

ABSTRACT
Assessing the process that gives rise to hybrid pathogens is central to understanding the evolution of emerging plant diseases. Phytophthora ×alni, a pathogen of alder, results from the homoploid hybridization of two related species, Phytophthora uniformis and Phytophthora ×multiformis. Describing the genetic characteristics of P. ×alni should help us understand how reproductive mechanisms and historical processes shaped the population structure of this emerging hybrid pathogen. The population genetic structure of P. ×alni and the relationship with its parental species were investigated using 12 microsatellites and one mitochondrial DNA (mtDNA) marker on a European collection of 379 isolates. Populations of P. ×alni were dominated by one multilocus genotype (MLG). The frequency of this dominant MLG increased after the disease emergence together with a decline in diversity, suggesting that it was favored by a genetic mechanism such as drift or selection. Combined microsatellite and mtDNA results confirmed that P. ×alni originated from multiple hybridization events that involved different genotypes of the progenitors. Our detailed analyses point to a geographic structure that mirrors that observed for P. uniformis in Europe. The study provides more insights on the contribution of P. uniformis, an invasive species in Europe, to the emergence of Phytophthora-induced alder decline.
IMPORTANCE Our study describes an original approach to assess the population genetics of polyploid organisms using microsatellite markers. By studying the parental subgenomes present in the interspecific hybrid P. ×alni, we were able to assess the geographical and temporal structure of European populations of the hybrid, shedding new light on the evolution of an emerging plant pathogen. In turn, the study of the parental subgenomes permitted us to assess some genetic characteristics of the parental species of P. ×alni, P. uniformis, and P. ×multiformis, which are seldom sampled in nature. The subgenomes found in P. ×alni represent a picture of the “fossilized” diversity of the parental species.
INTRODUCTION
Interspecific hybridization can be a rapid track for the evolution of new species in many phylogenetic groups, including fungi and oomycetes (1–3). It has been recognized as an important mechanism of plant disease emergence, with the description of many interspecific hybrid pathogens (4, 5). In particular, in the genus Phytophthora, interspecific hybridization is increasingly seen as a major force generating new taxa: hybrids have been successfully created under laboratory conditions, and more importantly, many natural hybrids have been reported (6–8). Thus, better knowledge of the processes that give rise to hybrid taxa is an important question in the evolutionary biology of emerging pathogens. Understanding the mechanisms that lead to speciation and to the establishment, spread, and evolution of hybrid taxa requires investigation of the identity and genetic variability of parental species and of the directionality and recurrence of the hybridization events (9, 10).
Analysis of allele and genotype frequencies using standard population genetic tools can at least in part answer these questions (11). However, the population genetics of polyploid organisms presents specific caveats: as allele copy numbers are ambiguous, computing allele frequencies and identifying multilocus genotypes can be complex (12). This has limited the use of these tools in the study of polyploid organisms. To tackle this problem, the development of specific primers from the contributing progenitor genomes may enable the analysis of population genetics of allopolyploids (13, 14). In this case, parental taxa must have been identified and their ploidy levels must be known. The identification of alleles from each parent and their separate treatment during the analysis can increase the robustness of the population genetic analysis (11).
The alder pathogen Phytophthora ×alni constitutes a good model to study the emergence and evolution of hybrids, since the parental taxa and their ploidy level are known. Phytophthora uniformis and Phytophthora ×multiformis have been identified as the parents of P. ×alni (15, 16). Recent work coupling flow cytometry and quantification of allele copy number determined the exact ploidy levels of the three species of the complex (17). Accordingly, P. ×alni was shown to be a triploid homoploid-type taxon that formed through the hybridization of a diploid taxon (P. uniformis) and a tetraploid taxon (P. ×multiformis) without chromosome doubling. Therefore, P. ×alni possesses one-half of the genome of the two parental species.
P. uniformis, one of the parental species of P. ×alni, is a diploid species and has been isolated both in Europe and in North America (18–20). Genetic characterization of European P. uniformis populations revealed low diversity and a clonal structure, while North American populations were more diverse and exhibited signs of outcrossing (19). These results suggest that P. uniformis is exotic to Europe and that North America could be its center of origin. The introduction of P. uniformis to Europe would have enabled its hybridization with the other parental species and could thus be a major factor in the emergence of P. ×alni. Less is known about the origin of the other parental species, P. ×multiformis. This species is infrequently isolated, and only few individuals are available for analysis. Husson et al. (17) showed that P. ×multiformis is a tetraploid organism, supporting the hypothesis raised by Ioos et al. (15) that P. ×multiformis could have emerged after an ancient interspecific hybridization of two currently undetermined Phytophthora species. Nevertheless, whether P. ×multiformis and/or its parental species are exotic or indigenous to Europe remains unknown.
It was suggested that the spread of P. ×alni within Europe is the product of clonal dispersion (21, 22). The first evidence confronting this hypothesis came from the studies of Ioos et al. (15), who studied mitochondrial and nuclear molecular markers, which under sexual reproduction are inherited uniparentally and biparentally, respectively. The authors showed that P. ×alni isolates exhibit different mitochondrial (mtDNA) and nuclear DNA patterns that are shared either with P. uniformis (multiple U mitotypes for the mtDNA and different P. uniformis allele types for each of the nuclear DNA genes studied) or with P. ×multiformis (multiple M mitotypes for the mtDNA and different P. ×multiformis allele types for each of the nuclear DNA genes studied). These multiple-mitotype patterns suggest that several sexual hybridization events have led to the genesis of the hybrid and that the spread of P. ×alni throughout Europe might not be attributable to a single clone (15).
In this article, we analyzed further the role of sexual reproduction in the genesis and genetic diversification of the hybrid species P. ×alni. Based on 12 microsatellite markers and one mitochondrial DNA marker, we assessed the level of genetic diversity of P. ×alni in Europe and the manner in which this diversity is structured in a geographical and a temporal scale. This was done by studying the parental subgenomes of P. uniformis and P. ×multiformis occurring in P. ×alni. In turn, the study of these subgenomes allowed to assess some genetic characteristics of the parental species, P. uniformis and P. ×multiformis, which are seldom sampled in nature.
MATERIALS AND METHODS
Isolate collection and DNA extraction.
For organisms with suspected low polymorphism, the choice of the sampling strategy is highly dependent on the objectives of the study. When the goal is to assess diversity in a population, as is the case here, the recommended strategy should be to sample along the whole distribution area of the target population, in order to attempt to collect more distinct and rare genotypes (23). This will minimize bias in the estimation of the diversity indices and deliver better approximations of the real population values. Here we also considered the temporal dimension. For this study, surveys of Alnus glutinosa and Alnus incana stands were conducted over 10 years (from 1999 to 2009) across France, Belgium, and Hungary to collect P. uniformis, P. ×multiformis, and P. ×alni samples. Sampling was performed from infected alder trees that exhibited typical Phytophthora collar canker symptoms. Small sections of necrotic bark pieces were placed on V8 juice or carrot agar medium selective for Phytophthora. As soon as detected, growing mycelium was transferred to fresh V8 juice agar containing rifampin (10 mg/liter) or nonamended carrot agar. To broaden sample collection, 57 additional isolates from 11 European countries were obtained from colleagues (see Table S1 in the supplemental material). Sampled locations are shown in Fig. 1. For P. uniformis, the European and North American isolates described by Aguayo et al. (19) were added as reference isolates to track the contribution of this invasive species to the emergence of P. ×alni. DNA extractions were performed from pure fresh or lyophilized mycelium using the BioSprint 96 DNA plant kit in combination with the BioSprint 96 automated workstation (Qiagen, Courtaboeuf, France), following the BS-96DNA-plant protocol, or using the Qiagen DNeasy Plant minikit according to supplier instructions. Isolate identity (P. ×alni, P. ×multiformis, or P. uniformis) was determined using the set of species-specific PCR primers TRP-PAU-F/-R, RAS-PAM1-F/-R, and RAS-PAM2-F/-R described by Husson et al. (17).
FIG 1.

