

Abstract
Damage-associated molecular patterns (DAMPs) are released in response to cell death and stress, and are potent triggers of sterile inflammation. Recent evidence suggests that DAMPs may also have a key role in the development of cancer as well as in the host response to cytotoxic anti-tumor therapy. As such, DAMPs may exert protective functions by alerting the immune system to the presence of dying tumor cells, thereby triggering immunogenic tumor cell death. On the other hand, cell death and release of DAMPs may also trigger chronic inflammation and thereby promote the development or progression of tumors. Here, we will review the contribution of candidate DAMPs and their receptors and discuss the evidence for DAMPs as tumor-promoting and anti-tumor effectors as well as unsolved questions such as DAMP release from non-tumor cells as well as the existence of tumor-specific DAMPs.
Keywords: Cell death, HMGB1, ATP, FPR1, Calreticulin, chemotherapy, radiation
Introduction
Cancers have been described as “wounds that do not heal”1, suggesting that multiple components of the wound healing process also contribute to carcinogenesis. This notion is supported by a landmark study by Bissel and colleagues, demonstrating that wounding strongly promotes the development of tumors following injection of Rous Sarcoma virus2. Likewise, clinical evidence suggest more common recurrence of tumors in resection margins, i.e. sites in which wound healing occurs, often with poorer differentiation and dismal prognosis3. Together, these studies suggest that injury and subsequent wound healing promote the development of cancer. Injured or stressed cells release a plethora of mediators, termed damage-associated molecular patterns (DAMPs), that potently trigger sterile inflammation. DAMPs represent a large range of chemically unrelated mediators entities such as High Mobility Group Box 1 (HMGB1), S100 proteins, hyaluronan, heat shock proteins, ATP and calreticulin that are retained inside the cell in the healthy state and only released following stress or cell death, thus allowing the host to sense and react to damage via specific DAMP receptors. While DAMPs were initially considered to be exclusively released from necrotic cells, recent evidence suggest that specific forms of programmed cell death such as necroptosis and immunogenic cell death (ICD) following anti-cancer therapies4–7 can also trigger DAMP emission into the extracellular space. DAMP-mediated sterile inflammation is an important component of a wide range of diseases including atherosclerosis, myocardial infarction, autoimmune diseases, and cancer8. As there are several excellent reviews on DAMPs and their receptors4, 8–10, we will selectively focus on their involvement in cancer in this review.
There are multiple parallels between innate immune responses to pathogens and cell death, such as the use of specific pattern recognition receptors (PRRs), and the occurrence of neutrophils and inflammation at sites of infection or injury. Hence, these pathways can be classified as a common danger response system that can be activated by either pathogen-associated molecular patterns (PAMPs) or DAMPs in order to combat danger and restore tissue homeostasis. DAMPs could not only warn the body about the presence of tissue injury in sterile conditions but also in the setting of infection, e.g. when pathogens induce cell death, and thereby trigger more profound immune responses. In analogy, death of premalignant or malignant cells might allow a more potent anti-tumor response or immunosurveillance. As such, there is accumulating evidence that anti-tumor therapies including radiation therapy and select chemotherapeutic agents not only trigger direct cytotoxic effects, but also contribute to subsequent priming of the immune system and immune-mediated anti-tumor responses7, 11, 12. On the other hand, inflammation is a double-edged sword that may not only trigger anti-tumor immune responses but also promote carcinogenesis in many settings (Table 1). Failure of DAMPs to elicit an effective anti-tumor response might turn DAMP-induced inflammation into a tumor-promoting mechanism13, 14 – similar to wound healing, which often becomes maladaptive when injury is chronic15. Here, we will discuss the possible roles of DAMPs in cancer, focusing not only on DAMPs as mediators of immunogenic tumor death but also the possible roles of DAMPs as triggers of tumor-promoting inflammation, as well as changes in the tumor microenvironment.
