.1083/jcb.200105110 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defranchi E. et al. Imaging and elasticity measurements of the sarcolemma of fully differentiated skeletal muscle fibres. Microscopy research and technique 67, 27–35, doi: 10.1002/jemt.20177 (2005). [DOI] [PubMed] [Google Scholar]
- Falcao-Pires I., Fontes-Sousa A. P., Lopes-Conceicao L., Bras-Silva C. & Leite-Moreira A. F. Modulation of myocardial stiffness by beta-adrenergic stimulation–its role in normal and failing heart. Physiological research/Academia Scientiarum Bohemoslovaca 60, 599–609 (2011). [DOI] [PubMed] [Google Scholar]
- Deacon R. M. Measuring the strength of mice. Journal of visualized experiments 76, e2610, doi: 10.3791/2610 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieoczym D., Socala K., Jedziniak P., Olejnik M. & Wlaz P. Effect of sildenafil, a selective phosphodiesterase 5 inhibitor, on the anticonvulsant action of some antiepileptic drugs in the mouse 6-Hz psychomotor seizure model. Progress in neuro-psychopharmacology & biological psychiatry 47, 104–110, doi: 10.1016/j.pnpbp.2013.08.009 (2013). [DOI] [PubMed] [Google Scholar]
- Canato M. et al. Mechanical and electrophysiological properties of the sarcolemma of muscle fibers in two murine models of muscle dystrophy: col6a1-/- and mdx. Journal of biomedicine & biotechnology 2010, 981945; doi: 10.1155/2010/981945 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter J. L. & Bechhoefer J. Calibration of atomice-force microscope tips. Rev. Sci. Instrum. 64, 1868–1873, doi: 10.1063/1.1143970 (1993). [DOI] [Google Scholar]
- Oliver W. C. & Pharr G. M. Measurement of hardness and elastic modulus by instrumented indentation: Advances in understanding and refinements to methodology. Journal of Materials Research 19, 3–20, doi: 10.1557/jmr.2004.19.1.3 (2004). [DOI] [Google Scholar]
- Miniaci M. C. et al. Cysteine Prevents the Reduction in Keratin Synthesis Induced by Iron Deficiency in Human Keratinocytes. Journal of cellular biochemistry 117, 402–412, doi: 10.1002/jcb.25286 (2016). [DOI] [PubMed] [Google Scholar]