

Abstract
Nucleoside or nucleotide inhibitors are a highly successful class of antivirals due to selectivity, potency, broad coverage, and high barrier to resistance. Nucleosides are the backbone of combination treatments for HIV, hepatitis B virus, and, since the FDA approval of sofosbuvir in 2013, also for hepatitis C virus (HCV). However, many promising nucleotide inhibitors have advanced to clinical trials only to be terminated due to unexpected toxicity. Here we describe the in vitro pharmacology of compound 1, a monophosphate prodrug of a 2′-ethynyluridine developed for the treatment of HCV. Compound 1 inhibits multiple HCV genotypes in vitro (50% effective concentration [EC50], 0.05 to 0.1 μM) with a selectivity index of >300 (50% cytotoxic concentration [CC50], 30 μM in MT-4 cells). The active triphosphate metabolite of compound 1, compound 2, does not inhibit human α, β, or γ DNA polymerases but was a substrate for incorporation by the human mitochondrial RNA polymerase (POLRMT). In dog, the oral administration of compound 1 resulted in elevated serum liver enzymes and microscopic changes in the liver. Transmission electron microscopy showed significant mitochondrial swelling and lipid accumulation in hepatocytes. Gene expression analysis revealed dose-proportional gene signature changes linked to loss of hepatic function and increased mitochondrial dysfunction. The potential of in vivo toxicity through mitochondrial polymerase incorporation by nucleoside analogs has been previously shown. This study shows that even moderate levels of nucleotide analog incorporation by POLRMT increase the risk of in vivo mitochondrial dysfunction. Based on these results, further development of compound 1 as an anti-HCV compound was terminated.
INTRODUCTION
Hepatitis C virus (HCV) chronically infects an estimated 184 million people worldwide (1). Complications of chronic HCV infection are severe and can include decompensated liver disease and the development of hepatocellular carcinoma, which may ultimately require liver transplantation. Until 2011, the standard of care for all HCV genotypes (1–7) was pegylated interferon and ribavirin; however, due to limited tolerability and efficacy, many patients were not treated. In recent years, focused efforts by multiple pharmaceutical companies have led to the development and eventual FDA approval of NS3 protease inhibitors (telaprevir, paritaprevir, and boceprevir), NS5A inhibitors (daclatasvir, ombitasvir, and ledipasvir), a nonnucleoside NS5B polymerase inhibitor (dasabuvir), and a nucleotide analog NS5B inhibitor (sofosbuvir) (Fig. 1) (2–4). Treatment with a combination of multiple direct-acting antivirals is proving to be very effective in patients. For example, Viekira Pak (Abbvie) combines ombitasvir, paritaprevir, ritonavir, and dasabuvir, while Harvoni (Gilead) is a combination of sofosbuvir and ledipasvir. Harvoni has been shown to achieve a 95% or higher sustained viral response rate after just 12 weeks of treatment (5).
FIG 1.
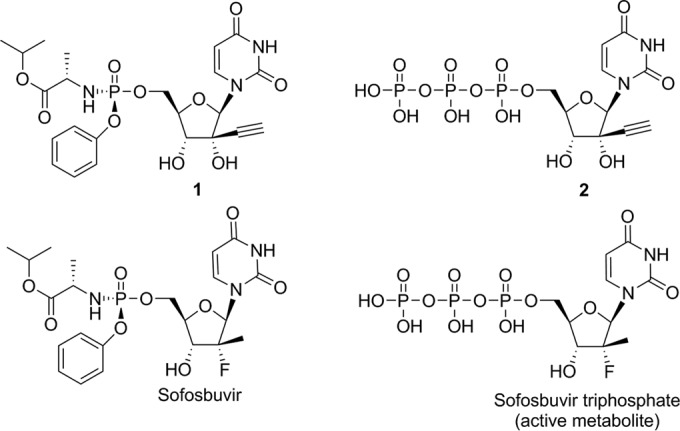
Chemical structures of compound 1, a phosphoroamidate prodrug, and its active triphosphate in the liver, compound 2 (top), and of sofosbuvir and its active triphosphate (bottom).
Nonnucleoside inhibitors, such as dasabuvir (3) and GS-9669 (6), bind to allosteric pockets on the HCV RNA polymerase. Unfortunately, these allosteric sites are not well conserved between the different HCV genotypes and can tolerate mutations without detrimental effects to the viral replication capacity (7). Nonnucleoside NS5B inhibitors therefore tend to lack pan-genotypic potency and have a low barrier to the development of resistance (8). In contrast, the polymerase active-site residues are highly conserved between the different HCV genotypes. Nucleotide inhibitors of NS5B therefore maintain pan-genotypic potency and show a high barrier to resistance (8).
Discovery efforts at several companies led to the development of multiple promising anti-HCV nucleotide inhibitors. However, with the exception of sofosbuvir, the use of all of these nucleotide inhibitors was stopped during clinical trials due to toxicities that were not detected in preclinical studies. Anti-HCV nucleotide analogs, including NM283 (prodrug of 2′-methylcytidine), RG1626 (prodrug of 4′-azidocytidine), PSI-938 (prodrug of 2′-deoxy-2′-fluoro-2′-methylguanosine), and BMS-986094 (prodrug of 2′-methylguanosine) were terminated for gastrointestinal toxicity, hepatotoxicity, or cardiotoxicity (9). GS-6620, a novel C-nucleoside, was well tolerated in phase I studies but showed highly variable human pharmacokinetics/pharmacodynamics (PK/PD), which limited its clinical potential (10). The failure to predict nucleotide toxicity in preclinical studies not only delayed the development of effective HCV therapies but also, in the case of BMS-986094, led to a fatality of one patient and severe cardiac and renal toxicity in others (11).
Mitochondrial effects are one possible mechanism of nucleoside analog toxicity. Some nucleoside inhibitors of HIV reverse transcriptase have the ability to chain terminate DNA elongation and inhibit the host mitochondrial DNA polymerase γ, which has been suggested to result in in vivo toxicities (12, 13). The incorporation of ribonucleotide analogs, such as the anti-HCV compounds, by mitochondrial DNA-dependent RNA polymerase (POLRMT) has been similarly put forward as a mechanism of toxicity (14). It has not been shown, however, that moderate incorporation by POLRMT can lead to mitochondrial dysfunction in vivo. While the failure of anti-HCV nucleotide clinical candidates such as NM283, RG1626, PSI-938, and BMS-986094 was suspected to be associated with mitochondrial toxicity, histopathological or electron microscopic analysis of the tissue was either not reported or not clear.
Compound 1 (prodrug of 2′-ethynyluridine) is a nucleotide inhibitor of NS5B that was under development by Novartis. Early during the preclinical phase, intensive in vitro and in vivo assessments of mitochondrial toxicity were performed. Moderate levels of incorporation of the nucleotide analog by POLRMT in vitro were predictive for in vivo toxicity. Significant mitochondrial gene expression alterations and ultrastructural mitochondrial swelling were associated with hepatic toxicity after 8-day repeat dosing in vivo. Further development of compound 1 was therefore terminated.