Table 1.
| DAMP | Receptor | Tumor | Main effects | Target cell | Effect on Tumor | Ref |
|---|---|---|---|---|---|---|
| Adenosine | A1 | Breast cancer | Cell proliferation | Tumor cells | Tumor growth | 19 |
| Glioblastoma multiforme | Anti-proliferative/pro-apoptotic effect | Cancer stem cells | Chemotherapy sensitivity | 118 | ||
| A2A | Immunosuppression | Treg | Likely to promote tumors | 76 | ||
| Treg expansion, immunosuppression | T cells and Treg | Likely to promote tumors | 117 | |||
| Inhibition of cytokines and chemokines release. | Teff and Treg | Likely to promote tumors | 116 | |||
| Lewis Lung carcinoma | Accumulation of intratumoral granulocytic MDSC | Hematopoietic cells | Tumor promotion | 130 | ||
| Lewis lung carcinoma | Suppression of NK cell maturation and activation | NK cells | Tumor immune evasion | 19 | ||
| A2B | Breast cancer | Tumor cell migration and metastasis | Tumor cells | Tumor promotion and metastasis | 79 | |
| Ovarian cancer Melanoma, lymphoma |
Decreased T cell infiltration | T cells | Tumor immune evasion | 77 | ||
| Glioblastoma multiforme | Anti-proliferative/pro-apoptotic effect | Cancer stem cells | Chemotherapy sensitivity | 118 | ||
| A3 | Breast cancer, Colorectal cancer melanoma, lymphoma | Proliferation | Tumor cells | Tumor promotion | 19 | |
| ATP | P2X7 | Sarcoma, lymphoma, colon cancer | NLRP3 inflammasome activation, cells recruitment, priming adaptive immune response, polarization CD8+ T cells | DC | Anti-tumor immune response, Tumor inhibition | 103 |
| Neuroblastoma | Immunosuppression | Monocytic MDSC | Tumor immune evasion | 73 | ||
| P2Y2 | Breast cancer, Leukemia | Recruitment, immunosuppression | DC, Macrophages | Anti-tumor immune response, Tumor inhibition | 113 | |
| Sarcoma, lymphoma, prostate cancer | Recruitment and differentiation | Myeloid cells | Anti-tumor immune response | 104 | ||
| HMGB1 | ? | Prostate cancer | Priming adaptive immune
response Proliferation |
T cells | Tumor promotion | 64 |
| ? | Endothelial cell migration and sprouting | Endothelial cells | Likely to promote tumors | 69 | ||
| RAGE? | Malignant mesothelioma | Migration and proliferation | Tumor cells | Tumor promotion | 33 | |
| RAGE | Hepatocellular carcinoma | Progenitor cell proliferation | Progenitor cells | Likely to promote tumors | 62 | |
| Neutrophil recruitment, injury amplification | Neutrophils | Not shown | 59 | |||
| RAGE | DC maturation, clonal expansion, T cells activation, Th1 polarization | DC | Not shown | 119 | ||
| Colon carcinoma Lung carcinoma |
Tumor regrowth and metastasis of remnant cancer cells following chemotherapy | Tumor cells | Tumor regrowth and metastasis | 67 | ||
| Pancreatic adenocarcinoma | Increased autophagy, decreased apoptosis | Tumor cells | Tumor survival (chemotherapy resistance) | 68 | ||
| RAGE/TLR4 | Prostate cancer | Induction of secretory/cytoplasmic clusterin, cell death inhibition | Tumor cells | Tumor survival (chemotherapy resistance) | 66 | |
| TLR4 | Mammary carcinoma, fibrosarcoma, lymphoma, colon carcinoma, osteosarcoma | Tumor Antigen processing and presentation | DC | Anti-tumor immune response | 101 | |
| TLR9 | Hepatocellular carcinoma | Proliferation, angiogenesis | Hypoxic tumor cells | Tumor progression | 31 | |
| TIM-3 | Melanoma, Colon carcinoma, Lewis lung carcinoma |
Decreased immunogenicity of nucleic acids | DC | Decreased tumor immune rejection | 70 | |
| Annexin A1 | Formyl peptide receptor (FPR) | Breast carcinoma, Lung carcinoma, Fibrosarcoma, |
Chemotherapy-induced antitumoral T cell response | DC | Chemotherapy-induced reduction of tumor growth | 105 |
| Calreticulin | Unknown | Colon carcinoma | Tumor cell uptake by dendritic cells and chemotherapy-induced anti-tumoral immune response | DC | Chemotherapy-induced reduction of tumor growth | 5 |
| Lymphoma | IL-6 and TNF-mediated Th17 priming | APC | Anti-tumor immune response (not shown) | 107 | ||
