

Abstract
Only one new class of antifungal drugs has been introduced into clinical practice in the last 30 years, and thus the identification of small molecules with novel mechanisms of action is an important goal of current anti-infective research. Here, we describe the characterization of the spectrum of in vitro activity and in vivo activity of AR-12, a celecoxib derivative which has been tested in a phase I clinical trial as an anticancer agent. AR-12 inhibits fungal acetyl coenzyme A (acetyl-CoA) synthetase in vitro and is fungicidal at concentrations similar to those achieved in human plasma. AR-12 has a broad spectrum of activity, including activity against yeasts (e.g., Candida albicans, non-albicans Candida spp., Cryptococcus neoformans), molds (e.g., Fusarium, Mucor), and dimorphic fungi (Blastomyces, Histoplasma, and Coccidioides) with MICs of 2 to 4 μg/ml. AR-12 is also active against azole- and echinocandin-resistant Candida isolates, and subinhibitory AR-12 concentrations increase the susceptibility of fluconazole- and echinocandin-resistant Candida isolates. Finally, AR-12 also increases the activity of fluconazole in a murine model of cryptococcosis. Taken together, these data indicate that AR-12 represents a promising class of small molecules with broad-spectrum antifungal activity.
INTRODUCTION
The treatment of life-threatening invasive fungal infections (IFIs) remains a significant challenge to modern medicine (1, 2). Two primary factors contribute to the difficult nature of IFI therapy. First, most IFIs affect people with compromised immune function, and therefore, IFI patients are more reliant on the efficacy of the antifungal drug than immunocompetent patients. Second, the development of effective antifungal drugs is difficult, because many fundamental biological processes are highly conserved between fungi and humans (3). Consequently, identifying molecules that kill the pathogen and spare the host is very challenging. Currently, only three primary classes of antifungal drugs are in clinical use: (i) azole ergosterol biosynthesis inhibitors (e.g., fluconazole [FLU]), (ii) polyene ergosterol binding agents (e.g., amphotericin B), and (iii) echinocandin 1,3-β-d-glucan synthase inhibitors (e.g., caspofungin). The number of antifungal drugs pales in comparison to the number of distinct classes of antimicrobial agents, and, in fact, there are currently more classes of antiretroviral agents available for the treatment of HIV/AIDS than classes of antifungal drugs (2).
The pace of antifungal drug development has been extremely slow. For example, the most recent addition to the antifungal pharmacopeia, the echinocandins, was discovered in the early 1970s and introduced into clinical practice in the early 2000s (3). Furthermore, no new drugs for the treatment of cryptococcal meningitis, one of the most important causes of infectious disease-related deaths in HIV/AIDS patients (4), have been introduced into clinical practice in more than 20 years. In fact, the current gold standard therapy is based on drugs (amphotericin B and 5-flucytosine) that are 50 years old (5). In principle, the low numbers of drugs and the slow pace of development would not be an issue if the current therapies were highly effective. Unfortunately, mortality rates associated with IFIs range from 20% to 100%, depending on the organism and the immune status of the host (1). Clearly, the pace of new antifungal drug development needs to increase to meet the current need.
As part of a program to identify and repurpose protein kinase inhibitors as potential antifungal drugs, we found that the anticancer clinical candidate OSU-03012/AR-12 is active against Candida albicans and Cryptococcus neoformans in vitro with a MIC of 4 μg/ml (6). In addition, we have shown that AR-12 is synergistic with FLU against C. neoformans (7) and is active against C. albicans biofilms (6). AR-12 is reported to be an inhibitor of the conserved protein kinase PDK1 in humans and other mammals (8). Reports from multiple groups, including our own, indicate that PDK1 may not be the target for AR-12 (9, 10). Indeed, we have recently found that AR-12 inhibits acetyl coenzyme A (acetyl-CoA) synthetase (Acs) in yeast species and that this mode of action is an important mechanism for its antifungal activity (10). Acs generates acetyl-CoA from acetate and coenzyme A in an ATP-dependent reaction (11). In most yeast species, acetate represents an important carbon source for acetyl-CoA generation (12), and thus Acs activity has been found to be essential in C. albicans (13) and required for virulence in C. neoformans (14). In contrast, the enzyme that generates the vast majority of acetyl-CoA in humans is ATP-citrate lyase (15). Recently, a number of studies have shown that cancer cells have increased dependence on acetate and that Acs activity correlates with outcomes in some cancer types (16–18). Based on these findings, Acs has been proposed as a possible anticancer target (16, 19). We propose that these same considerations indicate that Acs may also represent a potential antifungal target.
AR-12 is derived from the cyclooxygenase 2 (Cox2) inhibitor celecoxib and has been evaluated in phase I clinical trials as an anticancer agent (8; Clinical Trials registration no. NCT00978523). As such, it has a number of favorable pharmacological properties. In addition, serum levels of AR-12 that are comparable to the vitro MICs for fungi were achieved in humans and mouse models (20). Encouraged by these observations, we characterized the in vitro and in vivo antifungal activity of AR-12.
MATERIALS AND METHODS
Strains and media.
The clinical strains used for characterizing the activity of AR-12 against pathogenic fungi were from the collection at the Fungus Testing Laboratory, University of Texas Health Center at San Antonio. C. albicans strain SC5314 was obtained from Gus Haidaris (University of Rochester), and Cryptococcus neoformans strain H99 was a gift of Joseph Heitman (Duke University). C. albicans clinical isolates TWO7229, TWO7230, TWO7241, and TWO7243 were obtained from Ted White (UMKC). C. albicans clinical isolates (10B1A3A and 11A8A2A) and SC5314 derivatives containing gain-of-function alleles (mrr1P683S and tac1G980E) were obtained from P. David Rogers (University of Tennessee). C. albicans strains NC1010 and NCO-788, as well as C. glabrata strains NC1, NC102, and NC999, were obtained from Neil Clancy and Ryan Shields (University of Pittsburgh). SN250 and the mrr1Δ/Δ and tac1Δ/Δ derivatives were obtained from the Fungal Genetics Stock Center. Yeast medium was prepared using standard recipes. All yeast incubations were done at 30°C unless otherwise indicated.
In vitro antifungal susceptibility assays.
MIC values were determined using standard CLSI methods except for those noted for Histoplasma capsulatum, which were also performed using a recently published protocol (21). Antifungal susceptibility testing was performed in the Fungus Testing Laboratory by use of the CLSI M27-A3 and M38-A2 methods for yeast and molds, respectively. The interaction of AR-12 with fluconazole was characterized by checkerboard assays using CLSI M27-A3 methods. The fractional inhibitory concentration index was calculated as described previously (22). Pneumocystis susceptibility testing was performed as previously described (13).
Quantitative RT-PCR analysis of efflux pump expression.
The strains were grown overnight in yeast extract-peptone-dextrose (YPD) at 30°C and back-diluted to an optical density at 600 nm (OD600) of 0.1 in YPD. The cultures were treated with subinhibitory concentrations of AR-12 (4 μg/ml) in dimethyl sulfoxide (DMSO) (1%) or DMSO alone and incubated for 3 h at 30°C. The cells were harvested by centrifugation, and RNA was isolated using an Ambion Ribopure yeast kit. The RNA was processed for SYBR green reverse transcription (RT)-PCR using the iScript kit. Primers for the targets and conditions for RT-PCR were exactly as previously reported (23). Expression was internally normalized to ACT1 and compared to other strains or conditions using the ΔΔCT method.
Microscopy.
For assessment of the effect of AR-12 on mitochondrial activity, Saccharomyces cerevisiae BY4741 cells were grown to log phase, treated with AR-12 at concentrations indicated in the text for 4 h, and harvested. The mitochondria were visualized with MitoTracker red (Molecular Probes) by following previously described protocols (24). Lipid droplet characterization was performed using Nile red staining as described previously (25). Chitin staining was performed with calcofluor white (CFW) as described in reference 26. Images were collected with a Nikon ES80 epifluorescence microscope equipped with a CoolSnap charge-coupled device (CCD) camera using NIS-Elements software with constant exposure settings and were processed in the same way in PhotoShop. The percentage of cells showing clearly staining mitochondria was determined by counting at least 100 cells per replicate for two independent experiments. Propidium iodide staining was performed as previously described (27).
Glucose-induced acidification of culture medium by S. cerevisiae.
By following methods described by Soteropoulos et al. (28), logarithmic-stage S. cerevisiae BY4741 cells were harvested, suspended in 0.1 M KCl, and incubated at 30°C for 1 h to starve the cells of glucose. The suspensions were then stored overnight at 4°C at an OD600 of 2.3. The reaction mixtures contained 20 μl of cell suspension in 155 μl of buffer (0.1 M KCl, 50 μg/ml bromophenol blue, pH 5) with either 1% DMSO alone or the indicated amount of AR-12 in 1% DMSO. Medium acidification was initiated by the addition of 20 μl of 20% (wt/vol) glucose. The progress of the reaction was followed by measurement of A590 on a plate reader at 30-min intervals over a 4-h time course. The reduction in A590 for the untreated cells over this time course was essentially identical to that reported by Soteropoulos et al. (28).
Isolation and analysis of AR-12-resistant mutants.
S. cerevisiae strain BY4741 was passaged in culture flasks containing YP plus 2% acetate starting at 1 μg/ml (0.125× MIC; MIC, 4 μg/ml) and increasing 2-fold to 64 μg/ml. The flasks were incubated until saturated. Samples were harvested and used to inoculate a flask at a 2-fold-higher concentration in AR-12. At concentrations above the MIC (16, 32, and 64 μg/ml), a dilution of the culture was plated on YPD and single colonies were isolated. The colonies were passaged twice on nonselective YPD plates. Colonies from the passaged plates were incubated in microtiter plates containing the AR-12 concentration from which they were isolated and compared to the parental strain. Resistant isolates were then compared to the parental strain by growth curve analysis in the presence and absence of AR-12. Three isolates showed reproducible, stable resistance to AR-12 and were analyzed by whole-genome resequencing. Sequenced reads were cleaned according to a rigorous preprocessing workflow (Trimmomatic-0.32) before they were mapped to the S. cerevisiae genome (SGD) (EF4) with SHRiMP2.2.3 (http://compbio.cs.toronto.edu/shrimp/). SAMtools-1.0 and BCFtools-1.0 were used to perform the single-nucleotide polymorphism (SNP) and indel calling. A filter for low coverage (10×) and low quality (Clinical Trials registration no. NCT00978523) was then applied and marked in the filter column as either “Pass” or “LowQual.” Nonsynonymous mutations in confirmed open reading frames (ORFs) were identified using the UCSC Genome Bioinformatics website (http://genome.ucsc.edu) and the SGD (http://www.yeastgenome.org/). The relative susceptibilities of the mutants and reference strains to AR-12 and fluconazole were determined by incubating the strains in the presence of AR-12 overnight and then plating a 10-fold dilution series on nonselective YPD media to compare the number of viable cells in each culture.
