

Abstract
The glucose transporter PfHT is essential to the survival of the malaria parasite Plasmodium falciparum and has been shown to be a druggable target with high potential for pharmacological intervention. Identification of compounds against novel drug targets is crucial to combating resistance against current therapeutics. Here, we describe the development of a cell-based assay system readily adaptable to high-throughput screening that directly measures compound effects on PfHT-mediated glucose transport. Intracellular glucose concentrations are detected using a genetically encoded fluorescence resonance energy transfer (FRET)-based glucose sensor. This allows assessment of the ability of small molecules to inhibit glucose uptake with high accuracy (Z′ factor of >0.8), thereby eliminating the need for radiolabeled substrates. Furthermore, we have adapted this assay to counterscreen PfHT hits against the human orthologues GLUT1, -2, -3, and -4. We report the identification of several hits after screening the Medicines for Malaria Venture (MMV) Malaria Box, a library of 400 compounds known to inhibit erythrocytic development of P. falciparum. Hit compounds were characterized by determining the half-maximal inhibitory concentration (IC50) for the uptake of radiolabeled glucose into isolated P. falciparum parasites. One of our hits, compound MMV009085, shows high potency and orthologue selectivity, thereby successfully validating our assay for antimalarial screening.
INTRODUCTION
Malaria is a major threat to the human population in large areas of the world, affecting over 200 million people per year (1, 2). Beyond the effects of this disease on human life, malaria also cripples economic development and burdens the health care systems of countries where malaria is endemic (3). The emergence of parasites with resistance to even the most potent existing antimalarial drugs, such as the artemisinins (4), has made paramount the development of novel drugs that target essential pathways for parasite survival. Blood stage parasites utilize glucose for both biomass production and ATP synthesis (5). The malarial hexose transporter, Plasmodium falciparum hexose transporter (PfHT), first cloned by Woodrow et al. in 1999 (6), mediates parasite transport of glucose, fructose, mannose, and galactose (7). Since PfHT is essential for parasite survival (8), this protein is a highly promising molecular target for antimalarial drug development. This is supported by the ability of compound 3361, a glucose analogue that inhibits PfHT with high selectivity over the human orthologue GLUT1, to inhibit asexual intraerythrocytic growth in culture (9). Compound 3361 is also active against Plasmodium berghei liver and transmission stage parasites in infected mice (10), suggesting that PfHT is a promising target for full life cycle activity. However, while compound 3361 validates efforts to target PfHT, this compound is not itself considered drug-like and is therefore not a valid candidate for lead development (11). We have recently extended these earlier findings by identifying PfHT as a molecular target of the HIV protease inhibitor lopinavir, thus providing a link between lopinavir use and decreased malarial transmission in areas where HIV and malaria are both endemic (12). However, lopinavir has a relatively high half-maximal inhibitory concentration (IC50), 16 μM, in parasites and shows higher selectivity for the human insulin-responsive glucose transporter GLUT4 over PfHT (12). Therefore, novel therapeutics targeting PfHT with improved potency and selectivity are required.
The development of a robust and efficient high-throughput screening assay to identify novel PfHT inhibitors requires consideration of simplicity, sensitivity, scalability, cost, and reliability. Current assays for transporter inhibition in a high-throughput format generally employ radiolabeled substrate or cell death of a transporter-overexpressing cell line as a readout (13, 14). Both formats have significant limitations. Although measuring the uptake of radiolabeled substrate generally yields quantitative, highly reproducible results, the use and disposal of radiolabels are expensive and handling radioactive substances requires increased safety precautions. Alternatively, using cell death of an engineered cell line that requires transporter function for survival is an elegant way to simplify the readout. In both cases, these assays fail to discriminate between compounds that kill the cells through transporter inhibition and compounds that kill through other mechanisms, resulting in false-positive rates as high as 97.8% (15). Here, we report the development and validation of a novel assay for determination of compounds that efficiently and selectively block PfHT.
MATERIALS AND METHODS
Materials.
[14C]2-deoxyglucose (2-DOG) and [3H]2-DOG were purchased from American Radiolabels Inc. GLUT1 short hairpin RNA (shRNA) was obtained through the RNA interference (RNAi) core at Washington University School of Medicine. HEK293 cells were acquired from the American Type Culture Collection (ATCC). Lopinavir was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
HEK293 cell line generation.
HEK293 cells were transfected with pcDNA3.1 FLII12Pglu-700μδ6 (Addgene), containing the fluorescence resonance energy transfer (FRET) glucose sensor (HEK293-FLIP), using Optifect reagent (Life Technologies) according to the manufacturer's specifications. Cells that stably integrated the gene were selected using G418 (Sigma-Aldrich), and the highest expressers were identified using fluorescence-activated cell sorting (FACS). These cells were then stably transfected with PfHT, human GLUT1 (hGLUT1), hGLUT2, hGLUT3, or hGLUT4 DNA in the pcDNA 3.1(−) hygro plasmid (Life Technologies) as described previously (12). Single clones were selected by comparing their abilities to transport radiolabeled glucose. In all cell lines except for HEK293 cells overexpressing hGLUT1, native hGLUT1 was knocked down using shRNA as described elsewhere (12). PfHT expression levels in overexpressing cells were previously characterized (12).