MATERIALS AND METHODS
Compounds.
Nucleotide prodrug compound 1 and its triphosphate metabolite, compound 2, were synthesized at Novartis. Sofosbuvir was purchased from a commercial source (Acme Biosciences, Inc.), and its triphosphate metabolite was synthesized at Novartis (Fig. 1).
Cell culture.
Huh-7.5 cells are human hepatoma cells that are highly permissive for HCV replication (15) and were obtained from Apath LLC (Brooklyn NY). Huh-7.5 cells were maintained in complete medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 0.1 mM nonessential amino acids [NEAA], 1 mM sodium pyruvate, and 10% fetal bovine serum [FBS]). Stable HCV replicon cell lines were maintained in complete medium containing 500 μg/ml G418 (Corning).
Molt 4 (MT-4) is a T cell leukemia cell line that was obtained from the American Type Culture Collection (ATCC; CRL-1582, lot 58483180), Manassas, VA. glucose-conditioned MT-4 cells were cultured in Roswell Park Memorial Institute (RPMI) medium (Invitrogen, Carlsbad CA) containing 2.0 g/liter glucose. Galactose-conditioned MT-4 cells were cultivated in RPMI medium supplemented with 2.0 g/liter galactose. Both glucose- and galactose-conditioned media were supplemented with 10% FBS and 1× penicillin-streptomycin/glutamine; cells were cultivated at 37°C, in 5% CO2, at 98% humidity.
HCV replicon constructs.
The HCV genotype 1a (GT1a) (strain H77) replicon plasmid pH/SG-lucubineo-H77S was derived from pH/SG-Neo(L+I), obtained from Apath LLC, by replacing the NS3-NS5A coding region with the corresponding sequence from pH77-S (16), as described previously (17). This replicon was further modified to generate pH/SG-hRlucneo-H77S by replacing the firefly luciferase reporter cassette in the first cistron with the humanized Renilla luciferase (hRluc) gene in tandem with the neomycin phosphotransferase II gene (neo). The hRlucneo reporter gene cassette was obtained by PCR from pF9 cytomegalovirus (CMV) hRluc-Neo Flexi(R) plasmid (Promega, Madison WI), using primers encoding AscI and NotI restriction sites. A second NotI site within the coding sequencing of NS5B was removed by silent QuikChange mutagenesis (Agilent, Santa Clara CA).
The HCV GT1b (strain Con1) replicon plasmid pFKi389hRlucneo was derived from pFKi389lucubineo_3_3′_ET (obtained from R. Bartenschlager, University of Heidelberg) by replacing the reporter gene cassette in the first cistron with the hRlucneo cassette using AscI and NotI restrictions sites. This replicon encodes three cell culture adaptive mutations (E1202G and T1280I in NS3; K1846T in NS4B) (18).
The HCV GT2a (strain JFH1) replicon plasmid pSGhRlucneo-JFH1 was generated by replacing the neo gene in pSG-Neo-JFH1 (19) with the hRlucneo cassette. Briefly, a DNA fragment encoding the JFH-1 5′ untranslated region (UTR) in tandem with the hRlucneo reporter gene cassette was synthesized by Integrated DNA Technologies (IDT, Coralville, CA). The resulting DNA fragment was digested with AgeI and PmeI and ligated into pSG-Neo-JFH1 digested with the same enzymes.
Plasmids encoding subgenomic GT3a (pSGRlucneo-GT3a-S52-HDVR) and GT4a (pSGRlucneo-GT4a-ED43-HDVR) replicons were designed as previously described (20) and synthesized by GenScript. The plasmid pSGRlucneo-GT3a-S52-HDVR encodes a GT3a (strain S52; accession no. GU814264) bicistronic subgenomic replicon expressing the hRlucneo reporter gene in the first cistron and NS3-NS5B, driven by an encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES), in the second cistron. This replicon encodes three cell culture adaptive mutations, P1226S (NS3), D1437H (NS3), and S2210I (NS5A) (20). Similarly, the plasmid pSGRlucneo-GT4a-ED43-HDVR encodes a GT4a (strain ED43; accession no. GU814266) bicistronic subgenomic replicon expressing hRlucneo in the first cistron and encodes two cell culture adaptive mutations, T1369K (NS3) and R2882G (NS5B) (20). For GT3a and GT4a replicon plasmids, the hepatitis delta ribozyme (HDVR) was included to ensure proper generation of the 3′ end of the HCV genomic RNA during in vitro transcription. All plasmids were confirmed by restriction digestion and DNA sequencing.
In vitro transcription and electroporation.
HCV replicon plasmids were linearized with HpaI (GT1a), ScaI and PacI (GT1b), XbaI (GT2a and GT4a), or SpeI (GT3a) and purified with the MinElute PCR purification kit (Qiagen). HCV replicon RNA was generated from 1 μg DNA template using the T7 RiboMax Express large-scale RNA production system (Promega). Following transcription, reaction mixtures were treated with RNase-free DNase and purified using the RNeasy minikit (Qiagen); RNA quality was assessed by agarose gel electrophoresis and ethidium bromide staining. Stable HCV replicon cell lines were established essentially as previously described (21). Briefly, 4 × 106 Huh-7.5 cells were electroporated (960 μF and 270 V) with 10 μg in vitro-transcribed HCV replicon RNA using a Gene Pulser system (Bio-Rad, Hercules CA). Cells were resuspended in complete medium and incubated for 24 h at 37°C and 5% CO2 prior to the addition of G418 (500 μg/ml). Replicon-containing cells were selected in the presence of G418 for approximately 4 to 5 weeks, after which time surviving colonies were harvested and pooled.
HCV replicon assays.
Sofosbuvir and compound 1 were serially diluted in dimethyl sulfoxide (DMSO) and stamped into 384-well plates (0.5 μl/well) prior to the addition of 4 × 103 replicon-containing cells in complete medium (final DMSO concentration, 1%). Alternatively, 1 × 104 replicon-containing cells were seeded into 96-well plates prior to the addition of an equal volume of serially diluted compounds in complete medium to give a final DMSO concentration of 0.5%. Replicon cells were incubated for 72 h at 37°C in 5% CO2 prior to luciferase readout by using of the Renilla-Glo assay (Promega) in accordance with the manufacturer's instructions. Cytotoxicity was determined using the CellTiter-Glo luminescent cell viability assay (Promega) by following the manufacturer's instructions. The 50% effective (EC50) and 50% cytotoxic (CC50) concentrations were calculated using XLFit (IDBS) by nonlinear regression analysis using a four-parameter logistic equation (XLFit dose response one site, model 205).
Enzymes.
