

Abstract
We report here that GRL-10413, a novel nonpeptidic HIV-1 protease inhibitor (PI) containing a modified P1 moiety and a hydroxyethylamine sulfonamide isostere, is highly active against laboratory HIV-1 strains and primary clinical isolates (50% effective concentration [EC50] of 0.00035 to 0.0018 μM), with minimal cytotoxicity (50% cytotoxic concentration [CC50] = 35.7 μM). GRL-10413 blocked the infectivity and replication of HIV-1NL4-3 variants selected by use of atazanavir, lopinavir, or amprenavir (APV) at concentrations of up to 5 μM (EC50 = 0.0021 to 0.0023 μM). GRL-10413 also maintained its strong antiviral activity against multidrug-resistant clinical HIV-1 variants isolated from patients who no longer responded to various antiviral regimens after long-term antiretroviral therapy. The development of resistance against GRL-10413 was significantly delayed compared to that against APV. In addition, GRL-10413 showed favorable central nervous system (CNS) penetration properties as assessed with an in vitro blood-brain barrier (BBB) reconstruction system. Analysis of the crystal structure of HIV-1 protease in complex with GRL-10413 demonstrated that the modified P1 moiety of GRL-10413 has a greater hydrophobic surface area and makes greater van der Waals contacts with active site amino acids of protease than in the case of darunavir. Moreover, the chlorine substituent in the P1 moiety interacts with protease in two distinct configurations. The present data demonstrate that GRL-10413 has desirable features for treating patients infected with wild-type and/or multidrug-resistant HIV-1 variants, with favorable CNS penetration capability, and that the newly modified P1 moiety may confer desirable features in designing novel anti-HIV-1 PIs.
INTRODUCTION
Combination antiretroviral therapy (cART) has had a major impact on the AIDS epidemic in both developing and industrially advanced nations. In fact, recent analyses revealed that mortality rates for human immunodeficiency virus type 1 (HIV-1)-infected persons have become close to those for the general population (1–4). Moreover, the 2013 UNAIDS report on the global AIDS epidemic reports that an increase in the number of patients receiving cART has brought about >30% declines in the numbers of new infections, in particular in developing areas, including sub-Saharan countries (5). However, at present, no eradication of HIV-1 appears to be possible, in part due to the viral reservoirs remaining in blood and tissues in infected individuals. Furthermore, we have encountered a number of challenges in bringing about the optimal benefits of the currently available therapeutics for HIV-1 infection and AIDS to individuals receiving cART (6–8). These include (i) drug-related toxicities, (ii) an inability to fully restore normal immunologic functions once individuals develop full-blown AIDS, (iii) the development of various cancers as a consequence of survival prolongation, (iv) flaring up of inflammation in individuals receiving cART or the occurrence of immune reconstruction syndrome (IRS), (v) development of HIV-1-associated neurocognitive disorders (HAND) as a result of prolonged patient survival and poor antiretroviral drug penetration into the central nervous system (CNS), and (vi) the increased cost of antiviral therapy. Although recent first-line cART with boosted protease inhibitor (PI)-based and integrase inhibitor-based regimens has made the development of HIV-1 resistance relatively less likely over an extended period (9, 10), the various limitations and flaws of cART listed above are still exacerbated by persisting HIV-1 drug resistance (11–16).
Successful antiviral drugs, in theory, exert their virus-specific effects by interacting with viral receptors, virally encoded enzymes, viral structural components, and viral genes or their transcripts without disturbing cellular metabolism or function. However, at present, no antiretroviral drugs or agents are likely to be completely specific for HIV-1 or to be devoid of toxicity or side effects in the therapy of AIDS. This is a critical issue because patients with AIDS and its related diseases will have to receive antiretroviral therapy for long periods, perhaps for the rest of their lives. Thus, the identification of novel antiretroviral drugs which have greater antiviral activity, delay or prevent the emergence of variants resistant to them, and produce no or minimal side effects remains an important therapeutic objective.
We have been focusing on the design and synthesis of nonpeptidyl PIs that are active against HIV-1 variants resistant to the currently approved HIV-1 PIs. One such anti-HIV-1 agent, darunavir (DRV), which contains a structure-based designed privileged nonpeptidic P2 ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane (bis-THF) (17–19), has been approved as a first-line therapeutic agent for the treatment of individuals infected with HIV-1. In the present work, we examined and characterized a nonpeptidic HIV-1 PI, GRL-10413, which contains a 3-chloro-4-methoxy-phenylmethyl P1 moiety and a hydroxyethylamine sulfonamide isostere (Fig. 1). We found that GRL-10413 exerts strong activity against a wide spectrum of laboratory HIV-1 strains and primary clinical isolates, including multi-HIV-1 PI-resistant variants, with minimal cytotoxicity. We also attempted to select for HIV-1 variants resistant to GRL-10413 by propagating the laboratory wild-type strain HIV-1NL4-3 in MT-4 cells in the presence of increasing concentrations of GRL-10413, and we determined the amino acid substitutions that emerged in the protease region under the pressure of GRL-10413. Recently, we reported a few HIV PIs, GRL-04810, -05010, and -0739, that show good CNS penetration as tested in a blood-brain barrier (BBB) reconstruction model in vitro (20–22); structures and in vitro data for these three GRL-PIs are given in Table 1 and in Fig. S1 and Table S1 in the supplemental material. We therefore determined the CNS penetration properties of GRL-10413 by using the BBB reconstruction system. Finally, we conducted a crystallographic analysis to determine how GRL-10413 interacts with HIV-1 protease.
FIG 1.

Structures of GRL-10413, amprenavir, and darunavir. M.W., molecular weight.
TABLE 1.
Antiviral activity of GRL-10413 against HIV-1LAI and cytotoxicity against MT-2 cellsa
| Compound | EC50 (μM) against HIV-1LAI | CC50 (μM) | Selectivity index (CC50/EC50)b |
|---|---|---|---|
| GRL-10413 | 0.00035 ± 0.00003 | 35.7 ± 1.4 | 102,000 |
| GRL-04810c | 0.0008 ± 0.0002 | 17.5 ± 0.9 | 21,879 |
| GRL-05010c | 0.003 ± 0.001 | 37.0 ± 0.4 | 12,333 |
| GRL-0739c | 0.0019 ± 0.0007 | 21.0 ± 1.6 | 11,053 |
| APV | 0.034 ± 0.009 | 48.2 ± 9.9 | 1,418 |
| ATV | 0.0048 ± 0.0003 | 32.4 ± 1.0 | 6,750 |
| LPV | 0.036 ± 0.003 | 26.7 ± 4.2 | 742 |
| DRV | 0.0056 ± 0.0008 | 100.6 ± 8.8 | 17,964 |
MT-2 cells (104/ml) were exposed to 100 TCID50 of HIV-1LAI and cultured in the presence of various concentrations of each PI, and the EC50s were determined by MTT assay. All assays were conducted in duplicate, and the data shown represent mean values derived from the results of two or three independent experiments.
Each selectivity index denotes the CC50/EC50 ratio against HIV-1LAI.
MATERIALS AND METHODS
Cells and viruses.
MT-2 and MT-4 cells were grown in RPMI 1640-based culture medium supplemented with 10% fetal calf serum (FCS; JRH Biosciences, Lenexa, MD), 50 U/ml penicillin, and 100 μg/ml kanamycin. These cells were obtained from the NCI/NIH. The following HIV-1 strains were employed for the drug susceptibility assay (see below): HIV-1LAI, HIV-1NL4-3, HIV-1ERS104pre (23), clinical HIV-1 strains isolated from drug-naive patients with AIDS, and four HIV-1 clinical strains which were originally isolated from patients with AIDS who had received 9 or 10 anti-HIV-1 drugs over the past 34 to 83 months and which were genotypically and phenotypically characterized as multi-PI-resistant HIV-1 variants (24, 25). All primary HIV-1 strains were passaged once or twice in 3-day-old phytohemagglutinin-activated peripheral blood mononuclear cells (PHA-PBM), and the virus-containing culture supernatants were stored at −80°C until their use as sources of infectious virions.
Antiviral agents.
Roche Products Ltd. (Welwyn Garden City, United Kingdom) kindly provided saquinavir (SQV). Amprenavir (APV) was received as a courtesy gift from GlaxoSmithKline, Research Triangle Park, NC. Lopinavir (LPV) was kindly provided by Japan Energy Inc., Tokyo, Japan. Atazanavir (ATV) was a contribution from Bristol Myers Squibb (New York, NY). 3′-Azido-2′,3′-dioxythymidine (AZT) was purchased from Sigma-Aldrich (St. Louis, MO). Darunavir (DRV) was synthesized as previously described (26). HIV-192UG037 and HIV-197ZA003 were provided by the NIH AIDS Reagent Program.
Drug susceptibility assay.
