

ABSTRACT
Acetylation of small molecules is widespread in nature, and in some cases, cells use this process to detoxify harmful chemicals. Streptomyces species utilize a Gcn5 N-acetyltransferase (GNAT), known as Bar, to acetylate and detoxify a self-produced toxin, phosphinothricin (PPT), a glutamate analogue. Bar homologues, such as MddA from Salmonella enterica, acetylate methionine analogues such as methionine sulfoximine (MSX) and methionine sulfone (MSO), but not PPT, even though Bar homologues are annotated as PPT acetyltransferases. S. enterica was used as a heterologous host to determine whether or not putative PPT acetyltransferases from various sources could acetylate PPT, MSX, and MSO. In vitro and in vivo analyses identified substrates acetylated by putative PPT acetyltransferases from Deinococcus radiodurans (DR_1057 and DR_1182) and Geobacillus kaustophilus (GK0593 and GK2920). In vivo, synthesis of DR_1182, GK0593, and GK2920 blocked the inhibitory effects of PPT, MSX, and MSO. In contrast, DR_1057 did not detoxify any of the above substrates. Results of in vitro studies were consistent with the in vivo results. In addition, phylogenetic analyses were used to predict the functionality of annotated PPT acetyltransferases in Burkholderia xenovorans, Bacillus subtilis, Staphylococcus aureus, Acinetobacter baylyi, and Escherichia coli.
IMPORTANCE The work reported here provides an example of the use of a heterologous system for the identification of enzyme function. Many members of this superfamily of proteins do not have a known function, or it has been annotated solely on the basis of sequence homology to previously characterized enzymes. The critical role of Gcn5 N-acetyltransferases (GNATs) in the modulation of central metabolic processes, and in controlling metabolic stress, necessitates approaches that can reveal their physiological role. The combination of in vivo, in vitro, and bioinformatics approaches reported here identified GNATs that can acetylate and detoxify phosphinothricin.
INTRODUCTION
The Gcn5 N-acetyltransferase (GNAT, PF00583) superfamily of proteins is present in all domains of life and catalyzes the transfer of the acetyl group of acetyl coenzyme A (acetyl-CoA) to proteins or small molecules (reviewed in reference 1). These enzymes were first shown to acetylate and inactivate aminoglycoside antibiotics (2–6) and protect against cellular stressors (1, 7–9), providing a precedent for the role of GNAT-mediated detoxification via acetylation. Previous work identified a subset of GNAT enzymes annotated as phosphinothricin (PPT) acetyltransferases. These enzymes protect cells against the toxic effects of PPT (a glutamate analogue) and oxidized forms of methionine, e.g., methionine sulfoximine (MSX) and methionine sulfone (MSO) (7, 10–14) (Fig. 1).
FIG 1.
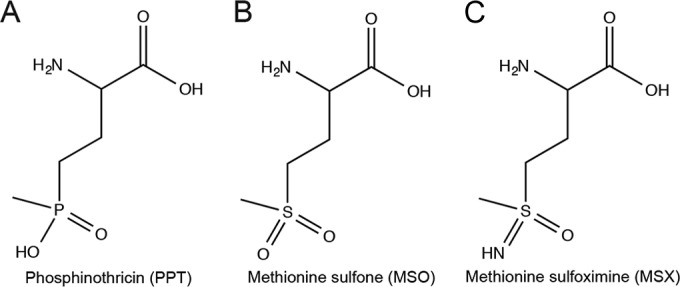
Chemical structure of PPT analogues. (A) Phosphinothricin (PPT); (B) methionine sulfone (MSO); (C) methionine sulfoximine (MSX).
PPT is a component of bialaphos, a tripeptide (phosphinothricyl-alanyl-alanine) with toxic properties produced by Streptomyces species (15). The toxic effect occurs when the tripeptide is cleaved, releasing PPT, which inhibits growth by irreversibly binding to glutamine synthetase (GlnA) (16, 17). A GNAT of Streptomyces spp., known as Bar (also known as Pat for phosphinothricin acetyltransferase), is responsible for the acetylation and detoxification of the endogenously produced PPT toxin in vivo (14). Once acetylated, acetyl-PPT can no longer bind to and inhibit GlnA. Bialaphos is a potent natural herbicide, and plants have been genetically engineered to be resistant by encoding the bar gene (14, 18).
A subgroup of GNATs has been classified as PPT acetyltransferases based on protein sequence similarity to the Bar protein of Streptomyces. Some Bar homologues have been incorrectly annotated as PPT acetyltransferases, including Pseudomonas aeruginosa PITA (gene locus PA4866) and Salmonella enterica MddA (gene locus STM1590) (7). Instead, these enzymes acetylate the structurally related toxic analogues MSX and MSO (Fig. 1), which are also inhibitors of glutamine synthetase.
Here we examine the selectivity of putative PPT acetyltransferases from Geobacillus kaustophilus and Deinococcus radiodurans. Using S. enterica as a heterologous host, we assayed for functions of the predicted PPT acetyltransferases in S. enterica mddA1::cat+ and mddA+ strains in culture medium containing MSX, MSO, or PPT. In vitro measurements of enzyme activity were consistent with the observed growth phenotypes. The data show diverse specificity of these enzymes for MSX, MSO, and PPT. Lastly, we investigated whether or not putative PPT acetyltransferases from Burkholderia xenovorans, Bacillus subtilis, Escherichia coli, Staphylococcus aureus, and Acinetobacter baylyi could acetylate PPT, MSX, and MSO. The data show that the above-mentioned enzymes acetylate MSX, MSO, and PPT in vivo, with the exception of EcYncA, which did not acetylate PPT, and Bxe_B1787, which did not acetylate MSO. Our results confirmed bioinformatics-based predictions and provided a reliable in vivo approach using S. enterica to identify PPT acetyltransferases.
MATERIALS AND METHODS
Culture media and chemicals.