RNA isolation and qPCR.
To quantify mRNA transcript levels in the four GLUT-overexpressing cell lines, total RNA was isolated using the TrizolW Plus RNA purification system (Invitrogen), and 1 μg of RNA was reverse transcribed using qScript cDNA Supermix (Quanta Biosciences). Quantitative reverse transcription-PCR (qRT-PCR) was performed using Power SYBR green PCR master mix (Applied Biosystems). Each reaction was run in triplicate using the primers listed in Table 1. Quantifications were performed with standard curves generated using plasmids containing each human GLUT (DNASU).
TABLE 1.
Human glucose transporter primers for qPCR
Assay optimization and high-throughput screening.
Several conditions were optimized to increase the sensitivity and reliability of the assay. Ninety-six-well plates were pretreated with 25 μg/ml of polyethylenimine (PEI; 750 kDa; Sigma-Aldrich) solution containing 150 mM NaCl to maintain cell adhesion during washing steps. After 20 min of incubation at room temperature, PEI was aspirated and wells were air dried for 5 min. PfHT-FLIP cells were plated 48 h prior to the assay in black opaque 96-well plates (Greiner Bio-One), which had been previously treated with PEI, at 30,000 cells/well. After cell plating, plates were left in the sterile hood at room temperature for 45 min before transfer to the incubator to reduce edge effects. Compound screening was performed using the integrated and automated screening system (Beckman Coulter) at the Washington University High Throughput Screening Core. We used SAMI EX software (Beckman Coulter) to design and execute the screening protocol. The Malaria Box (Medicines for Malaria Venture [MMV]) compound library was prediluted in glucose-free HEPES-buffered saline solution (HBSS) (146 mM NaCl, 4.7 mM KCl, 0.6 mM MgSO4, 1.6 mM NaHCO3, 0.13 mM NaH2PO4, 2.5 mM CaCl2, 20 mM HEPES [pH 7.3]) using a BiomekFX liquid handler (Beckman Coulter). We prepared a 1:100 dilution of 1 mM stock solution for a 10 μM final concentration.
To initiate the screening assay, cells were washed twice with 150 μl of HBSS per well using an ELx405 plate washer (Biotek). Cells were starved in HBSS for 30 min at room temperature, followed by aspiration of the HBSS from cell plates using the ELx405 plate washer. Cells were treated with 45 μl of diluted compound using the BiomekFX (with 3 replicate cell plates for each library plate) and incubated for 6 min at room temperature. To initiate the uptake, 5 μl of 100 mM glucose was added to the wells (final concentration, 10 mM) using a Multidrop384 dispenser (Thermo Fisher Scientific). After incubation at room temperature for 120 min, fluorescence was measured using a 2102 EnVision multilabel plate reader (PerkinElmer) at an excitation wavelength of 436 nm (cyan fluorescent protein [CFP]) and emission wavelengths of 485 nm (CFP) and 535 nm (yellow fluorescent protein [YFP]).
P. falciparum culture.
P. falciparum strain 3D7 was obtained from the Malaria Research and Reference Reagent Resource Center (MR4). P. falciparum parasites were cultured in a 2% suspension of human erythrocytes and RPMI 1640 medium (Sigma-Aldrich) supplemented with 27 mM sodium bicarbonate, 11 mM glucose, 5 mM HEPES, 1 mM sodium pyruvate, 0.37 mM hypoxanthine, 0.01 mM thymidine, 10 μg/ml of gentamicin, and 0.5% Albumax (Gibco) at 37°C in a 5% O2–5% CO2–90% N2 atmosphere as previously described (16, 17).
P. falciparum growth inhibition assays.
Asynchronous P. falciparum cultures were diluted to 1% parasitemia and were treated with compounds at concentrations ranging from 9.8 nM to 20 μM. Growth inhibition assays were performed in 100-μl cultures in opaque 96-well plates. Parasite growth was quantified after 3 days by measuring DNA content using PicoGreen (Life Technologies) (18). Fluorescence was measured by a FLUOstar Omega microplate reader (BMG Labtech) at a 485-nm excitation wavelength and a 528-nm emission wavelength. IC50s were calculated by nonlinear regression analysis using GraphPad Prism software.
Measurements of radiolabeled glucose uptake.
P. falciparum strain 3D7 was cultured in 100-mm tissue culture dishes (Techno Plastic Products) in a 2% suspension of human erythrocytes and RPMI 1640 medium until reaching >5% parasitemia. Cells were pelleted via centrifugation and resuspended in RPMI 1640 medium. In order to determine the uptake of radiolabeled glucose into the parasite, we isolated it from the erythrocytes while removing uninfected erythrocytes as described in references 19 and 20. Uptake of [14C]2-DOG into isolated parasites was determined at room temperature using the methods described previously (19). Test compounds were added 5 min prior to the addition of [14C]2-DOG (0.2-μCi/ml final concentration). Uptake was quenched after 2 min. Data were fit by nonlinear regression analysis using GraphPad Prism software. Uptake of [3H]2-DOG into HEK293-FLIP cells was measured in HEPES-buffered saline at room temperature for 4 min as described previously (20). Data were fit by nonlinear regression analysis using GraphPad Prism software.