| S100A8/9 | RAGE | Breast carcinoma, Fibrosarcoma, Neuroblastoma | NF-κB activation, cell growth | Tumor cells | Tumor growth | 84 |
| Colon carcinoma Colorectal adenocarcinoma |
Myeloid cells infiltration, inflammation, protumorigenic gene activation | Tumor cells | Tumor promotion and progression | 80 | ||
| Skin cancer | Infiltration, inflammation, epidermal hyperplasia | Immune cells | Tumor promotion and progression | 61 | ||
| S100A4 | ? | Melanoma | Secretion of paracrine factors and pro-inflammatory cytokines promoting angiogenesis and pro-tumor immune response | Tumor cells | Metastasis | 87 |
| Colon cancer | Metastasis, invasion, proliferation | Tumor cells | Metastasis and tumor progression | 86 | ||
| Uric acid | TLR2/TLR4 | NLRP3 inflammasome activation | Macrophage | Not shown | 92 | |
| ? | Neutrophil recruitment, inflammation | Neutrophil | Not shown | 93 | ||
| ? | Breast carcinoma | Migration | Tumor cells | Likely to promote tumors | 95 | |
| ? | Leukemia | Recruitment | Monocytes/macrophages | Anti-tumor immune response | 97 | |
| IL-1 | IL-1R1 | Cell activation, cytokine release | Endothelial cells, T cells Macrophages |
Tumor invasiveness Tumor-promoting inflammation |
38 | |
| IL-33 | IL-1R1 | Breast carcinoma | Intratumoral accumulation of immunosuppressive cells,
decreased innate antitumoral immunity Intratumoral cell proliferation Angiogenesis |
MDSC, NK, DC, macrophages Tumor cells Endothelial cells |
Tumor progression | 40 |
| Colorectal cancer | Cell activation, proliferation, apoptosis, angiogenesis | Stromal cell types, subepithelial myofibroblasts and mast cells | Tumor growth and progression | 41 | ||
| Colorectal Cancer | Invasion, growth, metastasis | Tumor cells | Tumor growth and progression | 42 |
1. Release of DAMPs in tumors or their environments
Although we are only starting to understand the functions of DAMPs in malignancy, it has become evident that DAMPs are released by a wide range of tumors (Table 1). DAMPs are released in response to different modes of cell death (apoptosis, necroptosis, necrosis) and their release is regulated by different mechanisms and at different stages of cell death. Although it is believed that necrotic and necroptotic cell death are more inflammatory than apoptotic cell death, this concept needs to be more rigorously tested in context-specific settings16. As tumors grow, metabolic demands increase and cancer cells are inevitably exposed to metabolic, hypoxic, genetic and/or mechanical stress, leading to the induction of cell death, often visible as a necrotic tumor core17. Whether this type of cell death is largely necrotic, necroptotic, apoptotic or a mix of all these remains to be determined and is likely to be tumor-specific. Although there is evidence for DAMP release in the setting of spontaneous tumor cell death, e.g. a strong increase of extracellular ATP and adenosine within tumors18, 19, the release of DAMPs is much better documented during anti-tumor therapy20–22. As such, therapeutic interventions such as chemo- and radiotherapy and oncolytic viruses trigger profound DAMP release12, 23, 24. The predominant form of cell death by these therapeutic interventions appears to be apoptosis11, 12, but other forms of cell death also participate17, 25, 26, and the mode of cell death likely determines the spectrum and activity of released DAMPs. Finally, cancer cells may also release DAMPs through stress pathways that either precede or are not directly related to cell death. Treatment with anthracyclins or photodynamic therapy result in the early translocation of calreticulin (CRT) to the cell surface before affected cells exhibit biochemical signs of apoptosis5, 6. CRT is not a classical DAMP as it is not secreted and appears to be selectively operating as an “eat me” signal to stimulate the engulfment of apoptotic cells by dendritic cells (DC)5, 27. Mechanistically, surface