Nutrient broth (NB; Difco) was used as rich medium to grow inocula. The minimal medium used in this study was no-carbon essential (NCE) minimal medium (19) containing MgSO4 (1 mM), sodium, ammonium phosphate (17 mM), Wolfe's trace minerals (1×) (20), and glycerol (22 mM) as the sole source of carbon and energy. When used, antibiotics were added at the following concentrations: chloramphenicol (20 μg ml−1), ampicillin (100 μg ml−1). All chemicals were purchased from Sigma-Aldrich unless noted otherwise; ampicillin, NaCl, and HEPES (Fisher Scientific); isopropyl β-d-1-thiogalactopyranoside (IPTG; IBI Scientific); and dithiothreitol (DTT; Gold BioTechnology). l-Methionine sulfoximine (MSX), l-methionine sulfone (MSO), and DL-phosphinothricin (PPT) were all purchased from Sigma-Aldrich.
Bacterial strains.
All S. enterica strains are derivatives of serovar Typhimurium strain LT2 (unless specified) and are listed in Table 1.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype | Reference(s) or sourcea |
|---|---|---|
| S. enterica strains | ||
| JE10079 | ara-9 mddA+ | Laboratory strain |
| Derivatives of JE10079 | ||
| JE18333 | mddA::cat+ | 10 |
| JE18961 | mddA::cat+/pMDD8 | 10 |
| JE20780 | pDR_1182-2 | |
| JE20781 | mddA::cat+/pDR1182-2 | |
| JE20782 | pGK0593-2 | |
| JE20783 | mddA::cat+/pGK0593-2 | |
| JE21642 | pDR_1057-7 | |
| JE21643 | mddA::cat+/pDR1057-2 | |
| JE20864 | mddA::cat+/pCV1 | |
| JE20865 | mddA::cat+/pGK2920-2 | |
| JE20866 | pGK2920-2 | |
| JE20973 | pCV1 | |
| JE21098 | mddA::cat+/pBXE_A2261-2 | |
| JE21597 | mddA::cat+/pBXE_B1787-2 | |
| JE21232 | mddA::cat+/pBSYwnH-1 | |
| JE21596 | mddA::cat+/pYNCA | |
| JE21595 | mddA::cat+/pSAYwnH-1 | |
| E. coli C41(λDE3) | ompT hsdS(rB mB) gal λ(DE3) including at least one noncharacterized mutation | 27, 28 |
| Plasmids | ||
| pMDD7 | S. enterica mddA+ cloned into pTEV16b | 10 |
| pMDD8 | S. enterica mddA+ cloned into pCV1b | 10 |
| pDR1057-8 | D. radiodurans DR_1057+ cloned into pTEV16 | |
| pDR1057-7 | D. radiodurans DR_1057+ cloned into pCV1 | |
| pDR1182-1 | D. radiodurans DR_1182+ cloned into pTEV16 | |
| pDR1182-2 | D. radiodurans DR_1182+ cloned into pCV1 | |
| pGK0593-3 | G. kaustophilus GK0593+ cloned into pTEV16 | |
| pGK0593-2 | G. kaustophilus GK0593+ cloned into pCV1 | |
| pGK2920-1 | G. kaustophilus GK2920+ cloned into pTEV16 | |
| pGK2920-2 | G. kaustophilus GK2920+ cloned into pCV1 | |
| pBXE_A2261-2 | B. xenovorans Bxe_A2261+ cloned into pCV1 | |
| pBXE_B1787-2 | B. xenovorans Bxe_B1787+ cloned into pCV1 | |
| pBSYwnH-1 | B. subtilis ywnH+ cloned into pCV1 | |
| pSAYwnH-1 | S. aureus SAUSA300_2468+ cloned into pCV1 | |
| pYNCA | E. coli yncA+ cloned into pCV1 | |
| pCV1 | Cloning vector | 22 |
All strains and plasmids were constructed during the course of this work, unless otherwise stated.
For details on pTEV16 and pCV1, see reference 22.
Plasmid construction for complementation and overexpression.
All plasmids used in this work are listed in Table 1. All primers used in this study were synthesized by IDT (Coralville, IA) and are listed in Table 2. We used cloning technology reported by Galloway et al. (21) to construct all plasmids in this study. Genes of interest were amplified from genomic DNA from D. radiodurans R1, G. kaustophilus HTA426, B. xenovorans LB400, E. coli MG1655, S. aureus USA3000, Acinetobacter baylyi ADP1, or B. subtilis SMY. D. radiodurans R1 genomic DNA was a gift from John Batista (Louisiana State University), A. baylyi ADP1 genomic DNA was a gift from Cory Momany (University of Georgia [UGA]), and S. aureus USA3000 genomic DNA was a gift from Alexander Horswill (University of Iowa). All other genomic DNA was obtained from strains available in our laboratory collection. DNA sequences were confirmed using BigDye (ABI PRISM) protocols, and sequencing reactions were resolved and analyzed at the Georgia Genomics Facility, UGA.
TABLE 2.
Primers used in this study
Plasmid pTEV16 (22), which directs the synthesis of the protein with a cleavable N-terminal hexahistidine tag, was used for overexpression. The resulting plasmids are referred to as pDR_1057-8, pDR_1182-1, pGK0593-3, and pGK2920-1.
For complementation purposes, each gene of interest was cloned into the l-(+)-arabinose-inducible vector pCV1 (22). The names and relevant genotypes of the resulting plasmids are shown in Table 1.
Site-directed mutagenesis was performed using primers designed from PrimerX (http://www.bioinformatics.org/primerx/) to mutate the asparagine (N114) of DR_1057WT to a glutamate residue (E114) to construct a variant (DR_1057N114E) in the pCV1 complementation vector (pDR_1057-3).
Growth behavior analyses.
Starter cultures were grown overnight at 37°C with shaking in NB containing the appropriate antibiotic, 2 μl of which was used to inoculate 198 μl of fresh medium in each well of a 96-well plate. Plates were incubated at 37°C with shaking for 20 to 48 h in a Powerwave Microplate Reader (Bio-Tek Instruments). Growth studies were performed in triplicate in three independent experiments, with a representative growth curve shown. Data were analyzed using Prism v6 (GraphPad) analytical software. Error bars represent standard deviations. Plasmid-borne genes of interest were induced with various concentrations of l-(+)-arabinose, as described in the figures and figure legends. Additional chemicals such as PPT, MSX, or MSO were added at the concentrations indicated in the figures and figure legends.