Geographical origins of P. ×alni (Pxa), P. ×multiformis (Pxm), and P. uniformis (Pu) isolates used in this study. 1, Sarre/Nied (France); 2, Rhine (France); 3, Meurthe/Moselle (France); 4, Saône (France); 5, Ognon (France); 6, Meuse (France and Belgium); 7, Loir (France); 8, Loire (France); 9, Sèvre Niortaise (France); 10, Salamanca (Spain); 11, England (United Kingdom); 12, Scotland (United Kingdom); 13, Rhineland (Germany); 14, Braunschweig (Germany); 15, Freising (Germany); 16, Oder (Germany); 17, Poland; 18, Répce (Hungary); 19, Balaton (Hungary); 20, Zala (Hungary).
Study of the P. alni complex by microsatellite and mitochondrial markers.
A set of microsatellite markers was generated independently from the two parental species. These markers were developed from microsatellite-enriched libraries sequenced with a Roche GS-FLX Titanium pyrosequencing platform following the procedure described by Malausa et al. (24) and Aguayo et al. (19). Markers obtained from the P. ×multiformis microsatellite-enriched library did not show polymorphism within any species of the P. alni complex; they were therefore discarded for further tests. Polymorphism was detected in P. ×alni, P. uniformis, and P. ×multiformis with markers obtained from the P. uniformis microsatellite-enriched library. This set of markers comprised five microsatellite markers developed by Aguayo et al. (19) and another five additional loci that exhibited polymorphism in P. ×alni and P. ×multiformis. Additionally, two polymorphic microsatellite loci derived from P. ×alni, PA17 and PA23, described by Ioos et al. (25), were added to the marker set. In all, we used 12 microsatellite markers to characterize our isolate collection. The PCR amplification conditions were those described by Aguayo et al. (19). The mitochondrial type of the P. ×alni isolates, either U or M mitotype (15), was studied using the cox1 gene primers (COXF4N/COXR4N) designed by Kroon et al. (26) with PCR mix and run conditions as described by Ioos et al. (15). Subsequently, 20 μl of the PCR product was digested using HaeIII for 1 h at 37°C, according to the manufacturer's instructions (Invitrogen, Saint-Aubin, France). Digested DNA patterns were resolved after 1 h of electrophoresis (0.6 V/cm) in a 1% agarose gel in 0.5% Tris-borate-EDTA (TBE) buffer. Gels were stained with ethidium bromide to visualize polymorphisms of DNA fragments.
Assignment of microsatellite alleles to species of the P. alni complex.
For each locus, we first assessed the different parental microsatellite profiles. As shown by Aguayo et al. (19), the diploid parent P. uniformis exhibited one or two alleles at all loci. For P. ×multiformis, we assumed the hypothesis of an ancient hybridization origin that formed a tetraploid hybrid as stated by Husson et al. (17). In the simplest cases, when one and four peaks were observed, we considered that each allele was present in four copies or one copy, respectively. If two or three peaks were observed, peak height ratios were used to assess allele copy numbers (27). In general, we observed two alleles with comparable peak sizes and we attributed two copies of each allele. Figure S1 in the supplemental material shows how chromatograms were used to determine the allele composition whenever 3 alleles where found in P. ×multiformis.
Microsatellite profiles for the triploid hybrid P. ×alni were then studied. First, at each locus we determined which specific alleles from each parental species could be found in P. ×alni. When alleles present in P. ×alni were found exclusively in P. uniformis and not in P. ×multiformis, we assumed that they represented the haploid contribution of P. uniformis (here named P. uniformis subgenome [PuSG]) to the genome of P. ×alni. When alleles in P. ×alni were found exclusively in P. ×multiformis and not in P. uniformis, we assumed that they represented the two haploid genomes' contribution of P. ×multiformis to P. ×alni, as described by Ioos et al. (15) and Husson et al. (17). The P. ×multiformis contribution will be referred to as P. ×multiformis subgenome (P×mSG) here. For the P. ×multiformis subgenome occurring in P. ×alni, different scenarios were considered: when only one P. ×multiformis allele was found in P. ×alni, these individuals were considered to be homozygous for the P. ×multiformis allele. Whenever two different P. ×multiformis alleles were found exclusively in P. ×alni, we considered that individuals might have inherited two different alleles coming from the double haploid contribution of P. ×multiformis. Another case occurred when P. uniformis and P. ×multiformis shared one allele. In such a case, we considered that the shared allele was a contribution of both P. uniformis and P. ×multiformis subgenomes in P. ×alni. In this case, the expected peak height ratio for the shared allele should be 2-fold compared to the remaining allele. The last case appeared when one allele was found exclusively in P. ×alni but not among parental isolates. In this case, alleles were assigned as belonging either to the P. uniformis or to the P. ×multiformis subgenome according to the mutation steps that separated the two subgenomes. To confirm reproducibility, a subset of 219 isolates (169 P. ×alni, 12 P. uniformis, and 38 P. ×multiformis isolates) were genotyped twice in independent PCR assays.
Characterization of the parental species P. uniformis and P. ×multiformis and comparisons with reconstructed subgenomes in P. ×alni.
Microsatellite profiles of P. uniformis and P. ×multiformis were compared to the reconstituted subgenomes in P. ×alni. Each locus in P. ×alni was converted into diploid profiles for the P. uniformis subgenome and to tetraploid profiles for the P. ×multiformis subgenome. The P. uniformis subgenome was compared to P. uniformis North American and European isolates, and the P. ×multiformis subgenome was compared to European P. ×multiformis samples. Multilocus genotypes (MLGs) were identified using Genotype (28), computing distances under the stepwise mutation model (SMM). The assignment was performed by computing a pairwise distance matrix and selecting a threshold that defines the maximum distance between two individuals at which they are still assigned to the same clonal lineage. The threshold was set at zero to differentiate individuals with small differences. Genotype can handle putative missing alleles by comparing the apparent difference in ploidy level in one locus to other loci of the same individual. Whenever an allele was missing due to potential amplification problems, it was considered a null allele. Poppr (29) was used to assess genotypic richness, genotypic diversity, and the evenness for each group of samples. Genotypic richness was assessed by computing the raw number of observed MLGs and the expected MLG (eMLG) corrected by the smallest sample size based on rarefaction. Genotypic diversity was assessed with the Simpson's diversity index (D), corrected by sample size across all samples. D, defined as the probability that two individuals taken at random belong to different MLGs, was equation 1 computed as
| 1 |
where N is the sample size, G the number of MLGs detected over all samples, and pi is the frequency of the ith MLG in the sample. Evenness (E5), an index that reflects equitability in the distribution of clonal membership among samples, was also estimated. E5 is the ratio of the number of abundant genotypes to the number of rarer genotypes (30). Evenness may range from 0 to 1, with 0 for a population dominated by a single MLG, and 1 for a population with equally abundant MLGs. Clonal richness (R), i.e., the ratio of the number of MLGs found over the sample size, was computed according to the formula (G − 1)/(N − 1). R ranges from 0, when all individuals share the same MLG, to 1, when all individuals have distinct MLGs. Minimum spanning networks (MSN) were constructed to assess genetic relationships among subgenomes and parental genomes. Poppr was used to compute the Bruvo pairwise genetic distance (31) between samples of the P. uniformis subgenome and P. uniformis and of the P. ×multiformis subgenome and P. ×multiformis. The Bruvo distance, adapted to polyploid organisms, is similar to band-sharing indices used with dominant data but takes into account the stepwise mutational distance between microsatellite alleles (12). The distance ranges from 0, indicating identical MLGs, to 1, indicating maximum dissimilarity. This distance is better adapted to autopolyploids but can be applied to allopolyploids, provided that when alleles present at one locus can originate from the two subgenomes, these alleles are separated by at least three mutation steps (32).
Last, we compared P. uniformis and P. ×multiformis subgenome profiles to the observed MLGs of the parental isolates to decipher their in natura contribution to the variability of P. ×alni. Simulated crosses between all parental P. uniformis and P. ×multiformis MLGs were performed under the hypothesis of a sexual origin of the hybrid to determine whether some of the possible mating combinations could generate a specific P. ×alni MLG. When a perfect match was found, the parental P. uniformis and P. ×multiformis MLGs were considered putative progenitors of P. ×alni.