The coding sequence of NS5B polymerase from HCV GT1b strain Con1 was cloned into pET21d(+), with a C-terminal truncation of 21 amino acids and a C-terminal His6 tag (termed NS5B Δ21 Con-1 His). It was purified as a recombinant protein from Escherichia coli (22). Recombinant human DNA polymerases α and β were purchased from Chimerx (Milwaukee, WI). The coding sequence of human DNA polymerase γ (30 to 1,239 amino acids [aa]) was synthesized by Genewiz with codon optimization. It was cloned into pDEST8 vector with an N-terminal His6 tag and tobacco etch virus (TEV) cleavage site and then purified from insect cells. Recombinant human mitochondrial RNA polymerase (POLRMT) was purchased from Enzymax (Lexington, KY).
NS5B polymerase assay.
The polymerase reaction mixtures were set up in 384-well white opaque plates (Corning) and contained 50 mM Tris-HCl (pH 7.5), 10 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.001% Triton X-100, 200 nM biotinylated double-stranded RNA template (23), and 100 nM NS5B Δ21 Con-1 His. The test compounds, compound 2 and sofosbuvir triphosphate (in 10-point, 3-fold serial dilutions, maximum concentration of 25 μM), were preincubated with this reaction mixture for 15 min at room temperature before the addition of 2.5 μM ATP, 2.5 μM CTP, 2 μM UTP, 2.5 μM GTP, and 0.5 μM [3H]UTP (55 Ci/mmol). Reaction mixtures were incubated at room temperature for 2 h, and reactions were stopped with an equal volume of buffer containing 100 mM Tris-HCl (pH 7.5), 50 mM EDTA (pH 8), 300 mM NaCl, and 1 mg/ml SPA beads (PerkinElmer). Reaction plates were sealed and shaken for 20 min at room temperature, followed by centrifugation at 2,000 rpm for 2 min, and read on a Trilux 1450 MicroBeta scintillation counter. The 50% inhibitory concentration (IC50) determination was done as described in reference 23.
Human DNA polymerase assays.
A 100 nM concentration of human DNA polymerase α or 50 nM human DNA polymerase β was added to a buffer containing 20 mM Tris, pH 7.5, 100 mM KCl, 10 mM MgCl2, 0.01% Tween 20, 0.5 mM EDTA, and 1 mM DTT. Test compounds (nucleotide triphosphates) were preincubated with the polymerase for 30 min at room temperature, after which reactions were initiated by the addition of 1.25 μM dATP, 1.25 μM dCTP, 1.25 μM dTTP, 1.25 μM dGTP, 200 nM primer B (5′-GAC GGG AAG-3′), and 100 nM molecular beacon (5′-5,6-FAM-CCT CTC CGT GTC TTG TAC TTC CCG TCA GAG AGG-BHQ1-3′). Reaction mixtures were incubated for 30 min (human DNA polymerase α) or 120 min (human DNA polymerase β) at room temperature and then read on a Perkin-Elmer EnVision 2101 plate reader (fluorescence mode; excitation at 480 nm and emission at 535 nm). For human DNA polymerase γ, 5 nM enzyme was added to a buffer containing 20 mM Tris, pH 7.5, 100 mM KCl, 10 mM MgCl2, 0.01% Tween 20, 0.5 mM EDTA, and 1 mM DTT. The test compound was preincubated with the polymerase for 30 min at room temperature before the addition of 0.25 μM dATP, 0.25 μM dCTP, 0.25 μM dTTP, 0.25 μM dGTP, 200 nM primer B, and 100 nM molecular beacon. Reaction mixtures were incubated for 30 min at room temperature before detection as described above.
Human mitochondrial RNA polymerase assay.
The human mitochondrial RNA polymerase (POLRMT) single-nucleotide incorporation assay was adapted from the study of Arnold et al. (14). Briefly, elongation complexes were formed by incubating 500 nM POLRMT with 250 nM RNA primer/DNA template duplex in assay buffer (25 mM Tris-HCl, pH 8, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, and 0.2 U/μl RNasin) for 1.5 min at room temperature, followed by rapid mixing with 500 μM nucleoside triphosphate substrate or test compounds, including compound 2, sofosbuvir triphosphate, GTP, 2′-C-methyl GTP, 3′ dGTP, or UTP. Purified POLRMT (Enzymax, Lexington, KY) was stored in 10 mM Tris-HCl (pH 8.0), 1 mM DTT, 100 mM NaCl, and 20% glycerol. The volume of POLRMT added to any reaction mixture was always less than or equal to 1/10 the total volume. Primer-template consisted of a 5′-FAM-labeled 8-mer RNA oligonucleotide primer (5′-UUUUGCCGCGCC-3′) annealed to the appropriate 18-mer DNA template (14). Reactions were allowed to proceed for 5 min at room temperature and were quenched by the addition of EDTA (50 mM). Products were resolved from substrates by denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. 2). Gels were visualized with a Typhoon Imager under fluorescence detection mode and quantified with the ImageQuant TL software (GE Healthcare, Piscataway, NJ). Percentage incorporations of the respective nucleoside triphosphates are calculated from the PAGE gel band intensities {[(band intensity of upper incorporated RNA product)/(total band intensities of upper incorporated RNA product and lower unincorporated RNA primer)] × 100}.
FIG 2.
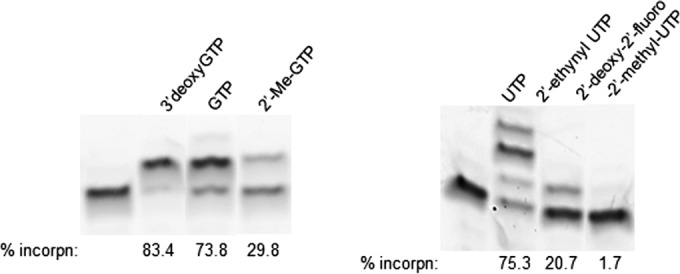
Nucleoside analog incorporation catalyzed by human POLRMT. Representative denaturing PAGE gels are shown indicating reaction products from POLRMT-catalyzed nucleotide incorporation with the indicated nucleoside analog triphosphate and RNA primer/DNA template. Reactions were allowed to proceed at room temperature for 5 min. RNA primer was extended to n + 1 in the presence of each nucleoside triphosphate with the exception of UTP (multiple misincorporations observed). Percentage incorporations of the respective nucleoside triphosphates are shown {based on [(band intensity of upper incorporated RNA product)/(total band intensities of upper incorporated RNA product and lower unincorporated RNA primer)] × 100}. The assay was repeated a minimum of three times to calculate the mean percentages of incorporation and standard deviations shown in Table 3.
Mitochondrial toxicity assay.