exposure of CRT is triggered by endoplasmic reticulum (ER) stress6. Another example is the release of ATP, which is mediated by active secretion from dying cancer cells preceding the post-mortem release of HMGB128. ATP release in this setting is triggered by activation of caspases, which contribute to the redistribution of ATP from lysosomes to autolysosomes as well as the opening of pannexin 1 channels28. Extracellular ATP can be converted into adenosine, which acts through distinct receptors that often result in immunosuppressive effects opposing the immunostimulatory effects of ATP. There is accumulating evidence on adenosine accumulation in the tumor environment, thus creating an immunosuppressed “tumor-friendly” niche19. Increased levels of extracellular adenosine are the result of increased expression of ectonucleotidases CD39 and CD7329. In addition, several tumors show altered purine metabolism, which facilitates the production of adenosine or reduces its degradation19. Moreover, some DAMPs such as HMGB1 cannot only passively leak from dying cells but also be actively secreted via mechanisms that require posttranslational modifications such as acetylation and translocation from the nucleus to the cytosol30. In hypoxic hepatocellular carcinoma, HMGB1 is almost exclusively located in the cytoplasm31 and hypoxia is sufficient to trigger HMGB1 release in carcinoma cell lines31, suggesting that cell death-independent HMGB1 secretion is also operative in cancer. Recent publications show lower nuclear HMGB1 staining in a large percentage of human tumor samples, which might – rather than indicating lower HMGB1 expression by the tumor – also indicate increased HMGB1 secretion32. Indeed, human malignant mesothelioma biopsies have been shown to display a variable degree of HMGB1 cytoplasmic staining, absent in normal pleura, that correlated with tumor stage33. Many members of S100 protein family, which serve as intracellular Ca2+ sensors under physiologic conditions but also as DAMPs under pathologic conditions, exhibit cancer-type-specific patterns of dysregulated expression, with most evidence pointing towards overexpression and cancer promotion22. S100 proteins lack a leader sequence, precluding secretion via the classical Golgi pathway. Whereas S100A8/A9 can be actively secreted in a microtubule-and proteinase kinase C-dependent manner, both S100A8/A9 and S100B can also be passively released by injured tissues34. However, the precise mechanisms, by which they are released in cancer, are currently unknown. Interleukin (IL)-1ɑ is released by many cell types upon activation or following necrosis and IL-1 signaling has been implicated in the response to necrotic cell death35. The precursor form of IL-1ɑ is upregulated and subsequently released from dying cells following hypoxia36. IL-1ɑ released by necrotic hepatocytes contributes to compensatory proliferation and carcinogenesis36. IL-1ɑ exists not only as a soluble form whose maturation and secretion is inflammasome and caspase-1 dependent37, but also as a cell-surface protein that is able to activate IL-1 receptor-like 1 on target cells such as T-cells and endothelial cells and potentiates induction of other cytokines38. While the secreted form of IL-1ɑ is highly pro-inflammatory in the tumor microenvironment and involved in tumor growth and invasiveness, its membrane form favors anti-tumor immunity, and leads to reduced tumor growth and invasiveness38. IL-33 is another cytokine released by necrotic cells and by a variety of tissue types under stress conditions39. Endogenous IL-33 expression is increased in tumor tissue and contributes to cancer progression40–42. IL-33 upregulation in tumor also correlates with increased expression of target receptor complex IL-1 receptor-like 1 in stromal cells41, 42, reflecting a paracrine effect of IL-33 as a result of crosstalk between tumor cells and surrounding stroma.