RESULTS
Assay development and optimization.
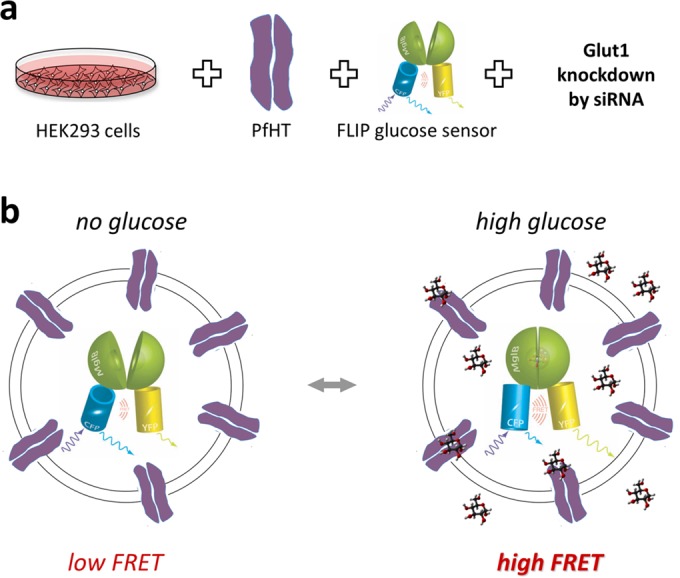
The ideal PfHT assay would evaluate transporter function as a direct, highly reproducible readout without the use of radiolabels. We therefore designed a cell line that transports glucose almost exclusively through PfHT and combined this with an intracellular glucose sensor protein that translates glucose concentration into a fluorescence signal as a readout. We used the glucose sensor FLII12Pglu-700μδ6 (FLIP), initially developed and characterized in the laboratory of Wolf Frommer, to qualitatively assess cellular glucose influx (21). This genetically encoded optical glucose sensor consists of three protein domains: a central glucose-binding domain that is coupled terminally to a CFP and a YFP. Upon excitation of CFP, energy is transferred to YFP through FRET. When glucose enters the cell, it binds to the glucose binding domain, leading to a conformational change that brings the two fluorescent proteins closer together and increases FRET (Fig. 1). Using the human embryonic kidney cell line HEK293, we created a reporter cell line stably expressing PfHT in conjunction with FLIP (PfHT-FLIP cells). HEK293 cells are known to exhibit relatively low endogenous glucose uptake (21). To further reduce background levels of glucose uptake, we knocked down the primary endogenous transporter isoform GLUT1 using shRNA, yielding a cell line that predominantly transports glucose through PfHT (12). We tested the function of our engineered cell line by measuring the time course of glucose uptake via readout of the YFP/CFP ratio (FRET ratio) in the presence and absence of known PfHT inhibitors. In the presence of the HIV protease inhibitor lopinavir (Fig. 2a), previously identified to block PfHT-mediated glucose uptake, PfHT-FLIP cells showed a decreased FRET ratio with addition of d-glucose, consistent with inhibition of radiolabeled d-glucose uptake in these cells (12). Additionally, we confirmed concentration-dependent inhibition of glucose uptake in PfHT-FLIP cells by the glucose transport inhibitor cytochalasin B (CB) as previously shown in Xenopus laevis oocytes (6). In PfHT-FLIP cells, CB causes a maximal decrease in FRET ratio of 75% at 50 μM, consistent with radiolabeled uptake inhibition in oocytes (6) and complete inhibition of d-glucose uptake at 200 μM (Fig. 2b). CB is therefore an ideal positive control for assay optimization and high-throughput screening.
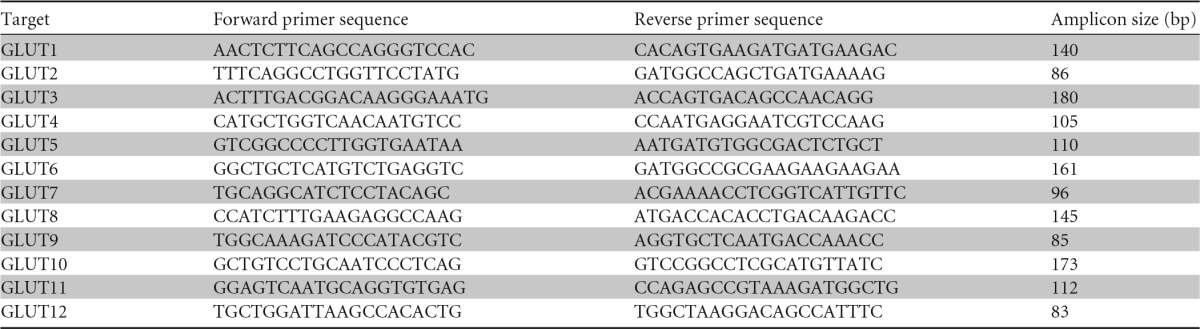
FIG 1.