The susceptibility of HIV-1LAI to various drugs was determined as previously described (19). Briefly, MT-2 cells (104/ml) were exposed to 100 50% tissue culture infective doses (TCID50) of HIV-1LAI in the presence or absence of various concentrations of drugs in 96-well microculture plates and were incubated at 37°C for 7 days. After incubation, 100 μl of the medium was removed from each well, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (10 μl; 7.5 mg/ml in phosphate-buffered saline) was added to each well in the plate, followed by incubation at 37°C for 2 h. After incubation to dissolve the formazan crystals, 100 μl of acidified isopropanol containing 4% (vol/vol) Triton X-100 was added to each well, and the optical density was measured in a kinetic microplate reader (Vmax; Molecular Devices, Sunnyvale, CA). All assays were performed in duplicate. For some experiments, MT-2 cells were chosen as target cells in the MTT assay, since these cells undergo greater HIV-1-elicited cytopathic effects than MT-4 cells. To determine the sensitivity of primary HIV-1 isolates to drugs, PHA-PBM (106/ml) were exposed to 50 TCID50 of each primary HIV-1 isolate and cultured in the presence or absence of various concentrations of drugs in 10-fold serial dilutions in 96-well microculture plates. In determining the drug susceptibility of certain laboratory HIV-1 strains, MT-4 cells were employed as target cells as previously described, with minor modifications. In brief, MT-4 cells (105/ml) were exposed to 100 TCID50 of drug-resistant HIV-1 strains in the presence or absence of various concentrations of drugs and were incubated at 37°C. On day 7 of culture, the supernatants were harvested, and the amounts of p24 (capsid [CA]) Gag protein were determined by using a fully automated chemiluminescence enzyme immunoassay system (Lumipulse F; Fujirebio Inc., Tokyo, Japan) (27, 28). Drug concentrations that suppressed the production of p24 Gag protein by 50% (50% effective concentrations [EC50s]) were determined by comparison with the p24 production level in a drug-free control cell culture. All assays were performed in duplicate. Each compound's EC50 shown in the current report is the average value for the data obtained from two to four independently conducted experiments. PHA-PBM were derived from a single donor for each independent experiment. Thus, two to four different healthy donors were recruited for collection of the data. The research protocol described above was approved by the Ethics Committee for Epidemiological and General Research at the Faculty of Life Sciences, Kumamoto University.
In vitro selection of protease inhibitor-resistant HIV-1 variants.
MT-4 cells (105/ml) were exposed to HIV-1NL4-3 (500 TCID50) and cultured in the presence of various HIV-1 PIs, initially at their EC50s. Viral replication was monitored by determination of the amount of p24 Gag produced by MT-4 cells. The culture supernatants were harvested on day 7 and were used to infect fresh MT-4 cells for the next round of culture in the presence of increasing concentrations of each drug. When the virus began to propagate in the presence of the drug, the drug concentration was increased, generally 2- to 3-fold. Proviral DNA samples obtained from the lysates of infected cells were subjected to nucleotide sequencing. This drug selection procedure was carried out until the drug concentration reached 5 μM, as previously described (29–32). In the experiments for selection of drug-resistant variants, MT-4 cells were also exploited as target cells, since HIV-1 generally replicates at greater levels in MT-4 cells than in MT-2 cells, as described above.
Determination of nucleotide sequences.
Molecular cloning and determination of the nucleotide sequences of HIV-1 strains passaged in the presence of anti-HIV-1 agents were performed as previously described (30). In brief, high-molecular-weight DNAs were extracted from HIV-1-infected MT-4 cells by use of InstaGene matrix (Bio-Rad Laboratories, Hercules, CA) and then subjected to molecular cloning followed by sequence determination. The primers used for the first round of PCR with the entire Gag- and protease-encoding regions of the HIV-1 genome were LTR F1 (5′-GAT GCT ACA TAT AAG CAG CTG C-3′) and PR12 (5′-CTC GTG ACA AAT TTC TAC TAA TGC-3′). The first-round PCR mixture consisted of 1 μl of proviral DNA solution, 10 μl of Premix Taq (Ex Taq version; TaKaRa Bio Inc., Otsu, Japan), and 10 pmol each of the first PCR primers in a total volume of 20 μl. The PCR conditions used were an initial 3 min at 95°C followed by 35 cycles of 40 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C. The first-round PCR products (1 μl) were used directly in the second round of PCR, using primers LTR F2 (5′-GAG ACT CTG GTA ACT AGA GAT C-3′) and KSMA2.1 (5′-CCA TCC CGG GCT TTA ATT TTA CTG GTA C-3′) and the following PCR conditions: an initial 3 min at 95°C followed by 35 cycles of 30 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C. The second-round PCR products were purified with spin columns (MicroSpin S-400 HR columns; Amersham Biosciences Corp., Piscataway, NJ), cloned directly using the pGEM-T Easy vector (Promega, Fitchburg, WI), and subjected to sequencing with a model 3130 automated DNA sequencer (Applied Biosystems, Foster City, CA).
Determination of viral growth kinetics of GRL-10413-resistant HIV-1NL4-3 variants and wild-type HIV-1NL4-3.
HIV-1NL4-3 selected in the presence of GRL-10413 over 50 passages (HIV-110413P50) was propagated in fresh MT-4 cells without GRL-10413 for 7 days, and aliquoted HIV-110413P50 viral stocks were stored at −80°C until use. MT-4 cells (3.2 × 105) were exposed to HIV-110413P50 or a wild-type HIV-1NL4-3 preparation containing 10 ng/ml p24 in 6-well culture plates for 3 h, and the newly infected MT-4 cells were washed with fresh medium, divided into 4 fractions, and each cultured with or without GRL-10413 (final concentration of MT-4 cells, 104/ml; drug concentration, 0, 0.001, 0.01, or 0.1 μΜ). The amounts of p24 were measured every 2 days for up to 7 days.
Determination of BBB Papp of GRL-10413 by use of a novel in vitro model.
A novel in vitro BBB model (BBB kit; PharmaCo-Cell Ltd., Nagasaki, Japan) incorporating a triple culture of rat-derived astrocytes, pericytes, and monkey-derived endothelial cells (33) was used to determine the BBB apparent permeability coefficients (Papp [cm/s]) of GRL-10413, AZT, SQV, APV, ATV, LPV, DRV, caffeine, and sucrose.
The BBB kit was kept at −80°C until thawing on day 0 of the experiments. Nutritional medium was added to both the brain and blood sides of the wells. This solution consists of Dulbecco's modified Eagle's medium (DMEM)–F-12 medium with 10% (vol/vol) FCS, 100 μg/ml heparin, 1.5 ng/ml basic fibroblast growth factor (bFGF), 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 500 nM hydrocortisone, and 50 μg/ml gentamicin. Fresh medium was added 3 h after thawing, following the manufacturer's instructions, and 24 h later. The plates were incubated at 37°C until day 4 of the experiment, when the condition of the astrocytes was checked under a light microscope. Following this, the integrity of the collagen-coated membrane was verified by measurement of the transendothelial electrical resistance (TEER) by use of an ohmmeter. Since the TEER increased over the days of the experiment, reaching optimal values between days 4 and 6 of the experiment, determinations were done during this period. Membranes were tested individually, and collagen-coated membranes displaying TEER values of >150 Ω/cm2 were suitable for execution of the drug BBB penetration assay. Detailed information regarding the components of the BBB kit as well as its mechanisms is available from the manufacturer (PharmaCo-Cell Ltd.).
Once the conditions of cell viability and membrane integrity were met, drug dilutions were performed from 20 mM dimethyl sulfoxide (DMSO) stocks of GRL-10413, AZT, SQV, APV, ATV, LPV, and DRV, while caffeine and sucrose were used as positive and negative controls, respectively. Standard curves were generated for each compound by use of a light spectrophotometer as previously described. Each compound (100 μM) was added to the luminal (blood) side of the wells and incubated at 37°C for 30 min, and then the amount of drug that crossed the in vitro BBB was collected and measured under a light spectrophotometer at 230 nm.
Papp was calculated using the following mathematical formula: Papp (cm/s) = VA/(A × [C]luminal) × Δ[C]abluminal/Δt, where VA is the volume of the abluminal chamber (0.9 cm3), A is the membrane surface area (0.33 cm2), [C]luminal is the initial luminal compound concentration (micromolar), Δ[C]abluminal is the abluminal compound concentration (micromolar), and Δt is the length of the experiment (seconds).
Determination of antiviral activities of GRL-10413 and other anti-HIV-1 drugs recovered from the brain side in the BBB assay.
To evaluate the efficacy of the drugs that successfully crossed the brain interface in the previous BBB assay (indicated by “drugbrain” names), the susceptibility of HIV-1LAI to GRL-10413brain, AZTbrain, DRVbrain, SQVbrain, APVbrain, ATVbrain, LPVbrain, and DRVbrain was determined in an MTT assay employing MT-2 cells as described above for the drug susceptibility assay. Remnants of each drug were recalled from the brain side of the wells, and stocks were generated. The assay was carried out using serially diluted brain-side stocks of compounds.
Expression, purification, and refolding of PRWT.