Protein overproduction and purification.
Plasmids encoding each protein of interest (DR_1182, DR_1057, GK0593, or GK2920M55-Stop) were transformed into E. coli C41(λDE3). Overnight cultures of the transformants were subcultured (1:100 [vol/vol, inoculum/medium]) into 1 liter of lysogeny broth (LB) containing ampicillin (100 μg ml−1). Cultures were grown at 37°C with shaking to an optical density at 600 nm (OD600) of 0.6, induced with IPTG (1 mM), and shaken overnight at ∼28°C. Cells were harvested by centrifugation at 6,000 × g in a Beckman Coulter Avanti J-20 XOI refrigerated centrifuge with a JLA-8.1000 rotor for 15 min at 4°C. The collected cell paste was resuspended in binding buffer A (HEPES buffer [50 mM, pH 7.2] containing NaCl [500 mM] and imidazole [20 mM] plus lysozyme [1 mg ml−1], DNase I [25 μg ml−1], and protease inhibitor phenylmethanesulfonyl fluoride, 0.5 mM]). Cells were lysed by sonication for 1 min (2 s, 50% duty) for two rounds on ice using a 550 Sonic Dismembrator (Fisher Scientific) at setting 4. Clarified cell lysates were obtained after centrifugation for 45 min at 4°C at 43,667 × g in a Beckman Coulter Avanti J-25I refrigerated centrifuge with a JA-25.50 rotor followed by filtration of the supernatant through a 0.45-μm filter (Millipore). Samples were loaded onto a 1-ml HisPur nickel-nitrilotriacetic acid (Ni-NTA) resin column (Thermo Scientific) at 4°C, preequilibrated with binding buffer. The Ni+ column was washed first with buffer B (HEPES buffer [50 mM, pH 7.2] with NaCl [500 mM]) that contained imidazole (40 mM) to remove nonspecifically bound proteins. Subsequently, the His6-tagged proteins eluted in the same buffer system that contained a high concentration of imidazole (500 mM). Proteins were dialyzed at 4°C and stored in HEPES buffer (50 mM, pH 7.2) containing NaCl (100 mM), tris-(2-carboxyethyl)phosphine hydrochloride (TCEP, 0.5 mM), and glycerol (10% vol/vol), drop-frozen in liquid nitrogen, and stored at −80°C. Proteins (DR_1182WT, DR_1057WT GK0593WT, and GK2920M55-Stop) were purified, and homogeneity ranged from 70 to 96%, as determined by densitometry calculations using Total Lab v2005 software (see Fig. S1 in the supplemental material).
Mass spectrometry analysis of proteins.
Proteins or peptides that required further analysis by mass spectrometry were excised from SDS-PAGE gels and analyzed by the Proteomics and Mass Spectrometry Core Facility at UGA. Peptide sequences were matched to proteins using the MASCOT database (Matrix Science), with protein coverage ranging from 55 to 77%. For details on mass spectrometry methods, see the Supplemental Materials and Methods in the supplemental material.
Kinetic analyses.
Apparent kinetic parameters (kcat, Km, kcat/Km) for DR_1057, DR_1182, GK0593, or GK2920M55-Stop when MSX, MSO, or PPT was used as the substrate were determined using a continuous spectrophotometric assay that employed 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB; Ellman's reagent) as described elsewhere (10). Briefly, assays were performed at 30°C in 100-μl reaction mixtures in 96-well plates, and reactions were monitored at 412 nm (23, 24). Reaction mixtures contained HEPES buffer (50 mM, pH 7.2), DTNB (0.3 mM), saturating levels of acetyl-CoA (1 mM), protein (DR_1057, DR_1182, GK0593, or GK2920M55-Stop) (100 to 200 nM), and substrate (PPT, MSO, or MSX) (0 to 2,000 μM). Data were acquired every 10 s over a 5-min time period and were reported using the averages of technical triplicates. Absorbance (415 nm) measurements of reaction mixtures were monitored over time (in seconds) using the Soft Max Pro 6.2 Spectramax software, and absorbance readings were corrected for path lengths for each well. Initial velocity was recorded from the slope of the linear range (ΔA415 s−1). ΔA415 s−1 was converted to micromolar per second units using Beer's Law (equation 1) with the molar extinction coefficient of the TNB2− thiolate anion (14,150 M−1 cm−1) (25). Because A415 readings were corrected for path lengths, a value of 1 cm was used for l and equation 1 was solved for concentration (c).
| (1) |
Graphs of initial velocity (micromolar per second) versus substrate concentration (micromolar) were plotted using Prism v6 (GraphPad) software. Curves were fitted to the model of Michaelis-Menton kinetics to determine Km and Vmax. The turnover number (kcat), was determined by equation 2, where [E] was the concentration of protein. The parameters are shown in Table 3, and the curves are shown in Fig. S2 in the supplemental material.
| (2) |
TABLE 3.
Kinetic parametersa of G. kaustophilus and D. radiodurans acetyltransferases
| Enzyme | Substrate | Km(app) (μM) | kcat (s−1) | kcat/Km(app) (M−1 s−1) |
|---|---|---|---|---|
| GK2920M55-Stop | MSX | 121 ± 26 | 9 ± 1 | 7 × 104 |
| MSO | 89 ± 16 | 10 ± 1 | 1 × 105 | |
| PPT | NDAb | NDA | NDA | |
| GK0593 | MSX | 17 ± 4 | 1 ± 0.1 | 6 × 104 |
| MSO | 4 + 2 | 1 ± 0.1 | 3 × 105 | |
| PPT | 5 + 3 | 1 ± 0.1 | 2 × 105 | |
| DR_1182 | MSX | 316 ± 23 | 11 ± 0.3 | 3 × 104 |
| MSO | 198 ± 30 | 11 ± 1 | 6 × 104 | |
| PPT | 721 + 79 | 5 + 0.2 | 7 × 103 |
Values represent means ± standard deviations.