Cells were seeded in 96-well U-bottom plates at 0.2 × 106 or 0.5 × 106 cells/well of glucose-conditioned and galactose-conditioned MT-4 cells, respectively. Test compounds (compound 1, sofosbuvir, or dideoxycytidine [ddC]) were diluted in a 10-step half-dilution series (highest final starting concentration, 1,000 μM) and applied to the cell culture; the final concentration of DMSO was 0.1% for all treatment conditions. Repeated treatments, performed at 2-day intervals, were applied until three cell doublings were achieved (typically 3 days in glucose-conditioned medium and 6 days in galactose-conditioned medium). Cell viability was assessed by ATP measurement using the CellTiter-Glo luminescent cell viability kit (Promega, Madison WI) as described by the manufacturer. The resulting luminescence was measured using the Envision 2104 luminescence plate reader (PerkinElmer, Waltham, MA).
Quantification of triphosphate levels in primary hepatocytes.
The in vitro formation of nucleoside triphosphate metabolite was investigated using dog and human primary hepatocytes. 14C-labeled compound 1 or sofosbuvir (50 μM) was incubated with hepatocytes for up to 24 h. Samples were collected at various time points into 4.5-ml cryotubes that contained 1 ml lysing solution (freshly made and kept on ice; 35% methanol, 100 mM EDTA, PhosSTOP tablet 1, and cOmplete tablet 2 [Roche Diagnostics, Germany] in phosphate-buffered saline [PBS, pH 7.4; Life Technologies, Grand Island, NY]). Following sample collection, each well was washed with 1 ml acetonitrile, and this was combined with the lysis solution. Samples were stored at −80°C until analysis.
4C-labeled test compounds and their triphosphate metabolites were analyzed by high-performance liquid chromatography (HPLC) with online radioactivity detection. Extracts were thawed, thoroughly mixed by vortexing, and concentrated ∼2- to 5-fold by evaporation before HPLC analysis. The analytical system consisted of a Waters Alliance (Waters, Milford, MA) HPLC equipped with a YMC-ODS AQ column (5 μm, 3.2 mm by 250 mm; YMC America, Inc., Allentown, PA). The column temperature was not controlled (room temperature). The mobile phases consisted of 10 mM ammonium acetate containing 0.02% ammonium hydroxide (vol/vol) (solvent A) and acetonitrile-methanol-water (45:45:10) with 0.02% ammonium hydroxide (vol/vol/vol/vol) (solvent B). The total run time was 40 min with a linear gradient elution program. Samples were analyzed after column chromatography by an online β-RAM radioactivity detector (LabLogic, Brandon, FL) equipped with a 250-μl liquid flow cell with a scintillant (FlowLogic U; LabLogic, Brandon, FL) flow rate of 1.5 ml/min. The resulting data were processed using the Laura data system (LabLogic, Inc., version 4.0.5).
In vivo toxicity studies.
In vivo experiments were performed in accordance with the National Animal Welfare guidelines of the U.S. National Research Council. A rodent toxicity study was performed using male Wistar Hannover [Crl:WI(Han)] rats obtained from Charles River Laboratories, Raleigh, NC. Animals were approximately 9 to 10 weeks of age and weighed 250 to 320 g at the initiation of dosing. Compound 1 was administered daily at 10, 50, and 300 mg/kg of free base (administered as the proline salt) for 15 doses. Control animals (n = 5) received the dosing vehicle only (30% [vol/vol] polyethylene glycol 300–69% [wt/vol] 1.4% polysorbate 80 solution in purified water; USP) at a volume of 10 ml/kg. Clinical observations, body weight and food consumption determinations, and clinical laboratory (hematology, clinical chemistry, coagulation, and blood gas analysis) evaluations were performed on all groups. At necropsy, gross pathology examinations and selected organ weight determinations were performed on all groups. Microscopic examinations were conducted on a standard list of organs and tissues from animals assigned to the control and high-dose groups. Gross lesions and ceca from all animals were examined microscopically.
Three separate studies were performed in beagle dogs. In these studies, pH-adjusted sofosbuvir and compound 1 (pH 3.5 ± 0.5 with 1 N HCl) were administered orally via gavage to male dogs (n = 3/group) as a formulation in vehicle (30% polyethylene glycol 300–69% [wt/vol] in 1.4% polysorbate 80 aqueous solution) at daily doses of 250 mg/kg/day (sofosbuvir) and 10, 50, 150, 250, and 1,000 mg/kg/day base (compound 1 administered as the proline salt) for 8 consecutive days. The control animals (n = 3/study) received vehicle without pH adjustment. Animals were obtained from Marshall BioResources, North Rose, NY, and at the initiation of dosing were approximately 9 to 24 months old and weighed approximately 7.0 to 10.2 kg. Clinical observations and body weight and food consumption determinations were performed for all animals. On day 8, prior to dosing, clinical pathology (hematology, clinical chemistry, coagulation, and blood gas analysis) assessments were performed on all groups. On day 8, 4 h after the last dose, complete necropsies were performed on all animals. Gross pathology examinations, organ weight determinations, and organ/tissue sampling for histopathology, electron microscopy, toxicokinetic (TK), and gene expression profiling were performed on all groups. The livers were not weighed due to the liver perfusion for TK sampling. Organs/tissues collected in 10% buffered formalin were processed to hematoxylin-and-eosin-stained tissue sections, and microscopic examinations were conducted. A panel of stains was performed on the livers of representative vehicle control and compound 1-treated animals (Long Ziehl-Neelsen staining for lipofuscin, Perl's staining for iron, Hall's staining for bile, Gomori's staining for reticulin, and Masson's trichrome for collagen). Transmission electron microscopy (TEM) evaluation was performed on the livers of representative vehicle control and compound 1-treated animals. A small piece of each liver from the left lateral lobe was collected in modified Karnovsky's fixative. Samples were routinely processed and were embedded in EMbed (Epon) 812. Semithin sections were stained with toluidine blue/basic fuchsin for light microscope evaluation. Ultrathin sections cut for TEM were double stained with uranyl acetate and lead citrate and examined using a FEI Tecnai G2 BioTwin electron microscope. Photomicrographs were captured using an Olympus-SIS Morada digital camera. Representative ultrastructural photomicrographs were taken and evaluated.
Quantification of triphosphates in dog biopsy liver samples.
Dog liver biopsy samples were homogenized using a Bullet Blender (Next Advance, Averill Park, NY) with approximately 1× volume of lysing solution (freshly made and kept on ice; 35% methanol, 100 mM EDTA, PhosSTOP tablet 1, and cOmplete tablet 2 [Roche Diagnostics, Germany] in PBS [pH 7.4; Life Technologies, Grand Island, NY]). Ten-microliter aliquots were counted using liquid scintillation counting. The remaining samples were transferred to a glass tube, and 0.5 to 2 ml of lysing solution was immediately added; samples were dissolved using a Focused-ultrasonicator (Covaris, Woburn, MA) at 15°C. Samples were centrifuged at 4,000 × g for 10 min before the addition of 1 to 2 ml of acetonitrile to the pellet and extraction using the Focused-ultrasonicator for 2 cycles. Supernatants were evaporated under nitrogen flow and reconstituted in 200 μl of an 80:20 (vol/vol) mixture of solvent A-solvent B. Samples were analyzed as described above.