Expression, purification, and refolding of the wild-type PR (PRWT) protein were performed as described previously (34). Briefly, inclusion bodies isolated from Escherichia coli containing PRWT were extracted with 3 M guanidine HCl (GndHCl) and centrifuged, and the supernatant was loaded on a Sephadex-200 column that was preequilibrated with 4 M GndHCl. Protease-containing fractions were pooled and were further purified by use of a reverse-phase column. Fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the purity was determined to be >95%. Lyophilized PRWT was dissolved in 1 ml of 50% acetic acid solution and then added dropwise to 29 ml of refolding buffer (50 mM sodium acetate, pH 5.2, 5% ethylene glycol, 10% glycerol, 5 mM dithiothreitol, and a 2-fold molar excess of GRL-10413) with stirring on ice. Refolding was continued at 4°C with constant stirring overnight. The refolded protease-drug complexes were concentrated using Amicon filters (3-kDa cutoff) and centrifugation at 4,800 × g. The final protease concentration was determined to be ∼2 mg/ml.
Crystallization of the PRWT–GRL-10413 complex.
The hanging-drop vapor diffusion method was used for cocrystallization. The PRWT–GRL-10413 complex (4 μl) was mixed with 4 μl of well solution per drop. Grid screens, such as ammonium sulfate, sodium chloride, and Quik screens (Hampton Research, CA), were used to obtain preliminary crystallization hits. Cocrystals of PRWT–GRL-10413 were obtained within 1 week at room temperature (298 K). Individual rod-shaped cocrystals of PRWT–GRL-10413 were obtained using ammonium sulfate as the precipitant in 0.1 M citric acid buffer at pH 5.0. The crystals were cryoprotected using their own mother liquor supplemented with 30% glucose and were flash frozen in liquid nitrogen.
X-ray diffraction data collection and processing.
X-ray diffraction data for PRWT–GRL-10413 were collected at Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID (insertion device) (wavelength, 1.0 Å) at the Advanced Photon Source (APS), Argonne National Labs, IL. A Rayonix MX300HS detector was used to record the diffraction data at a distance of 225 mm from the crystal. The exposure time for each frame was 1 s, with a frame width of 0.5°. Diffraction data were processed and scaled at a resolution of 1.8 Å by using HKL2000 (35). X-ray diffraction data processing details are given in Table S3 in the supplemental material.
Crystal structure solutions and refinement.
The phase problem was solved by molecular replacement with the program MOLREP (36) implemented in the CCP4 interface (37, 38), using the PRWT structure (PDB entry 4HLA) as a search model. Structure solutions were directly refined using REFMAC5 (39) through the CCP4 interface. The initial coordinates for GRL-10413 were prepared by modifying the structure of the PI TMC-126, taken from the crystal structure under PDB entry 2I4U. GRL-10413 was fit into the electron density by use of ARP/wARP ligands (40, 41) through the CCP4 interface. The initial refinement library for GRL-10413 was obtained from REFMAC. Solvent molecules were built using the ARP/wARP solvent-building module through the CCP4 interface. After the building of water molecules, the final model was refined using the simulated annealing method from phenix.refine (PHENIX, version 1.9-1692) (42) on the NIH Biowulf Linux cluster. The root mean square deviations of bond lengths and bond angles were improved significantly by generating a geometry-optimized library for GRL-10413 by use of a semiempirical quantum mechanical method of refinement (eLBOW-AM1) (43) during refinement in phenix.refine. Details of the refinement statistics are given in Table S3 in the supplemental material.
Structural analysis.
The final refined structure was used for structural analysis. Hydrogen bonds (H-bonds) were calculated by using cutoff values for distance (maximum distance between donor and acceptor heavy atoms of 3.0 Å) and angles (minimum donor angle of 90° and minimum acceptor angle of 60°). Hydrogen bonds with a distance of >3.0 Å were considered weak interactions. van der Waals (VdW) contacts between two atoms (one from GRL-10413 and one from PRWT) were calculated with a maximum distance cutoff of 3.5 Å.
Accession number(s).
The final refined coordinates for the crystal structure of PRWT in complex with GRL-10413 were deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) under accession number 5KAO.
RESULTS
Antiviral activity and cytotoxicity of GRL-10413 against wild-type HIV-1LAI.
We first examined the antiviral activity of GRL-10413 against a set of HIV-1 isolates. GRL-10413 was highly active against HIV-1LAI, with an EC50 of 0.00035 μM, compared to other clinically available Food and Drug Administration (FDA)-approved HIV-1 PIs examined, including DRV (Table 1), as assessed with the MTT assay using MT-2 target cells, while its cytotoxicity was evident only at high concentrations (50% cytotoxic concentration [CC50] = 35.7 μM). The selectivity index (SI) of GRL-10413 proved to be much more favorable (102,000 as assessed with MT-2 cells and HIV-1LAI) than even that of DRV, which had an SI of 17,964 (Table 1).
GRL-10413 exerts strong activity against highly PI-resistant clinical HIV-1 isolates.
In our previous work, we isolated highly multi-PI-resistant primary HIV-1 strains, HIV-1MDR/B, HIV-1MDR/C, HIV-1MDR/G, and HIV-1MDR/TM, from patients with AIDS who had failed then-existing anti-HIV regimens after receiving 9 or 10 anti-HIV-1 drugs over 34 to 83 months (24, 25). These primary strains contained 11 to 15 amino acid substitutions in the protease region which have reportedly been associated with HIV-1 resistance against various PIs (Table 2). The four different multidrug-resistant clinical isolates used in the assays reported in Table 2 contained various resistance-associated amino acid mutations in the RT (25) as well as the protease. All patients from whom these variants were isolated had received 6 different nucleoside reverse transcriptase inhibitors (NRTIs), and 1 patient had received 1 nonnucleoside reverse transcriptase inhibitor (NNRTI) (25). The potencies of APV, ATV, and LPV against such clinical multidrug-resistant HIV-1 strains were significantly compromised as examined with PHA-PBM as target cells, using p24 production inhibition as an endpoint (Table 2). However, GRL-10413 exerted strong antiviral activity, and its EC50s against these clinical variants were substantially low (0.0018 to 0.002 μM) and were indeed comparable to the EC50 of GRL-10413 against the wild-type clinical isolate HIV-1ERS104pre (0.0018 μM) (Table 2). GRL-10413 was more highly active against the multidrug-resistant clinical HIV-1 variants examined than the four FDA-approved HIV-1 PIs (APV, ATV, LPV, and DRV) were. We also examined the antiviral activity of GRL-10413 against a highly DRV-resistant variant, HIV-1DRVR20P (44). HIV-1DRVR20P was generated using a mixture of 8 highly multi-PI-resistant clinical isolates as a starting HIV-1 source and selection with increasing concentrations of DRV. GRL-10413 maintained its activity against HIV-1DRVR20P (EC50 = 0.0041 μM), although DRV lost its activity (73-fold less activity) (Table 2). Additionally, we determined the antiviral activity of GRL-10413 against an R5-tropic subtype A strain or subtype C strain in PHA-PBM, as shown in Table S2 in the supplemental material. GRL-10413 also effectively inhibited the replication of all such strains examined.
TABLE 2.
Antiviral activity of GRL-10413 against multidrug-resistant clinical isolates in PHA-PBM
| Virusa | EC50 (μM) (fold change)b |
||||
|---|---|---|---|---|---|
| GRL-10413 | APV | ATV | LPV | DRV | |
| HIV-1ERS104pre (wild type) | 0.0018 ± 0.0005 | 0.031 ± 0.011 | 0.0027 ± 0.0002 | 0.037 ± 0.005 | 0.0041 ± 0.0007 |
| HIV-1MDR/B | 0.0018 ± 0.0004 (1) | 0.50 ± 0.08 (16) | 0.46 ± 0.01 (170) | >1 (>27) | 0.026 ± 0.001 (6) |
| HIV-1MDR/C | 0.0019 ± 0.0004 (1) | 0.39 ± 0.01 (13) | 0.052 ± 0.016 (19) | 0.25 ± 0.17 (7) | 0.014 ± 0.002 (3) |
| HIV-1MDR/G | 0.0015 ± 0.0008 (1) | 0.48 ± 0.08 (15) | 0.034 ± 0.006 (13) | 0.36 ± 0.27 (10) | 0.023 ± 0.009 (6) |
| HIV-1MDR/TM | 0.002 ± 0.002 (1) | 0.34 ± 0.11 (11) | 0.16 ± 0.02 (59) | 0.42 ± 0.02 (11) | 0.014 ± 0.002 (3) |
| HIV-1DRVR20P | 0.0041 ± 0.0005 (2) | >1 (>32) | >1 (>370) | >1 (>27) | 0.30 ± 0.02 (73) |
The amino acid substitutions identified in the protease region compared to the consensus type B sequence cited from the Los Alamos database include the following: L63P in HIV-1ERS104pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1MDR/B; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, and L89M in HIV-1MDR/C; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90M in HIV-1MDR/G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, and I93L in HIV-1MDR/TM; and L10I, I15V, K20R, L24I, V32I, M36I, M46L, L63P, A71T, V82A, and L89M in HIV-1DRVR20P. HIV-1ERS104pre served as a source of wild-type HIV-1.
The EC50 values were determined by using PHA-PBM as target cells, and the inhibition of p24 Gag protein production by each drug was used as the endpoint. The numbers in parentheses represent the fold changes of EC50s for each isolate compared to the EC50s for HIV-1ERS104pre. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (±1 standard deviation [SD]) derived from the results of two to four independent experiments. PHA-PBM were derived from a single donor for each independent experiment.
GRL-10413 is active against various PI-selected laboratory HIV-1 variants.