Mouse model of disseminated cryptococcosis.
Male AJ/Cr mice (20 to 25 g) were purchased from the Frederick National Laboratory for Cancer Research (NCI, Frederick, MD). Animals were housed in the University of Rochester Medical Center vivarium and allowed food ad libitum. On day 0, mice were inoculated via lateral tail vein injection with 4.5 × 104 CFU/animal of C. neoformans H99 in phosphate-buffered saline (PBS) (100 μl). Beginning 24 h after inoculation and continuing daily for days 2 to 6, mice were sham treated (n = 8), treated with FLU by intraperitoneal injection (10 mg/kg in PBS; n = 8), treated with AR-12 by oral gavage (100 mg/kg suspended in PBS with 0.5% methylcellulose–0.1% Tween 80; n = 8), and treated with both FLU and AR-12 (n = 8). On day 7, all mice were euthanized, after which the brains were removed and homogenized in YPD (2 ml). Serial dilutions of the homogenates were inoculated on YPD agar plates containing vancomycin (10 μg/ml) and gentamicin (100 μg/ml). The number of CFU per gram of brain tissue was calculated and transformed into log10 units, and the differences between groups were analyzed by analysis of variance (ANOVA); statistical significance was set at a P value of <0.05 (SigmaPlot software). The experiment was performed twice with similar results for each replicate.
RESULTS
AR-12 has broad-spectrum antifungal activity against pathogenic yeasts, molds, dimorphic fungi, and Pneumocystis.
We initially identified AR-12 (Fig. 1) as an antifungal molecule by screening a series of molecules with activity against human PDK1 (6); as discussed above, subsequent studies have led to the conclusion that the biological activity of AR-12 is unlikely to be due to PDK1 inhibition. AR-12 is fungicidal against both C. albicans and C. neoformans (MIC or minimal fungicidal concentration [MFC], 4 μg/ml for both organisms), synergistic with FLU against C. neoformans, and has activity against C. albicans biofilms within 2-fold of its MIC toward planktonic cells (6, 7). The initial studies of AR-12 antifungal activity were limited to the standard laboratory strains of C. albicans (SC5314) and C. neoformans (H99, K99, JEC21). To further confirm and validate this activity, we tested a set of clinical isolates of C. albicans, non-albicans Candida spp., and C. neoformans (Table 1). Consistent with previous results, AR-12 is active against multiple clinical isolates of C. albicans (MIC, 4 μg/ml), including those with decreased susceptibility to FLU. AR-12 is similarly active against non-albicans Candida spp. (Table 1), including the relatively FLU-resistant species C. glabrata and C. krusei as well as C. parapsilosis, C. dubliniensis, and C. tropicalis. Finally, clinical isolates of Cryptococcus neoformans var. grubii were as susceptible as the standard laboratory strains. Consequently, AR-12 has consistent activity against clinical isolates of pathogenic yeast species. The MIC for these strains (4 μg/ml) is similar to AR-12 serum concentrations (8 μM or 3.7 μg/ml) observed in phase I clinical trials (S. Proniak, personal communication).
FIG 1.
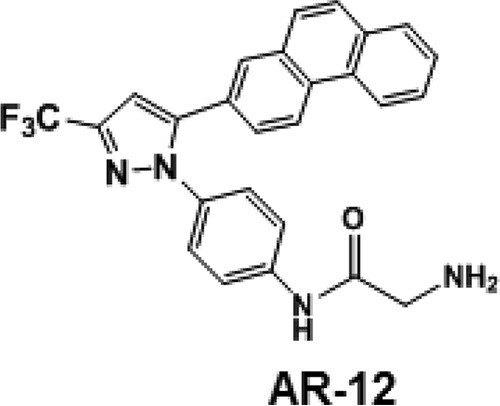
Chemical structure of AR-12.
TABLE 1.
MICs of AR-12 against fungal pathogens
| Species | No. of isolates | AR-12 MIC (μg/ml) | MIC range (μg/ml)a |
||
|---|---|---|---|---|---|
| FLU | VOR | POS | |||
| Candida albicans | 10 | 2–4 | 0.5 to >254 | ND | ND |
| C. glabrata | 14 | 4 | 2–64 | ND | ND |
| C. parapsilosis | 8 | 2–4 | 2–4 | ND | ND |
| C. tropicalis | 1 | 4 | ND | ND | ND |
| C. krusei | 1 | 4 | ND | ND | ND |
| C. guilliermondii | 7 | 2–4 | 1–8 | ND | ND |
| Cryptococcus neoformans | 4 | 4 | 2–64 | ND | ND |
| Fusarium oxysporum | 2 | 4 | ND | 16 | ND |
| F. solani | 1 | 4 | ND | ND | 8 |
| Scedosporium apiospermum | 2 | 2 | ND | 0.5–16 | ND |
| Paecilomyces variotii | 1 | 4 | ND | 0.25 | 0.125 |
| Lomentospora prolificans | 1 | 2 | ND | ND | 16 |
| Rhizopus oryzae | 6 | 4 | ND | ND | 0.125–1 |
| Apophysomyces | 2 | 4 | ND | ND | 0.125–1 |
| Blastomyces dermatiditis | 3 | 0.5–2b | ND | 0.03–0.125 | ND |
| Coccidioides immitis | 3 | 4–8 | ND | 0.03–0.125 | ND |
| Histoplasma capsulatum | 4 | 0.25–1b | ND | 0.03–0.125 | ND |
VOR, voriconazole; POS, posaconazole; ND, not determined.
The AR-12 MIC for these species is consistently 4 μg/ml based on a newly developed assay for H. capsulatum and B. dermatiditis (21).
One of the most pressing unmet clinical needs in medical mycology is the development of agents with activity against difficult-to-treat molds that cause aspergillosis, hyalohyphomycoses, phaeohyphomycoses, and mucormycosis. Therefore, we tested the activity of AR-12 against a panel of medically relevant molds (Table 1). AR-12 is active against Aspergillus fumigatus, and this activity is further characterized elsewhere. Mucormycosis has emerged as an increasingly prevalent infection, particularly within severely immunocompromised patients (29). AR-12 has activity against the most common cause of mucormycosis, Rhizopus oryzae, as well as two out of four isolates of the less commonly isolated species Apophysomyces. Except for the two isolates noted, the AR-12 MIC against all isolates was 4 μg/ml, which is lower than that observed for posaconazole.
AR-12 is also active against molds that cause phaeohyphomycosis (Table 1), including Fusarium solani and Fusarium oxysporum, Scedosporium apiospermum, Paecilomyces, and Lomentospora prolificans. Notably, AR-12 is more active against species that cause fusariosis than voriconazole, the recommended therapy for this difficult-to-treat mold infection. It is also more active than voriconazole against L. prolificans but less active against S. apiospermum and Paecilomyces. We also tested the activity of AR-12 against three medically important dimorphic fungi. Consistent with its activity against the other organisms, AR-12 showed activity in the 2- to 4-μg/ml range against H. capsulatum, Blastomyces dermatitidis, and Coccidioides spp. The activity against H. capsulatum varied somewhat with the methodology; the MIC obtained with a recently developed method optimized for the yeast phase rather than hyphae was higher, with consistent values of 4 μg/ml (21).
Finally, we determined the activity of AR-12 against Pneumocystis, an opportunistic fungal pathogen not susceptible to standard antifungal agents. Mammals are infected by host-specific species of Pneumocystis which are not culturable in vitro under normal laboratory conditions. Therefore, AR-12 was tested against rat (Pneumocystis carinii)- and mouse (Pneumocystis murina)-specific species using an ATP-based viability assay (13). AR-12 showed moderate activity against both species, with 72-h 50% inhibitory concentrations (IC50s) of 4.8 μg/ml for P. carinii and 1.8 μg/ml for P. murina. IC50s for pentamidine, an agent used for treatment and prophylaxis, are typically less than 1 μg/ml (30). AR-12 has a remarkably broad spectrum of antifungal activity at concentrations that are similar to those obtained in humans during a phase I anticancer clinical trial. These data strongly suggest that AR-12 and molecules of this class may be promising candidates for additional development and optimization.
AR-12 modulates the susceptibility of Candida strains with decreased fluconazole susceptibility.
The survey of C. albicans clinical isolates indicated that AR-12 is active against fluconazole (FLU)-resistant isolates or isolates with reduced FLU susceptibility. In order to explore this observation in more detail, we tested the activity of AR-12 against FLU-resistant isolates that had been characterized with respect to the mechanism of reduced susceptibility to FLU (31). We first examined the activity of AR-12 toward a set of strains isolated from a single AIDS patient over the course of FLU therapy (Table 2, entries 2 to 5). Over this time, the FLU MIC of the isolates increased. White characterized these strains and found that TWO7230 had increased MDR expression, TWO7241 added increased ERG11 expression, while the final isolate (TWO7243) also showed increased CDR expression (31). The MIC for AR-12 against all four isolates tested (4 μg/ml), including the strain with a FLU MIC of 128 μg/ml, was identical to those for FLU-susceptible strains (Table 2, entries 2 to 5). This indicates that AR-12 is active against C. albicans strains that are resistant to FLU by common molecular mechanisms. To further confirm this observation, we tested AR-12 against C. albicans clinical isolates with gain-of-function mutations in two transcription factors that regulate efflux pump expression in C. albicans MRR1 and TAC1 mutants (23). As shown in Table 2 (entries 6 and 7), the AR-12 MIC was unchanged from that of the reference strain (4 μg/ml). Similar results were obtained for derivatives of SC5314 in which the gain-of-function alleles had been introduced genetically (Table 2, entries 8 and 9). The deletion of neither TAC1 nor MRR1 affected the susceptibility of strains to AR-12 (Table 2, entries 10 and 11). A strain with a gain-of-function mutation in UPC2 (32), a regulator of sterol and fatty acid biosynthesis, is slightly more susceptible to AR-12 (Table 2, entry 18), while the UPC2 homozygous deletion mutant has the same susceptibility as the parental strain (Table 2, entries 17 and 19). These data indicate that AR-12 susceptibility is not affected by alteration in the expression of the efflux pumps that mediate FLU resistance or increased expression of ergosterol biosynthesis genes.