In addition to DAMPs secreted within tumors, it is also conceivable that DAMP secretion from cells in the tumor microenvironment can modulate tumor biology and development. However, this area requires further investigation.
While there is ample evidence for DAMP release in multiple settings, much of the current data is based on cell lines and animal models. Further studies are required to understand DAMP release in human cancer patients, to what degree therapeutic inventions alter DAMP release and whether DAMP levels correlate with therapeutic prognosis or clinical outcome. In systemic therapies, it is likely that DAMPs are not only released from tumors but that a large proportion of DAMPs are derived from other cell types that are typically affected by chemotherapy or irradiation such as intestinal epithelia. Hence, DAMPs from these cells rather than tumor-derived DAMPs might also affect inflammation and immune responses.
2. Contribution of DAMPs to tumor promotion via inflammation or immunosuppression
Whereas acute inflammation is often beneficial for the host and an essential component of pathogen defense and tissue repair, failure to eliminate the causative agent leads to chronic unresolved inflammation that promotes mal-adaptive wound healing and increased risk to develop cancer as demonstrated in animal models13, 14, 43 and humans13. Mechanistically, inflammation predisposes to malignant transformation via multiple mechanisms, including (i) genetic damage caused by inflammation-associated reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI), (ii) promotion of proliferation via cytokine-induced growth factors, and (iii) resistance to cell death via activation of anti-apoptotic cell death pathways such as NF-κB. Accordingly, the most prevalent conditions predisposing to cancer all have significant inflammatory components, such as chronic infections (i.e., H. pylori, HBV, HCV, HPV), exposure to inhalative pathogens (including cigarette smoke, asbestos, silica) as well as obesity13, 44, 45. In addition to inflammatory conditions that promote the development of tumors, inflammation is also an essential component of established tumors. Of note, inflammation is not merely an epiphenomenon but an essential driver of tumor growth, whose significance for the progression of malignant disease is highlighted by their emergence as a novel “hallmark of cancer”46. The role of tumor-associated inflammation is highly context- and stage- and tumor-specific. While inflammation in early stages may contribute to anti-tumor responses, immune cell exhaustion and loss of neoantigens after initially successful immunoediting may switch inflammation into a tumor-promoting response once anti-tumor immunity has started failing47, 48. DAMPs including HMGB1, S100 and heat shock proteins, act via potent PRRs that are also employed by PAMPs such as Toll-like receptors (TLRs), formyl peptide receptor (FPR), C-type lectins and the receptor for advanced glycation endproducts (RAGE). These receptors activate a shared set of inflammatory pathways, including NF-κB, p38, ERK, inflammasome assembly and IL-1β and IL-18 release, as well as secretion of IL-6, TNF, LT-β, IFNγ and TGF-β, and promote the recruitment of inflammatory cells. As such, IL-1, IL-6 and LT-β are well-known promoters of carcinogenesis14, 43, 49, 50; recent evidence suggests that recruited inflammatory cells, in particular “ectopic lymphoid structures” – which are often in close proximity to damaged tissue – promote the development of cancer51. Not only is there a strong correlation between DAMP expression and carcinogenesis in multiple tumors, but a plethora of DAMPs have been implicated in promoting inflammation and tumor development both in early stages of carcinogenesis as well as in established tumors (Fig. 1; Table 1). In addition to promoting tumor growth via increased inflammation, there is accumulating evidence that multiple DAMPs also exert immunosuppressive effects that ultimately promote the development or progression of tumors (Fig. 1; Table 1).
Fig 1. DAMPs mediate tumor progression.