Geographical and temporal structure of P. ×alni.
For the whole P. ×alni collection (269 samples), MLGs were identified using Genotype as explained previously. Genotypic richness, genotypic diversity, and evenness were computed using Poppr. Clonal richness corrected by sample size was also estimated. Two approaches were used to study the geographical population structure of P. ×alni. The first approach used principal-component analysis (PCA) to assess genetic relationships among samples of P. ×alni. Multivariate analyses such as PCA are exploratory methods that can be used to cluster individuals without making strong assumptions about an underlying genetic model, allowing to summarize genetic variability to reveal structure (33). POLYSAT (12) was used to compute the Bruvo distance between P. ×alni samples. The distance matrix was visualized after scaling using PCA with FactoMineR (34). An analysis of variance (ANOVA) test was used to assess whether P. ×alni clusters observed in PCA could be explained by the geographical coordinates and by the putative P. uniformis and P. ×multiformis parents assigned to P. ×alni after the simulated sexual crosses. Likewise, the homogeneity of mtDNA type (M or U) distribution in Europe was tested by ANOVA, using the geographical coordinates of each sampling location as independent variables.
The second approach used conventional population genetics tools based on allele frequency. For this approach, isolates were assigned to 20 watershed/country populations. The number of isolates per site was in many cases limited because of low isolation rates in some locations. We thus grouped isolates at a larger geographical scale (minimum group size of 4). As P. ×alni is able to disperse over long distances by river water, isolates were grouped by watershed whenever the exact sampling locations were known. Seventeen watershed populations could be defined (see Table 3). Three additional populations (from England, Scotland, and Poland) were also defined for isolates with unknown or incomplete geographical coordinates. These three populations were arbitrarily located at the country's barycenter. The number of P. ×alni isolates that could be assigned to watershed/country populations (populations with at least four P. ×alni individuals) was 254. The remaining 15 isolates could not be assigned to a watershed or a country population because it was not possible to group more than 4 isolates together. For each population, we computed the number of observed MLGs, clonal richness, and genotypic diversity as described previously. Population differentiation was studied by estimation of the genetic differentiation indexes Fst and Rst, using SPAGeDi 1.3a (35). The Fst and Rst indexes measure the apportionment of genetic variability between population and individual levels, based on allelic states and allele sizes, respectively. Polyploids with disomic inheritance, such as allopolyploids, can be analyzed using these two estimators when alleles from the different subgenomes can be distinguished in the hybrid genome. Both indexes were tested for significance by 10,000 permutations. Diversity indexes and genetic differentiation were assessed for P. ×alni isolates and for their reconstructed P. uniformis and P. ×multiformis subgenomes for each population.
TABLE 3.
Characteristics of P. ×alni populations in Europe
| ID no. | Country | Population | No. of samples | M/U typea |
P. ×alni |
PuSG |
P×mSG |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MLG | R | D | MLG | R | D | MLG | R | D | |||||
| 1 | France | Sarre/Nied | 37 | 37/0 | 4 | 0.08 | 0.16 | 2 | 0.03 | 0.05 | 3 | 0.06 | 0.11 |
| 2 | France | Rhine | 10 | 9/1 | 2 | 0.11 | 0.2 | 1 | 0 | 0 | 2 | 0.11 | 0.2 |
| 3 | France | Meurthe/Moselle | 8 | 6/2 | 2 | 0.14 | 0.25 | 1 | 0 | 0 | 2 | 0.14 | 0.25 |
| 4 | France | Saône | 21 | 20/1 | 2 | 0.05 | 0.1 | 1 | 0 | 0 | 1 | 0 | 0 |
| 5 | France | Ognon | 5 | 4/1 | 2 | 0.25 | 0.4 | 1 | 0 | 0 | 2 | 0.25 | 0.4 |
| 6 | France-Belgium | Meuse | 19 | 17/2 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 7 | France | Loir | 12 | 0/10 (2 NA) | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 8 | France | Loire | 8 | 5/3 | 2 | 0.14 | 0.25 | 1 | 0 | 0 | 2 | 0.14 | 0.25 |
| 9 | France | Sèvre Niortaise | 14 | 13/1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 10 | Spain | Salamanca | 6 | 6/0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 11 | United Kingdom | England | 5 | 3/2 | 4 | 0.75 | 0.9 | 2 | 0.25 | 0.4 | 3 | 0.5 | 0.8 |
| 12 | United Kingdom | Scotland | 4 | 3/1 | 1 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 |
| 13 | Germany | Rhineland | 6 | 5/0 (1 NA) | 4 | 0.6 | 0.87 | 1 | 0 | 0 | 4 | 0.6 | 0.87 |
| 14 | Germany | Braunschweig | 4 | 2/2 | 4 | 1 | 1 | 3 | 0.67 | 0.83 | 3 | 0.67 | 0.83 |
| 15 | Germany | Freising | 6 | 0/6 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 16 | Germany | Oder | 9 | 9/0 | 3 | 0.25 | 0.42 | 1 | 0 | 0 | 3 | 0.25 | 0.42 |
| 17 | Poland | Poland | 9 | 2/7 | 2 | 0.13 | 0.5 | 2 | 0.13 | 0.5 | 1 | 0 | 0 |
| 18 | Hungary | Répce | 26 | 19/7 | 6 | 0.2 | 0.65 | 3 | 0.08 | 0.52 | 3 | 0.08 | 0.22 |
| 19 | Hungary | Balaton | 9 | 8/1 | 2 | 0.13 | 0.22 | 1 | 0 | 0 | 2 | 0.13 | 0.22 |
| 20 | Hungary | Zala | 36 | 36/0 | 3 | 0.06 | 0.11 | 3 | 0.06 | 0.11 | 2 | 0.03 | 0.06 |
Abbreviations: M/U type, frequency of samples with M or U mtDNA type; NA, information not available; MLG, number of multilocus genotypes per population; R, clonal richness; D, genotypic diversity (Simpson's index); PuSG, P. uniformis subgenome in P. ×alni isolates; P×mSG, P. ×multiformis subgenome in P. ×alni isolates.
For the temporal structure of P. ×alni, the evolution of clonal richness over the years was compared by regression with a linear model. To account for the strong unbalanced sampling between years and locations, we simulated samples by randomly selecting one isolate per year for each location. Estimation of the mean clonal richness per year was computed after 1,000 repetitions. By using the same procedure, we studied the evolution of the mtDNA type, either M or U, over the years. Temporal population differentiation of P. ×alni was assessed by estimation of global Fst and Rst using SPAGeDi 1.3a as described above for geographical structure. Collection years were known for 266 P. ×alni samples, which were used to determine temporal structure.
RESULTS
Microsatellite analyses and allele assignment to P. uniformis and P. ×multiformis subgenomes.
A total of 379 isolates were genotyped (269 P. ×alni, 39 P. ×multiformis, and 71 P. uniformis isolates), including all the 71 P. uniformis isolates previously studied by Aguayo et al. (19). Repeatability of the allele peak height ratios was consistent between replicates (R2 = 0.76; P < 0.001). This information was therefore used to estimate the allele copy number present in each isolate whenever the number of alleles was lower than expected and to assign alleles to P. uniformis and P. ×multiformis subgenomes. Polymorphism of the microsatellite markers was low, although all loci exhibited polymorphism for at least one species of the P. alni complex. In total, 34 alleles were amplified for the 12 microsatellite loci. The number of alleles at each locus for each species ranged from 1 to 4 (Table 1). Consistent with the hypothesis used for ploidy levels, 1 or 2, 1 to 4, and 1 to 3 alleles per isolate were amplified at each locus in P. uniformis, P. ×multiformis, and P. ×alni, respectively. Four microsatellite loci amplified exclusively in P. uniformis and P. ×alni, and one exclusively in P. ×multiformis and P. ×alni (Table 1). The Venn diagram in Fig. 2 displays the allelic distribution within the three species. Most alleles were specific either to both P. uniformis and P. ×alni or to both P. ×multiformis and P. ×alni. Only one allele was shared among the three species (allele 68 at locus M-PAU14).
TABLE 1.