TABLE 2.
Fractional inhibitory concentrations for AR-12 and FLU against C. albicans and C. glabrata strains
| Entry | Species | Strain or mutanta | MORb | MIC (μg/ml)c |
FICId | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AR-12 | CAS | FLU | AR-12 comb | CAS comb | FLU comb | |||||
| 1 | C. albicans | SC5314 | 1 | 4 | NA | 0.25 | 2 | NA | 0.125 | 1 |
| 2 | C. albicans | TWO7229 | 1 | 4 | NA | 0.25 | 2 | NA | 0.125 | 1 |
| 3 | C. albicans | TWO7230 | 2 | 4 | NA | 2 | 2 | NA | 0.5 | 0.75 |
| 4 | C. albicans | TWO7241 | 2,3 | 4 | NA | 16 | 2 | NA | 0.5 | 0.53 |
| 5 | C. albicans | TWO7243 | 2,3 | 4 | NA | 128 | 2 | NA | 16 | 0.63 |
| 6 | C. albicans | 10B1A3A | 2 | 2 | NA | 32 | 1 | NA | 2 | 0.56 |
| 7 | C. albicans | 11A8A2A | 2 | 2 | NA | 4 | 1 | NA | 1 | 0.75 |
| 8 | C. albicans | SCmrr1P683S mutant | 2 | 2 | NA | 4 | 1 | NA | 2 | 1 |
| 9 | C. albicans | SCtac1G980E mutant | 2 | 4 | NA | 2 | 2 | NA | 1 | 1 |
| 10 | C. albicans | SNmrr1Δ/Δ mutant | NA | 4 | NA | 2 | 0.25 | NA | 0.5 | 0.31 |
| 11 | C. albicans | SNtac1Δ/Δ mutant | NA | 2 | NA | 2 | 0.25 | NA | 0.5 | 0.38 |
| 12 | C. albicans | NC1010 | 4 | 4 | 16 | NA | 2 | 4 | NA | 0.75 |
| 13 | C. albicans | NCO-788 | 4 | 6 | 64 | NA | 2 | 4 | NA | 0.56 |
| 14 | C. glabrata | NC1 | 4 | 4 | 4 | NA | 1 | 1 | NA | 0.50 |
| 15 | C. glabrata | NC102 | 4 | 8 | 64 | NA | 4 | 0.03 | NA | 0.50 |
| 16 | C. glabrata | NC999 | 4 | 8 | 32 | NA | 2 | 2 | NA | 0.31 |
| 17 | C. albicans | SN250 | 1 | 4 | NA | 1 | 2 | NA | 0.5 | 1 |
| 18 | C. albicans | SCupc2GOF mutant | 5 | 2 | NA | 16 | 2 | NA | 16 | 2 |
| 19 | C. albicans | SNupc2Δ/Δ mutant | NA | 4 | NA | 0.125 | 2 | NA | 0.06 | 0.98 |
Clinical isolates are in bold; mutants derived from SC5314 have SC prefixes; derivatives of SN250 have SN prefixes.
MOR, mechanism of resistance: 1, none; 2, elevated efflux pump expression; 3, elevated ERG11 expression; 4, FKS hot spot mutation; 5, UPC2 gain-of-function mutation. NA, not applicable.
CAS, caspofungin; FLU, fluconazole; comb, combination.
FICI, fractional inhibitory concentration index.
The emergence of azole-resistant Candida strains is an increasingly troubling clinical problem (33). One potential approach to addressing this problem is to identify adjuvants that modulate FLU susceptibility (34). Therefore, we examined the activity of AR-12 combined with FLU against C. albicans and C. glabrata strains with altered FLU susceptibility. In standard checkerboard assays with SC5314 and the initial isolate of the resistant series, AR-12 and FLU are additive, with a fractional inhibitory concentration index (FICI) of 1 (Table 2, entries 1 and 2). As shown in Table 2 (entries 3, 4, and 5), a subinhibitory concentration of AR-12 (2 μg/ml) reduced the FLU concentration required to inhibit growth of FLU-resistant strains between 4- and 32-fold. Neither TWO7241 nor TWO7243 (Table 2, entries 4 and 5) is susceptible to FLU alone (CLSI cutoff for C. albicans, 8 μg/ml; MIC for the strain, 16 μg/ml), but in the presence of a subinhibitory concentration of AR-12, the inhibitory concentration of FLU is within the susceptible range for TWO7241 (FIC of FLU, 0.5 μg/ml) and within one dilution of the cutoff for susceptibility for TWO7243.
AR-12 also modulated the FLU susceptibility of two additional FLU-resistant C. albicans clinical isolates that have been determined to contain gain-of-function mutations in MRR1 and TAC1 transcription factors regulating drug efflux pump expression (Table 2, entries 6 and 7). Similar additive FLU–AR-12 interactions were also observed with SC5314 derivatives that were generated by integrating gain-of-function alleles of MRR1 and TAC1 plasmids (Table 2, entries 8 and 9). Interestingly, deletion of either MRR1 or TAC1 increased the strength of the interaction between FLU and AR-12. Specifically, the combination of AR-12 and FLU was synergistic against both deletion mutants (Table 2, entries 10 and 11) compared to the reference strain for the SN background (Table 2, entry 10), and neither deletion mutant had significantly altered AR-12 susceptibility. Gain-of-function mutations in UPC2, a transcription factor involved in the regulation of ergosterol gene expression (32), have also been shown to mediate FLU resistance. In contrast to strains with FLU resistance mediated by altered efflux pump expression, AR-12 had no effect on the FLU susceptibility of a C. albicans strain harboring a gain-of-function mutation in UPC2 (Table 2, entry 18) nor did it affect AR-12 susceptibility. A homozygous deletion mutant of UPC2 showed no change in FICI relative to the reference strain, although it was more susceptible to FLU as expected (Table 2, entry 19). These observations indicate that AR-12 modulates efflux-mediated FLU resistance but has no effect on Upc2-associated FLU resistance.
AR-12 modulates the susceptibility of Candida strains with decreased echinocandin susceptibility.
In addition to the emergence of azole resistance in Candida spp., the rates of resistance to the echinocandins are also increasing (35). Particularly worrisome is the fact that this is most common in strains of C. glabrata, a species with high rates of azole resistance. Thus, azole/echinocandin-resistant C. glabrata strains are becoming a significant therapeutic problem that clinicians must face. The predominant mechanism of echinocandin resistance is due to the presence of so-called “hot spot” mutations in the FKS gene, which encodes the β-1,3-glucan synthase target of the echinocandins (36). We first tested whether AR-12 was active against C. albicans and C. glabrata strains harboring hot spot mutations. Because the mechanism of AR-12 is distinct from that of the echinocandins, it was not surprising to observe that the activity of AR-12 was not significantly affected by the presence of FKS hot spot mutations (Table 2, entries 10 to 14). We next examined the effect of AR-12 on the MIC of caspofungin by use of a checkerboard assay. As with FLU, subinhibitory concentrations of AR-12 modulated the activity of caspofungin (Table 2) toward echinocandin-resistant C. albicans and C. glabrata. Additive interactions were observed with three isolates (Table 2, entries 10 to 13). In all cases, subinhibitory concentrations of AR-12 reduced the caspofungin component of the FICI at least 4-fold. The strains we used contained four specific hot spot mutations: CgFKSD632Y (D-to-Y mutation at position 632 encoded by C. glabrata FKS), CgFKSF659del (deletion of F659 in CgFKS), CgFKSS663P, and CaFKSS645P. In addition, three of the tested strains were also resistant to azoles, namely, C. glabrata NC102 and NC999 and C. albicans NC1010. Thus, AR-12 also modulates the activity of caspofungin toward strains with reduced susceptibility to echinocandins.
AR-12 reduces mitochondrial function and cellular lipid content but does not inhibit efflux pump expression.
As discussed above, an important target of AR-12 is acetyl-CoA synthetase (Acs) (10). Acs plays a role in multiple cellular functions in yeast that could, in principal, affect FLU susceptibility. These include cellular energetics, gene expression through histone acetylation, and lipid biosynthesis (12). One of the most important mechanisms for the development of azole resistance in clinics is through the acquisition of gain-of-function mutations in the transcription factors (TAC1 and MRR1) that regulate the expression of three efflux pumps (MDR1, CDR1, and CDR2). For example, the SAGA/Ada complex has been shown to be important for upregulation of MDR1 expression in C. albicans (13). The SAGA complex and nuclear acetyl-CoA synthetase 2 are required for histone acetylation in yeast (37). We have shown that AR-12 blocks histone acetylation in C. albicans (10). We therefore hypothesized that AR-12 may negatively regulate the expression of the efflux pumps that mediate FLU resistance and, thereby, modulate FLU resistance. To test this hypothesis, we examined the effect of AR-12 on the expression of MDR1, CDR1, and CDR2 using quantitative RT-PCR. For this set of experiments, the reference strain SC5314 was treated with AR-12 (4 μg/ml) for 3 h; propidium iodide staining indicated that >95% of the cells were viable at the time of harvest. Under these conditions, the expression of MDR1 and CDR2 was reduced slightly or was not significantly changed relative to that of untreated cells (Fig. 2). In contrast, CDR1 expression was increased significantly (∼5-fold). AR-12 did not have a significant effect on the increased expression of MDR1, CDR1, or CDR2 in strains with gain-of-function mutations in TAC1 or MRR1. Although we cannot rule out that higher concentrations of AR-12 may have an effect on efflux pump expression, these data suggest that AR-12 does not reduce the expression of efflux pumps as part of its mechanism for FLU resistance modulation.
FIG 2.