NDA, no detectable activity.
Bioinformatics analyses.
An alignment of the primary amino acid sequence of various annotated PPT acetyltransferases was generated using the NCBI COBALT multiple alignment tool (http://www.st-va.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi). A phylogenetic tree was generated using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/).
RESULTS
Phosphinothricin delays the onset of exponential growth of S. enterica.
Previous work from our research group characterized the MddA (formerly YncA, STM1590) enzyme from S. enterica as an MSX and MSO acetyltransferase (10). Although the addition of PPT delayed the onset of exponential growth, the effect was not dependent on the presence or absence of MddA (Fig. 2D). As shown in Fig. 2C and D, PPT affected growth of S. enterica only when present at a high concentration (e.g., 100 μM), delaying the onset of exponential growth by ∼15 h. The onset of growth was not due to the acquisition of mutations, since reinoculation of fresh medium with cells from cultures that experienced the lag phase did not result in any reduction of the lag time (data not shown). We used the phenotypes shown in Fig. 2 to identify PPT acetyltransferases from other microorganisms.
FIG 2.

Growth of the S. enterica mddA1::cat+ strain under conditions containing MSX, MSO, or PPT. Growth and complementation of the S. enterica mddA1::cat+ strain in NCE minimal medium (glycerol, 22 mM) was examined in the presence of 10 μM MSX (A), 50 μM MSO (B), 20 μM PPT (C), or 100 μM PPT (D). Vectors were induced with 10 μM l-(+)-arabinose. Growth curves were performed using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. Strains analyzed: ara-9/vector (JE20973, squares), ara-9 mddA1::cat+ (JE18333, triangles), ara-9 mddA1::cat+/vector (JE20864, inverted triangles), ara-9 mddA1::cat+/mddA+ (pMDD8; JE18961, diamonds). Error bars represent standard deviations. Symbols in panel D apply to all panels.
Deinococcus radiodurans and Geobacillus kaustophilus encode PPT acetyltransferases with different substrate preferences.
To simplify the interpretation of our results, the activities of putative PPT acetyltransferases were assessed using an S. enterica strain devoid of MddA (mddA1::cat+, JE18333). Ectopic synthesis of Deinococcus radiodurans DR_1182 or Geobacillus kaustophilus GK0593 restored growth of the S. enterica mddA1::cat+ strain to wild-type levels at low levels of induction, 10 μM l-(+)-arabinose, in medium supplemented with either MSX or MSO (Fig. 3). In contrast, expression of GK2920 restored growth at concentrations of inducer ranging between 0.2 and 1 mM, suggesting that MSX and MSO were poor substrates for the enzyme. Synthesis of DR_1057 protein failed to correct the phenotype of the mddA1::cat+ strain caused by MSX or MSO, even when the medium contained high levels of inducer, i.e., 1 mM l-(+)-arabinose (open squares, Fig. 3A or C, respectively).
FIG 3.

Complementation studies using genes encoding putative PPT acetyltransferases from D. radiodurans and G. kaustophilus in S. enterica. Growth of an S. enterica mddA1::cat+ strain carrying a plasmid encoding DR_1057, DR_1182, GK0593, or GK2920 under the control of an l-(+)-arabinose inducible promoter was examined in the presence of 10 μM MSX (A and C) or 50 μM MSO (B and D) in glycerol (22 mM) minimal medium. Plasmids were induced with various concentrations of l-(+)-arabinose (0.01 to 1 mM), as indicated. Strains analyzed: ara-9/vector (JE20973, squares), ara-9 mddA1::cat+/vector (JE20864, circles), ara-9 mddA1::cat+/pmddA+ (JE18961, triangles), ara-9 mddA1::cat+/pDR_1057-2 (JE21642, open squares in panels A and C), ara-9 mddA1::cat+/pDR_1182-2 (JE20781, open diamonds in panels A and C), ara-9 mddA1::cat+/pGK0593-2 (JE20783, open diamonds in panels B and D), ara-9 mddA1::cat+/pGK2920-2 (JE20865, open squares in panels B and D). Growth curves were performed using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. Error bars represent standard deviations. Symbols in panel B apply to panels A and B; symbols in panel D apply to panels C and D.
Direct correlation between enzyme level and degree of protection.
Because S. enterica MddA does not acetylate PPT, we used the wild-type S. enterica strain to screen for phenotypes at higher concentrations of MSX, MSO, and PPT. Growth of wild-type S. enterica was negatively affected but not abolished by MSX (20 μM), MSO (500 μM), and PPT (100 μM) (10). Plasmids carrying DR_1057+, DR_1182+, GK0593+, or GK2920+ placed under the control of an l-(+)-arabinose-inducible promoter were each introduced into an S. enterica mddA+ strain. The resulting strains were grown in minimal medium containing MSX (20 μM), MSO (500 μM), or PPT (100 μM) and various levels of inducer [10 to 1,000 μM l-(+)-arabinose]. When induced, cultures expressing DR_1182+ or GK2920+ grew with substantially shorter lag times than the S. enterica mddA+ strain carrying the vector only (VOC, vector-only control) (Fig. 4). These data were consistent with the idea that increased protein levels provided greater protection against the toxic effects of MSX, MSO, and PPT. Low levels of induction [i.e., 10 μM l-(+) arabinose] generated sufficient GK0593 enzyme to completely reduce the growth lag of the S. enterica mddA+ strain in medium containing 100 μM PPT (Fig. 4E). In contrast, the same level of induction of GK0593+ provided limited protection against MSX (20 μM) or MSO (500 μM). A 10- and 100-fold-higher level of inducer was needed to express sufficient GK0593 protein to reduce the lag phase in the presence of MSX or MSO, respectively (Fig. 4D and F). The DR_1057 enzyme did not improve growth of the mddA+ S. enterica strain in medium containing MSX, MSO, or PPT even at high induction [1 mM l-(+) arabinose], suggesting that this enzyme may not recognize MSX, MSO, or PPT as substrates (Fig. 4A to C). The GK2920+ and DR_1182+ alleles corrected all phenotypes at low induction [10 μM l-(+) arabinose] (Fig. 4A to F).