Characteristics of the microsatellite markers used for this study
| Locus | GenBank accession no. | Primerd sequence (5′–3′) | Repeat motif | Size (bp) | Allele(s) |
||
|---|---|---|---|---|---|---|---|
| P. uniformis | P. ×multiformis | P. ×alni | |||||
| M-PAU3a | JX462795 | F, TAAGAGACCTCCGGCAGAGA | (GA)10 | 110 | 105/107/113 | 107/113 | |
| R, AAAGCGAACACGAAGTCCAC | |||||||
| M-PAU9a | JX462796 | F, TCATGGCGCTGATCAAGTAG | (AC)9 | 95 | 93/95 | 83 | 83/93 |
| R, TAGTGGAGACTTACGGGGTT | |||||||
| M-PAU11c | JX462790 | F, AGGTGGAGTGCTAGAGGCAA | (CAT)7, C(TTC)9 | 189 | 151 | 188/191 | 151/185/188/191 |
| R, GCGACCTTTGAGTGACCAAT | |||||||
| M-PAU14c | JX462791 | F, GAAGGCTACGTAACTTGCTTTT | (GT)9 | 80 | 68 | 62/68/75/77 | 62/68/75/77 |
| R, ATCGAACTTCTCTTCCTTCACG | |||||||
| M-PAU15c | JX462792 | F, CCCGTCCTTCATCAACAAAA | (CT)9 | 80 | 74 | 71 | 71/74 |
| R, GAGGCTCTGCGATGCAATAG | |||||||
| M-PAU32a | JX462797 | F, TCAGCTCCTGTATCATCAATCG | (CA)9 | 99 | 90/92 | 86 | 86/92 |
| R, AAGTTGCCGGTGAGTTGG | |||||||
| M-PAU53a | JX462798 | F, TCTGACGAAGACCTCGACCT | (CT)8 | 187 | 202/204 | 204/206 | |
| R, CTCGAGATTGCCTTGCTGTC | |||||||
| M-PAU55a | JX462799 | F, ACATTGCTCATTCAGATGCG | (GT)8 | 232 | 243/245 | 243/245 | |
| R, GTGGAGGAGCACTTCATGGT | |||||||
| M-PAU56c | JX462793 | F, GCTGGTGGATAATTCGTCGT | (GA)7 | 81 | 71 | 74/78 | 71/74/78 |
| R, CAAAAGCGATCCTCTTCACC | |||||||
| M-PAU72c | JX462794 | F, GTTCTCGAGACTCAGCAGCC | (CAA)7 | 146 | 140 | 152/156 | 140/152/156 |
| R, CAGAGGGATACCCGAGTGAA | |||||||
| PA17b | DQ665905 | F, AGCGACAATGCAGGAAGC | (GTC)4(…) (GC)4 | 317 | 317 | 317/322 | |
| R, CTGTCTGGGCATTCATGTCG | |||||||
| PA23b | DQ665906 | F, GGAGATAGCCACGAGACACC | (GAA)7 | 155 | 135/150 | 135/150 | |
| R, CAAGCATCGCTGTAAACGAC | |||||||
FIG 2.

Venn diagram representing the allelic distribution within P. ×alni, P. ×multiformis, and P. uniformis. Numbers in the circles represent the number of alleles found in one specific species or in more than one of the species simultaneously.
Concerning the parental species P. uniformis, 4 private alleles were observed. Three were observed only in North American samples (allele 90 at locus M-PAU32, allele 95 at locus M-PAU9, and allele 202 at locus M-PAU53), while one was shared by American and European samples (allele 105 at locus M-PAU3). No private P. ×multiformis alleles were observed. Concerning P. ×alni, all alleles could be assigned either to a P. uniformis or to a P. ×multiformis subgenome (Table 2). One single allele was found at each exclusive P. uniformis and P. ×alni locus. For these loci, the observed allele was thus considered to represent the haploid contribution of the P. uniformis subgenome. Similarly, 1 or 2 alleles were present in P. ×alni at loci exclusive to P. ×multiformis and P. ×alni, and they were considered to represent the haploid contribution of the P. ×multiformis subgenome. For loci that cross-amplified in the three species, assignment was usually straightforward since they generally differed between P. uniformis and P. ×multiformis. The only shared allele (allele 68 at locus M-PAU14) was found in all P. ×alni samples. For 99% of the P. ×alni isolates that presented 3 alleles at locus M-PAU14, the alleles exhibited similar height ratios, and assignment was straightforward. For the 1% of the P. ×alni isolates that exhibited only two alleles at locus M-PAU14, peak height ratios enabled us to determine that allele 68 was present in two copies, and it was assumed that one copy originated from P. uniformis and the other from P. ×multiformis. Three private alleles were observed exclusively in P. ×alni, though present in few individuals. These alleles were at one mutation step away from their putative parental alleles in P. ×multiformis (allele 185 at locus M-PAU11) and P. uniformis (allele 206 at locus M-PAU53 and allele 322 at locus PA17). Eleven P. ×alni isolates (4.09%) presented missing alleles at least at one locus, which were considered null alleles. It is interesting that private P. uniformis alleles observed only in North American samples were very seldom observed in P. ×alni. The only exception was allele 243 at locus M-PAU55, which was observed in one Hungarian isolate. Multilocus allelic profiles for all isolates used in this study are presented in Table S1 in the supplemental material.
TABLE 2.
Multilocus genotypes of P. ×alni and their putative parents after computed mating simulationsa
| P. ×alni MLG | P. uniformis subgenome MLG | P. ×multiformis subgenome MLG | n | P. uniformis putative parentsb | P. ×multiformis putative parents | Cluster |
|---|---|---|---|---|---|---|
| P×a-1 | Pu-SG1 | P×m-SG1 | 216 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
| P×a-2 | Pu-SG3 | P×m-SG1 | 10 | Pu-E2 | P×m-1, P×m2 | B |
| P×a-3 | Pu-SG3 | P×m-SG7 | 7 | Pu-E2 | P×m-2, P×m-4 | B |
| P×a-4 | Pu-SG1 | P×m-SG9 | 4 | Pu-E1, Pu-E3 | nf | A |
| P×a-5 | Pu-SG1 | P×m-SG4 | 3 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
| P×a-6 | Pu-SG1 | P×m-SG5 | 3 | Pu-E1, Pu-E3 | P×m-1, P×m-2, P×m-5 | A |
| P×a-7 | Pu-SG1 | P×m-SG6 | 3 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
| P×a-8 | Pu-SG1 | P×m-SG2 | 3 | Pu-E1, Pu-E3 | P×m-1, P×m-2, P×m-5 | A |
| P×a-9 | Pu-SG1 | P×m-SG7 | 3 | Pu-E1, Pu-E3 | P×m-2, P×m-4 | A |
| P×a-10 | Pu-SG1 | P×m-SG2 | 1 | nfc | nf | C |
| P×a-11 | Pu-SG1 | P×m-SG3 | 1 | Pu-E1, Pu-E3 | P×m-2, P×m-4 | A |
| P×a-12 | Pu-SG2 | P×m-SG6 | 1 | nf | P×m-1, P×m-2 | C |
| P×a-13 | Pu-SG2 | P×m-SG1 | 1 | nf | P×m-1, P×m-2 | A |
| P×a-14 | Pu-SG3 | P×m-SG8 | 1 | Pu-E2 | P×m-2, P×m-4 | B |
| P×a-15 | Pu-SG3 | P×m-SG5 | 1 | Pu-E2 | P×m-1, P×m-2, P×m-5 | B |
| P×a-16 | Pu-SG4 | P×m-SG1 | 1 | nf | P×m-1, P×m-2 | A |
| P×a-17 | Pu-SG3 | P×m-SG1 | 1 | Pu-E2 | P×m-1, P×m-2 | B |
| P×a-18 | Pu-SG3 | P×m-SG10 | 1 | Pu-E2 | P×m-3 | B |
| P×a-19 | Pu-SG1 | P×m-SG11 | 1 | Pu-E1, Pu-E3 | P×m-1, P×m-2, P×m-5 | A |
| P×a-20 | Pu-SG1 | P×m-SG8 | 1 | Pu-E1, Pu-E3 | P×m-2, P×m-4 | A |
| P×a-21 | Pu-SG1 | P×m-SG1 | 1 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
| P×a-22 | Pu-SG1 | P×m-SG12 | 1 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
| P×a-23 | Pu-SG5 | P×m-SG1 | 1 | nf | P×m-1, P×m-2 | A |
| P×a-24 | Pu-SG1 | P×m-SG1 | 1 | nf | P×m-1, P×m-2 | A |
| P×a-25 | Pu-SG1 | P×m-SG6 | 1 | nf | nf | C |
| P×a-26 | Pu-SG1 | P×m-SG13 | 1 | Pu-E1, Pu-E3 | P×m-1, P×m-2 | A |
Relationship between P. ×alni and parental species P. uniformis and P. ×multiformis.