Effect of AR-12 on expression of efflux pumps involved in fluconazole resistance. The indicated strains were treated with either DMSO or AR-12 (2 μg/ml; 1/4 MIC) for 3 h in YPD and harvested. The expression of CDR1, CDR2, and MDR1 was determined by quantitative RT-PCR. Bars show the mean fold change for AR-12-treated cells relative to untreated cells, and error bars indicate standard deviations of two or three biological replicates determined with triple technical replicates. The strain names indicate the allele of MRR1 or TAC1 integrated into the reference strain SC5314.
A second key function of Acs is to maintain proper carbon-based metabolism and energetics. Drug efflux pumps require ATP as well as appropriate electromotive forces and gradients for function (38). We therefore wondered whether disruption of the acetyl-CoA pool through inhibition of Acs activity might interfere with mitochondrial function. MitoTracker red is a dye that stains mitochondria in a manner dependent upon the presence of an appropriate proton motive force across the membrane. To examine the effect of AR-12 on the mitochondria, C. albicans SC5314 cells were treated with a range of concentrations of AR-12 and then stained with MitoTracker red. As shown in Fig. 3A and B, AR-12 exposure causes a dose-dependent reduction in MitoTracker red-stained mitochondria. This suggests that there may be a disruption in the proton motive force across the mitochondrial membrane in the presence of AR-12.
FIG 3.

AR-12 effects mitochondrial function and lipid synthesis. (A) C. albicans SC5314 was treated with DMSO or AR-12 and stained with MitoTracker red. (B) Percentage of cells with visualized mitochondria. The bars represent the mean result of two experiments with a minimum of 100 cells counted per replicate, and error bars indicate standard deviations. (C) The ability of S. cerevisiae cells to acidify the extracellular medium in response to glucose was determined in the presence and absence of AR-12 using the decreased absorbance of bromophenol blue at A590 as an indicator of decreasing pH. (D) SC5314 cells were grown overnight to stationary phase in the presence of AR-12 (8 μg/ml) or DMSO. The cells were harvested and stained with Nile red as described in Materials in Methods. The micrographs are representative of multiple fields and were taken with identical exposure and contrast settings. (E) Quantitation of the number of cells with Nile red staining. For each experiment, at least 100 cells were counted for each indicated concentration. The bars represent the mean result of two independent experiments, with error bars indicating standard deviations. Two-sided Student's t tests were used to analyze data, and an asterisk indicates a P value of <0.05 relative to untreated samples; double asterisks indicate a P value of <0.005. BF indicates bright field.
Based on this observation, we hypothesized that an AR-12-mediated reduction in mitochondrial function might lead to reduced cellular ATP and, thereby, reduced efflux pump function. If that were the case, this would not represent a specific effect on drug efflux pumps but rather would lead to a global reduction in all ATP-dependent pumps. The most abundant membrane pump in yeast is Pma1, a plasma membrane H+-ATPase that acidifies the medium in response to glucose exposure (28). To determine the effect of AR-12 on Pma1 function, we used a well-established whole-cell assay to measure the ability of starved cells to acidify the medium upon exposure to glucose (28). The lethal concentration of AR-12 under the conditions of the assay was 8 μg/ml. At 2- and 4-fold-lower concentrations than the lethal concentration of AR-12, the rate at which treated cells acidified the medium was indistinguishable from that of untreated cells (Fig. 3C). This observation is inconsistent with the hypothesis that the effect of AR-12 on fluconazole resistance is due to decreased mitochondrial function, which, in turn, indirectly leads to decreased efflux pump function. Since Pma1 function is also dependent on an intact plasma membrane and electrochemical gradients, these data suggest that AR-12 does not induce global membrane derangements which impair pump function.
Acs is also critical for fatty acid and sterol biosynthesis since it regulates the cytoplasmic pool of acetyl-CoA. Acetyl-CoA is converted to malonyl-CoA through the action of Acc1, and this provides the building block for other lipids (12). Therefore, another potential mechanism by which AR-12 might modulate fluconazole susceptibility is through an overall reduction in carbon flux through pathways required for lipid biosynthesis. A convenient measure of lipid homeostasis in yeast is the assessment of the number and clustering of lipid droplets (25); lipid droplets are organelles containing a triacylglycerol core and sterol esters and are readily detected using the vital stain Nile red. Previous studies in C. parapsilosis have shown that strains lacking genes required for fatty acid synthesis have reduced numbers of lipid droplets (39). Lipid droplets accumulate in stationary phase, and we therefore compared the number of lipid droplets in cells treated with subinhibitory concentrations of AR-12 to that in untreated cells. AR-12-treated cells show a concentration-dependent reduction in lipid droplet formation (Fig. 3D and E). This observation indicates that AR-12-treated cells are relatively lipid depleted. We propose that AR-12 may deplete the cellular pool of acetyl-CoA, which in turn reduces flux through lipid biosynthetic pathways. In this setting, lower concentrations of fluconazole or less effective inhibition may be sufficient to inhibit growth. This model is supported by the observation that the ability of AR-12 to modulate FLU susceptibility is lost in a strain with an activating UPC2 mutation. Upc2 regulates the expression of fatty acid and ergosterol biosynthesis genes, and increased activity of this pathway would be expected to compensate for the decreased availability of acetyl-CoA induced by AR-12.
AR-12 modulates the fungiolytic activity of caspofungin against echinocandin-resistant C. albicans and C. glabrata.
One of the features of the echinocandins in the treatment of candidiasis is that this class of drugs is directly fungicidal and causes the lysis of yeast cells by inhibition of cell wall synthesis. In principle, the ability of AR-12 to lower the inhibitory concentration of caspofungin in resistant strains could be due to a fungistatic or fungicidal effect. To distinguish between these possibilities, we used an assay developed in our laboratory to measure fungal cell lysis (27). The assay detects the leakage of the intracellular enzyme adenylate kinase (AK) into the culture medium that occurs when cells lose cellular integrity; fungiolytic drugs such as caspofungin induce AK release, while fungistatic drugs such as FLU do not. As shown in Fig. 4A, concentrations of AR-12 and caspofungin that do not cause lysis by themselves lead to cell lysis in combination. This indicates that AR-12 reduces the concentration at which caspofungin causes yeast cell lysis.
FIG 4.
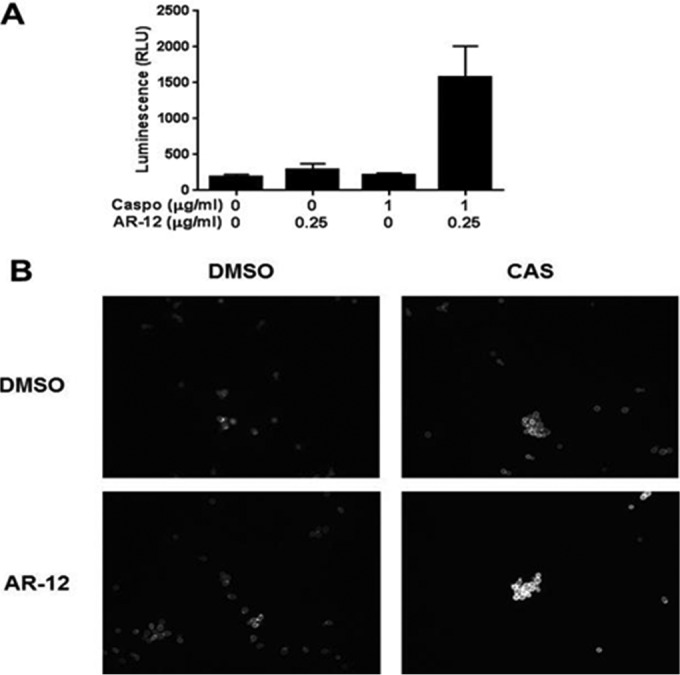
AR-12 restores fungiolytic activity of caspofungin against echinocandin-resistant C. albicans and synergizes with caspofungin to cause cell wall stress. (A) Echinocandin-resistant C. albicans strain NC1010 was grown into exponential phase overnight in YPD and treated with the indicated concentrations of AR-12 and caspofungin (Caspo) for 5 h. The amount of adenylate kinase released into the medium was measured as an indicator of cell lysis (see Materials and Methods). The mean raw luminescence from the AK reaction (biological duplicates measured in technical triplicate) is represented by bars, with standard deviations represented by error bars. RLU, relative light units. (B)The cells were exposed as described for panel A. The cells were harvested, fixed, and stained with calcofluor white as described in Materials and Methods. CAS, caspofungin.
In principle, AR-12 could either interfere with cell wall biosynthesis or inhibit the cell wall stress response induced by caspofungin. To explore these possibilities, we examined the effect of the molecules on cell wall chitin content using the microscopy-based calcofluor white (CFW) staining assay developed by Walker et al. (26). In the presence of cell wall stress, the cell wall integrity signaling pathway as well as other stress response pathways leads to increased cell wall chitin. Molecules or mutations that cause cell wall stress lead to increased cell wall chitin and CFW staining; in contrast, perturbations of the cell wall stress response blunt the increase in chitin. Doses of AR-12 and caspofungin that individually do not alter cell wall chitin staining, in combination, in contrast, lead to a dramatic increase in chitin staining relative to that of DMSO-treated cells (Fig. 4B). These observations suggest that the combination of AR-12 and caspofungin synergize to cause increased cell wall stress and, ultimately, cell lysis.
Spontaneous resistance to AR-12 occurs at a low rate and may require multiple mutations.
An important characteristic of any new anti-infective small molecule is the rate at which resistant mutants emerge. We therefore plated C. albicans, C. neoformans, and S. cerevisiae cells on solid agar containing inhibitory concentrations of AR-12. Although apparently resistant colonies occasionally emerged, none of these were stably resistant to AR-12 based on the fact that passaging them on nonselective medium led to strains with wild-type (WT) susceptibility for all isolates. From these experiments, we estimate the rate of spontaneous mutation to be <107.
As an alternative approach (40), we serially passaged S. cerevisiae BY4741 in increasing concentrations of AR-12 (0.5× and 16× MIC). Three strains that showed stable (Fig. 5A), reproducible resistance to AR-12 were isolated: two were isolated at 16 μg/ml (16-14 and 16-77) and one at 64 μg/ml (64-77). As shown in Fig. 5A, strains 16-77 and 64-77 were viable at 64 μg/ml while strain 16-77 was tolerant to 16 μg/ml. All three strains were slightly less susceptible to high concentrations of FLU, suggesting that the mutations may not be specific to AR-12 (Fig. 5B). Backcross of the mutant strains to a wild type of the opposite mating type led to diploid derivatives with susceptibility identical to that of the diploid derived from two wild-type strains, indicating that the AR-12 resistance phenotype was recessive (Fig. 5C). The AR-12-resistant haploid strains as well as the parental wild type were analyzed by whole-genome sequencing to identify mutations.