FIG 4.

Overexpression provides resistance to higher levels of PPT, MSX, and MSO. Growth of wild-type S. enterica (JE10079) carrying a plasmid encoding DR_1057, DR_1182, GK0593, or GK2920 under the control of an l-(+)-arabinose-inducible promoter was examined in the presence of 100 μM PPT (B and E), 20 μM MSX (A and D), or 500 μM MSO (C and F) in glycerol (22 mM) minimal medium with various concentrations of l-(+)-arabinose (0.01 to 1 mM), as indicated. Strains analyzed: ara-9/vector (JE20973, squares), ara-9 mddA1::cat+/vector (JE20864, triangles), ara-9 mddA1::cat+/pmddA+ (JE18961, inverted triangles), ara-9/pDR_1057-2 (JE21642, circles, panels A to C), ara-9/pDR_1182-2 (JE20780, diamonds, panels A to C), ara-9/pGK0593-2 (JE20782, circles, panels D to F), ara-9/pGK2920-2 (JE20866, diamonds, panels D to F). Growth curves were performed using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. Error bars represent standard deviations. Symbols in panel C apply to panels A to C; symbols in panel F apply to panels D to F.
In vitro activity analyses of putative PPT acetyltransferases.
To study the substrate preference of the annotated PPT acetyltransferases in D. radiodurans (DR_1057, DR_1182) and G. kaustophilus (GK0593, GK2920), the proteins were isolated to 70 to 95% homogeneity (see Fig. S1 in the supplemental material), and enzyme kinetic parameters were determined to gain insights into their substrate preference (Table 3; see also Fig. S2 in the supplemental material). The catalytic efficiencies of Deinococcus enzymes correlated with in vivo data, with DR1182 having similar kcat/Km values for MSX, MSO, and PPT as substrates (Table 3). Given that DR_1057 failed to acetylate any of the substrates in vivo, it was not surprising that this enzyme did not acetylate MSX, MSO, or PPT under the in vitro conditions used (data not shown).
Acetyltransferases from Geobacillus showed similar catalytic efficiencies for MSX and MSO as substrates, with GK0593 having a higher catalytic efficiency for PPT than GK2920.
We noted that during the purification of GK2920, large amounts of protein were collected in flowthrough fractions. We posited that during overproduction, the protein was cleaved into two peptides, resulting in one cleavage product unable to interact with the Ni-NTA resin column. The GK2920 protein fragments were excised from the gel and analyzed by liquid chromatography-tandem mass spectroscopy (LC-MS/MS), which revealed that the first 55 amino acids of the N terminus (including the His6 tag) of GK2920 were cleaved by an unknown mechanism (Fig. 5). The allele encoding GK2920M55-Stop was cloned into an overexpression vector and subsequently purified. Results from kinetic analyses showed that GK2920M55-Stop acetylated MSX and MSO but had no activity when PPT was the substrate (Table 3). This result was unexpected, as the in vivo data shown in Fig. 4E (diamonds) demonstrated that low levels of induction of GK2920 substantially improved growth of the S. enterica mddA+ strain in medium containing PPT. Our interpretation of this discrepancy is considered below in Discussion.
FIG 5.
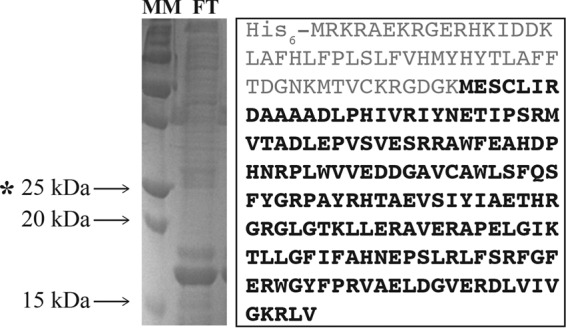
Isolation of a truncated form of GK2920. (Left) SDS-PAGE gel of the flowthrough (FT) fraction from the Ni-NTA affinity chromatography column used to purify GK2920. Arrows indicate relevant mass markers (MM; Bio-Rad Precision Plus Blue Standard). The asterisk indicates the predicted size of GK2920WT. (Right) LC/MS of the 17-kDa protein revealed the amino acid sequence highlighted in bold.
Bioinformatics analyses of putative PPT acetyltransferases.
An alignment was generated for the active sites of putative PPT acetyltransferases across a range of bacterial species (NCBI COBALT multiple alignment tool). From this alignment, we noted that DR_1057 from D. radiodurans lacked the putative catalytic glutamate of GNATs and encoded an asparagine at that position (Fig. 6A). To determine whether or not the lack of in vivo function against MSX, MSO, and PPT was due to the absence of the glutamate residue, we engineered a variant of DR_1057 in which the asparagine residue was replaced by a glutamate (DR_1057N114E). The resulting variant did not restore growth of an S. enterica mddA::cat+ strain in medium containing MSX or MSO and did not improve growth of the S. enterica mddA+ strain in medium containing PPT (data not shown). Therefore, it was concluded that the absence of the alluded glutamate residue in DR_1057 was not responsible for the lack of enzyme activity.
FIG 6.

Bioinformatics analyses of annotated PPT acetyltransferases. (A) Alignments were generated using the NCBI COBALT multiple alignment tool. The arrow points at the predicted active-site glutamate of acetyltransferases. Numbers on the left of the sequences indicate the location of the first residue in the native protein; boxed residues show regions of at least 70% similarity; a conserved glycyl residue is identified by white letters in a black background. (B) A phylogenetic tree of 19 annotated PPT acetyltransferases was generated using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/). For DR_1057, only the sequence of the GNAT domain that aligned to the other putative PPT acetyltransferases (residues 1 to 180) was used for the phylogenetic tree. When gene names were unavailable, locus tags were used (i.e., for P. putida, PP_1924); 0.06 designates the scale for tree length.