(a) Generation of cell lines for high-throughput screening. HEK293 cells were stably transfected with the P. falciparum hexose transporter (PfHT) and the glucose sensor FLII1212Pglu-700μδ6 and treated with small interfering RNA (siRNA) directed at hGLUT1 to reduce background glucose uptake. (b) Glucose FRET sensor expressed in PfHT-expressing cells. In the absence of glucose, CFP and YFP are further apart and the amount of energy transferred from the donor CFP to the acceptor YFP that is emitted as light (FRET signal) is low (figure adapted from reference 21; used with permission). Glucose binding to the glucose/galactose binding domain (MgIB) leads to a conformational change that brings CFP and YFP closer together, resulting in an increased FRET signal.
FIG 2.
(a) Time-dependent change in FRET ratio (YFP emission/CFP emission) after addition of glucose to PfHT-FLIP cells in the presence or absence of lopinavir. (b) Time-dependent change in FRET ratio after addition of glucose to PfHT-FLIP cells in the presence or absence of cytochalasin B (CB).
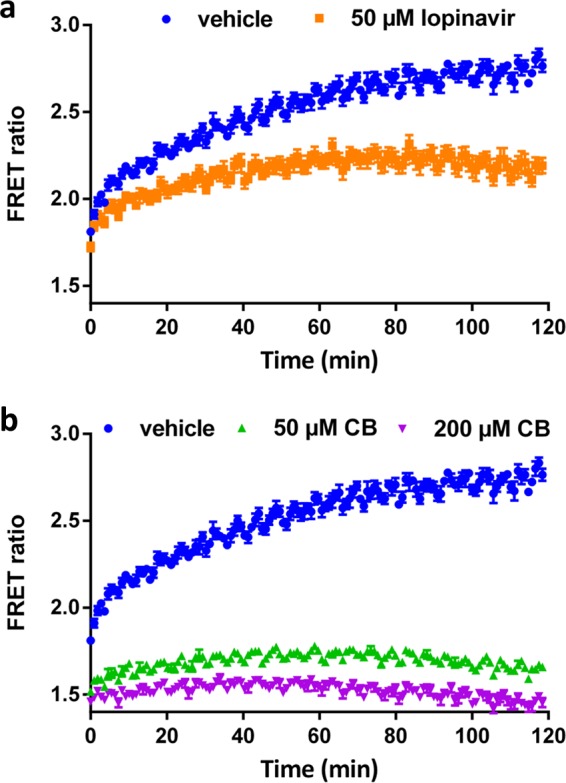
We optimized the assay for high-throughput application in a 96-well format using the Z′ factor and the coefficient of variation (CV) as measures of assay robustness (22). For these measurements, half of the plate was treated with 200 μM CB as a positive control and the other half with a vehicle prior to the addition of d-glucose. Cells adhered tightly to the plate bottom after treatment of the plate with the highly branched polymer PEI, which acted as an adhesive, thereby preventing cells from dislodging during washing and buffer exchange steps. After optimization of assay temperature, fluorescence read mode, cell density, and cell plating protocol, we were able to routinely obtain a Z′ factor of >0.8 (a perfect assay would have a Z′ factor of 1.0) and a CV of ∼2% (Fig. 3).
FIG 3.
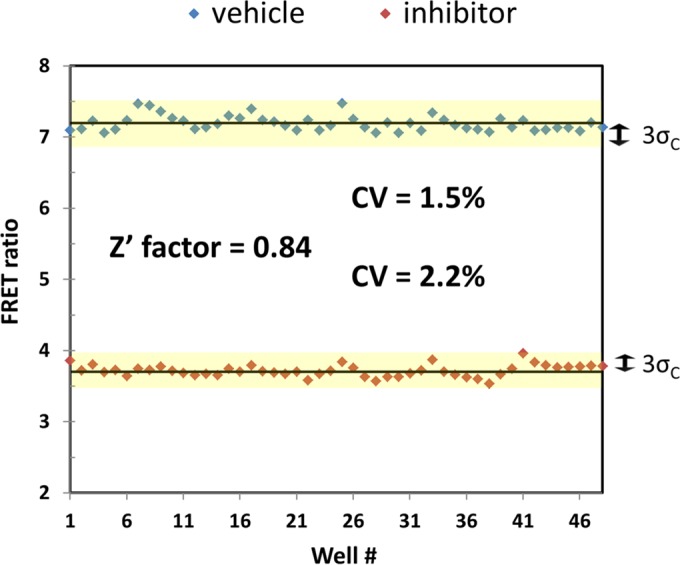
FRET ratio per well of a 96-well plate, with 48 wells treated with 200 μM CB (inhibitor) and 48 wells treated with a vehicle. Z′ factor and the coefficient of variation (CV) were determined from the average and SD of each set of wells according to reference 20.
Screening of the Malaria Box compound library.