FIG 5.
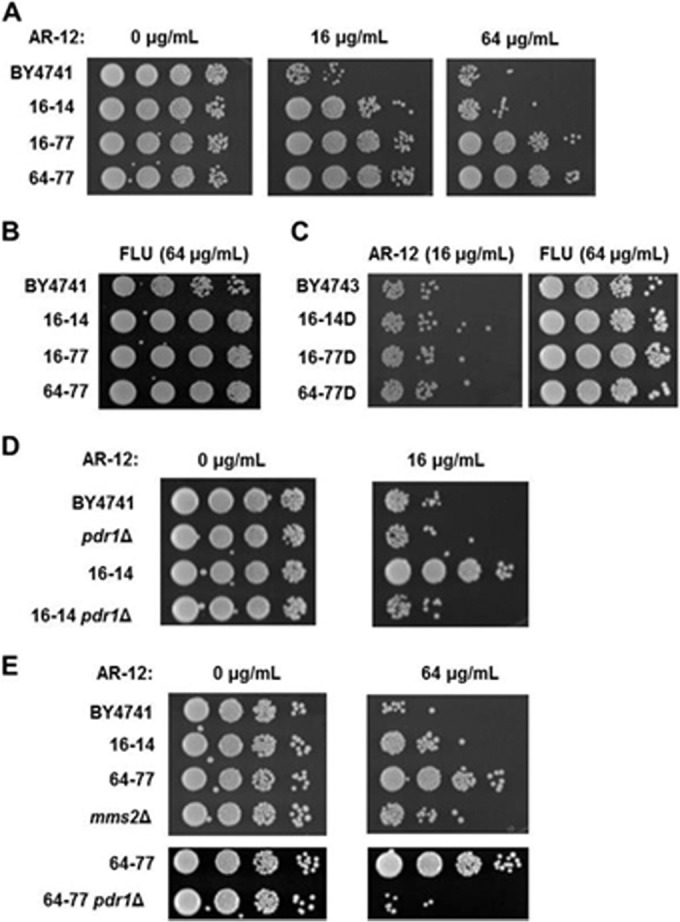
Serial passaging of S. cerevisiae in increasing concentrations of AR-12 yields resistant strains with multiple mutations. (A to E) The viability of the indicated strains was determined by harvesting cells from liquid cultures containing the indicated concentrations of AR-12 or FLU (fluconazole) and spotting a 10-fold dilution series on YPD plates. BY4743 or strain name followed by a delta denotes a diploid derived from mating the indicated strain with BY4742.
As summarized in Table 3, the resistant strains contained multiple nonsynonymous mutations. Consistent with the sequential passaging from a single ancestor clone, the strains all contained the same mutations in YPR137-B, a transposon element, and TRA1, a gene encoding an essential scaffold protein in the SAGA complex. In addition, strains 16-14 and 64-77 contained identical mutations in PDR1, which encodes a transcriptional regulator of drug efflux (41), and in SVL3, which encodes a vacuolar protein. Strain 16-77 contains a distinct mutation in PDR1. The PDR1 mutations are in the activation domain of the protein (41). Mutations in this region of Pdr1 are associated with decreased susceptibility to FLU. However, the three strains with PDR1 mutations show only modestly decreased FLU susceptibility (Fig. 5B); the decreased FLU susceptibility is also recessive (Fig. 5C). Strain 64-77, which appears closely related to strain 16-14, has a mutation in the start codon of MMS2, which encodes a ubiquitin ligase involved in DNA repair. MMS2 is one of the relatively few genes in the S. cerevisiae genome containing an intron. Broomfield et al. have shown that translation of MMS2 from the alternative start codon leads to a nonfunctional protein (42), indicating that the MMS2 mutation in strain 64-77 should lead to a loss of Mms2 function.
TABLE 3.
Nonsynonymous mutations in AR-12-resistant S. cerevisiae strains
| Gene | Chromosome/position | Codon changea | Amino acid change | Presence of mutation in strain: |
||
|---|---|---|---|---|---|---|
| 16-14 | 16-77 | 64-77 | ||||
| TRA1 | VIII/304575 | AAC-AAA | N605K | + | + | + |
| YPR137-C | XVI/805261 | GTC-ATG | V1670 M | + | + | + |
| YPR137-C | XVI/805267 | CAC-TAC | H1668Y | + | + | + |
| SVL3 | XVI/489657 | GGC-AGC | G570S | + | − | + |
| YIL169C | IX/25418 | GAT-GGT | D230G | + | + | − |
| PDR1 | VII/469717 | TCT-TAT | S861Y | + | − | + |
| PDR1 | VII/469849 | ACA-AAA | T817K | − | + | − |
| KCC4 | III/81761 | AGT-AAT | S867N | + | − | − |
| WHI2 | XV/411309 | TCA-TAA | S147Stop | + | − | − |
| YLL066W-B | VIII/5683 | CCA-ACA | P27T | − | + | − |
| YRF1-6 | VIII/5871 | TTA-TCA | L27S | − | + | − |
| MMS2 | VII/346902 | ATG-ATT | M1I | − | − | + |
| HPF1 | VIII/31103 | GAA-GCA | E169A | − | − | + |
Nucleotide changes are indicated by underlining.
We assessed the contributions of PDR1 and MMS2 mutations to the resistant phenotypes of the isolated mutants. First, we performed an exhaustive search of the literature for similar mutations in PDR1. This search revealed that Anderson et al. had previously isolated PDR1 mutations identical to one of the AR-12-induced mutations, T817K, and only one residue away from S861Y, at C862W (43). These PDR1 mutations were isolated as fixed mutations from evolution experiments using low-dose FLU (16 μg/ml). Both of the mutations were shown to lead to increased expression of the efflux pump Pdr5 and, consistent with our observations, modestly decreased FLU susceptibility. High-level FLU resistance with strains harboring these PDR1 mutations was found to require the presence of at least one additional mutation, but that mutation was not identified by Anderson et al. (43). Finally, these mutations were also found to be recessive in that hybrids derived from crossing with parental WT strains showed susceptibility identical to that of WT diploids (43). Consistent with the observations of Anderson et al., we also found that the hybrids of our AR-12-resistant strains lost their resistance to both AR-12 and FLU (Fig. 5C).
These observations suggested that PDR1 mutations may contribute to AR-12 resistance in our isolates. To test this hypothesis, we deleted PDR1 in strain 16-14 as well as in the parental strain and compared their susceptibilities to AR-12. Deletion of PDR1 in the parental strain BY4741 did not significantly change AR-12 susceptibility (Fig. 5D). In contrast, strain 16-14 pdr1Δ had the same susceptibility as both BY4741 and the pdr1Δ derivative of BY4741. This indicates that the PDR1 mutation mediates a significant portion of the AR-12 resistance of 16-14. Strain 64-77 has the same pdr1S861Y mutation as strain 16-14 but is significantly more resistant than 16-14 (Fig. 4A). This indicates that pdr1S861Y is necessary but not sufficient to induce high-level resistance to AR-12. The difference between the 16-14 and 64-77 genotypes is that 64-77 contains two additional mutations, mms2M1I and hpf1E169A. As noted above, the mms2 mutation removes the normal start codon and leads to the production of a nonfunctional protein from an alternative start codon (42). We hypothesized that this mutation may contribute to the high-level resistance of 64-77. To explore this possibility further, we characterized the role of the MMS2 mutation in resistance to AR-12 by comparing the susceptibility of a MMS2 deletion strain to that of 64-77 and the parental BY4741. As shown in Fig. 4E, the mms2Δ mutant is less susceptible to AR-12 than the parental strain BY4741 but is more susceptible than strain 64-77. These data suggest that the mms2M1I mutation contributes to the AR-12 resistance of strain 64-77. However, loss of Mms2 function does not recapitulate the resistance of 64-77, indicating that the PDR1 mutation is required. Consistent with that hypothesis, deletion of PDR1 in 64-77 abolishes AR-12 resistance. Taken together, these data are consistent with a model whereby high-level resistance to AR-12 can emerge through a PDR-mediated mechanism but appears to require the presence of additional mutations.
AR-12 improves the activity of fluconazole in a murine model of disseminated cryptococcosis.
Since AR-12 has been evaluated in a phase I clinical trial as a potential cancer therapy (Clinical Trials registration no. NCT00978523) and this molecule represents a novel structural class of antifungal agents, we examined its activity in a mouse model as an initial proof-of-principle evaluation of the scaffold. Preclinical studies with mice indicated that serum concentrations up to 1.5 μg/ml were obtained after a single oral dose of 40 mg/kg (S. Proniak, personal communication). Based on our in vitro data and these pharmacokinetics/pharmacodynamics (PK/PD) data, it seemed that the combination of FLU and AR-12 would be the most likely to show efficacy in the mouse model. One of the most pressing unmet clinical needs in medical mycology is the identification of an oral, fungicidal therapy for cryptococcal meningitis (4, 44). As such, there is interest in developing novel molecules as stand-alone therapies as well as adjuvants that yield fungicidal combinations with FLU (22). With these considerations in mind, we tested the efficacy of AR-12 in combination with FLU in a mouse model of cryptococcosis as a proof-of-principle test of whether the in vitro antifungal activity of AR-12 could be observed in vivo.
In accordance with an established model of disseminated cryptococcosis, mice were inoculated with C. neoformans H99 by tail vein inoculation, a mode that rapidly establishes a central nervous system infection (45). One day after inoculation, the mice were treated with carrier, FLU (10 mg/kg), AR-12 (100 mg/kg), or AR-12 plus FLU for 6 days. The combination of FLU and AR-12 reduced the brain fungal burden ∼1 log10 relative to FLU alone, while treatment with AR-12 alone had no effect (Fig. 6). In our experience, fungal brain burden reductions of greater than 1 log10 are generally required if a survival benefit is to be observed. Therefore, we did not evaluate the activity of AR-12 plus FLU using that endpoint because we felt it would be unethical. The activity of FLU plus AR-12 is comparable to that observed with a 3-mg/kg dose of amphotericin B as reported by other groups (46). In vitro fungicidal activity for FLU plus AR-12 in vitro is seen at 2 μg/ml of AR-12, while the MIC is 4 μg/ml for AR-12 alone. Thus, it is likely that multiple dosing leads to higher concentrations and, in turn, is sufficient for activity of the AR-12–FLU combination but is not sufficient for AR-12 alone.