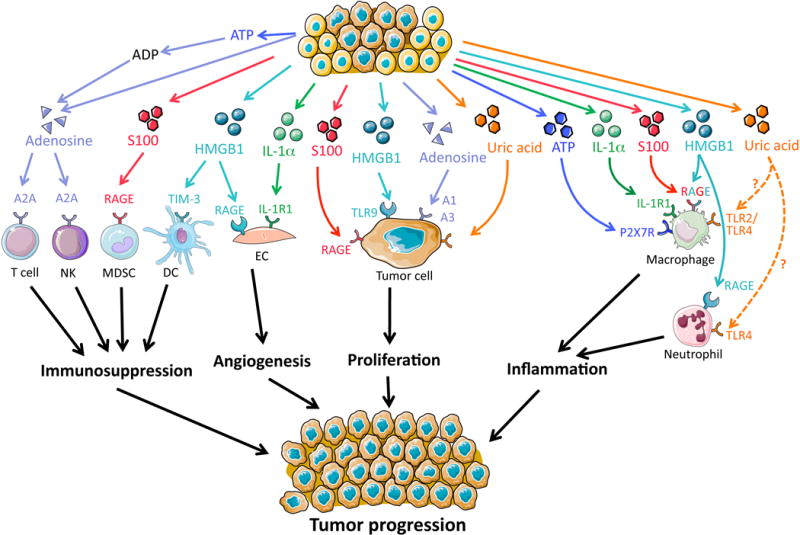
Cellular release of DAMPs such as uric acid, HMGB1, S100 proteins, IL-1α and adenosine can promote tumor progression via distinct mechanisms and target cells. Adenosine and HMGB1 may contribute to immunosuppression, HMGB1 and IL-1α to angiogenesis; uric acid, HMGB1, S100 proteins and adenosine to tumor cell proliferation; and ATP, IL-1α, S100 proteins, HMGB1 and uric acid to inflammation. NK, natural killer cell; MDSC, myeloid-derived suppressor cell; DC, dendritic cell; EC, endothelial cell.
HMGB1
HMGB1 is overexpressed in precancerous states such as liver cirrhosis and gastric dysplasia52, 53 as well as in a wide range of tumors54–58 and may trigger a number of inflammatory responses that promote tumor development and/or progression. HMGB1 triggers the recruitment of neutrophils, subsequent inflammation and amplification of injury in multiple injury models59, 60. Ablation of HMGB1 receptor RAGE suppresses carcinogenesis in multiple cancer models, including inflammation-induced liver and skin cancer as well as a xenotransplant glioma model61–63. As such, HMGB1 promotes the recruitment and activation of intratumoral T cells in prostate cancer, which in turn recruit tumor-promoting macrophages64. Moreover, HMGB1 promotes the growth of hepatocellular carcinoma in concert with mitochondrial DNA via activation of TLR931. TLR4, another receptor for HMGB1, is also expressed by tumor cells; activation of the TLR4 pathway has been associated with tumor cell survival, chemoresistance and tumor progression and metastasis65. Of note, the HMGB1-TLR4/RAGE pathway has recently been involved in chemoresistance to docetaxel by inducing the secretory/cytoplasmic clusterin in prostate tumor cells, a potent anti-apoptotic protein that mediates BAX sequestration, preventing caspase 3 activation66. Chemotherapy-induced release of HMGB1 from necrotic colon cancer cells enhances the growth and metastasis of remnant cancer cells through RAGE67 while blockade of the HMGB1-RAGE axis inhibits tumor growth and metastasis63 and enhances sensitivity to chemotherapy68. Moreover, HMGB1 released from hypoxic cells triggers endothelial cell proliferation in vitro and neoangiogenesis in vivo69, and expression levels of HMGB1 in tumors are associated with invasion and metastasis53.
Interactions between HMGB1 and TIM-3, expressed on DC, result in suppression of the therapeutic efficacy and of DNA vaccination and chemotherapy by decreasing immunogenicity of nucleic acids released from dying tumor cells70. Hence, HMGB1 may not only promote carcinogenesis via the activation of inflammatory pathway but also through immunosuppressive pathways.