Seven MLGs were observed in North American P. uniformis samples. Although clonal richness was low (R = 0.23), genotypic diversity (D = 0.70) and evenness (E5 = 0.63) were moderate. Diversity in European P. uniformis was low, with only 3 observed MLGs. One dominant MLG (Pu-E1) included 91% of the isolates (see Table S1 in the supplemental material). This influenced the low levels of clonal richness (R = 0.05), genotypic diversity (D = 0.18), and evenness (E5 = 0.14) exhibited by European isolates. The number of expected MLGs (eMLG) at the smallest sample size (27 North American isolates) based on rarefaction was 2.58. Concerning the P. uniformis subgenome, 5 MLGs were observed. Two MLGs were shared between European P. uniformis isolates and the P. uniformis subgenome (Pu-E1 and PuSG-1 and Pu-E2 and PuSG-3). As for P. uniformis isolates, a dominant P. uniformis subgenome MLG (PuSG-1) grouped 91% of the isolates. The eMLG index for the P. uniformis subgenome data set was 2.2, a value close to the one observed for the European P. uniformis isolates, assuming equal sample sizes. Similar to what was seen for European P. uniformis, the P. uniformis subgenome data set presented low levels of clonal richness (R = 0.01), genotypic diversity (D = 0.17), and evenness (E5 = 0.14). The minimum spanning network (MSN) clearly differentiated two groups of isolates (Fig. 3a). A first group included exclusively North American P. uniformis isolates (MLGs Pu-A1 to Pu-A7). A second group included European P. uniformis isolates and P. uniformis subgenome MLGs, indicating their straight relationship. Within this group, PuSG-1 MLG was placed together with Pu-E1 and Pu-E3 MLGs, which were found as its putative parents after simulated crosses (see Table S1 in the supplemental material). Similarly, PuSG-3 was placed together with Pu-E2, its putative P. uniformis parent (see Table S1 in the supplemental material). PuSG-2, PuSG-4, and PuSG-5 grouped 2.6% of the isolates characterized by nonsampled P. uniformis putative parents.
FIG 3.

Minimum spanning network (MSN) performed on Bruvo's distances. (a) Phytophthora uniformis isolates (Pu) and P. uniformis subgenome (PuSG) found in P. ×alni isolates. (b) Phytophthora ×multiformis isolates (P×m) and P. ×multiformis subgenome (P×mSG) found in P. × alni isolates.
Five MLGs were identified among P. ×multiformis isolates, with one major MLG (P×m-1) including 56% of the isolates. P. ×multiformis clonal richness was low (R = 0.11), and genotypic diversity was moderate (D = 0.59). The evenness index was estimated at 0.72, indicating some dissimilarity among samples. For the P. ×multiformis subgenome, 13 MLGs were observed, with clonal richness and genotypic diversity levels of 0.04 and 0.25, respectively. Estimated P. ×multiformis subgenome eMLG rarefied to 39 samples was 5.12, indicating that genotypic richness was similar to that of P. ×multiformis when considering equal samples. MSN did not exhibit a clear structure pattern because polymorphism was low and several P. ×multiformis subgenome MLGs shared putative parents (Fig. 3b). This is specially the case for putative parents P×m-1 and P×m-2, which were found as putative progenitors for 89% of P. ×multiformis subgenome MLGs (Table 3; see also Table S1 in the supplemental material). The percentage of isolates for which putative progenitors could not be attributed was 2.6%.
General genetic characteristics of P. ×alni.
Twenty-six MLGs were observed in P. ×alni samples. One dominant MLG (P×a-1) included 80% of the isolates. Minor MLGs, comprising 3 to 10 isolates, encompassed 13% of the isolates. Seventeen MLGs (6% of the isolates) were sampled once. Clonal richness was low (R = 0.09), and genotypic diversity was moderate (D = 0.35). Evenness was low (E5 = 0.288), indicating low diversity within P. ×alni isolates. Mitochondrial types studied for 265 P. ×alni isolates showed that most (80%) had an M mtDNA profile while 20% exhibited a U mtDNA. In four microsatellite MLGs, both U and M mitotypes were present, adding a new level of polymorphism. When microsatellite and mtDNA data were combined, the number of MLGs increased to 30. This means that microsatellite MLGs with both mtDNAs are not true lineages but the product of at least two hybridization events. Under a conservative estimation (considering only known assigned parents to P. ×alni MLGs), the number of hybridization events was at least 13.
Geographical structure of P. ×alni populations.
The conditions required to use Bruvo's distance for P. ×alni were fulfilled, except for allele 68 at locus M-PAU14, which was shared by the two parental species. However, this allele was present in all P. ×alni samples and did not impact the estimation of genetic distances between isolates. The first two axes of the PCA explained 98.5% of the total variation and revealed a strong structure with three distinct clusters (Fig. 4). A first cluster, here called A, grouped most of the samples (91% of the isolates and 17 of the 26 P. ×alni MLGs). Consistently with the MSN built for P. uniformis and PuSG, cluster A was characterized by isolates that had either Pu-E1, Pu-E3, or a nonsampled MLG as putative P. uniformis parent. A second cluster, here called B, grouped samples with the Pu-E2 MLG as the putative P. uniformis parent. The third cluster (C) is less clear, was characterized by samples with missing alleles at some loci, and therefore had nonsampled P. uniformis putative parents. As for the MSN computed for P. ×multiformis and P×mSG, the P. ×multiformis putative parents did not show any relationship with the structure of the P. ×alni clusters. Significant differences were found between samples of the different clusters when tested against latitude (F = 16.6; P < 0.001) and longitude (F = 12.8; P < 0.001), with clusters B and C located further east and north than cluster A (Fig. 5). Neither latitude nor longitude was found to be significantly related to the mtDNA type of samples (F = 0.07, P = 0.78; and F = 3.74, P = 0.053, respectively).
FIG 4.
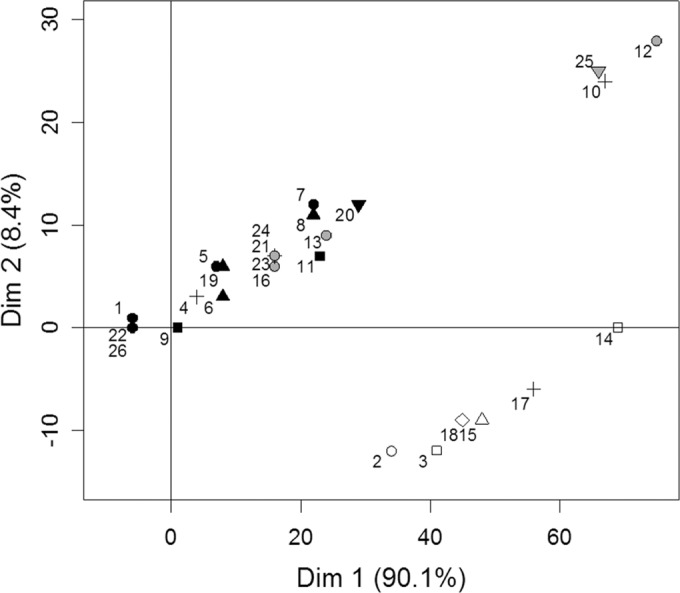
Principal component analysis (PCA) performed on Bruvo's distances on the whole collection of P. ×alni (269 isolates). Symbols indicate P. ×multiformis and P. uniformis putative parents: ○, P×m-1 or P×m-2; □, P×m-2 or P×m-4; △, P×m-1, P×m-2, or P×m-5; ▽, P×m-2; ♢, P×m-4; +, undetermined. Black filled symbols, Pu-E1 or Pu-E3; empty symbols, Pu-E2; gray filled symbols, undetermined P. uniformis putative parents. The numbers indicate the P. ×alni MLGs.