FIG 6.
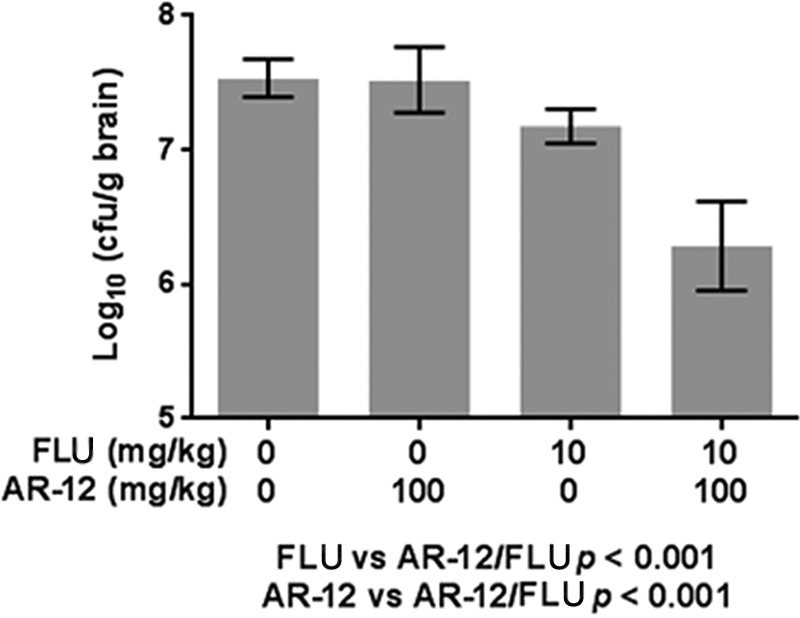
AR-12 improves the activity of fluconazole in a mouse model of cryptococcosis. C. neoformans brain burdens (in CFU/g of brain) are shown for animals treated with the indicated doses of AR-12 and/or FLU for 6 days after tail vein inoculation with 4.5 × 104 CFU/animal. P values are from ANOVA.
We also examined the activity of AR-12 alone and in combination with FLU in a mouse model of disseminated candidiasis. The combination of AR-12 and FLU reduced the kidney burden relative to either agent alone, but the difference did not reach statistical significance despite multiple experiments (data not shown). The maximum serum level of AR-12 observed in single-dose PK studies in mice was 2.4 μM (1.1 μg/ml). We suspect that insufficient AR-12 concentrations are achieved during the rather short duration of the candidiasis treatment model (3 to 4 days). Our data provide proof-of-principle that AR-12 has in vivo antifungal activity when combined with FLU. Further optimization of dosing or additional experiments in mammals with drug metabolism profiles more like those of humans will be required to establish the potential of AR-12 itself as an antifungal therapy. Nonetheless, AR-12 is the first example of a novel structural class of antifungal small molecules and has bona fide drug-like properties. As a lead molecule, AR-12 is extremely attractive, given its broad spectrum of activity, good pharmacology, and in vivo activity. Given its modest in vivo activity, additional improvements in activity or pharmacology are likely to be required.
DISCUSSION
The antifungal pipeline has delivered only one new structural or mechanistic class of drugs to clinical practice in the last 30 years (1–3). During that same time period, the incidence of invasive fungal infections has increased globally. Currently, the antifungal drugs used to treat invasive fungal infections are limited to three structural and mechanistic classes (2, 3). New additions to the antifungal pharmacopeia in the last 15 years have been limited to new variations of established scaffolds targeting either ergosterol (azoles) or glucan (echinocandins) biosynthesis. As discussed in a recent review of the antifungal pipeline (2), only a few molecules with novel mechanisms or targets have progressed in development as antifungal agents. Thus, an important task for antifungal drug development in general is to identify new targets that are druggable and that are either not essential in humans or present opportunities for selective inhibition. Ideally, new agents will also have broad-spectrum activity; the overall incidence of any one fungal infection is generally too low to make development of agents tailored to specific organisms economically or logistically viable.
AR-12 has progressed as a targeted anticancer therapy and has been found to be well tolerated in a phase I clinical trial (Clinical Trials registration no. NCT00978523). In this trial, a serum concentration of 8 μM or 3.7 μg/ml was achieved and well tolerated by subjects (S. Proniak, Arno Pharmaceuticals, personal communication). This serum level is similar to the MIC for AR-12 against a wide range of human fungal pathogens (Fig. 1B). Although serum levels do not necessarily correlate with antifungal activity, AR-12 and related molecules appear to have good drug-like properties and toxicity profiles. The general chemical structure represented by AR-12 is a scaffold well suited for additional optimization of its antifungal properties as part of a repurposing approach. As has been well discussed in the literature (47), it is generally more efficient to optimize on-target activity within a pharmacologically suitable chemical scaffold than to engineer away from toxicity or poor pharmacology.
The spectrum of in vitro activity for AR-12 is remarkably broad. It is active against pathogenic yeast, molds, and dimorphic fungi, with MICs generally between 2 and 4 μg/ml for multiple isolates of each species. AR-12 is active against species of Candida that are intrinsically less susceptible to FLU (C. glabrata and C. krusei) as well as clinical isolates of C. albicans and C. glabrata resistant to FLU, indicating that the mechanisms responsible for FLU resistance do not affect susceptibility to AR-12. Similarly, AR-12 is active against isolates of the basidiomycete C. neoformans, including an isolate with an elevated FLU MIC. No new classes of antifungal drugs with clinically useful activity against this pathogen have been developed in over 30 years, and the current therapy is based on drugs that are over 50 years old (5). As such, activity against Cryptococcus is an important feature of AR-12's spectrum.
One of the most exciting features of AR-12 is its in vitro activity against mold species that are either extremely difficult or impossible to treat using currently available drugs. The two most important examples are Fusarium and Scedosporium (48). Fusarium spp. are relatively resistant to most agents and are generally treated with either amphotericin B or voriconazole. Scedosporium spp. are largely regarded as being resistant to all available drugs, although some isolates are susceptible to voriconazole in vitro. AR-12 is also active against members of the order Mucorales, such as R. oryzae, which also represent difficult-to-treat species with variable susceptibility to currently available drugs (49). Finally, AR-12 is active against the three most important dimorphic fungal pathogens in North America: H. capsulatum, B. dermatitidis, and Coccidioides spp. As a whole, the spectrum of antifungal activity of AR-12 is comparable to that of amphotericin B in that AR-12 is active against a wide range of yeast and mold species with variable responses to any antifungal drug. The spectrum of activity alone suggests that the scaffold is worthy of continued optimization and development as a potential antifungal drug.
AR-12 is active against strains of Candida with resistance to azoles and echinocandins, the two most clinically important types of antifungal resistance. Furthermore, AR-12 appears to modulate the FLU susceptibility of FLU-resistant strains in that subinhibitory concentrations of AR-12 reduce the concentrations of FLU needed to inhibit growth. This occurs in strains with mutations increasing the expression of both efflux pumps and ERG11, the target of FLU. The mechanism of this effect is not related to decreased pump expression, and indeed, AR-12 induces the expression of one of the pumps (CDR2). Two alternative mechanisms are (i) AR-12 inhibits pump function or (ii) AR-12 decreases carbon flux through the ergosterol biosynthesis pathway, reducing the amount of FLU needed to achieve an antifungal effect. Our observations are most consistent with the latter mechanism in that lipid content of the cells is reduced and that gain-of-function mutations in UPC2, a regulator of lipid biosynthesis, reduce the ability of AR-12 to modulate FLU susceptibility. It must, however, be emphasized that other factors may also contribute.
In order to gain information regarding the development of resistance toward AR-12, we performed selection experiments designed to isolate AR-12-resistant mutants. Traditional plating experiments using concentrations above the MIC did not yield stably resistant mutants, indicating that the rate of spontaneous resistance is low. We then used an in vitro evolution approach in which S. cerevisiae strains were passaged for multiple generations in the presence of increasing concentrations of AR-12. This led to the isolation of three lineages that showed decreased susceptibility to AR-12. None of the lineages contained mutations in the putative target genes ACS1 or ACS2. Two strains showed a 4-fold increase in MIC (strains 16-14 and 16-77), while strain 64-77 showed high-level resistance (16-fold). All three strains contained mutations in PDR1, the transcription factor that regulates multidrug efflux pump expression and FLU susceptibility (41). The fixation of PDR1 mutations in our lineages may explain why mutations within the putative target genes, ACS1 and ACS2, were not observed. One of the mutations, PDR1T817K (strain 16-77), was isolated by Anderson et al. using the same evolution experiment with FLU, while the other mutation (PDR1S861Y in strains 16-14 and 64-77) is one position removed from a second mutation isolated by the same group (43). Anderson et al. found that these mutations led to increased PDR gene expression (43). They also observed that the mutations were associated with modest effects on FLU susceptibility that were augmented by unidentified second-site mutations and were recessive. The FLU-associated phenotypes of our pdr1 mutants are consistent with these findings.
The most informative pair of mutants in our set are strains 16-14 and 64-77, because each strain contains the same pdr1 mutation but they have 4-fold differences in susceptibility. Deletion of PDR1 returns both strains to wild-type susceptibility. This observation indicates that pdr1S861Y is necessary for resistance but is not sufficient for the high-level resistance observed with strain 64-77. This strain contains a loss-of-function mutation in MMS2 (42) which appears to contribute to the resistance phenotype of strain 64-77, although we cannot rule out the possibility that a mutation in HPF1 makes a contribution as well.
Taken together, our data regarding the role of efflux pumps in the susceptibility of yeast strains to AR-12 paint a complex picture. First, well-characterized transcription factor gain-of-function mutations in laboratory and clinical isolates that lead to increased pump activity and FLU resistance have no effect on AR-12 susceptibility. Second, deletion of these same transcription factors (e.g., S. cerevisiae PDR1 and C. albicans TAC1) hypersensitize cells to FLU; these deletions have no effect on AR-12 susceptibility. Third, gain-of-function mutations in S. cerevisiae PDR1 clearly play a role in the evolution of AR-12 resistance. Thus, it appears that under some circumstances AR-12 activity can be influenced by efflux pump activity. However, this is not consistently the case and may be dependent on either specific conditions or the presence of additional mutations in the strains. A full understanding of the role of efflux pumps in the activity of AR-12 will require additional work specifically focused on this question.