We also examined GRL-10413 against an array of HIV-1NL4-3 variants which had been selected by propagating HIV-1NL4-3 in the presence of increasing concentrations (up to 5 μM) of each of 3 FDA-approved HIV-1 PIs (ATV, LPV, and APV) in MT-4 cells (22, 29). Such variants had acquired various HIV-1 PI resistance-associated amino acid substitutions encoded in the protease-encoding region of the viral genome (Table 3). Each variant was highly resistant to the PI by which the variant was selected and showed significant resistance, with an EC50 of >1 μM. GRL-10413 was active against all such variants, with EC50s of 0.0021 to 0.0023 μM (Table 3). Overall, GRL-10413 exerted significantly more favorable antiviral activity against various wild-type HIV-1 strains and drug-resistant HIV-1 variants than those of other, conventional HIV-1 PIs (Tables 1 to 3).
TABLE 3.
Antiviral activity of GRL-10413 against highly conventional-PI-resistant laboratory variants
| Virusa | EC50 (μM) (fold change)b |
||||
|---|---|---|---|---|---|
| GRL-10413 | APV | ATV | LPV | DRV | |
| HIV-1NL4-3 | 0.00037 ± 0.00001 | 0.029 ± 0.006 | 0.002 ± 0.001 | 0.014 ± 0.009 | 0.0034 ± 0.0002 |
| HIV-1ATVR5μM | 0.0023 ± 0.0001 (6) | 0.37 ± 0.06 (13) | >1 (>500) | 0.69 ± 0.05 (49) | 0.030 ± 0.08 (9) |
| HIV-1LPVR5μM | 0.0023 ± 0.0001 (6) | >1 (>34) | 0.041 ± 0.006 (21) | >1 (>71) | 0.034 ± 0.004 (10) |
| HIV-1APVR5μM | 0.0021 ± 0.0001 (6) | >1 (>34) | 0.42 ± 0.06 (210) | >1 (>71) | 0.42 ± 0.01 (124) |
The amino acid substitutions identified in the protease region compared to that of wild-type HIV-1NL4-3 include the following: L23I, E34Q, K43I, M46I, I50L, G51A, L63P, A71V, V82A, and T91A in HIV-1ATVR5μM; L10F, M46I, I54V, and V82A in HIV-1LPVR5μM; and L10F, V32I, M46I, I54 M, A71V, and I84V in HIV-1APVR5μM.
The EC50 values were determined by using MT-4 cells as target cells. MT-4 cells (105/ml) were exposed to 100 TCID50 of each HIV-1 strain, and the inhibition of p24 Gag protein production by each drug was used as the endpoint. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (±1 SD) derived from the results of two to four independent experiments.
In vitro selection of HIV-1 variants resistant to GRL-10413.
We next attempted to select HIV-1 variants resistant to GRL-10413 by propagating a laboratory HIV-1 strain (HIV-1NL4-3) in MT-4 cells in the presence of increasing concentrations of GRL-10413, as previously described (29). HIV-1NL4-3 was initially exposed to 0.0005 μM GRL-10413 and underwent 50 passages, after which it was found to have acquired only a 6-fold increase (0.003 μM) in GRL-10413 concentration compared to that at the initiation of the selection. Compared to the kinetics of the emergence of variants resistant to APV, the emergence of GRL-10413-resistant variants was apparently significantly delayed (Fig. 2). The protease-encoding region of the proviral DNA isolated from infected MT-4 cells was cloned and sequenced at passages 10, 20, 30, 40, and 50 under GRL-10413 selection. The sequences of the cloned region and the percent frequency of identical sequences at each passage are depicted in Fig. 3. All clones examined at 10, 20, 30, 40, and 50 passages had a G16E substitution. The percentages of clones containing the G16E substitution alone were 86% (12 of 14 clones), 54% (7 of 13 clones), 62% (8 of 13 clones), 74% (14 of 19 clones), and 74% (28 of 38 clones) at 10, 20, 30, 40, and 50 passages, respectively. Some sporadic substitutions were noted, but no accumulation of substitutions was identified except for G16E, as shown in Fig. 3. The locations of G16 in the protease (PR) dimer are illustrated in Fig. S2 in the supplemental material. Additionally, HIV-1 selected with GRL-10413 at passage 50 acquired the following Gag amino acid substitutions: P48H in the matrix (p17) region, M68I, G116E, and P123T in the capsid (p24) region. None of them were at or near the Gag cleavage site (see Fig. S3 in the supplemental material).
FIG 2.

In vitro selection of HIV-1 protease inhibitor-resistant HIV-1 variants. HIV-1NL4-3 was propagated in MT-4 cells in the presence of increasing concentrations of APV (■), LPV (△), ATV (▲), DRV (○), or GRL-10413 (●). Each passage of virus was conducted in a cell-free manner. The p24 concentrations of GRL-10413 selection culture supernatants from passages 10, 20, 30, 40, and 50 were 206, 397, 360, 189, and 361 ng/ml, respectively. *, the data on LPV, ATV, and DRV are from references 29 and 30 and are shown for comparison.
FIG 3.

Amino acid sequences of the protease regions of HIV-1NL4-3 variants selected in the presence of GRL-10413. The amino acid sequence of protease deduced from the nucleotide sequence of the protease-encoding region of each proviral DNA isolated at each indicated time is shown. The amino acid sequence of the wild-type HIV-1NL4-3 protease is illustrated at the top as a reference.
HIV-110413P50 remains sensitive to GRL-10413 and other conventional PIs.
Since the viral growth kinetics of HIV-110413P50 was thought to be reasonably well maintained despite the presence of GRL-10413, as mentioned above, we determined the viral growth kinetics of HIV-110413P50 and HIV-1NL4-3. As shown in Fig. 4A, HIV-1NL4-3 failed to grow fully in the presence of as little as 0.001 μM GRL-10413 during the entire culture period of 7 days, and the amounts of p24 produced in the culture medium were almost half the amount without GRL-10413 on days 5 and 7. However, the growth of HIV-110413P50 in the presence of 0.001 μM GRL-10413 was comparable to that in the absence of GRL-10413, and the amounts of p24 produced in the culture medium in the presence of 0.001 μM GRL-10413 were the same with and without the compound on day 7 (Fig. 4B). These data strongly suggest that the long-term GRL-10413-selected strain HIV-110413P50 is still susceptible to GRL-10413. Moreover, we determined the antiviral activities of various conventional PIs against HIV-110413P50. All tested conventional PIs also proved to be active against HIV-110413P50 (Table 4). Taken together, the data strongly suggest that GRL-10413 does not allow HIV-1 to acquire resistance to GRL-10413 or other conventional PIs, in line with the observation that no specific amino acid substitutions were seen over 50 passages of selection with GRL-10413 (Fig. 3).
FIG 4.

Viral growth kinetics of HIV-1NL4-3 and HIV-110413P50. MT-4 cells (3.2 × 105) were exposed to an HIV-1NL4-3 (A) or HIV-110413P50 (B) preparation containing 10 ng/ml p24 in 6-well culture plates for 3 h and then washed with fresh medium, divided into 4 fractions, and cultured with or without GRL-10413 (final concentration of MT-4 cells, 104/ml; drug concentration, 0, 0.001, 0.01, or 0.1 μM). The amount of p24 in each culture flask was measured every 2 days for up to 7 days, once at each time point. All p24 values are single point determinations. HIV-110413P50 contains a G16E substitution in the PR region, a P48H substitution in the Gag matrix (p17) region, and M68I, G116E, and P123T substitutions in the Gag capsid (p24) region.
TABLE 4.
Antiviral activities of GRL-10413 and various PIs against a GRL-10413-selected HIV-1 variant, HIV-11041350P
| Virus | EC50 (μM) (fold change)a |
|||||
|---|---|---|---|---|---|---|
| SQV | APV | LPV | ATV | DRV | GRL-10413 | |
| HIV-1NL4-3 | 0.019 ± 0.001 | 0.029 ± 0.006 | 0.014 ± 0.009 | 0.002 ± 0.001 | 0.0034 ± 0.0002 | 0.00037 ± 0.00001 |
| HIV-11041350P | 0.015 ± 0.002 (1) | 0.027 ± 0.003 (1) | 0.030 ± 0.004 (2) | 0.0045 ± 0.0007 (2) | 0.007 ± 0.005 (2) | 0.0023 ± 0.0005 (6) |
MT-4 cells (104) were exposed to 100 TCID50 of each HIV-1 isolate, and the inhibition of p24 Gag protein production by each drug was used as the endpoint. The numbers in parentheses represent the fold changes of EC50s for HIV-11041350P compared to the EC50s for HIV-1NL4-3. All assays were conducted in duplicate, and the data shown represent mean values (±1 SD) derived from the results of two to four independent experiments.
GRL-10413 effectively penetrates the blood-brain barrier as tested in an in vitro assay.