Genomic analysis of the toxicity study liver samples.
Dog liver samples were collected for gene expression profiling. Total RNA was obtained by TRIzol extraction from frozen tissue, purified by RNeasy (Qiagen), and quantified by absorbance at 260 nm. Samples were prepared on Rat Genome 230 2.0 arrays (Affymetrix, Inc., Santa Clara, CA), and labeling of nucleic acid for hybridization was performed with 100 ng of total RNA using the 3′-IVT express kit and poly-A control reagent. An aliquot of biotin-labeled cRNA was hybridized for approximately 16 to 18 h at 45°C on an array. The array was then washed and stained using an Affymetrix Fluidics Workstation 450 and scanned on the Affymetrix GeneChip scanner 3000. The scanned image was analyzed using Affymetrix MAS 5.0 software.
RESULTS
Compound 1 inhibits HCV replication with minimal cytotoxicity.
The activity of a novel nucleotide analog, compound 1 (Fig. 1), against various HCV genotypes was determined using the stable replicon system. Compound 1 showed potency against genotype 1a, 1b, 2a, 3a, and 4a replicons, with EC50s ranging from 0.052 to 0.10 μM (Table 1). The compound was also active against full-length infectious HCV GT2a. Compound 1 was not cytotoxic (CC50 > 50 μM) in any of the HCV replicon cell lines tested. Cytotoxicity was similarly undetectable in liver-derived cell lines not bearing the HCV replicon (data not shown). In contrast, compound 1 was mildly cytotoxic in MT-4 cells (CC50 = 30.7 μM), a human lymphocyte-derived line. The MT-4 cell CC50 was used to determine an in vitro selectivity index (SI = MT-4 cell CC50/genotype 1b EC50) of 300.
TABLE 1.
Activity and cytotoxicity of compound 1
| Cell line | Replicon genotype (strain) | EC50 (μM) | CC50 (μM) |
|---|---|---|---|
| Huh-7.5 | 1a (H77) | 0.061 ± 0.03 | >50 |
| 1b (Con1) | 0.10 ± 0.03 | >50 | |
| 2a (JFH-1) | 0.074 ± 0.02 | >50 | |
| 2a (JFH-1, infectious virus) | 0.077 ± 0.03 | >50 | |
| 3a (S52) | 0.052 ± 0.01 | >50 | |
| 4a (ED43) | 0.069 ± 0.02 | >50 | |
| MT-4 | NAa | 30.7 ± 1.27 |
NA, not applicable.
The triphosphate of compound 1, compound 2, inhibits the HCV NS5B polymerase.
To investigate the specificity of compound 1 for the viral polymerase, in vitro activity of the active triphosphate (compound 2) was tested against recombinant NS5B (GT1b) and human DNA polymerases. Compound 2 inhibited NS5B RNA elongation with an IC50 of 0.23 μM (Table 2), and sofosbuvir triphosphate inhibited NS5B RNA elongation with an IC50 of 0.12 μM. Against recombinant human DNA polymerases α, β, and γ, compound 2 showed no inhibition up to the highest concentration tested (250 μM). We next investigated whether compound 2 was a substrate for human mitochondrial RNA polymerase (POLRMT). UTP and 3′-dGTP, which are known substrates for POLRMT, incorporated into the RNA product at 70% (±7.7%) and 71.5% (±17%), respectively (Table 3). In contrast, the triphosphate metabolite of sofosbuvir, which is not a substrate for POLRMT, incorporated at a rate of 1.7% (±0.25%), equivalent to background signal. 2′-C-methyl GTP, the active triphosphate metabolite of BMS-986094 and IDX184, both of which induce toxicity in vivo, was incorporated into 27.4% (±3.4%) of the RNA. Compound 2 showed a POLRMT incorporation rate of 17% (±5.5%), which is lower than that of the natural UTP substrate but well above background.
TABLE 2.
Inhibition of HCV NS5B polymerase by compound 2
| Polymerase assay and inhibitor | IC50 (μM) |
|---|---|
| HCV NS5B polymerase | |
| Sofosbuvir triphosphate | 0.12 |
| Compound 2 | 0.23 |
| Human polymerases (compound 2) | |
| Human polymerase α | >250 |
| Human polymerase β | >250 |
| Human polymerase γ | >250 |
TABLE 3.
Human mitochondrial polymerase (POLRMT) nucleotide analog incorporation assay
| Nucleotide triphosphate | % single-nucleotide incorporation (mean ± SD) |
|---|---|
| GTP | 67 ± 7.2 |
| 2′-Me-GTP (IDX184 and BMS-986094 triphosphate) | 27.4 ± 3.4 |
| 3′-dGTP | 71.5 ± 17 |
| UTP | 70 ± 7.7 |
| 2 (2′-ethynyl UTP, 1 triphosphate) | 17 ± 5.5 |
| 2′-deoxy-2′-fluoro-2′-methyl-UTP (sofosbuvir triphosphate) | 1.7 ± 0.25 |
In vitro mitochondrial toxicity assay.
Given the ability of compound 2 to be incorporated by POLRMT, the potential of compound 1 to induce mitochondrial toxicity was evaluated in cell culture. MT-4 cells were treated with compound 1 and comparators, and cell viability was measured in glucose-rich versus galactose-only medium. Cancer cell lines such as MT-4 cultured in standard, glucose-rich cell culture media can tolerate a significant loss of mitochondrial function because they have a high rate of glycolysis and therefore do not rely on mitochondrial respiration/oxidative phosphorylation for survival (24). Consequently, MT-4 cells cultured in the presence of glucose are more resistant to mitochondrial toxicity, while cells cultivated in galactose are more sensitive (25). A known mitochondrion-toxic nucleoside, 2′-3′-dideoxycytidine (ddC), was 3-fold more cytotoxic in galactose-rich medium than in glucose-rich medium (Fig. 3), while sofosbuvir showed minimal cytotoxicity under both conditions. Compound 1 was <1.5-fold more cytotoxic in MT-4 cells cultured in galactose than in those cultured in glucose-rich medium. Since a 3-fold or greater shift between glucose and galactose cultures is considered biologically relevant (26), this result does not confirm a mitochondrial mechanism of toxicity for compound 1.
FIG 3.

Mitochondrial toxicity in MT-4 cell culture. ATP concentrations in MT-4 cells cultured in galactose-only medium (filled circles) and glucose medium (empty circles) after three cell population doublings in the presence of nucleoside/nucleotide. *, 95% confidence interval for the CC50.
Concentration of compound 2 (triphosphate of compound 1) in cell culture and dog liver.