ATP and adenosine
Although there are unusually high concentrations of extracellular ATP in the tumor interstitium as well as increased expression levels of the ATP receptor P2X7R on a variety of human cancers18, 71, there is only limited evidence in supporting their role in tumor promotion. Although P2X7 promotes inflammation, largely mediated by activation of the inflammasome72, and modulates myeloid-derived suppressor cell (MDSC) immunosuppressive functions73, the majority of studies show a protective role (see next section). The finding that P2X7R-deficiency exacerbated tumor development in a colitis-associated colon cancer model despite decreased inflammation74 suggests only a limited role in P2X7R-mediated inflammation in promoting carcinogenesis. Adenosine is a potent anti-inflammatory DAMP that acts on a variety of cell types and contributes to limiting inflammatory response following injury. Increased adenosine levels in the tumor microenvironment contribute to tumor progression via suppression of T cells and natural killer (NK) cells19. Stimulation of A2A receptor decreases maturation and cytotoxic function of NK cells, leading to promotion of metastasis75. Moreover, regulatory T (Treg) cells express ectonucleotidases CD39 and CD73 in a variety of tumors19, 76, resulting in increased generation of pericellular adenosine and A2A receptor-mediated inhibition of effector T cells. CD73 is also expressed on tumor cells or stroma, which further increases adenosine levels and contributes to immune evasion by suppressing T cell recruitment and activation as well as chemoresistance to doxorubicin in an A2A-dependent manner76–78. Accordingly, blockade of CD73 inhibits breast tumor growth and metastasis79 and increases doxorubicin-mediated anti-tumor immune response78. Moreover, a large number of studies have shown that adenosine triggers tumor proliferation via A1 and A3 receptors19, suggesting that this pathway may contribute to tumor progression. Moreover, adenosine promotes chemotaxis and metastasis via A2B receptors79.
S100 proteins
Dysregulated expression of various members of the S100 protein family has been observed in various cancers, with tumor-stage and –subtype specific expression profiles. Similarly to HMGB1, S100A8/A9 and S100A12 circulating levels are upregulated in a number of chronic inflammatory diseases and types of tumors and strongly correlate with disease course and/or severity34. In addition, the S100A8/A9-RAGE axis links inflammation and tumor promotion through activation of MAPK and NF-κB pathways61, 80. S100 proteins have been ascribed with diverse DAMP functions following release from necrotic cells, mostly through interaction with RAGE, although interaction with TLR4 has been demonstrated in other settings60, 81. Interference with their expression or signaling pathways has subsequently revealed a complex role for the various S100 proteins in the growth and dissemination of established tumors, with effects both on cancer cell growth as well as on concomitant inflammation22. As such, the expression S100A8 and S100A9 is induced by distant primary tumors, resulting in the attraction of myeloid cells in the premetastatic lung82. This is mediated by the induction of S100A8- and S100A9-induced production of SAA3, which in turn activated TLR4 and facilitated metastasis83. In addition, S100A8 and S100A9 act directly on tumor cells, activating p38 MAPK, thereby promoting tumor cell migration82. Similarly, it has been shown that low concentrations of extracellular S100A8/9 also enhance NF-κB activation in tumor cells and promotes their growth through interactions with RAGE84. S100A4 expression is significantly increased in prostate cancer cell lines compared to normal prostate epithelial cells, and expression correlates with increased tumor grade85. A causative relationship between S100A4 expression and tumor progression has been demonstrated in colon cancer cells, where pharmacologic targeting of S100A4 via the anti-helmintic niclosamide profoundly inhibited growth and metastatic spread86. S100A4, however, apparently exhibits both tumor cell-intrinsic as well as –extrinsic effects, as demonstrated by the ability of S100A4 from metastases-associated stroma to bind to RAGE on tumor cells, leading to the secretion of paracrine factors and pro-inflammatory cytokines such as IL-8, CCL2, IL-6 and IL-1β, which in turn promote angiogenesis and pro-tumor immune responses87. Accordingly, both intracellular and extracellular S100A4 may represent promising therapeutic targets to prevent progression of neoplastic diseases.