We also attempted to evaluate whether GRL-10413 had optimal BBB apparent permeability coefficients by employing an in vitro model using a triple-cell coculture system with rat astrocytes and pericytes and monkey endothelial cells. This model (BBB kit; PharmaCo-Cell Ltd.) is thought to represent an in vitro BBB model for drug transport assays, permitting adequate cross talk of the cell lines involved and providing a way to test the apparent passage of small molecules across the BBB, as previously described by Nakagawa et al. (33). GRL-10413 (100 μM) was added to the luminal interface (blood side) of microtiter culture wells under the optimal conditions for TEER determination. The concentration of each compound that permeated into the abluminal interface (brain side) was determined using a spectrophotometer 30 min after the addition of each drug to the blood-side wells. As shown in Table 4, the amounts of caffeine and sucrose, serving as the most and least lipophilic substances, in the abluminal interface were 5.04 and 0.07 μM, respectively. Six conventional anti-HIV-1 drugs, AZT, SQV, APV, LPV, ATV, and DRV, were also used as controls in the assay, giving concentrations of 1.05, 0.33, 0.70, 0.95, 1.02, and 0.65 μM, respectively. GRL-10413 yielded the highest concentration (1.4 μM) in the abluminal interface of the microtiter culture wells among the compounds we tested (Table 5).
TABLE 5.
Determination of BBB apparent permeability coefficients of GRL-10413 and other agents by use of a novel in vitro modela
| Compound | Class | Initial luminal tracer concn (μM) | Final abluminal tracer concn (μM) | Papp (10−6 cm/s) |
|---|---|---|---|---|
| AZT | NRTI | 100 | 1.05 ± 0.06 | 15.8 ± 0.9 |
| SQV | PI | 100 | 0.33 ± 0.03* | 4.9 ± 0.4* |
| APV | PI | 100 | 0.70 ± 0.14 | 10.6 ± 2.1 |
| LPV | PI | 100 | 0.95 ± 0.07 | 14.4 ± 1.1 |
| ATV | PI | 100 | 1.02 ± 0.10* | 15.4 ± 1.4* |
| DRV | PI | 100 | 0.65 ± 0.23* | 9.9 ± 4.2* |
| GRL-10413 | PI | 100 | 1.40 ± 0.21 | 21.1 ± 3.1 |
| Caffeine (positive control) | 100 | 5.04 ± 0.16 | 76.4 ± 2.4 | |
| Sucrose (negative control) | 100 | 0.07 ± 0.03 | 1.1 ± 0.4 |
In an in vitro model using a triple coculture of rat astrocytes, pericytes, and monkey endothelial cells, AZT, SQV, APV, ATV, LPV, DRV, GRL-10413 (all at 100 μM), and the positive and negative controls (caffeine and sucrose) were added to the luminal interface (blood side) of duplicate wells. The mathematical formula used for the calculation of Papp is given in Materials and Methods. Results show average values ± 1 SD for duplicate determinations. *, our previously reported data.
The apparent permeability coefficient (Papp), referred to as the brain uptake index (BUI), is a way to determine the penetration efficiency of a compound across a BBB model quantitatively and qualitatively (35). The Papp value of GRL-10413 (21.1 × 10−6 cm/s) was greater than that of DRV (9.9 × 10−6 cm/s) and those of the other antiviral drugs tested, i.e., AZT (15.8 × 10−6 cm/s), SQV (4.9 × 10−6 cm/s), APV (10.6 × 10−6 cm/s), LPV (14.4 × 10−6 cm/s), and ATV (15.4 × 10−6 cm/s) (Table 5). Compounds with apparent permeability coefficients of >20 × 10−6 cm/s are thought to have reasonably efficient penetration across the BBB, those with values of 10 × 10−6 to 20 × 10−6 cm/s are thought to have a moderate degree of penetration, and those with values of <10 × 10−6 cm/s are thought to not effectively penetrate the BBB (34).
GRL-10413 recovered from the brain interface in the BBB model retained antiviral activity compared to that of DRV.
In order to test the antiviral activity of the drugs used in the BBB assay that were able to penetrate the barrier to the brain side of the kit, we generated drug stocks designated GRL-10413brain, DRVbrain, AZTbrain, SQVbrain, APVbrain, ATVbrain, and LPVbrain. We used the standard protocol for the drug susceptibility MTT assay as described in Materials and Methods. Serial dilutions (20-, 200-, and 2,000-fold) were performed using MT-2 cells and the viral strain HIV-1LAI. Note that the antiviral activity of GRL-10413 was superior to that of DRV and the rest of the tested drugs. GRL-10413brain was able to suppress 89% of HIV-1LAI at the 200× dilution, while the other control drugs, DRVbrain, AZTbrain, SQVbrain, APVbrain, ATVbrain, and LPVbrain, suppressed less than 20% of the viral replication at the 20× dilution (Fig. 5A). We also performed the antiviral assay using different dilutions (10-, 50-, 100-, 500-, 1,000-, and 5,000-fold) for GRL-10413brain and DRVbrain (Fig. 5B). DRVbrain inhibited 50% of viral replication only at the 10× dilution, while GRL-10413brain showed the same antiviral effect at the 500× dilution, strongly suggesting that GRL-10413 penetrated the in vitro BBB more effectively than DRV did (Fig. 5B).
FIG 5.

Antiviral activities of GRL-10413brain, AZTbrain, SQVbrain, APVbrain, ATVbrain, LPVbrain, and DRVbrain against HIV-1LAI. Brain-side stocks of the compounds employed in the in vitro BBB assay were generated for use in downstream viral inhibition assays. The brain-side stocks, termed GRL-10413brain, AZTbrain, DRVbrain, ATVbrain, LPVbrain, and SQVbrain, were tested with the wild-type laboratory strain HIV-1LAI by MTT assay. (A) Percent inhibition of serial dilutions (20-, 200-, and 2,000-fold) of brain-side stocks. (B) We also examined antiviral activity by using different dilutions (10-, 50-, 100-, 500-, 1,000-, and 5,000-fold) for GRL-10413brain and DRVbrain.
X-ray crystallographic analysis of PRWT in complex with GRL-10413.
The X-ray crystal structure of PRWT in complex with GRL-10413 was solved in the space group P212121, with one protease dimer per asymmetric unit (RCSB PDB accession number 5KAO). The binding mode and structural interactions are shown in Fig. 6. In order to analyze the hydrogen bonds (H-bonds) and van der Waals (VdW) contacts, hydrogen atoms were added, and their orientations were optimized by sampling the crystallographic water molecules through the protein preparation wizard in Maestro (v9.0; Schrodinger LLC). GRL-10413 shows multiple H-bonds and VdW contacts with PRWT in the active site. As shown in Fig. 6B, the P2 bis-THF moiety of GRL-10413 shows three strong H-bonds: two with the backbone amide hydrogen atom of D29 (interatomic distances of 1.9 Å and 2.6 Å) and one with the backbone amide hydrogen atom of D30 (interatomic distance of 2.2 Å). One strong H-bond was seen with the backbone carbonyl oxygen atom of G27 (interatomic distance of 2.2 Å). The transition-state-mimic hydroxyl group of GRL-10413 showed at least one strong H-bond each with the side chain δ-oxygen atoms of D25 and D25′ (with interatomic distances ranging from 1.5 Å to 2.4 Å). The oxygen atom from the P2′ methoxybenzene moiety of GRL-10413 showed one strong H-bond with the backbone amide hydrogen atom of D30′ (interatomic distance of 2.4 Å). One conserved crystallographic water molecule was seen bridging GRL-10413 and the backbone amide hydrogen atoms of I50 and I50′ (with interatomic distances ranging from 1.8 Å to 2.1 Å). The overall profile of VdW contact for GRL-10413 in the active site of PRWT looks similar to that of darunavir (DRV) (PDB entry 4HLA). There was strong electron density for GRL-10413, as evident from the 2|Fo|-|Fc| density shown in Fig. 7. There was also strong density for the chlorine atom in two distinct locations, as seen in the 2|Fo|-|Fc| map (Fig. 7B). Hence, the P1 moiety was fit at two alternate positions. After refinement, the relative occupancies were determined to be 0.55 (B factor = 24.49 Å2) and 0.45 (B factor = 23.84 Å2) (Fig. 7B). In fact, when the P1 moiety was fitted in only one orientation with full occupancy, the B factor for the chlorine atom (after refinement) was found to be almost 40 Å2. Note that in the major configuration (occupancy of 0.55), the chlorine atom forms a structurally significant bidentate halogen bond with the positively charged guanidinium group of R8′, as shown in Fig. 8. Moreover, in the minor configuration (occupancy of 0.45), the chlorine atom is directed toward the amide plane formed between G49 and I50. A statistical analysis of halogen bonding indicated that glycine is the most common residue taking part in halogen–main-chain interactions (45). Instances of significant chlorine-arginine halogen bonding were also reported for the inhibition of hepatitis C virus polymerase (46).
FIG 6.