FIG 5.

Geographical distribution of P. ×alni clusters determined by principal component analysis (PCA). Isolates from clusters B and C are distributed mostly in the north and the east of Europe, in contrast to isolates from cluster A.
Diversity and population structure were further studied for 254 samples that could be assigned to the 20 watershed/country populations. Diversity indices for each population are listed in Table 3. No genetic diversity was found for six populations (Freising, Loir, Sèvre Niortaise, Meuse, Salamanca, and Scotland), and only the P×a-1 MLG was present. In contrast, the Braunschweig population exhibited different MLGs for the 4 sampled isolates. Although limited in size, the England and Rhineland populations exhibited relatively high diversity, with levels of clonal richness and genotypic diversity close to 1. Moderate diversity indices were estimated for the Oder, Ognon, and Répce populations. For the remaining populations, diversity indices were low. In most populations, the dominant P×a-1 MLG was present in frequencies ranging from 0.20 to 1, except in the Oder population, where P×a-1 was absent. P×a-3 was observed only in the Oder population, while P×a-4 was observed only in the Ognon population. Rare MLGs (only one isolate) were observed in England, Braunschweig, Oder, Répce, Rhin, Rhineland, Saône, Sarre and Nied, and Zala populations. It must be taken into account that three rare MLGs (P×a-24, P×a-25, and P×a-26) were not included in this section of the study because these isolates could not be assigned to any watershed/country population. The P. ×alni populations presented low, albeit significant, genetic differentiation, with estimated Fst of 0.024 (P < 0.001) and Rst of 0.004 (P < 0.001). Genetic diversity within populations was significantly lower for the P. uniformis subgenome than for the P. ×multiformis subgenome. Indeed, no genetic diversity was found for 14 of 20 populations when they were analyzed for the P. uniformis subgenome. Despite this low population diversity, when evaluated for the P. uniformis subgenome, strong genetic differentiation was found with high Fst and Rst values (Fst = 0.51, P < 0.001; Rst = 0.56, P < 0.001). When populations were analyzed for the P. ×multiformis subgenome, genetic differentiation estimates were similar to those obtained for P. ×alni, with low but significant Fst and Rst (Fst = 0.02, P < 0.001; Rst = 0.021, P < 0.001).
Temporal structure of P. ×alni isolates.
Figure 6 summarizes the assessment of the temporal changes of the clonal richness for P. ×alni. The regression showed a significant decrease of the clonal richness from 1996 to 2010 (F = 12.1, R2 = 0.559, P = 0.013). P×a-1 had little presence at the beginning of the sampling period, and was strongly dominant in the late collection years (after 2000). Changes in the ratio of the mtDNA haplotypes, M or U, over the years did not show any significant trend, staying stable during the evaluated period (P = 0.07). Moreover, while samples of clusters A and B were collected at about the same time (respective means of 2006 ± 0.5 and 2007 ± 1.1), samples of cluster C were collected much earlier (1999 ± 3.9). Fst and Rst estimated values suggest a low but significantly different from zero temporal differentiation (Fst = 0.003, P = 0.04; and Rst = 0.001, P = 0.003).
FIG 6.

Temporal changes of clonality in P. ×alni. Linear regression shows that clonal richness decreased with time.
DISCUSSION
In our study, we were able to assign alleles from parental subgenomes to most P. ×alni samples by combining the analysis of microsatellite profiles and peak height ratios. Microsatellites revealed low polymorphism in P. ×alni. Yet, the genotypic richness and diversity within P. ×alni were consistent with those of its parental species. Moreover, our approach allowed us to explore the diversity of both parental species, by studying the extra genetic variability that is found in P. ×alni subgenomes. In the case of P. uniformis and P. ×multiformis, this is interesting because both species are seldom isolated, likely due to the capacity of P. ×alni to replace its parents under most environmental conditions (22, 36).
Comparing the genetic characteristics of P. uniformis isolates to those of the P. uniformis subgenome highlighted interesting information. All diversity indexes used in this study, including estimations within watershed/country populations, were extremely low for the European P. uniformis isolates and for the reconstructed P. uniformis subgenome. These results differ from diversity indexes estimated for North American isolates, which were consistently higher. This indicates that P. uniformis is present in Europe as very few genotypes dispersed throughout the continent. Despite this low diversity, P. uniformis seems to have presented a strong geographic differentiation in Europe, as shown by MSN, PCA, and by Fst and Rst, estimated for the P. uniformis subgenome. However, it appears that there are genotypes other than those described for P. uniformis by Aguayo et al. (19). In fact, some P. ×alni isolates exhibited P. uniformis subgenomes that were not attributable to any P. uniformis isolate sampled so far. A hypothesis would be that they were not recovered because of too-low sampling pressure. It has, for example, been suggested that P. uniformis is well adapted to cold climates, which explains its frequent isolation from Swedish rivers (20), as under these environmental conditions P. ×alni may not be able to develop and replace P. uniformis. This is consistent with the widespread presence of P. uniformis in cold regions, such as Alaska and Oregon (18, 37). P. uniformis from colder locations could hide new diversity, and other genotypes may be discovered. The characterization and comparison of new P. uniformis individuals from these locations could bring new insights into the origins of both P. uniformis and the hybrid P. ×alni.
The genetic characteristics of P. ×multiformis and of the P. ×multiformis subgenome were also compared. P. ×multiformis is an extremely rare species, and only a few European strains have been sampled so far. However, after intense sampling in France and Belgium and the contribution of European colleagues, we were able to gather a P. ×multiformis collection of 39 isolates. Overall, P. ×multiformis exhibited low polymorphism. However, diversity indexes within the species showed that there is some diversity, and 5 P. ×multiformis MLGs were observed. Concerning the P. ×multiformis subgenome, the number of P. ×multiformis subgenome MLGs increased to 13. However, diversity indexes computed for the P. ×multiformis subgenome were smaller than for P. ×multiformis. When genotypic richness (estimated by eMLG rarefying to 39 samples) was computed for the P. ×multiformis subgenome, it was found to be similar to that of P. ×multiformis, suggesting that some diversity within P. ×multiformis effectively exists but has not been sampled. It must be considered that P. ×multiformis has been described as a polyploid product of an interspecific hybridization between two unknown Phytophthora species (15, 17) and that it may present diversity levels close to those of P. ×alni. Sampling complications, added to the low polymorphism exhibited by the microsatellite markers, make the characterization of P. ×multiformis a difficult task. P. ×multiformis diversity is far from being well understood, and if possible, more samples should be included to make more-robust genetic inferences and determine its putative origin. We can, however, suggest that this sampling effort should be performed in areas were P. ×alni is not present, including samples from other European alder species and integrating asymptomatic or nondeclining trees, as was the case for the Durance and Marais-Poitevin populations.