Compound 1 must be metabolized to its active triphosphate form (compound 2) in the target cell at high enough concentrations and with a long enough intracellular half-life to maintain anti-HCV activity. Therefore, intracellular levels of compound 2 and sofosbuvir triphosphate were investigated in various cell types. HCV (GT1b) Huh-7.5 replicon cells, as well as human and dog primary hepatocytes, were incubated with 50 μM compound 1 or sofosbuvir for 24 h (Table 4). In human primary hepatocytes, the compound 2 intracellular triphosphate area under the concentration-time curve (AUC) was approximately 3.6-fold lower than that for the sofosbuvir triphosphate. In dog primary hepatocytes, the intracellular compound 2 AUC was similar to that of the sofosbuvir triphosphate. In HCV GT1b replicon cells, the AUC of compound 2 was ∼2-fold lower than that of the sofosbuvir triphosphate. The levels of triphosphate in the liver were then determined after a single 50-mg/kg oral dose of compound 1 or sofosbuvir in dogs. In the comparative dog liver laparoscopy, the AUC of compound 2 was 159 ng · h/ml, while the AUC of the sofosbuvir triphosphate metabolite was 3-fold higher, at 485 ng · h/ml (Table 4).
TABLE 4.
Intracellular and hepatic tissue triphosphate concentrations (AUC)
| Cell type and/or assay | AUC (ng · h/ml) |
|
|---|---|---|
| Compound 2 | Sofosbuvir triphosphate | |
| In vitroa | ||
| Huh-7 cell culture | 132 | 239 |
| Dog primary hepatocytes | 54.8 | 59.5 |
| Human primary hepatocytes | 72.3 | 262 |
| In vivo (liver biopsy)b | 159 | 485 |
For in vitro AUC determinations, cells and hepatocytes were incubated with 50 μM compound 1 or sofosbuvir before compound 2 or sofosbuvir triphosphate AUCs, respectively, were determined.
For in vivo AUC determinations, dogs were dosed with 50 mg/kg compound 1 or sofosbuvir before triphosphate levels were determined.
High doses of compound 1 result in liver toxicity in dog.
A 2-week toxicity study in rats showed no evidence of toxicity at doses up to 300 mg/kg/day. Rats (and most rodents) have higher esterase activity than humans and higher-order species and may therefore metabolize the esterase-sensitive prodrug of compound 1 more rapidly than they do.
In dogs, low doses of compound 1 (10 and 50 mg/kg/day) showed no evidence of liver toxicity based on liver enzyme levels and histopathology results. Doses of ≥150 mg/kg/day yielded compound 1-related alterations in clinical chemistry and/or microscopic hepatic changes within 8 days of the administration of compound 1, though without a clear dose relationship. By day 8, animals dosed with 250 and 1,000 mg/kg/day of compound 1 had non-dose-dependent, moderate to marked increases in aspartate transaminase (AST) (3.4- to 29-fold) and alanine aminotransferase (ALT) (10- to 59-fold) levels in comparison to pretest values. Alkaline phosphatase (ALP) activity also tended to be increased (4.2- to 51-fold), often accompanied by a minimal increase in serum bilirubin concentrations (up to 2.7-fold). At 150 mg/kg/day, a 2-fold increase in serum ALT activity was present in one animal. Macroscopically, pale discoloration of the liver was noted in individual animals dosed at ≥250 mg/kg/day. Minimal mixed inflammatory cell infiltrates with multifocal single-cell hepatocellular necrosis were present in one animal at a dose of 150 mg/kg compound 1, consistent with the serum ALT elevation in the same animal. At doses of ≥250 mg/kg/day, centrilobular to diffuse micro- and/or macrovesicular vacuolation and centrilobular hepatocellular single-cell necrosis and centrilobular/central-vein fibrosis were present. The changes were often minimal to slight; however, necrosis was often accompanied by centrilobular hepatocellular loss, widened sinusoids, trabecular disorganization, and parenchymal collapse and, in the affected centrilobular areas, by mixed cell inflammation. Increased hepatocellular mitosis was present in one dog dosed at ≥250 mg/kg/day; minimal biliary hyperplasia was observed at 1,000 mg/kg/day.
Electron microscopy confirmed that compound 1 administration to dogs for 8 consecutive days at ≥150 mg/kg/day resulted in the compound 1-related ultrastructural findings in the liver. At 150 mg/kg/day, an increase in cytoplasmic lipid vacuoles (steatosis) in individual hepatocytes was the only ultrastructural finding noted in the single animal with increased serum ALT.
At ≥250 mg/kg/day, markedly increased lipid vacuoles and changes compatible with mitochondrial toxicity were noted in the cytoplasm of hepatocytes in most treated animals. The mitochondrial toxicity was characterized primarily by mitochondrial enlargement/swelling (megamitochondria) and less consistently by mitochondrial pleomorphism, crystolysis, and/or paracrystalline inclusions (Fig. 4). Secondary hepatocellular ultrastructural changes described as increased autophagolysosomes (sometimes containing degenerated mitochondria), hydropic degeneration (dilated endoplasmic reticulum and/or nuclear membrane), and/or necrosis (karyolysis, pyknosis, and dilated mitochondria with swollen cristae) were noted in the compound 1-treated hepatocytes at these doses. In the affected centrilobular areas, deposition of collagen was noted in the Disse's space and increased lysosome-containing Kupffer cells/macrophages and neutrophils were occasionally seen in the sinusoidal lumen or Disse's space.
FIG 4.

Transmission electron micrographs of liver sections collected from dogs receiving vehicle control or 250 mg/kg daily dosing of drug compound 1. (A) Animals receiving vehicle control have normal-sized mitochondria. (B) In contrast to panel A, the mitochondria in drug compound 1-treated animals are enlarged (megamitochondria). (C and D) Higher magnification of the hepatocyte mitochondria of drug compound 1-dosed animals (D) shows clear swelling relative to those of the control (C). M, mitochondrion; M*, megamitochondrion; n, nucleus.
Eight doses of sofosbuvir (250 mg/kg/day) in dogs resulted in no evidence of liver toxicity, as evidenced by clinical chemistry and liver histopathology results.
Gene expression analysis of liver samples.
Gene signatures consistent with hepatocellular loss and vacuolization, macrophage infiltration, increased mitosis, and bile ductule proliferation were observed following 8 daily doses of compound 1 at 250 and 1,000 mg/kg in dogs (Table 5). Gene expression analysis indicated a dose-dependent, selective depletion of mitochondrion-encoded transcripts starting at the 150-mg/kg/day dosing of compound 1, though only limited histopathologic changes were observed in the liver at this dose. The loss of mitochondrion-encoded transcripts positively correlates with the gene signature of hepatocyte loss. In addition to these changes, gene signatures of altered lipid metabolism and increased expression of perilipin transcripts were identified, suggesting altered lipid metabolism starting at the 150-mg/kg/day dose of compound 1. Animals dosed with 250 mg/kg/day sofosbuvir had no significant changes in gene expression signatures relative to those of the control animals (Table 5).