In order to test our assay conditions in an automated setting for high-throughput application, we selected the MMV Malaria Box, a small library of 400 compounds previously shown to have a cytotoxic effect on malaria parasites (23). This library contains chemically diverse compounds, with half of the compounds having drug-like properties and the other half having been selected as molecular probes. As this compound set was selected for their cytotoxic activity against blood stage malaria parasites, the molecular drug targets remain to be determined. The rationale for choosing this library was to identify compounds that inhibit parasite growth mainly or partially through blockade of PfHT-mediated glucose uptake, since deorphanization of these compounds (i.e., identification of the direct molecular targets of anti-antimalarial action) will aid in further target-directed drug development.
We screened the Malaria Box at 10 μM drug concentrations in triplicate and selected hits that decrease the FRET ratio by more than 30% with vehicle-treated cells set to 100% and cells treated with 200 μM CB set to 0%. Since fluorescent compounds have the potential to generate false-positive hits, the FRET ratio of compounds without cells present was determined. After false-positive elimination, we identified 5 compounds as primary hits. Hit confirmation was ascertained by determining the IC50 for glucose uptake into isolated P. falciparum parasites from blood culture using radiolabeled d-glucose. Four of the five primary hits could be confirmed to inhibit glucose uptake into P. falciparum parasites in the low micromolar range (Table 2), with compound MMV665941 showing only partial inhibition at the highest concentration tested (Fig. 4). We included compound MMV020548 for comparison as it was previously identified by Ortiz et al. (15) from the same library. The similar IC50 observed in the free-parasite glucose uptake assay and in vitro growth inhibition assay for MMV009085 (23) is consistent with the interpretation that this compound acts through PfHT blockage to inhibit parasite growth (Table 2), although alternative targets in the parasite might be inhibited as well. Since the measurement of 2-DOG uptake involves both the transport of this sugar and phosphorylation to 2-DOG-6-phosphate (2-DOG-6-P), we cannot fully exclude the possibility that the identified compounds also inhibit the malarial hexokinase. However, the observations that they inhibit 2-DOG uptake with different potencies and that compound MMV009085 shows a widely different IC50 when measured in HEK293 cells overexpressing different GLUTs (see Fig. 6) suggest that hexokinase is not directly targeted.
TABLE 2.
Comparison of hit IC50s for different assaysa
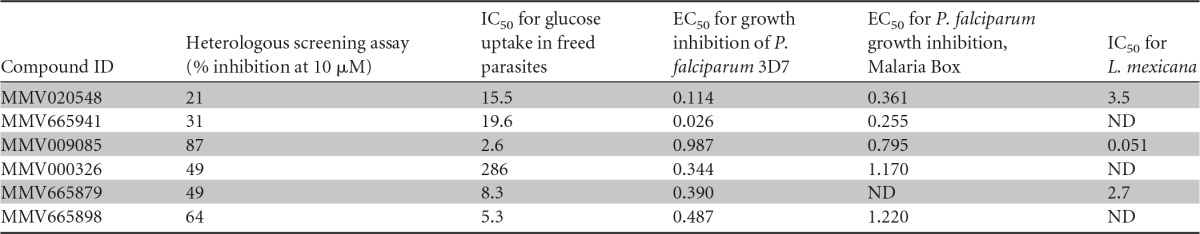
IC50s for glucose uptake in freed parasites were determined from dose-response curves shown in Fig. 4. IC50s for inhibition of growth of P. falciparum strain 3D7 intraerythrocytic forms by each compound were determined via a growth inhibition assays as described in Materials and Methods. Half-maximal effective concentrations (EC50s) provided with the malaria box and IC50 values determined by Ortiz et al. (15) for inhibition of glucose uptake into PfHT-overexpressing L. mexicana parasites are also tabulated. ID, identifier; ND, not determined.
FIG 4.

Uptake of [14C]2-DOG by isolated P. falciparum trophozoites at increasing concentrations of hit compounds. Distribution ratios (i.e., the ratio of intracellular concentration of radiolabel relative to the extracellular concentration) were calculated as described previously (14). IC50s were calculated using nonlinear regression analysis and are tabulated in Table 2. Uptake data are expressed as means ± SEMs (n = 3). Chemical structures of the compounds tested are shown for comparison.
FIG 6.

Specificity of hit compounds for PfHT over human orthologues. (a) Uptake of [3H]2-DOG by HEK293 cells overexpressing hGLUT1 to hGLUT4 in the presence of hit compounds at their IC50 for PfHT (Table 2). (b) Uptake of [3H]2-DOG by HEK293 cells overexpressing PfHT or hGLUT1 to hGLUT4 at increasing concentrations of compound MMV009085. Uptake data are expressed as means ± SEMs (n = 3). IC50s were calculated using nonlinear regression analysis.