Although AR-12 has activity against a number of medically important fungi, we focused our initial in vivo study on cryptococcosis. As discussed above, new therapies for this globally important pathogen are urgently needed (4, 5, 44). AR-12 has a number of features that make it, or molecules derived from it, attractive for further development as a potential anticryptococcal agent. First, AR-12 is fungicidal, a property this has been shown to be especially important for the treatment of cryptococcal meningitis (50, 51). Second, AR-12 combines with FLU to yield a fungicidal cocktail against C. neoformans in vitro. Third, AR-12 has oral bioavailability, which is ideal for a cryptococcal agent since resource-limited regions have among the highest burden of this disease (44). The combination of FLU and AR-12 was more effective than FLU alone, with an ∼1-log10 CFU/g reduction in brain burden for FLU plus AR-12 relative to FLU alone. This is consistent with our in vitro data for the combination. These data provide proof-of-principle for the fact that AR-12 is able to show antifungal activity in vivo in combination with FLU.
The lack of activity of AR-12 by itself is likely a function of insufficient levels in the brain achieved at this dosing in mice. The maximum concentration of drug in plasma (Cmax) observed in single-oral-dose PK/PD experiments in mice is very close to the concentrations required for synergistic activity with FLU. We suspect that upon multiple dosing over the 6-day experiment, levels in tissue sufficient for synergy are achieved; however, it appears that the levels are insufficient for activity as a single agent. As a comparison, the addition of flucytosine to amphotericin B reduces the brain burden by ∼0.5 log10 CFU/g compared to amphotericin B alone in a murine model (46); clinically, this combination is superior to amphotericin B alone (51). Furthermore, the combination of sertraline and FLU was recently reported to give a 2-fold reduction in cryptococcal brain burden (52), and it is now being studied in a phase III clinical trial (53). Therefore, the activity of AR-12 in combination with FLU supports the possibility that further optimization of dosing or the development of more active analogs could lead to clinically useful combination.
The fact that AR-12 has also been in human clinical trials indicates that it is, in principle, a suitable scaffold from a pharmacological point of view (Clinical Trials registration no. NCT00978523). Based on the PK/PD data and in vivo experiments in mice, it seems that further medicinal chemistry optimization of the general class represented by AR-12 will be needed to generate a clinical candidate. It should be noted, however, that mouse drug metabolism differs significantly from that of humans; for example, voriconazole is not active in mouse models due to rapid clearance. Data from the AR-12 phase I clinical trial indicates that higher concentrations are achievable in humans than in mice. Therefore, additional testing of AR-12 in models of fungal infections in animals with drug metabolism similar to that of humans may be warranted to fully evaluate the potential of AR-12 itself. Whether or not AR-12 itself is sufficiently active, these data certainly support the notion that AR-12 represents an important new lead scaffold with attractive antifungal activity and favorable pharmacologic properties and that further optimization of this scaffold may lead to even better candidates.
As we have recently reported, AR-12 inhibits yeast acetyl-CoA synthetase in vitro (10), and based on a variety of cellular and genetic evidence, this inhibition appears to contribute significantly to the mechanism of its antifungal activity. Acetyl-CoA synthetase has been shown to be essential in C. albicans (13) and S. cerevisiae (54). Although studies in other pathogens have not been carried out, deletion of one of the three isoforms in C. neoformans has been shown to reduce virulence in vivo (14). Acetyl-CoA synthetase converts acetic acid to acetyl-CoA in an ATP-dependent process (11). In humans, the majority of acetyl-CoA is generated from glucose by the action of the enzyme ATP-citrate lyase (15, 16). Recent studies have found that tumor cells show an increased dependence on acetate metabolism relative to that of normal cells and, accordingly, an increased proportion of acetyl-CoA arising from acetate through acetyl-CoA synthetase (16–19). Indeed, increased expression of acetyl-CoA synthetase in tumors appears to correlate with poor outcomes (17). These observations have led to the hypothesis that acetyl-CoA synthetase may be a good anticancer target since it is not crucial for normal cells (19); deletion of acetyl-CoA synthetase in mice is possible, but deletion of ATP-citrate lyase is lethal (55). These same considerations also apply to acetyl-CoA synthetase as a potential antifungal target. Regardless of whether AR-12 itself is sufficiently active to be clinically useful, the broad-spectrum antifungal activity of the molecule and its drug-like properties and novel target suggest that further optimization of the scaffold is warranted.
ACKNOWLEDGMENTS
This work was supported by National Institute of Allergy and Infectious Diseases (NIAID) grant 1R01AI097142 (to D.J.K.) and 5K12HD068373 (to J.G.). In addition, this project utilized preclinical services funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHS272201000018I. Chemical synthesis of AR-12 was provided by SRI International under NIAID contract no. N01-AI-60011.
We thank Stefan Proniak and Alexander Zukiwski (Arno Pharmaceuticals) for providing additional samples of clinical AR-12 material and for sharing pharmacology data and assistance in arranging antifungal drug testing through the NIH contract.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 2.Denning DW, Bromley MJ. 2015. How to bolster the antifungal pipeline. Science 347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- 3.Roemer T, Krysan DJ. 1 May 2014. Antifungal drug therapy: challenges, un-met clinical needs, and approaches. Cold Spring Harb Persp Med doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong-James D, Meintjes G, Brown GD. 2014. A neglected epidemic: fungal infections in HIV/AIDS. Trends Microbiol 22:120–127. doi: 10.1016/j.tim.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Krysan DJ. 2015. Toward improved anti-cryptococcal drugs: novel molecules and repurposed drugs. Fungal Genet Biol 78:93–98. doi: 10.1016/j.fgb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Baxter BK, DiDone L, Oga D, Schor S, Krysan DJ. 2011. Identification, in vitro activity and mode of action of phosphoinositide-1 kinase inhibitors as antifungal molecules. ACS Chem Biol 6:502–510. doi: 10.1021/cb100399x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabrier-Rosello Y, Gerik K, Koselny K, DiDone L, Lodge JK, Krysan DJ. 2013. Cryptococcus neoformans phosphoinositide-dependent kinase 1 (PDK1) ortholog is required for stress tolerance. Eukaryot Cell 12:12–22. doi: 10.1128/EC.00235-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu J, Huang J-W, Tseng P-H, Yang Y-Z, Fowble J, Shiau C-W, Shaw YS, Kulp SK, Chen CS. 2004. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent kinase-1 inhibitors. Cancer Res 64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 9.Nagashima K, Shumway SD, Sathyanarayanan S, Chen AH, Dolinski B, Xu Y, Keilhack H, Nguyen T, Wiznerowicz M, Li L, Lutterbach BA, Chi A, Paweletz C, Allison T, Yan Y, Munshi SK, Klippel A, Kraus M, Bobkova EV, Deshmukh S, Xu Z, Mueller U, Szewczak AA, Pan BS, Richon V, Pollock R, Blume-Jensen P, Northrup A, Andersen JN. 2011. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric inhibitor. J Biol Chem 286:6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koselny K, Green J, Favazzo L, Glazier VE, DiDone L, Ransford S, Krysan DJ. 2016. Antitumor/antifungal celecoxib derivative AR-12 is a non-nucleoside inhibitor of the ANL-family adenylating enzyme acetyl CoA synthetase. ACS Infect Dis 2:268–280. doi: 10.1021/acsinfecdis.5b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starai VJ, Escalante-Semerena JCS. 2004. Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci 61:2020–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strijbis K, Distel B. 2010. Intracellular acetyl unit transport and fungal carbon metabolism. Eukaryot Cell 9:1809–1815. doi: 10.1128/EC.00172-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carman AJ, Vylkova S, Lorenz MC. 2008. Role of acetyl coenzyme A synthesis and breakdown in alternative carbon source utilization in Candida albicans. Eukaryot Cell 7:1733–1741. doi: 10.1128/EC.00253-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu G, Cheng PY, Sham A, Perfect JR, Kronstad JW. 2008. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol Microbiol 69:1456–1475. doi: 10.1111/j.1365-2958.2008.06374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellen KE, Hatzivassilious G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP. 2014. Acetate dependence of tumors. Cell 159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM. 2014. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, James D, Shanks E, Kalna G, Saunders RE, Jiang M, Howell M, Lassailly F, Thin MZ, Spencer-Dene B, Stamp G, van den Broek NJ, Mackay G, Bulusu V, Kamphorst JJ, Tardito S, Strachan D, Harris AL, Aboagye EO, Critchlow SE, Wakelam MJ, Schulze A, Gottlieb E. 2015. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyssiotis CA, Cantley LC. 2014. Acetate fuels the cancer engine. Cell 159:1492–1494. doi: 10.1016/j.cell.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Gao M, Yeh PY, Lu Y-S, Hsu C-H, Chen K-F, Lee W-C, Feng W-C, Chen C-S, Kuo M-L, Cheng A-L. 2008. OSU-03012, a novel celecoxib derivative, induces reactive oxygen species-related autophagy in hepatocellular carcinoma. Cancer Res 68:9348–9357. doi: 10.1158/0008-5472.CAN-08-1642. [DOI] [PubMed] [Google Scholar]