TABLE 5.
Hepatic genomic expression analysis in dog in response to 8 consecutive days of dosing
| Compound or vehicle | Dose (mg/kg) | Fold shift change in gene expression signatures in liver (mean ± SD) |
||||||
|---|---|---|---|---|---|---|---|---|
| Hepatocyte function | Macrophage | Mitosis | Bile duct | Lipid metabolism | Nuclear encoded genes | Mitochondrion-encoded genes | ||
| Vehicle | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | |
| Compound 1 | 10 | −1.1 ± 0.0 | −1.2 ± 0.1 | 1.0 ± 0.0 | −1.1 ± 0.1 | 1.0 ± 0.0 | 1.1 ± 0.0 | 1.0 ± 0.0 |
| 50 | −1.2 ± 0.1 | −1.1 ± 0.1 | 1.0 ± 0.1 | −1.0 ± 0.1 | −1.1 ± 0.0 | 1.1 ± 0.0 | −1.1 ± 0.0 | |
| 150 | −2.3 ± 1.0 | 1.2 ± 0.25 | 1.1 ± 0.1 | 1.5 ± 0.6 | −1.5 ± 0.3 | 1.1 ± 0.0 | −1.4 ± 0.3 | |
| 250 | −4.9 ± 1.3 | 2.7 ± 0.1 | 1.7 ± 1.3 | 3.1 ± 1.1 | −2.1 ± 0.2 | 1.2 ± 0.0 | −1.5 ± 0.0 | |
| 1,000 | −3.3 ± 2.3 | 1.8 ± 1.1 | 1.7 ± 0.6 | 2.7 ± 0.9 | −1.7 ± 0.3 | 1.2 ± 0.0 | −1.4 ± 0.0 | |
| Sofosbuvir | 250 | −1.1 ± 0.1 | −1.1 + 0.6 | 1 ± 0.1 | 1.0 ± 0.0 | −1.0 ± 0.0 | 1.1 ± 0.0 | −1.2 ± 0.1 |
DISCUSSION
The monophosphate amidate prodrug compound 1 of 2′-ethynyluridine is a potent pan-genotype HCV inhibitor with minimal cytotoxicity in various cell lines; an in vitro selectivity index of 300 was calculated in MT-4 cells. In a human mitochondrial RNA polymerase (POLRMT) assay, however, compound 2 showed incorporation in 17% of the available RNA primers. The rate at which POLRMT incorporated compound 2 was lower than that for UTP but about 10-fold higher than the rate for active triphosphate of sofosbuvir, a safe, FDA-approved anti-HCV nucleotide analog. The ability to be incorporated by recombinant POLRMT indicates that compound 1 has a higher potential for mitochondrial toxicity than sofosbuvir (14). However, it was not known if a 17% POLRMT incorporation level predicted an increased potential for mitochondrial toxicity in vivo. For comparison, the active triphosphate metabolite (2′-Me-GTP) shared by the monophosphate prodrugs BMS-986094 and IDX14184 is incorporated by POLRMT at a rate of 27.4%; both BMS-986094 and IDX14184 induced severe in vivo toxicity (11, 27). The mechanisms of in vivo toxicity for BMS-986094 and IDX14184 were not conclusive. A retrospective study on the pathology findings from the BMS-986094 phase II clinical study did not support a conclusion of mitochondrial toxicity (27). The authors of an IDX14184 rat and monkey toxicity study also concluded that the metabolite 2′-Me-GTP was not toxic to a mitochondrial mechanism, despite the observation of mitochondrial swelling in the liver and kidney sections of two rats that received 300 mg/kg/day IDX14184 for 50 and 84 days (11). Since the in vitro incorporation level of compound 2 was lower than that of 2′-Me-GTP, the development of compound 1 has proceeded with caution and there has been a focused effort to define the mitochondrial toxicity risk in vivo as early as possible.
An MT-4 cell-based assay was used to further quantitate mitochondrial toxicity in vitro. In this assay, toxicity involving the mitochondria is amplified in galactose- versus glucose-containing medium (25). For example, ddC becomes ∼3-fold more toxic when cultured in galactose-only medium. Compound 1 showed a CC50 of 30.7 μM in standard glucose-rich medium but became 1.5-fold more cytotoxic in galactose. In contrast, sofosbuvir showed minimal cytotoxicity in glucose-rich or galactose-only medium. The small CC50 drop observed for compound 1 in the galactose-only medium was still inconclusive for mitochondrial toxicity.
Preclinical studies were used to investigate the safety of compound 1 in vivo. A 2-week rat study showed no evidence of toxicity at doses up to 300 mg/kg/day (data not shown). Most rodents metabolize the esterase-sensitive prodrug of compound 1 significantly differently than humans and other mammals, and we therefore speculate that compound 1 is too rapidly metabolized in the rat gastrointestinal tract and plasma and that therefore the rat is unable to load compound 2 in the liver to toxic concentration levels. In 8-day toxicity studies in dogs, however, increases in serum liver enzymes and hepatic macroscopic, microscopic, and ultrastructural changes were present at doses of ≥150 mg/kg/day of compound 1. The microscopic steatohepatitis and ultrastructural hepatocellular changes, such as increased cytoplasmic lipid vacuoles and mitochondrial swelling/degeneration, suggested mitochondrial hepatopathy. Similar findings have been described with other nucleotide/nucleoside analogs (28, 29). The mitochondrial toxicity mechanism of compound 1 was confirmed by gene expression signature analyses. In the 150-, 250-, and 1,000-mg/kg/day dosing groups of compound 1, gene signatures associated with hepatic dysfunction were dose dependent and correlated with the observed toxicity. These results indicate that a positive result in the POLRMT mitochondrial incorporation assay can be predictive for in vivo mitochondrial toxicity. Animals dosed with sofosbuvir at 250 mg/kg/day for 8 days did not have any signs of hepatotoxicity or gene expression changes, in full agreement with the excellent clinical safety profile (5).
At lower doses (10 and 50 mg/kg/day for 8 days), compound 1 did not induce hepatotoxicity; hepatic function and mitochondrial gene expression signals were also unaltered. However, due to the narrow therapeutic index and given that HCV therapy would require extended dosing, compound 1 therapy might lead to mitochondrial toxicity. Based on these findings, further development efforts for compound 1 were terminated.
ACKNOWLEDGMENTS
We thank Jennifer Cunliffe for bioanalytical analysis and Catherine Jones for editorial assistance.
This study was funded by Novartis.
All authors are, or were at the time the study was conducted, employees of Novartis and/or shareholders of Novartis stock.