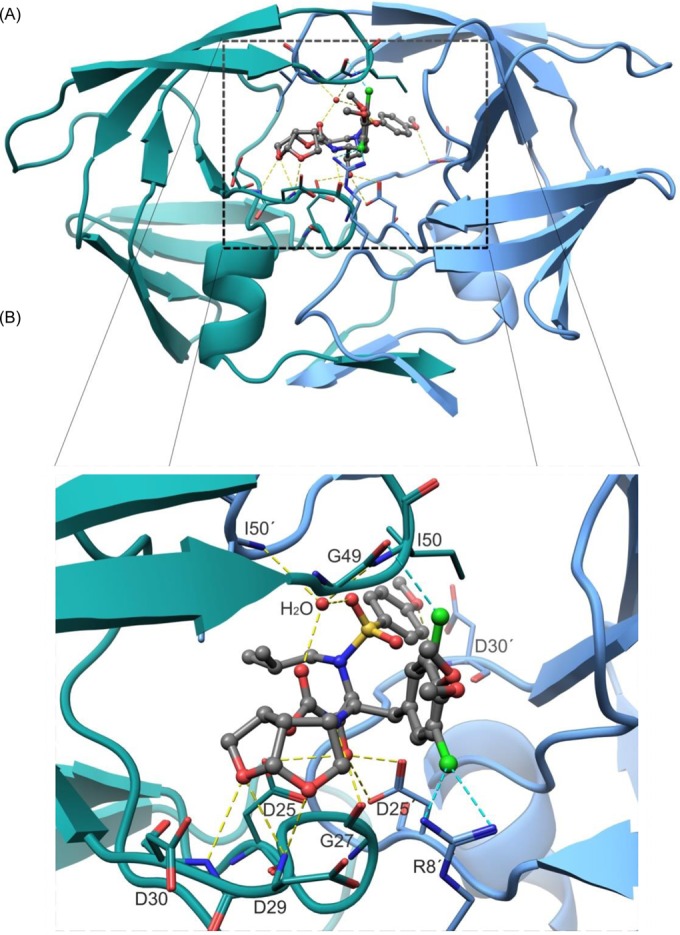
Overall structural presentation (A) and detailed views of ligand binding sites (B). (A) Structure of the PRWT/GRL-10413 complex in cartoon mode. The subunits comprising HIV-1 protease are depicted in blue and green and form a tunnel-shaped binding pocket that sequesters the inhibitor from the exterior. (B) Zoomed view of the binding pocket. GRL-10413 forms multiple hydrogen bonds (H-bonds) with PRWT. The protease side chains are shown as wire models, and GRL-10413 is shown in ball-and-stick mode (gray). The H-bonds are shown as yellow dashed lines, and halogen bonds are shown as cyan dashed lines. The backbone atoms of protease are shown in green or blue, and those of GRL-10413 are shown in gray. The nitrogen, oxygen, and sulfur atoms are shown in blue, red, and yellow, respectively. Protease residues are labeled numerically from 1 to 99 and 1′ to 99′ for subunits 1 and 2 of the protease dimer, respectively. The P2 bis-THF moiety of GRL-10413 accepts three strong H-bonds: one from the backbone amide hydrogen atom of D30 and two from the backbone amide hydrogen atom of D29. One direct H-bond is seen with the backbone carbonyl oxygen atom of G27. The hydroxyl group of GRL-10413 shows one and two H-bonds with the δ-oxygen atoms from the side chains of D25 and D25′, respectively. The P2′ methoxybenzene moiety of GRL-10413 forms one H-bond with the backbone amide hydrogen atom of D30′. A conserved water molecule is seen bridging H-bonds between GRL-10413 and the backbone amide hydrogen atoms of I50 and I50′.
FIG 7.

Electron density maps of GRL-10413. (A and B) 2|Fo|-|Fc| electron density maps for GRL-10413, at a contour level of 2.0 σ. In both panels, GRL-10413 is shown in stick representation, with carbon, nitrogen, oxygen, and chlorine atoms shown in gray, blue, red, and green, respectively. Panel B shows a magnified image of the P1 moiety of GRL-10413 with alternate conformations of the chlorine atom after structure refinement. The occupancy of the chlorine atom pointing toward the right is 0.55 (B factor = 24.49 Å2), and that of the chlorine atom pointing toward the left is 0.45 (B factor = 23.84 Å2).
FIG 8.

Halogen bonding of the P1 moiety of GRL-10413 with PRWT. (A) The P1 moiety (top view) is shown in ball-and-stick mode with two different conformations within the binding pocket of PRWT. Distances (cyan dashed lines) are given in angstroms. While in one conformation (black) the chlorine atom forms a bidentate halogen bond with R8′, in the other conformation (gray) the chlorine atom forms a halogen bond with the carbonyl group of G49. (B) Side view of the P1 moiety, with halogen bonds and their corresponding angles highlighted. Protease residues L23′, V82′, and P81′ are shown in the upper part and are involved in multiple van der Waals (VdW) contacts with the P1 moiety. Those interactions apparently lock the P1 moiety in one stable conformation.
Halogen bond formation between the meta-chlorine of the P1 moiety and PRWT residues is shown in Fig. 8. The distances of the bifurcated halogen bonds formed between C-Cl and the η1- and η2-nitrogen moieties of R8′ are both 3.3 Å, with angles of 169° and 145.8°, respectively. On the opposite side, the C-Cl—O halogen bond is 3.3 Å long, with an angle of 134.5°. Although in ideal halogen bonds the angles are close to 180°, for interactions in the protein environment the chlorine-mediated halogen angles were found to be distributed in the range of 120° to 180° (55). The hydrogen atoms from the Cβ and Cγ atoms of the P81 side chain were also found to be within interatomic distances of 2.9 Å and 3.2 Å, respectively, from the oxygen atom of the P1 moiety of GRL-10413. Taken together, these observations show that GRL-10413 has an improved binding profile in the active site of PRWT compared to that of DRV (PDB entry 4HLA), thus supporting GRL-10413's greater antiviral activity.
DISCUSSION
GRL-10413, which contains a 3-chloro-4-methoxybenzene P1 moiety and a sulfonamide isostere, suppressed the replication of wild-type HIV-1 and HIV-2, with extremely low EC50s (Table 1). GRL-10413 maintained its favorable antiviral activity against a variety of multidrug-resistant clinical HIV-1 isolates, with EC50s ranging from 0.0015 to 0.002 μM, while the existing FDA-approved HIV-1 PIs examined either failed to suppress the replication of those isolates or required much higher concentrations for reasonable viral inhibition (Table 2). GRL-10413 also inhibited the replication of HIV-1 PI-selected HIV-1 laboratory variants, with low EC50s (Table 3). Previously reported GRL-labeled PIs (30–32), including DRV, were generated based on the structure of APV, so they were generally less active against APV-resistant HIV-1 variants. However, GRL-10413 effectively inhibited the replication of HIV-1APVR5 μM, with an EC50 of 0.0021 μM (6-fold difference from the EC50 against wild-type HIV-1), indicating that GRL-10413 should have a unique antiviral feature compared to such previously reported GRL-labeled PIs.
It should be noted that the population size of HIV-1 in the present selection culture system is relatively small, that the appearance of mutations can relatively easily be affected by stochastic phenomena, and that rates of appearance of mutations in culture may not be reliable. The profiles of development of drug-resistant HIV-1 variants can be defined only when the very drug is administered to HIV-1-infected individuals in carefully monitored clinical research. However, in our HIV-1NL4-3 selection experiment using GRL-10413, the emergence of GRL-10413-resistant variants was significantly delayed compared to the analogous case with APV selection. HIV-1 cultured with GRL-10413 for a long period acquired solely a G16E substitution in the PR region, strongly suggesting that GRL-10413 has a significantly high genetic barrier against the emergence of HIV-1 drug resistance. Reportedly, APV-resistant HIV-1 variants contain V32I, I50V, I54L/M, L76V, I84V, and L90M substitutions (47, 48). However, no such APV resistance-associated amino acid substitutions emerged during the present GRL-10413 selection (Fig. 2 and 3). V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, and L89V are known as DRV resistance-associated substitutions. However, none of these substitutions were observed in our long-term selection with GRL-10413, suggesting that the resistance profiles for DRV and GRL-10413 are distinctly different. It is particularly noteworthy that the A28S amino acid substitution did not emerge in the present selection experiment with GRL-10413. In our previous studies, selection experiments with strong HIV-1 PIs, such as TMC-126 and GRL-1398, containing a paramethoxy group as the P2′ moiety, led to the selection of resistant variants with the A28S substitution (24, 31). Intriguingly, when HIV-1NL4-3 was selected with GRL-0519, which contains the same paramethoxy group in the P2′ site, the A28S substitution did not appear in passages up to passage 37 (29). GRL-0519 has tris-tetrahydrofuranylurethane (tris-THF) as the P2 ligand and the paramethoxy moiety at the P2′ site, suggesting that the presence of tris-THF prevented the selection of the A28S substitution. In this regard, the combination of the 3-chloro-4-methoxy-attached P1 moiety and the para-methoxy moiety at P2′ in GRL-10413 potentially prevented the selection of the A28S substitution.
Considering that the EC50s of GRL-10413 are favorably low (Tables 1 to 3) and that GRL-10413's selectivity index of 102,000 is considerably higher than those of the other conventional HIV-1 PIs examined in this study (Table 1), both the anti-HIV activity and safety of GRL-10413 may be desirable, although the efficacy and emergence of adverse effects should ultimately be tested by controlled clinical trials.