Our study allowed us to determine some genetic characteristics of the hybrid P. ×alni. Simulated sexual crosses between the two parental species allowed us to identify P. uniformis and P. ×multiformis putative progenitors for 89% of the P. ×alni isolates and to assess its diversity, revealing at least 30 different MLGs. As both U and M mtDNA patterns could be found in isolates exhibiting the same microsatellite MLG, these are not likely to be true clonal lineages but rather products of several hybridization events involving the same parental MLGs. Moreover, this constitutes supplementary pieces of evidence that the sexual hybridization took place in both directions, with both parents potentially acting as the antheridial strain, as previously shown by Ioos et al. (15). In this regard, the predominance of the M over the U mtDNA in P. ×alni is also consistent with the pattern found by Ioos et al. (15). Despite the fact that during sexual hybridization both parents could act as the antheridial strain, it seems that one is often favored, leading to an unequal transmission of the mtDNA (38, 39). Bringing together this evidence, we estimated that at least 13 different hybridization events occurred during the genesis of P. ×alni. These multiple hybridization events have shaped the geographical structure of P. ×alni populations. Although no obvious geographical pattern could be demonstrated for the mtDNA and for the P. ×multiformis subgenome, a clear spatial pattern was shown for the P. uniformis subgenome, indicating that P. uniformis, described as an invasive species (19), had a major role in shaping this structure. At least two clusters differentiated by their putative P. uniformis parents exist within P. ×alni. While cluster A, with Pu-E1 and Pu-E3 putative progenitors, is found throughout Europe, cluster B, with the Pu-E2 MLG progenitor, is preferentially found in Eastern Europe. Although P×a-1 MLG, which belongs to cluster A, became dominant in Europe, MLGs deriving from the Pu-E2 subgenome were not replaced by this MLG in Eastern Europe. This suggests that the different hybridization events that occurred in several European areas shaped the P. ×alni population structure and that more than one single hybridization center occurred, from which the different MLGs arose and then dispersed. This pattern is not unexpected for an infertile hybrid like P. ×alni (40) and can be explained by restricted gene flow between populations, founder effects resulting from the introduction of a limited number of parental genotypes, absence of recombination, and spread of few genotypes within its populations.
Overall, diversity within P. ×alni was low. In particular, a dominant microsatellite MLG, including more than 80% of the isolates (including both U and M types), was observed. This largely explains the low clonal richness estimated for P. ×alni. Although genetic differentiation over the years was not evident by Fst or Rst estimations, we observed a declining tendency of clonal richness with an increased frequency of this dominant MLG over time. This could be explained by genetic drift, which can eliminate rare genotypes from local populations (41), especially if the populations experience bottlenecks. These can be associated with human intervention such as eradication: however, management of alders in Europe is limited (42). Adverse climatic events that negatively affect the pathogen population may better explain the occurrence of bottlenecks. Indeed, population crashes after an unfavorable season offer an advantage to already well established genotypes during the recolonization process and may thus cause the extinction of rare genotypes (43). This applies to P. ×alni, as the pathogen has been shown to be highly susceptible to both high summer and low winter temperatures (44, 45). Another explanation for the decrease in clonal richness could be selection, which results in shifts in the frequency of genotypes over time when fitness differences are large (46). This may result from differences in adaptation to their host and/or environment. For example, Chandelier et al. (47) found that, while little variability in aggressiveness toward A. glutinosa exists within P. ×alni, the sporulation capacity is isolate dependent. However, whether genetic drift or selection is the mechanism that enables one MLG to finally predominate is currently not clear. Resolving this question would require an analysis of life history traits determining the fitness of P. ×alni (48). Nevertheless, our results illustrate how the diversity of an emerging oomycete may decrease during or shortly after the invasion process.
Finally, this study illustrates the benefits of studying the parental subgenomes present in an interspecific hybrid when the parental species are difficult to sample, rare, or even extinct. These subgenomes represent a picture of the “fossilized” diversity of the parental species.
Supplementary Material
ACKNOWLEDGMENTS
We thank our European and American colleagues for sharing their isolates. We thank Renaud Ioos (ANSES) for helpful comments on early versions of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02221-16.
REFERENCES
- 1.Giraud T, Refrégier G, Le Gac M, de Vienne DM, Hood ME. 2008. Speciation in fungi. Fungal Genet Biol 45:791–802. doi: 10.1016/j.fgb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Schardl CL, Craven KD. 2003. Interspecific hybridization in plant-associated fungi and oomycetes: a review. Mol Ecol 12:2861–2873. doi: 10.1046/j.1365-294X.2003.01965.x. [DOI] [PubMed] [Google Scholar]
- 3.Stukenbrock EH. 2016. The role of hybridization in the evolution and emergence of new fungal plant pathogens. Phytopathology 106:104–112. doi: 10.1094/PHYTO-08-15-0184-RVW. [DOI] [PubMed] [Google Scholar]
- 4.Desprez-Loustau M-L, Robin C, Buée M, Courtecuisse R, Garbaye J, Suffert F, Sache I, Rizzo DM. 2007. The fungal dimension of biological invasions. Trends Ecol Evol 22:472–480. doi: 10.1016/j.tree.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Stukenbrock EH, McDonald BA. 2008. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol 46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- 6.Bertier L, Leus L, D'hondt L, de Cock AWAM, Höfte M. 2013. Host adaptation and speciation through hybridization and polyploidy in Phytophthora. PLoS One 8:e85385. doi: 10.1371/journal.pone.0085385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Man in't Veld WA, Rosendahl K, Hong C. 2012. Phytophthora Xserendipita sp. nov. and P. Xpelgrandis, two destructive pathogens generated by natural hybridization. Mycologia 104:1390–1396. doi: 10.3852/11-272. [DOI] [PubMed] [Google Scholar]
- 8.Burgess TI. 2015. Molecular characterization of natural hybrids formed between five related indigenous clade 6 phytophthora species. PLoS One 10:e0134225. doi: 10.1371/journal.pone.0134225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop-Esteban M, Segarra-Moragues JG, González-Candelas F. 2011. Polyploid origin, genetic diversity and population structure in the tetraploid sea lavender Limonium narbonense Miller (Plumbaginaceae) from eastern Spain. Genetica 139:1309–1322. doi: 10.1007/s10709-012-9632-2. [DOI] [PubMed] [Google Scholar]
- 10.Robertson A, Rich TCG, Allen A, Houston L, Roberts C, Bridle JR, Harris SA, Hiscock SJ. 2010. Hybridization and polyploidy as drivers of continuing evolution and speciation in Sorbus. Mol Ecol 19:1675–1690. doi: 10.1111/j.1365-294X.2010.04585.x. [DOI] [PubMed] [Google Scholar]
- 11.Dufresne F, Stift M, Vergilino R, Mable BK. 2014. Recent progress and challenges in population genetics of polyploid organisms: an overview of current state-of-the-art molecular and statistical tools. Mol Ecol 23:40–69. doi: 10.1111/mec.12581. [DOI] [PubMed] [Google Scholar]
- 12.Clark LV, Jasieniuk M. 2011. POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11:562–566. doi: 10.1111/j.1755-0998.2011.02985.x. [DOI] [PubMed] [Google Scholar]
- 13.Korbecka G, Rymer PD, Harris SA, Pannell JR. 2010. Solving the problem of ambiguous paralogy for marker loci: microsatellite markers with diploid inheritance in allohexaploid Mercurialis annua (Euphorbiaceae). J Heredity 101:504–511. doi: 10.1093/jhered/esq026. [DOI] [PubMed] [Google Scholar]