A phylogenetic tree was generated by comparing 19 PPT acetyltransferase homologues (FigTree, Fig. 6B). Clustering of enzymes that acetylated MSX and MSO but not PPT was observed, with the exception of DR_1182, which clustered more closely with the non-PPT-utilizing enzymes. Organisms containing two annotated PPT acetyltransferases clustered separately in different nodes of the tree (e.g., D. radiodurans, B. xenovorans, and G. kaustophilus). A recent report examining the specificity of the two annotated PPT acetyltransferases from Pseudomonas putida, which were also clustered in separate nodes of the tree, demonstrated that each enzyme was specific for either PPT or MSX and MSO (26). From the in vivo data reported here for G. kaustophilus, we see that enzyme functions overlap and each of the two enzymes can acetylate PPT, MSX, and MSO, although why an organism would encode two enzymes with overlapping functions is still not clear.
Can PPT acetyltransferases be identified by computational approaches? The goal of this work was to expedite the validation of putative of PPT acetyltransferases by using the above-mentioned in vivo, in vitro, and bioinformatics approaches. As proof of principle, we focused on putative PPT acetyltransferases from B. xenovorans (Bxe_A2261, Bxe_B1787), B. subtilis (YwnH), Staphyloccocus aureus (SAUSA300_2468), and E. coli (YncA). We wished to determine whether or not there was divergence in the phylogeny that correlated with the ability of a particular organism to acetylate PPT. Based on the phylogenetic tree and proximity to known PPT-utilizing enzymes, the following predictions were made. We predicted that Bxe_A2661, Bxe_B1787, and BsYwnH would acetylate PPT while SaYwnH and EcYncA would not.
B. xenovorans Bxe_A2261 and Bxe_B1781, B. subtilis ywnH, S. aureus SAUSA300_2468, and E. coli yncA genes were cloned under the control of an l-(+)-arabinose-inducible promoter and expressed in trans in an S. enterica mddA1::cat+ strain grown in the presence of MSX (10 μM), MSO (50 μM), or PPT (100 μM) (Table 3). Each gene was induced with various concentrations of l-(+)-arabinose (10 μM, 100 μM, or 1 mM) (Fig. 7). Acetylation of MSO or MSX correlated with the growth profile of the S. enterica mddA::cat+/pmddA+ strain (JE18961) rather than to that of the mddA1::cat+/pVOC strain (JE20864). Acetylation of PPT correlated with a higher growth rate and faster entry into stationary phase than the mddA+/pVOC strain.
FIG 7.

In vivo functional analysis of putative PPT acetyltransferases from B. xenovorans, B. subtilis, S. aureus, E. coli, and A. baylyi. Growth behaviors of an S. enterica mddA1::cat+ strain carrying a plasmid carrying Bxe_A2261, Bxe_B1787, BsYwnH, SaYwnH, EcYncA, or ACIAD1637 under the control of an l-(+)-arabinose-inducible promoter. Growth was examined in the presence of 10 μM MSX (panels A, D, G, J, M, P), 100 μM PPT (panels B, E, H, K, N, Q), or 50 μM MSO (panels C, F, I, L, O, R), in glycerol (22 mM) minimal medium with various concentrations of l-(+)-arabinose (0.01 to 1 μM), as indicated. Strains analyzed: ara-9/vector (JE20973, squares), ara-9 mddA1::cat+/vector (JE20864, circles), ara-9 mddA1::cat+/pMDD8 (JE18961, diamonds), ara-9 mddA1::cat+/pBxe_A2261 (JE21098), ara-9 mddA1::cat+/pBxe_B1787 (JE21597), ara-9 mddA1::cat+/pBsYwnH (JE21232), ara-9 mddA1::cat+/pSaYwnH (JE21595), ara-9 mddA1::cat+/pEcYncA (JE21596), and ara-9 mddA1::cat+/pACIAD1637 (JE21982). All strains that detoxified PPT are identified by gray diamonds. Growth curves were performed using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. Error bars represent standard deviations. Symbols in panel C apply to panels A to C; symbols in panel F apply to panels D to F; symbols in panel I apply to panels G to I, symbols in panel L apply to panels J to L; symbols in panel O apply to panels M to O; symbols in panel R apply to panels P to R.
As predicted, Bxe_A2661 acetylated PPT in vivo, as seen by the reduction in lag phase at high concentrations of inducer; Bxe_A2661 also acetylated MSX and MSO (Fig. 7A to C). Bxe_B1787 acetylated PPT in vivo, and MSX acetylation was limited (as demonstrated by the extended lag phase compared to what was seen with the mddA::cat+/pmddA+ strain) and was observed only at a high concentration of inducer, while MSO was not acetylated even at a high inducer concentration (Fig. 7D to F). BsYwnH acetylated MSX, MSO, and PPT as predicted, but only at a high inducer concentration (Fig. 7G to I). Surprisingly, SaYwnH acetylated PPT, MSX, and MSO even at low inducer concentrations (Fig. 7J to L). Lastly, EcYncA acetylated MSX and MSO, but not PPT, as predicted (Fig. 7M to O).
It was intriguing that SAUSA300_2468 (encoding SaYwnH) clustered closely with A. baylyi ACIAD1637, yet the latter acetylated only MSX and MSO, whereas SaYwnH acetylated all three substrates. Studies that reported the crystallization and characterization of ACIAD1637 revealed that ACIAD1637 preferred MSX and MSO and that PPT was not considered a valid substrate for ACIAD1637 (12). To resolve the discrepancies of the bioinformatics prediction, we sought to examine the substrates of ACIAD1637 in vivo. ACAID1637 was cloned into pCV1 and was induced with various levels of arabinose in an mddA1::cat+ strain to determine whether or not its substrate preference in vivo was similar to that SAUSA300_2468. In vivo analysis showed that synthesis of ACIAD1637 blocked the deleterious effects of MSX, MSO, and PPT (Fig. 7P to R), in support of the prediction that SaYwnH was a PPT acetyltransferase.