- 21.Goughenour KD, Balada-Llasat JM, Rappleye CA. 2015. Quantitative microplate-based growth assay for determination of antifungal susceptibility of Histoplasma capsulatum yeasts. J Clin Microbiol 53:3286–3295. doi: 10.1128/JCM.00795-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukherjee PK, Sheehan DJ, Hitchcock CA, Ghannoum MA. 2005. Combination treatment of invasive fungal infections. Clin Microbiol Rev 18:163–194. doi: 10.1128/CMR.18.1.163-194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasicek EM, Berkow EL, Flowers SA, Barker KS, Rogers PD. 2014. UPC2 is universally essential for azole antifungal resistance in Candida albicans. Eukaryot Cell 13:933–946. doi: 10.1128/EC.00221-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata T, Takahashi T, Yamada E, Kimura A, Nishikawa H, Hayakawa H, Nomura N, Mitsuyama J. 2012. T-2307 causes collapse of mitochondrial membrane potential in yeast. Antimicrob Agents Chemother 56:5892–5897. doi: 10.1128/AAC.05954-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sitepu IR, Ignatia L, Franz AK, Wong DM, Faulina SA, Tsui M, Kanti A, Boundy-Mills K. 2012. An improved high-throughput Nile red fluorescence assay for estimating intracellular lipids in a variety of yeast species. J Microbiol Methods 91:321–328. doi: 10.1016/j.mimet.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, Gow NAR. 2008. Stimulation of chitin synthesis rescues Candida albicans from echinocandins. PLoS Pathog 4:e1000040. doi: 10.1371/journal.ppat.1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krysan DJ, Didone L. 2008. A high-throughput screening assay for small molecules that disrupt yeast cell integrity. J Biomol Screen 13:657–664. doi: 10.1177/1087057108320713. [DOI] [PubMed] [Google Scholar]
- 28.Soteropoulos P, Vaz T, Santangelo R, Paderu P, Huang DY, Tamás MJ, Perlin DS. 2000. Molecular characterization of the plasma membrane H(+)-ATPase, an antifungal target in Cryptococcus neoformans. Antimicrob Agents Chemother 44:2349–2355. doi: 10.1128/AAC.44.9.2349-2355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis RE, Kontoyiannis DP. 2013. Epidemiology and treatment of mucormycosis. Future Microbiol 8:1163–1175. doi: 10.2217/fmb.13.78. [DOI] [PubMed] [Google Scholar]
- 30.Cushion MT, Walzer PD, Collins MS, Rebholz S, Vanden Eynde JJ, Mayence A, Huang TL. 2004. Highly active anti-Pneumocystis carinii compounds in a library of novel piperazine-linked bisbenzamidines and related compounds. Antimicrob Agents Chemother 48:4209–4216. doi: 10.1128/AAC.48.11.4209-4216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White TC. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother 41:1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whaley SG, Caudle KE, Vermitsky JP, Chadwick SG, Toner G, Barker KS, Gygax SE, Rogers PD. 2014. UPC2A is required for high-level azole antifungal resistance in Candida glabrata. Antimicrob Agents Chemother 58:4543–4554. doi: 10.1128/AAC.02217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. 2014. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med 5:a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youngsaye W, Dockendorff C, Vincent B, Hartland CL, Bittker JA, Dandapani S, Palmer M, Whitesell L, Lindquist S, Schreiber SL, Munoz B. 2012. Overcoming fluconazole resistance in Candida albicans clinical isolates with tetracyclic indoles. Bioorg Med Chem Lett 22:3362–3365. doi: 10.1016/j.bmcl.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shields RK, Nguyen MH, Clancy CJ. 2015. Clinical perspectives on echinocandin resistance among Candida species. Curr Opin Infect Dis 28:514–522. doi: 10.1097/QCO.0000000000000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramírez-Zavala B, Mogavero S, Schöller E, Sasse C, Rogers PD, Morschhäuser J. 2014. SAGA/ADA complex subunit Ada2 is required for Cap1- but not Mrr1-mediated upregulation of the Candida albicans multidrug efflux pump MDR1. Antimicrob Agents Chemother 58:5102–5110. doi: 10.1128/AAC.03065-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas E, Roman E, Claypool S, Manzoor N, Pla J, Panwar SL. 2013. Mitochondria influence CDR1 efflux pump activity, Hog1-mediated oxidative stress pathway, iron homeostasis, and ergosterol levels in Candida albicans. Antimicrob Agents Chemother 57:5580–5599. doi: 10.1128/AAC.00889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen LN, Nosanchuk JD. 2011. Lipid droplet formation protects against gluco/lipotoxicity in Candida parapsilosis: an essential role of fatty acid desaturase Ole1. Cell Cycle 10:3159–3167. doi: 10.4161/cc.10.18.16932. [DOI] [PubMed] [Google Scholar]
- 40.Anderson JB, Sirjusingh C, Parsons AB, Boone C, Wickens C, Cowen LE, Kohn K. 2003. Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 163:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carvajal E, van den Hazel HB, Cybularz-Kolaczkowska A, Balzi E, Goffeau A. 1997. Molecular and phenotypic characterization of yeast PDR1 mutants that show hyperactive transcription of various ABC multidrug transporter genes. Mol Gen Genet 256:406–415. doi: 10.1007/s004380050584. [DOI] [PubMed] [Google Scholar]
- 42.Broomfield S, Chow BL, Xiao W. 1998. MMS2, encoding an ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway. Proc Nat Acad Sci U S A 95:5678–5683. doi: 10.1073/pnas.95.10.5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson JB, Ricker N, Sirjusingh C. 2006. Antagonism between two mechanisms of antifungal drug resistance. Eukaryot Cell 5:1243–1251. doi: 10.1128/EC.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sloan DJ, Dedicoat MJ, Lalloo DG. 2009. Treatment of cryptococcal meningitis in resource-limited settings. Curr Opin Infect Dis 22:455–463. doi: 10.1097/QCO.0b013e32832fa214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livermore J, Howard SJ, Sharp AD, Goodwin J, Gregson L, Felton T, Schwartz JA, Walker C, Moser B, Müller W, Harrison TS, Perfect JR, Hope WW. 2014. Efficacy of an abbreviated induction regimen of amphotericin B deoxycholate for cryptococcal meningoencephalitis: 3 days of therapy is equivalent to 14 days. mBio 5:e00725-13. doi: 10.1128/mBio.00725-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarz P, Dromer F, Lortholary O, Dannaoui E. 2006. Efficacy of amphotericin B in combination with flucytosine against flucytosine-susceptible or flucytosine-resistant isolates of Cryptococcus neoformans during disseminated murine cryptococcosis. Antimicrob Agents Chemother 50:113–120. doi: 10.1128/AAC.50.1.113-120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strittmatter SM. 2014. Overcoming drug development bottlenecks with repurposing. Nat Med 20:590–591. doi: 10.1038/nm.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tortorano AM, Richardson M, Roilides E, van Diepeningen A, Caira M, Munoz P, Johnson E, Meletiadis J, Pana ZD, Lackner M, Verweij P, Freiberger T, Cornely OA, Arikan-Akdagli S, Dannaoui E, Groll AH, Lagrou K, Chakrabarti A, Lanternier F, Pagano L, Skiada A, Akova M, Arendrup MC, Boekhout T, Chowdhary A, Cuenca-Estrella M, Guinea J, Guarro J, de Hoog S, Hope W, Kathuria S, Lortholary O, Meis JF, Ullmann AJ, Petrikkos G, Lass-Flörl C, European Society of Clinical Microbiology and Infectious Diseases Fungal Infection Study Group, European Confederation of Medical Mycology. 2014. ESCMID and ECMM joint guidelines on diagnosis and management of hyalohyphomycosis: Fusarium spp., Scedosporium spp. and others. Clin Microbiol Infect 20(Suppl 3):27–46. doi: 10.1111/1469-0691.12465. [DOI] [PubMed] [Google Scholar]
- 49.Marty FM, Ostrosky-Zeichner L, Cornely OA, Mullane KM, Perfect JR, Thompson GR III, Alangaden GJ, Brown JM, Fredricks DN, Heinz WJ, Herbrecht R, Klimko N, Klyasova G, Maertens JA, Melinkeri SR, Oren I, Pappas PG, Ráčil Z, Rahav G, Santos R, Schwartz S, Vehreschild JJ, Young JH, Chetchotisakd P, Jaruratanasirikul S, Kanj SS, Engelhardt M, Kaufhold A, Ito M, Lee M, Sasse C, Maher RM, Zeiher B, Vehreschild MJ, VITAL and FungiScope Mucormycosis Investigators. 2016. Isavuconazole treatment for mucormycosis: a single-arm open-label trial and case-control analysis. Lancet Infect Dis 16:828–837. doi: 10.1016/S1473-3099(16)00071-2. [DOI] [PubMed] [Google Scholar]
- 50.Bicanic T, Muzoora C, Brouwer AE, Meintjes G, Longley N, Taseera K, Rebe K, Loyse A, Jarivis J, Bekker LG, Wood R, Limmathurostakul D, Chierakul W, Stepniewska K, White NJ, Jaffar S, Harrison TS. 2009. Independent association of clearance of infection and clinical outcome of HIV-associated cryptococcal meningitis: analysis of combined cohort of 262 patients. Clin Infect Dis 49:702–709. doi: 10.1086/604716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Day JN, Chau TT, Wolbers M, Mai PP, Dung NT, Mai NH, Phu NH, Nghia HD, Phong ND, Thai CQ, Thai LH, Chuong LV, Sinh DX, Duong VA, Hoang TN, Diep PT, Campbell JI, Sieu TP, Baker SG, Chau NV, Hien TT, Lalloo DG, Farrar JJ. 2013. Combination therapy for cryptococcal meningitis. N Engl J Med 368:1291–1302. doi: 10.1056/NEJMoa1110404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai B, Wu C, Wang L, Sachs MS, Lin X. 2012. The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrob Agents Chemother 56:3758–3766. doi: 10.1128/AAC.00212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhein J, Morawski BM, Hullsiek KH, Nabeta HW, Kiggundu R, Tugume L, Musubire A, Akampurira A, Smith KD, Alhadab A, Williams DA, Abassi M, Bahr NC, Velamakanni SS, Fisher J, Nielsen K, Meya DB, Boulware DR, ASTRO-CM Study Team . 2016. Efficacy of adjunctive sertraline for the treatment of HIV-associated cryptococcal meningitis: an open-label dose-ranging study. Lancet Infect Dis 16:809–818. doi: 10.1016/S1473-3099(16)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Jong-Gubbels P, van den Berg MA, Steensma HY, van Dijken JP, Pronk JT. 1997. The Saccharomyces cerevisiae acetyl CoA synthetase encoded by the ACS1 gene but not the ACS2-encoded enzyme is subject to catabolite inactivation. FEMS Microbiol Lett 153:75–81. doi: 10.1111/j.1574-6968.1997.tb10466.x. [DOI] [PubMed] [Google Scholar]
- 55.Beigneux AP, Kosinski C, Gavino B, Harton JD, Skarness WC, Young SG. 2004. ATP-citrate lyase deficiency in the mouse. J Biol Chem 279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]