To establish selectivity of these hits for PfHT over human orthologues, we cross-validated the inhibition of GLUT1, -2, -3, and -4 by the confirmed hits. Using the same HEK293-FLIP cell line, we overexpressed each of the individual class I transporters. With the exception of GLUT1-FLIP, the other GLUT cell lines were also treated with GLUT1-specific shRNA to reduce background glucose uptake. We confirmed that the overexpressed GLUT was the main glucose transporter expressed in each cell line by comparing the cDNA copy number of all human SLC2A genes via qPCR (Fig. 5). Total transcript numbers differed between various GLUT isoform-overexpressing cell lines as the construct was integrated randomly into the genome of the host cell as discussed in Materials and Methods. We determined the inhibitory effects of the five confirmed hits on all four human class I GLUTs at the IC50 for PfHT-mediated glucose uptake inhibition. Only compound MMV009085, the most potent hit, with an IC50 of 2.6 μM for PfHT-mediated glucose uptake, showed significantly less inhibition of the human GLUTs than for PfHT (Fig. 6a). Comparison of the IC50s for glucose uptake inhibition, mediated by either the human GLUTs or PfHT, revealed a >10-fold-higher selectivity of MMV009085 for PfHT over human orthologues (Fig. 6b). Although MMV009085 is considered a probe-like molecule, its high potency in inhibiting both glucose uptake and growth of the parasites as well as its high selectivity for PfHT over human orthologues makes this compound a potential candidate for lead optimization. Additionally, it demonstrates that our newly developed assay can identify glucose transporter-specific inhibitors and further distinguish compounds with high selectivity for their target over its orthologues or isoforms.
FIG 5.

Copies of transcript per nanogram of cDNA for each glucose transporter SLC2A family member in HEK293-FLIP cells overexpressing (OE) hGLUT1 (a), hGLUT2 (b), hGLUT3 (c), and hGLUT4 (d) in comparison to HEK293 wild-type cells and/or HEK293 wild-type cells treated with siRNA targeting hSLC2A1 (hGLUT1 kd).
DISCUSSION
We describe here the development of a novel assay system adaptable to high-throughput screening for small molecules that inhibit glucose uptake. The ability to identify compounds with selectivity for the malarial glucose transporter PfHT over human GLUT transporters provides a powerful new means to identify safe and effective antimalarial agents. Our results show high reproducibility, with a CV of ∼2% and good separation of hits from background, with a Z′ factor of >0.8, indicative of a high-quality assay for high-throughput screening (22). Additionally, by not requiring radioactive labels, this assay increases throughput and ease of use with substantially reduced cost. This is especially important in the field of infectious diseases affecting people in the developing world (24). The assay employs cells that genetically encode the glucose transporter and a glucose sensor utilizing a direct fluorescent readout. This eliminates the need for time-consuming protein purification steps and expensive radiolabeled or fluorescently labeled substrates. Every step in our protocol was successfully tested in a fully automated setup in order to test the true scalability to high throughput. Our initial screen of a 400-compound library resulted in four verified hits. Although the rate of false-positive compounds identified after the initial round of screening is significantly lower than in a previously reported PfHT inhibitor assay (15), further improvements can be made by selecting a higher threshold of FRET ratio reduction, especially when screening a larger library or using different detection methods like fluorescence lifetime (25, 26). The hits identified in this screen include three compounds (MMV009085, MMV020548, and MMV665879) that were previously identified and characterized by Ortiz et al. (15) to selectively inhibit glucose uptake, but not proline transport, in PfHT-overexpressing Leishmania mexicana. However, the L. mexicana assay system showed noncorrelated results for the IC50 of compound MMV009085 (IC50 of 0.99 μM in the malaria parasite growth assay but 0.051 μM in the L. mexicana PfHT overexpression assay). This 20-fold difference in potency by the two assays indicates that in the cross-expression system (PfHT in the Leishmania parasite) MMV009085 might have an alternative target, originating from L. mexicana. In our screen, reduction in FRET ratio correlated with inhibitory potency of the compound in the freed-parasite glucose uptake assay (R2 = 0.83, excluding outlier MMV000326), with the most potent inhibitor, MMV009085, showing the strongest decrease in FRET ratio.
The quality of hits in drug discovery projects is crucial in improving the odds of developing a successful candidate for clinical application, especially as downstream investments into the hits that are selected for lead development are significant in terms of both funding and time (24). Therefore, target-based screens have advantages over phenotypic screens, as they allow lead identification and optimization that are directed toward the malarial target protein versus human orthologues. We adapted our assay system to include counterscreening of PfHT-specific hits against human glucose transporters. The facilitative glucose transporters GLUT1 to -4 are fundamentally important for human glucose homeostasis, as they are primary glucose transporters in most tissues (27). PfHT and the human transporters share a major facilitator superfamily (MFS) fold and show a sequence similarity of close to 50%. The significance of the human GLUTs and their close homology to the malarial glucose transporter make it crucial, but potentially challenging, to identify drugs with high parasite orthologue selectivity. By replacing PfHT with human GLUTs, our HEK293 cell-based assay system successfully distinguished PfHT-specific from orthologue-nonspecific inhibitors and identified a compound that showed a 19- to >250-fold high selectivity for PfHT over the human GLUTs.
An additional advantage of target-directed screening is the opportunity for structure-based rational drug design to aid in lead compound optimization, particularly with respect to selectivity for PfHT over human GLUTs. For the mammalian glucose transporters, recent progress has been made in solving the crystal structures of several isoforms in both inward and outward conformations (28–30). Furthermore, our prior work has provided evidence for the binding pocket that mediates transporter inhibition by HIV protease inhibitors (12, 20, 31). This has been used to model lopinavir binding to a model of PfHT (12). Taken together, these data provide a framework for iterative structure-activity analysis of compounds identified via our newly developed high-throughput screening assay.