DISCUSSION
The combination of in vivo and in vitro approaches used in this study allowed us to validate the function of putative PPT acetyltransferases identified by bioinformatics and phylogenetic analyses. From their position in the phylogenetic tree (Fig. 6), several predictions were made about putative PPT acetyltransferases and were experimentally confirmed.
The combination of phenotypic analyses using a heterologous system and kinetic analyses offers the means to assign PPT acetyltransferase function.
Results obtained with the G. kaustophilus GK2920 protein were intriguing. For as-yet-unknown reasons, GK2920 was isolated as a cleaved protein (GK2920M55-stop [Fig. 5]) that retained biochemical activity when MSO or MSX was used as the substrate but not when PPT was the substrate. The results of in vitro assays with MSO and MSX indicate that the active site of the GK2920M55-stop enzyme remained functional but appeared to be sufficiently altered to prevent PPT acetylation. One explanation for the lack of activity with PPT may be that the N-terminal 54 residues of GK2920 are needed for PPT binding. Clearly, the GK2920 protein efficiently detoxifies PPT in vivo (Fig. 4E), but whether or not the protein remains intact or is truncated is unclear at this time. We also do not yet understand why GK2920 is cleaved during isolation given that a protease inhibitor cocktail is included during cell breakage to decrease the possibility of degradation. Purified GK2920M55-stop showed higher catalytic efficiency for MSO than for MSX (Table 3), a result that was consistent with those obtained in vivo (Fig. 3C and D). Taken together, these data suggest that MSO may be the preferred substrate for GK2920.
Kinetic analyses of GK0593 were also consistent with in vivo data, in that this enzyme acetylated all three substrates in vitro, while also complementing Salmonella phenotypes associated with toxic levels of MSX, MSO, and PPT. The catalytic efficiency of GK0593 for MSO and PPT was higher than that for MSX, which may reflect the levels of these substrates found in the environment of Geobacillus. When comparing GK2920 and GK0593, the catalytic efficiencies were similar for both MSX and MSO, raising questions about why this organism encodes two acetyltransferases with overlapping functions. Because GK2920 could not acetylate PPT in vitro, no conclusions can be drawn when comparing kinetic parameters to GK0593.
Synthesis of D. radiodurans DR_1057 failed to prevent the inhibitory effect of PPT even when the gene encoding it was expressed with high levels of inducer (Fig. 4B). These results may suggest that DR_1057 may have diverted from the protein conformation that allows it to acetylate PPT. We note that the D. radiodurans genome contains one additional gene encoding a putative PPT acetyltransferase (i.e., DR_1182). In contrast to DR_1057, synthesis of DR_1182 blocked the inhibitory effect of PPT (Fig. 4B). Additionally, DR_1182 showed higher catalytic efficiencies for MSX and MSO as substrates than for PPT. D. radiodurans encodes two putative phosphinothricin acetyltransferases, although only one of these enzymes (DR_1182) carries out its annotated function. Either DR_1057 lost the ability to acetylate MSX, MSO, and PPT, or this enzyme is an acetyltransferase with alternative target(s). Further analysis of this enzyme is needed.
Bioinformatics analyses provide additional means for the identification of PPT acetyltransferases.
From the phylogenetic tree in Fig. 6, we can determine where the limit of an enzyme's ability to acetylate PPT may lie. It appears as though enzymes clustering near Streptomyces coelicolor Bar PPT acetyltransferase can acetylate PPT, while enzymes clustering near S. enterica MddA cannot. In vivo analyses validated PPT acetyltransferase activities of several proteins of interest. That is, synthesis of genes clustered near bar (B. xenovorans Bxe_A2261 and Bxe_B1787, B. subtilis YwnH, and S. aureus SAUSA300_2468) clearly blocked the deleterious effect of PPT on the indicator strain (Fig. 7B, E, H, K, respectively). The lack of protection against PPT by E. coli YncA was not surprising, given the phylogenetic relatedness of this protein to the S. enterica MddA enzyme, which has been shown to lack PPT acetyltransferase activity (10).
Correction of a functional assignment in the literature.
Our results with A. baylyi ACIAD1637 (Fig. 7Q) were inconsistent with those reported by Davies et al., who concluded that A. baylyi ACIAD1637 did not have PPT acetyltransferase activity. These authors reached their conclusion because wild-type and ACIAD1637-devoid strains of A. baylyi were equally sensitive to PPT (12). We note, however, that although A. baylyi ACIAD1637 blocked PPT inhibition of the indicator strains, a high level of inducer (1 mM) was needed to afford the observed protection, suggesting that A. baylyi ACIAD1637 may have a low level of PPT acetylation activity. Regardless, these results highlight the usefulness of the in vivo system that we report here for the assessment of PPT acetyltransferase function.
Usefulness of heterologous systems in the assignment of gene function.
The use of model organisms with sophisticated genetic systems that allow investigators to rapidly construct strains in diverse genetic backgrounds is a powerful, yet simple, way to probe for gene function. Results obtained with such a system guide the use of phylogenetic, bioinformatics, and biochemical approaches to further cement the assignment of protein function. One factor to consider, however, is that a heterologous system could produce false negatives depending on protein solubility or degradation in a heterologous environment. It is important to note that this heterologous system will not identify substrate preferences but will provide a tool to narrow down enzymes to be kinetically characterized. Results from this study demonstrate the effectiveness of utilizing S. enterica mddA::cat+ strains in the assessment of PPT acetyltransferase function from a variety of bacteria. With the rapidly increasing number of genomes available in databases, the need for experimental validation of predicted gene function becomes more important than ever, especially since in many cases homologous proteins share a great deal of identity yet lack the predicted function. For example, the MddA protein of S. enterica and E. coli are currently annotated as PPT acetyltransferases, and they lack such an activity.
Supplementary Material
ACKNOWLEDGMENTS
We do not have any conflict of interest. We acknowledge Dennis Phillips and Chau-Wen Chou at the Proteomics and Mass Spectrometry Core Facility (UGA) for assistance with mass spectrometry.