As described above, GRL-10413 inhibited the replication of wild-type HIV-1 and various drug-resistant HIV-1 variants at lower concentrations than those required for DRV. To clarify the reason for such differences, we performed a structural analysis using the crystal data for HIV-1 protease–GRL-10413 or –DRV complexes. The P2 moiety of GRL-10413 was seen to form a strong hydrogen bond network with backbone atoms of D29 and D30. In addition, as illustrated in Fig. 8, the modified P1 moiety of GRL-10413 has greater VdW contacts with more protease active site amino acids than the P1 phenyl ring of DRV. Such greater VdW (hydrophobic) contacts of GRL-10413 should be responsible for GRL-10413's higher anti-HIV-1 activity than that of DRV. In addition, the modified P1 moiety possibly confers on GRL-10413 desirable BBB penetration properties as well, considering that GRL-10413's P1 moiety is significantly different from DRV's P1 moiety (Fig. 1), although further clarification of the structure-activity relationships is required.
Recently, the impact of halogen bonding on favorable protein-ligand interactions has received increased attention, with several successful applications (49, 50). In particular, fluorine and chlorine substitutions not only enhance binding affinity and specificity but also facilitate membrane permeability and metabolic stability (51, 52). Halogen–main-chain interactions have been found to be more common (∼65%) than halogen–side-chain interactions (∼35%) (45). In the present structural study of GRL-10413 complexed with HIV-1 protease, the halogen bond between a chlorine atom and the G49 residue was formed via the backbone carbonyl. In the higher-occupancy configuration, the chlorine in the P1 moiety interacted with the R8′ residue.
Zidovudine (ZDV) is the only ARV agent which has been shown to be effective for the treatment of certain HIV-1-associated CNS abnormalities. Considering that HIV-1 causes various CNS abnormalities, ranging from HIV-1-associated neurocognitive disorders (HAND) and the milder but also serious presentation of HIV-1-associated minor cognitive/motor disorder (MCMD) to the devastating HIV-1-associated encephalopathy, more effective ARV regimens with agents exerting efficient penetration of the BBB are urgently needed. In this regard, cell culture-based models have greatly contributed to the understanding of the physiology, pathology, and pharmacology of the blood-brain barrier (33). In fact, certain in vitro BBB models have proven to serve as useful tools that permit estimations of the apparent penetration of molecules into the CNS. A well-characterized in vitro BBB cell model may also provide a valuable tool for studying mechanistic aspects of transport as well as biological and pathological processes related to the BBB (53). For any in vitro BBB cell model to be used successfully, it needs to fulfill a number of criteria, such as reproducible permeation by reference compounds, good screening capacity, the display of complex tight junctions, adequate expression of BBB phenotypic transporters, and transcytotic activity. The BBB model employed in the present study complies with all of these parameters and provides an additional advantage by incorporating a trilayer of cells consisting of astrocytes, pericytes, and brain endothelial cells, thus increasing its anatomical and physiological reliability. Molecules and compounds that reach Papp values of >20 × 10−6 cm/s are deemed to be favorable in terms of relative penetration across the BBB. Conversely, those that display values between 2 × 10−6 and 10 × 10−6 cm/s are defined as compounds with low BBB penetration. We determined the BBB penetration of various currently available anti-HIV-1 drugs (partial data are displayed in Table 5) by using the in vitro BBB model, and GRL-10413 showed favorable features suggesting potentially favorable penetration ability across the BBB compared to DRV and other anti-HIV-1 drugs examined in the present study, including AZT, IDV, SQV, and ATV. In the present work, GRL-10413 showed the highest Papp value (21.1 × 10−6 cm/s) among the anti-HIV-1 drugs examined (Table 5). The brain-side stocks of GRL-10413 inhibited the replication of HIV-1LAI more potently than those of other conventional anti-HIV-1 drugs, suggesting that GRL-10413 may effectively suppress the replication of HIV-1 in a “sanctuary” CNS and control chronic inflammation, which is thought to be a primary factor in the onset and progression of HAND/MCMD induced by HIV-1 infection in the CNS.
In conclusion, the present data demonstrate that GRL-10413 has desirable features as a candidate drug for treating patients infected with wild-type and/or multidrug-resistant HIV-1 variants and that the newly generated modified P1 moiety containing O-methoxy and chlorine groups combined with P2 bis-THF and P2′ methoxybenzene moieties should be critical for the strong binding of GRL-10413 to HIV-1 protease. Note that in the MONET trial, recently conducted in order to reduce the toxicities and costs of current cART (54), although switching from therapy with DRV/r plus 2 NRTIs (DRV/r-plus-2NRTI therapy) to DRV/r monotherapy showed noninferior efficacy among patients with HIV-1 RNA levels of <50 copies/ml at baseline, HIV-1 RNA elevations were seen by week 144 in 16% and 11% of patients receiving DRV/r monotherapy and DRV/r-plus-2NRTI therapy, respectively, suggesting that DRV/r monotherapy may have a less potent efficacy than that of DRV/r-plus-2NRTI therapy. In this regard, GRL-10413, which has stronger and more favorable antiviral profiles than those of DRV, may serve as a potential candidate for PI monotherapy. GRL-10413 possesses a number of desirable features as a candidate drug for HIV-1 infection and AIDS, although various other parameters, including oral bioavailability, pharmacokinetics/pharmacodynamics, and biodistribution, remain to be determined, and further investigation is warranted.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a Grant for Development of Novel Drugs for Treating HIV-1 Infection and AIDS from the Japan Agency for Medical Research and Development (AMED) and the Ministry of Health, Welfare, and Labor of Japan, a grant from the National Center for Global Health & Medicine, a grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re-Emerging Infectious Diseases (Renkei Jigyo no. 78; Kumamoto University) of Monbu-Kagakusho (H.M.), a grant from the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.), and a grant from the National Institutes of Health (GM53386) (A.K.G.). We thank the Southeast Regional Collaborative Access Team (SER-CAT), Advanced Photon Source (APS) for X-ray diffraction data collection. Supporting institutions for the SER-CAT may be found at www.ser-cat.org. Use of APS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract W-31-109-Eng-38.
This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://hpc.nih.gov). We also appreciate Miho Matsumoto for her technical support.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01428-16.
REFERENCES
- 1.Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F. 2011. The effect of highly active antiretroviral therapy on the survival of HIV-infected children in a resource-deprived setting: a cohort study. PLoS Med 8:e1001044. doi: 10.1371/journal.pmed.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohse N, Hansen AB, Gerstoft J, Obel N. 2007. Improved survival in HIV-infected persons: consequences and perspectives. J Antimicrob Chemother 60:461–463. doi: 10.1093/jac/dkm241. [DOI] [PubMed] [Google Scholar]
- 3.Mitsuya H, Maeda K, Das D, Ghosh AK. 2008. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv Pharmacol 56:169–197. doi: 10.1016/S1054-3589(07)56006-0. [DOI] [PubMed] [Google Scholar]
- 4.Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackman BR, Sax PE, Weinstein MC, Freedberg KA. 2006. The survival benefits of AIDS treatment in the United States. J Infect Dis 194:11–19. doi: 10.1086/505147. [DOI] [PubMed] [Google Scholar]
- 5.UNAIDS. 2013. 2013 report on the global AIDS epidemic. http://www.unaids.org/en/resources/campaigns/globalreport2013/globalreport.