In addition to selecting orthologue-specific PfHT inhibitors, the GLUT-expressing FLIP cell lines can be used to identify GLUT-specific inhibitors with isoform selectivity with applications beyond antimalarial therapy. Several types of tumors have been shown to overexpress specific GLUT isoforms that mediate the transport of high levels of glucose to malignant cells that have undergone metabolic rearrangement and primarily metabolize glucose through aerobic glycolysis (known as the Warburg effect) (32, 33). Many cancer cells express transporter isoforms that are not found in these tissues under nonmalignant conditions. Among the class 1 facilitative glucose transporters, GLUT1 is most abundantly expressed in breast, brain, lung, colorectal, and bladder cancers (34). Inhibition of GLUT1 has already been shown to exert antineoplastic effects both in vitro and in vivo (35, 36), making it a promising target for cancer therapy. Expression of the glucose transporters GLUT2, GLUT3, and GLUT4 is also upregulated in various tumors, among them several types that are difficult to treat and are associated with low survival rates, like lung, pancreatic, and liver tumors (32, 34, 37). Our cell-based, target-specific screen is designed to be easily adaptable to mediate glucose transport through selectively expressed transporter isoforms. This versatility combined with a robust, easily detectable readout significantly increases the feasibility of screening large compound libraries to identify isoform-specific GLUT inhibitors that can be further developed into drugs for either stand-alone or adjunct cancer therapy.
ACKNOWLEDGMENTS
We thank Kurt Peterson and Prachi Bawaskar from the Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, for productive discussions and their work on the PfHT screening assay using fluorescence lifetime measurement.
The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
T.E.K. conceived and designed the research, with oversight by P.W.H. Creation and optimization of the cell lines and screening conditions were performed by T.E.K. together with M.P. IC50 determinations for parasite growth inhibition and parasite culture were performed by R.L.E., supervised by A.R.O. Radiolabel uptake experiments with HEK293 cells were performed by M.R.H. Radiolabel uptake experiments with parasites were performed by T.E.K. together with M.R.H. qPCR experiments were performed by M.A.P. A.R.O. provided expertise for parasite culture and drug mechanism. High-throughput screening protocols were designed and executed by M.X.G.I. and T.E.K. Data were analyzed by T.E.K. The paper was written by T.E.K. with contributions from all coauthors, and T.E.K. produced the figures.
We declare no competing financial interests.
REFERENCES
- 1.World Health Organization. 2014. World malaria report 2014, p 165–176. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Wells TNC, van Huijsduijnen RH, Van Voorhis WC. 2015. Malaria medicines: a glass half full? Nat Rev Drug Discov 14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 3.Sachs J, Malaney P. 2002. The economic and social burden of malaria. Nature 415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 4.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olszewski KL, Llinás M. 2011. Central carbon metabolism of Plasmodium parasites. Mol Biochem Parasitol 175:95–103. doi: 10.1016/j.molbiopara.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodrow CJ, Penny JI, Krishna S. 1999. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem 274:7272–7277. doi: 10.1074/jbc.274.11.7272. [DOI] [PubMed] [Google Scholar]
- 7.Blume M, Hliscs M, Rodriguez-Contreras D, Sanchez M, Landfear S, Lucius R, Matuschewski K, Gupta N. 2011. A constitutive pan-hexose permease for the Plasmodium life cycle and transgenic models for screening of antimalarial sugar analogs. FASEB J 25:1218–1229. doi: 10.1096/fj.10-173278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slavic K, Straschil U, Reininger L, Doerig C, Morin C, Tewari R, Krishna S. 2010. Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol Microbiol 75:1402–1413. doi: 10.1111/j.1365-2958.2010.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joet T, Eckstein-Ludwig U, Morin C, Krishna S. 2003. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci U S A 100:7476–7479. doi: 10.1073/pnas.1330865100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slavic K, Delves MJ, Prudêncio M, Talman AM, Straschil U, Derbyshire ET, Xu Z, Sinden RE, Mota MM, Morin C, Tewari R, Krishna S, Staines HM. 2011. Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite's life cycle. Antimicrob Agents Chemother 55:2824–2830. doi: 10.1128/AAC.01739-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Staines HM, Derbyshire ET, Slavic K, Tattersall A, Vial H, Krishna S. 2010. Exploiting the therapeutic potential of Plasmodium falciparum solute transporters. Trends Parasitol 26:284–296. doi: 10.1016/j.pt.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Kraft TE, Armstrong C, Heitmeier MR, Odom AR, Hruz PW. 2015. The glucose transporter PfHT1 is an antimalarial target of the HIV protease inhibitor lopinavir. Antimicrob Agents Chemother 59:6203–6209. doi: 10.1128/AAC.00899-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JB, Mallorga PJ, Lemaire W, Williams DL, Na S, Patel S, Conn JP, Pettibone DJ, Austin C, Sur C. 2003. Development of a scintillation proximity assay for analysis of Na+/Cl−-dependent neurotransmitter transporter activity. Anal Biochem 321:31–37. doi: 10.1016/S0003-2697(03)00431-7. [DOI] [PubMed] [Google Scholar]