Funding Statement
This work was funded by HHS | NIH | National Institute of General Medical Sciences (NIGMS) (R01GM062203), awarded to Jorge C. Escalante-Semerena.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02604-16.
REFERENCES
- 1.Hentchel KL, Escalante-Semerena JC. 2015. Acylation of biomolecules in prokaryotes: a widespread strategy for the control of biological function and metabolic stress. Microbiol Mol Biol Rev 79:321–346. doi: 10.1128/MMBR.00020-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies J, Wright GD. 1997. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240. doi: 10.1016/S0966-842X(97)01033-0. [DOI] [PubMed] [Google Scholar]
- 3.Wright GD, Ladak P. 1997. Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium. Antimicrob Agents Chemother 41:956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. 1998. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 94:439–449. doi: 10.1016/S0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 5.Draker KA, Northrop DB, Wright GD. 2003. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry 42:6565–6574. doi: 10.1021/bi034148h. [DOI] [PubMed] [Google Scholar]
- 6.Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS. 2004. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol 11:565–573. doi: 10.1016/j.chembiol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 7.Davies AM, Tata R, Beavil RL, Sutton BJ, Brown PR. 2007. L-Methionine sulfoximine, but not phosphinothricin, is a substrate for an acetyltransferase (gene PA4866) from Pseudomonas aeruginosa: structural and functional studies. Biochemistry 46:1829–1839. doi: 10.1021/bi0615238. [DOI] [PubMed] [Google Scholar]
- 8.Carper SW, Willis DG, Manning KA, Gerner EW. 1991. Spermidine acetylation in response to a variety of stresses in Escherichia coli. J Biol Chem 266:12439–12441. [PubMed] [Google Scholar]
- 9.Liang W, Malhotra A, Deutscher MP. 2011. Acetylation regulates the stability of a bacterial protein: growth stage-dependent modification of RNase R. Mol Cell 44:160–166. doi: 10.1016/j.molcel.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hentchel KL, Escalante-Semerena JC. 2015. In Salmonella enterica, the Gcn5-related acetyltransferase MddA (formerly YncA) acetylates methionine sulfoximine and methionine sulfone, blocking their toxic effects. J Bacteriol 197:314–325. doi: 10.1128/JB.02311-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies AM, Tata R, Chauviac FX, Sutton BJ, Brown PR. 2008. Structure of a putative acetyltransferase (PA1377) from Pseudomonas aeruginosa. Acta Crystallogr Sect F Struct Biol Cryst Commun 64:338–342. doi: 10.1107/S1744309108007665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies AM, Tata R, Snape A, Sutton BJ, Brown PR. 2009. Structure and substrate specificity of acetyltransferase ACIAD1637 from Acinetobacter baylyi ADP1. Biochimie 91:484–489. doi: 10.1016/j.biochi.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Wu GB, Yuan MR, Wei L, Zhang Y, Lin YJ, Zhang LL, Liu ZD. 2014. Characterization of a novel cold-adapted phosphinothricin N-acetyltransferase from the marine bacterium Rhodococcus sp strain YM12. J Mol Catal B-Enzym 104:23–28. doi: 10.1016/j.molcatb.2014.03.001. [DOI] [Google Scholar]
- 14.Deblock M, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Vanmontagu M, Leemans J. 1987. Engineering herbicide resistance in plants by expression of a detoxifying enzyme. EMBO J 6:2513–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogawa Y, Tsuruoka T, Inoue S, Niida T. 1973. Studies on a new antibiotic SF-1293. II. Chemical structure of antibiotic SF-1293. Sci Rep Meijii Kaisha 13:42–48. [Google Scholar]
- 16.Circello BT, Eliot AC, Lee JH, van der Donk WA, Metcalf WW. 2010. Molecular cloning and heterologous expression of the dehydrophos biosynthetic gene cluster. Chem Biol 17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maughan SC, Cobbett CS. 2003. Methionine sulfoximine, an alternative selection for the bar marker in plants. J Biotechnol 102:125–128. doi: 10.1016/S0168-1656(03)00028-2. [DOI] [PubMed] [Google Scholar]
- 18.Murakami T, Anzai H, Imai S, Satoh A, Nagaoka K, Thompson CJ. 1986. The bialaphos biosynthetic genes of Streptomyces hygroscopicus—molecular cloning and characterization of the gene cluster. Mol Gen Genet 205:42–50. doi: 10.1007/BF02428031. [DOI] [Google Scholar]
- 19.Berkowitz D, Hushon JM, Whitfield HJ Jr, Roth J, Ames BN. 1968. Procedure for identifying nonsense mutations. J Bacteriol 96:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balch WE, Wolfe RS. 1976. New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere. Appl Environ Microbiol 32:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galloway NR, Toutkoushian H, Nune M, Bose N, Momany C. 2013. Rapid cloning for protein crystallography using type IIS restriction enzymes. Crystal Growth Design 13:2833–2839. doi: 10.1021/cg400171z. [DOI] [Google Scholar]
- 22.VanDrisse CM, Escalante-Semerena JC. 2016. New high-cloning-efficiency vectors for complementation studies and recombinant protein overproduction in Escherichia coli and Salmonella enterica. Plasmid 86:1–6. doi: 10.1016/j.plasmid.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 24.Griffith KL, Wolf RE Jr. 2002. Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem Biophys Res Commun 290:397–402. doi: 10.1006/bbrc.2001.6152. [DOI] [PubMed] [Google Scholar]
- 25.Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol 91:49–60. doi: 10.1016/S0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 26.Paez-Espino AD, Chavarria M, de Lorenzo V. 2015. The two paralogue phoN (phosphinothricin acetyl transferase) genes of Pseudomonas putida encode functionally different proteins. Environ Microbiol 17:3330–3340. doi: 10.1111/1462-2920.12798. [DOI] [PubMed] [Google Scholar]
- 27.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 28.Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. 2008. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59:231–237. doi: 10.1016/j.plasmid.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.