Funding Statement
This study was funded by Novartis. All authors are, or were at the time the study was conducted, employees of Novartis and/or shareholders of Novartis stock.
REFERENCES
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.Trivella JP, Gutierrez J, Martin P. 2015. Dasabuvir: a new direct antiviral agent for the treatment of hepatitis C. Expert Opin Pharmacother 16:617–624. doi: 10.1517/14656566.2015.1012493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Clercq E. 2014. Current race in the development of DAAs (direct-acting antivirals) against HCV. Biochem Pharmacol 89:441–452. doi: 10.1016/j.bcp.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Lawitz E, Jacobson IM, Nelson DR, Zeuzem S, Sulkowski MS, Esteban R, Brainard D, McNally J, Symonds WT, McHutchison JG, Dieterich D, Gane E. 31 July 2015. Development of sofosbuvir for the treatment of hepatitis C virus infection. Ann N Y Acad Sci doi: 10.1111/nyas.12832. [DOI] [PubMed] [Google Scholar]
- 5.Gentile I, Scotto R, Zappulo E, Buonomo AR, Pinchera B, Borgia G. 2016. Investigational direct-acting antivirals in hepatitis C treatment: the latest drugs in clinical development. Expert Opin Investig Drugs 25:557–572. doi: 10.1517/13543784.2016.1161023. [DOI] [PubMed] [Google Scholar]
- 6.Fenaux M, Eng S, Leavitt SA, Lee YJ, Mabery EM, Tian Y, Byun D, Canales E, Clarke MO, Doerffler E, Lazerwith SE, Lew W, Liu Q, Mertzman M, Morganelli P, Xu L, Ye H, Zhang J, Matles M, Murray BP, Mwangi J, Zhang J, Hashash A, Krawczyk SH, Bidgood AM, Appleby TC, Watkins WJ. 2013. Preclinical characterization of GS-9669, a thumb site II inhibitor of the hepatitis C virus NS5B polymerase. Antimicrob Agents Chemother 57:804–810. doi: 10.1128/AAC.02052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng JY, Cheng G, Perry J, Barauskas O, Xu Y, Fenaux M, Eng S, Tirunagari N, Peng B, Yu M, Tian Y, Lee YJ, Stepan G, Lagpacan LL, Jin D, Hung M, Ku KS, Han B, Kitrinos K, Perron M, Birkus G, Wong KA, Zhong W, Kim CU, Carey A, Cho A, Ray AS. 2014. Inhibition of hepatitis C virus replication by GS-6620, a potent C-nucleoside monophosphate prodrug. Antimicrob Agents Chemother 58:1930–1942. doi: 10.1128/AAC.02351-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCown MF, Rajyaguru S, Le Pogam S, Ali S, Jiang WR, Kang H, Symons J, Cammack N, Najera I. 2008. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother 52:1604–1612. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerber L, Welzel TM, Zeuzem S. 2013. New therapeutic strategies in HCV: polymerase inhibitors. Liver Int 33(Suppl 1):S85–S92. [DOI] [PubMed] [Google Scholar]
- 10.Cho A, Zhang L, Xu J, Lee R, Butler T, Metobo S, Aktoudianakis V, Lew W, Ye H, Clarke M, Doerffler E, Byun D, Wang T, Babusis D, Carey AC, German P, Sauer D, Zhong W, Rossi S, Fenaux M, McHutchison JG, Perry J, Feng J, Ray AS, Kim CU. 2014. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J Med Chem 57:1812–1825. doi: 10.1021/jm400201a. [DOI] [PubMed] [Google Scholar]
- 11.Luo S, Rush R, Standring D. 2015. Single- and repeat-dose toxicity of IDX14184, a nucleotide prodrug with antiviral activity for hepatitis C viral infection, in mice, rats, and monkeys. Hum Exp Toxicol doi: 10.1177/0960327115592939. [DOI] [PubMed] [Google Scholar]
- 12.Birkus G, Hitchcock MJ, Cihlar T. 2002. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother 46:716–723. doi: 10.1128/AAC.46.3.716-723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem 276:40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 14.Arnold JJ, Sharma SD, Feng JY, Ray AS, Smidansky ED, Kireeva ML, Cho A, Perry J, Vela JE, Park Y, Xu Y, Tian Y, Babusis D, Barauskus O, Peterson BR, Gnatt A, Kashlev M, Zhong W, Cameron CE. 2012. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog 8:e1003030. doi: 10.1371/journal.ppat.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi M, Lemon SM. 2004. Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J Virol 78:7904–7915. doi: 10.1128/JVI.78.15.7904-7915.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton-Davies J, Wilson CJ, Myer V, Cornellataracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither LA. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol 83:10058–10074. doi: 10.1128/JVI.02418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77:3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 20.Saeed M, Scheel TK, Gottwein JM, Marukian S, Dustin LB, Bukh J, Rice CM. 2012. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells. Antimicrob Agents Chemother 56:5365–5373. doi: 10.1128/AAC.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 22.Barnes-Seeman D, Boiselle C, Capacci-Daniel C, Chopra R, Hoffmaster K, Jones CT, Kato M, Lin K, Ma S, Pan G, Shu L, Wang J, Whiteman L, Xu M, Zheng R, Fu J. 2014. Design and synthesis of lactam-thiophene carboxylic acids as potent hepatitis C virus polymerase inhibitors. Bioorg Med Chem Lett 24:3979–3985. doi: 10.1016/j.bmcl.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 23.Hung M, Gibbs CS, Tsiang M. 2002. Biochemical characterization of rhinovirus RNA-dependent RNA polymerase. Antiviral Res 56:99–114. doi: 10.1016/S0166-3542(02)00101-8. [DOI] [PubMed] [Google Scholar]
- 24.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. 2004. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64:985–993. doi: 10.1158/0008-5472.CAN-03-1101. [DOI] [PubMed] [Google Scholar]
- 25.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 26.Eakins J, Bauch C, Woodhouse H, Park B, Bevan S, Dilworth C, Walker P. 2016. A combined in vitro approach to improve the prediction of mitochondrial toxicants. Toxicol In Vitro 34:161–170. doi: 10.1016/j.tiv.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad T, Yin P, Saffitz J, Pockros PJ, Lalezari J, Shiffman M, Freilich B, Zamparo J, Brown K, Dimitrova D, Kumar M, Manion D, Heath-Chiozzi M, Wolf R, Hughes E, Muir AJ, Hernandez AF. 2015. Cardiac dysfunction associated with a nucleotide polymerase inhibitor for treatment of hepatitis C. Hepatology 62:409–416. doi: 10.1002/hep.27488. [DOI] [PubMed] [Google Scholar]
- 28.Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. 2011. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol 54:773–794. doi: 10.1016/j.jhep.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Cheville NF. 1994. Ultrastructural pathology: an introduction to interpretation. Iowa State University Press, Ames, IA. [Google Scholar]