- 14.Gui C, Obaidat A, Chaguturu R, Hagenbuch B. 2010. Development of a cell-based high-throughput assay to screen for inhibitors of organic anion transporting polypeptides 1B1 and 1B3. Curr Chem Genomics 4:1–8. doi: 10.2174/1875397301004010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ortiz D, Guiguemde WA, Johnson A, Elya C, Anderson J, Clark J, Connelly M, Yang L, Min J, Sato Y, Guy RK, Landfear SM. 2015. Identification of selective inhibitors of the Plasmodium falciparum hexose transporter PfHT by screening focused libraries of anti-malarial compounds. PLoS One 10:e0123598. doi: 10.1371/journal.pone.0123598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 17.Zhang B, Watts KM, Hodge D, Kemp LM, Hunstad DA, Hicks LM, Odom AR. 2011. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry 50:3570–3577. doi: 10.1021/bi200113y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbett Y, Herrera L, Gonzalez J, Cubilla L, Capson TL, Coley PD, Kursar TA, Romero LI, Ortega-Barria E. 2004. A novel DNA-based microfluorimetric method to evaluate antimalarial drug activity. Am J Trop Med Hyg 70:119–124. [PubMed] [Google Scholar]
- 19.Saliba KJ, Krishna S, Kirk K. 2004. Inhibition of hexose transport and abrogation of pH homeostasis in the intraerythrocytic malaria parasite by an O-3-hexose derivative. FEBS Lett 570:93–96. doi: 10.1016/j.febslet.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 20.Hresko RC, Kraft TE, Tzekov A, Wildman SA, Hruz PW. 2014. Isoform-selective inhibition of facilitative glucose transporters: elucidation of the molecular mechanism of HIV protease inhibitor binding. J Biol Chem 289:16100–16113. doi: 10.1074/jbc.M113.528430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou B-H, Takanaga H, Grossmann G, Chen L-Q, Qu X-Q, Jones AM, Lalonde S, Schweissgut O, Wiechert W, Frommer WB. 2011. Optical sensors for monitoring dynamic changes of intracellular metabolite levels in mammalian cells. Nat Protoc 6:1818–1833. doi: 10.1038/nprot.2011.392. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J-H. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 23.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. 2013. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsuno K, Burrows JN, Duncan K, van Huijsduijnen RH, Kaneko T, Kita K, Mowbray CE, Schmatz D, Warner P, Slingsby BT. 2015. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat Rev Drug Discov 14:751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 25.Muretta JM, Kyrychenko A, Ladokhin AS, Kast DJ, Gillispie GD, Thomas DD. 2010. High-performance time-resolved fluorescence by direct waveform recording. Rev Sci Instrum 81:103101. doi: 10.1063/1.3480647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen KJ, Peterson KC, Muretta JM, Higgins SE, Gillispie GD, Thomas DD. 2014. Fluorescence lifetime plate reader: resolution and precision meet high-throughput. Rev Sci Instrum 85:113101. doi: 10.1063/1.4900727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorens B, Mueckler M. 2010. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab 298:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. 2014. Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 29.Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W, Hirata K, Yamamoto M, Fan S, Yan N. 2015. Molecular basis of ligand recognition and transport by glucose transporters. Nature 526:391–396. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 30.Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, Hussien SA, Qureshi AA, Coincon M, Sato Y, Abe H, Nakada-Nakura Y, Hino T, Arakawa T, Kusano-Arai O, Iwanari H, Murata T, Kobayashi T, Hamakubo T, Kasahara M, Iwata S, Drew D. 2015. Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526:397–401. doi: 10.1038/nature14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hresko RC, Hruz PW. 2011. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS One 6:e25237. doi: 10.1371/journal.pone.0025237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szablewski L. 2013. Expression of glucose transporters in cancers. Biochim Biophys Acta 1835:164–169. [DOI] [PubMed] [Google Scholar]
- 33.Galluzzi L, Kepp O, Heiden Vander MG, Kroemer G. 2013. Metabolic targets for cancer therapy. Nat Rev Drug Discov 12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 34.Medina RA, Owen GI. 2002. Glucose transporters: expression, regulation and cancer. Biol Res 35:9–26. [DOI] [PubMed] [Google Scholar]
- 35.Gautier EL, Westerterp M, Bhagwat N, Cremers S, Shih A, Abdel-Wahab O, Lütjohann D, Randolph GJ, Levine RL, Tall AR, Yvan-Charvet L. 2013. HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J Exp Med 210:339–353. doi: 10.1084/jem.20121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J, Chen X. 2012. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 11:1672–1682. doi: 10.1158/1535-7163.MCT-12-0131. [DOI] [PubMed] [Google Scholar]
- 37.NIH National Cancer Institute. SEER cancer stat fact sheets. National Cancer Institute, Bethesda, MD: http://seer.cancer.gov/statfacts/ Accessed 2 December 2015. [Google Scholar]