- 14.Pairon M, Jacquemart A-L, Potter D. 2008. Detection and characterization of genome-specific microsatellite markers in the allotetraploid Prunus serotina. J Am Soc Hortic Sci 133:390–395. [Google Scholar]
- 15.Ioos R, Andrieux A, Marcais B, Frey P. 2006. Genetic characterization of the natural hybrid species Phytophthora alni as inferred from nuclear and mitochondrial DNA analyses. Fungal Genet Biol 43:511–529. doi: 10.1016/j.fgb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Ioos R, Panabieres F, Industri B, Andrieux A, Frey P. 2007. Distribution and expression of elicitin genes in the interspecific hybrid oomycete Phytophthora alni. Appl Environ Microbiol 73:5587–5597. doi: 10.1128/AEM.00721-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Husson C, Aguayo J, Revellin C, Frey P, Ioos R, Marçais B. 2015. Evidence for homoploid speciation in Phytophthora alni supports taxonomic reclassification in this species complex. Fungal Genet Biol 77:12–21. doi: 10.1016/j.fgb.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Adams GC, Catal M, Trummer LM. 2010. Distribution and severity of alder phytophthora in Alaska, p 29–49. In U.S. Department of Agriculture Forest Service (ed), Gen Tech Rep PSW-GTR-229, Proceedings of the sudden oak death, fourth science symposium, Albany, CA. Pacific Southwest Research Station, Albany, CA. [Google Scholar]
- 19.Aguayo J, Adams GC, Halkett F, Catal M, Husson C, Nagy ZA, Hansen EM, Marcais B, Frey P. 2013. Strong genetic differentiation between North American and European populations of Phytophthora alni subsp. uniformis. Phytopathology 103:190–199. doi: 10.1094/PHYTO-05-12-0116-R. [DOI] [PubMed] [Google Scholar]
- 20.Redondo MA, Boberg J, Olsson CHB, Oliva J. 2015. Winter conditions correlate with Phytophthora alni subspecies distribution in southern Sweden. Phytopathology 105:1191–1197. doi: 10.1094/PHYTO-01-15-0020-R. [DOI] [PubMed] [Google Scholar]
- 21.Brasier CM, Rose J, Gibbs JN. 1995. An unusual Phytophthora associated with widespread alder mortality in Britain. Plant Pathol 44:999–1007. doi: 10.1111/j.1365-3059.1995.tb02658.x. [DOI] [Google Scholar]
- 22.Nagy ZÁ, Bakonyi J, Érsek T. 2003. Standard and Swedish variant types of the hybrid alder Phytophthora attacking alder in Hungary. Pest Manag Sci 59:484–492. doi: 10.1002/ps.681. [DOI] [PubMed] [Google Scholar]
- 23.Arnaud-Haond S, Duarte CM, Alberto F, Serrão EA. 2007. Standardizing methods to address clonality in population studies. Mol Ecol 16:5115–5139. doi: 10.1111/j.1365-294X.2007.03535.x. [DOI] [PubMed] [Google Scholar]
- 24.Malausa T, Gilles A, Meglécz E, Blanquart H, Duthoy S, Costedoat C, Dubut V, Pech N, Castagnone-Sereno P, Délye C, Feau N, Frey P, Gauthier P, Guillemaud T, Hazard L, Le Corre V, Lung-Escarmant B, Malé P-JG, Ferreira S, Martin J-F. 2011. High-throughput microsatellite isolation through 454 GS-FLX Titanium pyrosequencing of enriched DNA libraries. Mol Ecol Resour 11:638–644. doi: 10.1111/j.1755-0998.2011.02992.x. [DOI] [PubMed] [Google Scholar]
- 25.Ioos R, Barrès B, Andrieux A, Frey P. 2007. Characterization of microsatellite markers in the interspecific hybrid Phytophthora alni ssp. alni, and cross-amplification with related taxa. Mol Ecol Notes 7:133–137. [Google Scholar]
- 26.Kroon L, Bakker FT, van den Bosch G, Bonants P, Flier WG. 2004. Phylogenetic analysis of Phytophthora species based on mitochondrial and nuclear DNA sequences. Fungal Genet Biol 41:766–782. doi: 10.1016/j.fgb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Esselink GD, Nybom H, Vosman B. 2004. Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting—peak ratios) method. Theor Appl Genet 109:402–408. [DOI] [PubMed] [Google Scholar]
- 28.Meirmans PG, Van Tienderen PH. 2004. Genotype and Genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794. doi: 10.1111/j.1471-8286.2004.00770.x. [DOI] [Google Scholar]
- 29.Kamvar ZN, Tabima JF, Grünwald NJ. 2014. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grünwald NJ, Goodwin SB, Milgroom MG, Fry WE. 2003. Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93:738–746. doi: 10.1094/PHYTO.2003.93.6.738. [DOI] [PubMed] [Google Scholar]
- 31.Bruvo R, Michiels NK, D'Souza TG, Schulenburg H. 2004. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol 13:2101–2106. doi: 10.1111/j.1365-294X.2004.02209.x. [DOI] [PubMed] [Google Scholar]
- 32.Clark LV. 2013. Polysat version 1.3 tutorial manual. Department of Crop Sciences, University of Illinois, Urbana-Champaign, IL. [Google Scholar]
- 33.Jombart T, Pontier D, Dufour AB. 2009. Genetic markers in the playground of multivariate analysis. Heredity 102:330–341. doi: 10.1038/hdy.2008.130. [DOI] [PubMed] [Google Scholar]
- 34.Lê S, Josse J, Husson F. 2008. FactoMineR: an R package for multivariate analysis. J Stat Software 25:1–18. [Google Scholar]
- 35.Hardy OJ, Vekemans X. 2002. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. doi: 10.1046/j.1471-8286.2002.00305.x. [DOI] [Google Scholar]
- 36.Brasier CM, Kirk SA. 2001. Comparative aggressiveness of standard and variant hybrid alder phytophthoras, Phytophthora cambivora and other Phytophthora species on bark of Alnus, Quercus and other woody hosts. Plant Pathol 50:218–229. doi: 10.1046/j.1365-3059.2001.00553.x. [DOI] [Google Scholar]
- 37.Sims LL, Sutton W, Reeser PW, Hansen EM. 2015. The Phytophthora species assemblage and diversity in riparian alder ecosystems of western Oregon, USA. Mycologia 107:889–902. doi: 10.3852/14-255. [DOI] [PubMed] [Google Scholar]
- 38.Man In't Veld WA, Veenbaas-Rijks WJ, Ilieva E, de Cock AW, Bonants PJ, Pieters R. 1998. Natural hybrids of Phytophthora nicotianae and Phytophthora cactorum demonstrated by isozyme analysis and random amplified polymorphic DNA. Phytopathology 88:922–929. doi: 10.1094/PHYTO.1998.88.9.922. [DOI] [PubMed] [Google Scholar]
- 39.Goodwin SB, Fry WE. 1994. Genetic analyses of interspecific hybrids between Phytophthora infestans and Phytophthora mirabilis. Exp Mycol 18:20–32. doi: 10.1006/emyc.1994.1003. [DOI] [Google Scholar]
- 40.Delcán J, Brasier CM. 2001. Oospore viability and variation in zoospore and hyphal tip derivatives of the hybrid alder Phytophthoras. Forest Pathol 31:65–83. doi: 10.1046/j.1439-0329.2001.00223.x. [DOI] [Google Scholar]
- 41.Vercauteren A, De Dobbelaere I, Grünwald NJ, Bonants P, Van Bockstaele E, Maes M, Heungens K. 2010. Clonal expansion of the Belgian Phytophthora ramorum populations based on new microsatellite markers. Mol Ecol 19:92–107. doi: 10.1111/j.1365-294X.2009.04443.x. [DOI] [PubMed] [Google Scholar]
- 42.Gibbs J. 2003. Management and control of Phytophthora disease of alder, p 73–78 In Gibbs JN, Van Dijk C, Webber JF (), Phytophthora disease of alder in Europe. Forestry Commission Bulletin no. 126. Forestry Commission, Edinburgh, United Kingdom. [Google Scholar]
- 43.Mascheretti S, Croucher PJP, Vettraino A, Prospero S, Garbelotto M. 2008. Reconstruction of the Sudden Oak Death epidemic in California through microsatellite analysis of the pathogen Phytophthora ramorum. Mol Ecol 17:2755–2768. doi: 10.1111/j.1365-294X.2008.03773.x. [DOI] [PubMed] [Google Scholar]
- 44.Aguayo J, Elegbede F, Husson C, Saintonge F-X, Marçais B. 2014. Modeling climate impact on an emerging disease, the Phytophthora alni-induced alder decline. Global Change Biol 20:3209–3221. doi: 10.1111/gcb.12601. [DOI] [PubMed] [Google Scholar]
- 45.Cerny K, Strnadova V. 2012. Winter survival of Phytophthora alni subsp. alni in aerial tissues of black alder. J Forest Sci 58:328–336. [Google Scholar]
- 46.Goodwin SB. 1997. The population genetics of Phytophthora. Phytopathology 87:462–473. doi: 10.1094/PHYTO.1997.87.4.462. [DOI] [PubMed] [Google Scholar]
- 47.Chandelier A, Husson C, Druart P, Marçais B. 2016. Assessment of inoculation methods for screening black alder resistance to Phytophthora ×alni. Plant Pathol 65:441–450. doi: 10.1111/ppa.12418. [DOI] [Google Scholar]
- 48.Pariaud B, Ravigné V, Halkett F, Goyeau H, Carlier J, Lannou C. 2009. Aggressiveness and its role in the adaptation of plant pathogens. Plant Pathol 58:409–424. doi: 10.1111/j.1365-3059.2009.02039.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.