- 6.De Clercq E. 2002. Strategies in the design of antiviral drugs. Nat Rev Drug Discov 1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 7.Siliciano JD, Siliciano RF. 2004. A long-term latent reservoir for HIV-1. J Antimicrob Chemother 54:6–9. doi: 10.1093/jac/dkh292. [DOI] [PubMed] [Google Scholar]
- 8.Simon V, Ho DD. 2003. HIV-1 dynamics in vivo: implications for therapy. Nat Rev Microbiol 1:181–190. doi: 10.1038/nrmicro772. [DOI] [PubMed] [Google Scholar]
- 9.Dow DE, Bartlett JA. 2014. Dolutegravir, the second-generation of integrase strand transfer inhibitors (INSTIs) for the treatment of HIV. Infect Dis Ther 3:83–102. doi: 10.1007/s40121-014-0029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naggie S, Hicks C. 2010. Protease inhibitor-based antiretroviral therapy in treatment-naive HIV-1-infected patients: the evidence behind the options. J Antimicrob Chemother 65:1094–1099. doi: 10.1093/jac/dkq130. [DOI] [PubMed] [Google Scholar]
- 11.Carr A. 2003. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov 2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- 12.Fumero E, Podzamczer D. 2003. New patterns of HIV-1 resistance during HAART. Clin Microbiol Infect 9:1077–1084. doi: 10.1046/j.1469-0691.2003.00730.x. [DOI] [PubMed] [Google Scholar]
- 13.Grabar S, Weiss L, Costagliola D. 2006. HIV infection in older patients in the HAART era. J Antimicrob Chemother 57:4–7. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch HH, Kaufmann G, Sendi P, Battegay M. 2004. Immune reconstitution in HIV-infected patients. Clin Infect Dis 38:1159–1166. doi: 10.1086/383034. [DOI] [PubMed] [Google Scholar]
- 15.Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. 2002. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med 347:385–394. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- 16.Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC. 2016. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat Rev Neurol 12:234–248. doi: 10.1038/nrneurol.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. 1998. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg Med Chem Lett 8:687–690. doi: 10.1016/S0960-894X(98)00098-5. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK, Krishnan K, Walters DE, Cho W, Cho H, Koo Y, Trevino J, Holland L, Buthod J. 1998. Structure based design: novel spirocyclic ethers as nonpeptidal P2-ligands for HIV protease inhibitors. Bioorg Med Chem Lett 8:979–982. doi: 10.1016/S0960-894X(98)00139-5. [DOI] [PubMed] [Google Scholar]
- 19.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. 2003. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother 47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salcedo-Gómez PM, Amano M, Yashchuk S, Mizuno A, Das D, Ghosh AK, Mitsuya H. 2013. GRL-04810 and GRL-05010; difluoride-containing nonpeptidic HIV-1 protease inhibitors (PIs) that inhibit the replication of multi-PI-resistant HIV-1 in vitro and possess favorable lipophilicity that may allow blood-brain barrier penetration. Antimicrob Agents Chemother 57:6110–6121. doi: 10.1128/AAC.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Yashchuk S, Mizuno A, Chakraborty N, Agniswamy J, Wang YF, Aoki M, Salcedo-Gómez PM, Amano M, Weber IT, Mitsuya H. 2015. Design of gem-difluoro-bis-tetrahydrofuran as P2 ligand for HIV-1 protease inhibitors to improve brain penetration: synthesis, X-ray studies, and biological evaluation. Chem Med Chem 10:107–115. doi: 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amano M, Tojo Y, Salcedo-Gómez PM, Parham GL, Nyalapatla PR, Das D, Ghosh AK, Mitsuya H. 2015. A novel tricyclic ligand-containing nonpeptidic HIV-1 protease inhibitor, GRL-0739, effectively inhibits the replication of multidrug-resistant HIV-1 variants and has a desirable central nervous system penetration property in vitro. Antimicrob Agents Chemother 59:2625–2635. doi: 10.1128/AAC.04757-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirasaka T, Kavlick MF, Ueno T, Gao WY, Kojima E, Alcaide ML, Chokekijchai S, Roy BM, Arnold E, Yarchoan R, Mitsuya H. 1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci U S A 92:2398–2402. doi: 10.1073/pnas.92.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. 2002. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J Virol 76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshimura K, Kato R, Yusa K, Kavlick MF, Maroun V, Nguyen A, Mimoto T, Ueno T, Shintani M, Falloon J, Masur H, Hayashi H, Erickson J, Mitsuya H. 1999. JE-2147: a dipeptide protease inhibitor (PI) that potently inhibits multi-PI-resistant HIV-1. Proc Natl Acad Sci U S A 96:8675–8680. doi: 10.1073/pnas.96.15.8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh AK, Leshchenko S, Noetzel M. 2004. Stereoselective photochemical 1,3-dioxolane addition to 5-alkoxymethyl-2(5H)-furanone: synthesis of bis-tetrahydrofuranyl ligand for HIV protease inhibitor UIC-94017 (TMC-114). J Org Chem 69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- 27.Maeda K, Yoshimura K, Shibayama S, Habashita H, Tada H, Sagawa K, Miyakawa T, Aoki M, Fukushima D, Mitsuya H. 2001. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J Biol Chem 276:35194–35200. doi: 10.1074/jbc.M105670200. [DOI] [PubMed] [Google Scholar]
- 28.Nakata H, Amano M, Koh Y, Kodama E, Yang G, Bailey CM, Kohgo S, Hayakawa H, Matsuoka M, Anderson KS, Cheng YC, Mitsuya H. 2007. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob Agents Chemother 51:2701–2708. doi: 10.1128/AAC.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amano M, Tojo Y, Salcedo-Gómez PM, Campbell JR, Das D, Aoki M, Xu C, Rao KV, Ghosh AK, Mitsuya H. 2013. GRL-0519, a novel oxatricyclic-ligand-containing nonpeptidic HIV-1 protease inhibitor (PI), potently suppresses the replication of a wide spectrum of multi-PI-resistant HIV-1 variants in vitro. Antimicrob Agents Chemother 57:2036–2046. doi: 10.1128/AAC.02189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. 2007. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother 51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ide K, Aoki M, Amano M, Koh Y, Yedidi RS, Das D, Leschenko S, Chapsal B, Ghosh AK, Mitsuya H. 2011. Novel HIV-1 protease inhibitors (PIs) containing a bicyclic P2 functional moiety, tetrahydropyrano-tetrahydrofuran, that are potent against multi-PI-resistant HIV-1 variants. Antimicrob Agents Chemother 55:1717–1727. doi: 10.1128/AAC.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tojo Y, Koh Y, Amano M, Aoki M, Das D, Kulkarni S, Anderson DD, Ghosh AK, Mitsuya H. 2010. Novel protease inhibitors (PIs) containing macrocyclic components and 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane that are potent against multi-PI-resistant HIV-1 variants in vitro. Antimicrob Agents Chemother 54:3460–3470. doi: 10.1128/AAC.01766-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M. 2009. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54:253–263. doi: 10.1016/j.neuint.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Yedidi RS, Maeda K, Fyvie WS, Steffey M, Davis DA, Palmer I, Aoki M, Kaufman JD, Stahl SJ, Garimella H, Das D, Wingfield PT, Ghosh AK, Mitsuya H. 2013. P2′ benzene carboxylic acid moiety is associated with decrease in cellular uptake: evaluation of novel nonpeptidic HIV-1 protease inhibitors containing P2 bis-tetrahydrofuran moiety. Antimicrob Agents Chemother 57:4920–4927. doi: 10.1128/AAC.00868-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Vagin A, Teplyakov A. 1997. MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30:1022–1025. doi: 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 37.Collaborative Computational Project Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Lamzin VS, Wilson KS. 1993. Automated refinement of protein models. Acta Crystallogr D Biol Crystallogr 49:120–147. doi: 10.1107/S0907444992006747. [DOI] [PubMed] [Google Scholar]
- 41.Zwart PH, Langer GG, Lamzin VS. 2004. Modelling bound ligands in protein crystal structures. Acta Crystallogr D Biol Crystallogr 60:2230–2239. doi: 10.1107/S0907444904012995. [DOI] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moriarty NW, Grosse-Kunstleve RW, Adams PD. 2009. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. 2010. In vitro selection of highly darunavir-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J Virol 84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Z, Yang Z, Liu Y, Lu Y, Chen K, Zhu W. 2014. Halogen bond: its role beyond drug-target binding affinity for drug discovery and development. J Chem Inf Model 54:69–78. doi: 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]
- 46.Biswal BK, Cherney MM, Wang M, Chan L, Yannopoulos CG, Bilimoria D, Nicolas O, Bedard J, James MNG. 2005. Crystal structures of the RNA dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem 280:18202–18210. doi: 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- 47.Marcelin AG, Affolabi D, Lamotte C, Mohand HA, Delaugerre C, Wirden M, Voujon D, Bossi P, Ktorza N, Bricaire F, Costagliola D, Katlama C, Peytavin G, Calvez V. 2004. Resistance profiles observed in virological failures after 24 weeks of amprenavir/ritonavir containing regimen in protease inhibitor experienced patients. J Med Virol 74:16–20. doi: 10.1002/jmv.20140. [DOI] [PubMed] [Google Scholar]
- 48.Young TP, Parkin NT, Stawiski E, Pilot-Matias T, Trinh R, Kempf DJ, Norton M. 2010. Prevalence, mutation patterns, and effects on protease inhibitor susceptibility of the L76V mutation in HIV-1 protease. Antimicrob Agents Chemother 54:4903–4906. doi: 10.1128/AAC.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. 2013. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem 56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 50.Tiefenbrunn T, Forli S, Happer M, Gonzalez A, Tsai Y, Soltis M, Elder JH, Olson AJ, Stout CD. 2014. Crystallographic fragment-based drug discovery: use of a brominated fragment library targeting HIV protease. Chem Biol Drug Des 83:141–148. doi: 10.1111/cbdd.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Müller K, Faeh C, Diederich F. 2007. Fluorine in pharmaceuticals: looking beyond intuition. Science 317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 52.Li CM, Chen J, Lu Y, Narayanan R, Parke DN, Li W, Ahn S, Miller DD, Dalton JT. 2011. Pharmacokinetic optimization of 4-substituted methoxybenzoyl-aryl-thiazole and 2-aryl-4-benzoyl-imidazole for improving oral bioavailability. Drug Metab Dispos 39:1833–1839. doi: 10.1124/dmd.110.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. 2007. Modelling of the blood brain barrier in drug discovery and development. Nat Rev Drug Discov 6:650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 54.Arribas JR, Clumeck N, Nelson M, Hill A, van Delft Y, Moecklinghoff C. 2012. The MONET trial: week 144 analysis of the efficacy of darunavir/ritonavir (DRV/r) monotherapy versus DRV/r plus two nucleoside reverse transcriptase inhibitors, for patients with viral load <50 HIV-1 RNA copies/ml at baseline. HIV Med 13:398–405. doi: 10.1111/j.1468-1293.2012.00989.x. [DOI] [PubMed] [Google Scholar]
- 55.Auffinger P, Hays FA, Westhof E, Ho PS. 2004. Halogen bonds in biological molecules. Proc Natl Acad Sci U S A 101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.