In addition to promoting inflammation, recruitment of macrophages and tumor migration, S100 proteins also affect anti-tumor immune responses. As such, S100A9 enhances the production of myeloid-derived suppressor cells, thereby suppression anti-tumor responses. S100A9-deficient mice rejected implanted tumors, which was reversed by administration of wild-type MDSCs from tumor-bearing mice to S100A9-deficient mice88. Notably, RAGE null mice exhibit reduced tumor growth in experimental skin and colon cancers61, 62, 89, and display reduced numbers of MDSC in tumor stroma, pointing at a central role for RAGE in the mediation of S100A8/A9-mediated MDSC recruitment90, 91.
Uric acid
Dying cells release their intracellular uric acid stores, and additional uric acid is generated post mortem during enzymatic degradation of nucleic acids. Extracellular uric acid triggers inflammatory responses to cell death, possibly through TLR4-mediated NLRP3 inflammasome activation92, by mediating neutrophil activation93 as well as DC maturation and T cell differentiation94. Moreover, cancer cells themselves respond to uric acid by increasing migratory activity95. Accordingly, elevated uric acid levels in patients have been associated with an excess cancer risk96. However, uric acid released from tumors subject to chemotherapy or immune rejection accelerates tumor regression97.
3. Contribution of DAMPs to tumor inhibition/rejection via immunogenic cell death and other mechanisms
Physiological cell death, such as apoptosis, has long been considered non- or low-inflammatory due to the rapid removal of apoptotic cells by phagocytic cells, whereas pathological cell death, induced by physicochemical stress or noxious stimuli, such as necrosis, necroptosis and pyroptosis, has been described as inherently immunogenic and highly inflammatory. Since cancer therapies often induce cell death via apoptosis and additionally can be immunosuppressive either on their own or in combination with the commonly co-administered corticosteroids, the concept that tumor cell death triggered by cytostatic therapies might be immunogenic has long been ignored98. However, this traditional perspective of cell death has been challenged by the finding that in response to specific anti-cancer agents, tumor cells can undergo an immunogenic cell death (ICD) that combines modalities of apoptosis with the emission of DAMPs, fostering a potent, therapeutic reinforcing anti-tumor immune response (Fig 2). Moreover, tumor cell death is not selectively apoptotic as other death modalities including necrosis and necroptosis are also potently induced by cytostatic therapies and necrosis is even commonly found in untreated tumors, often visible as necrotic tumor center17, 25, 26. Although the contribution of non-apoptotic forms of cell death including necrosis, necroptosis and pyroptosis to ICD is not as well characterized, it is likely that non-apoptotic cell death commonly occurs in anti-cancer therapy strategies such as chemotherapy and irradiation17, 25, 26. There is accumulating evidence that DAMPs exert a key role in ICD. ICD strongly relies on the induction of an ER stress response triggered or accentuated by ROS production6, 99. The combined action of ER stress and ROS promotes the activation of DAMP signaling pathways, involving the pre-apoptotic exposure of the ER chaperone CRT on the cell surface (ecto-CRT)5, early apoptotic secretion of ATP100, and post-apoptotic release of HMGB1101. Engagement of these DAMPs with various target receptors present on immune cells, leads to the elicitation of a potent anti-tumor immunity (Fig. 2; Table 1). Several studies demonstrated that interfering with the emission of these DAMPs compromised the anti-tumor immune response5, 23, 100, providing evidence for its critical role in shaping cancer cell immunogenicity. However, a recent study using spontaneous mammary tumor models demonstrated that the adaptive immune system is dispensable for the therapeutic efficacy of oxaliplatin, doxorubicin and cisplatin102, raising concerns about experimental models used for ICD studies. In fact, most landmark studies on ICD rely on functional data from cell line-based models5, 6, 70, 100, 103–105. Transplanted cell lines are likely to differ substantially in their genetic profile to endogenously arising tumors and thus may induce immune responses that cannot be triggered by endogenously arising tumors. In addition to more profoundly altered genetic profiles, endogenous tumors undergo constant immunoediting106, whereas transplanted cell lines lack this selection and are most likely much more immunogenic due to a higher load of tumor antigens to which the host immune system can respond. Some of the concerns are alleviated the inclusion of human data in recent studies, showing poorer survival in patients with loss of function of FPR1105 or more rapid development of metastasis in patients with loss of function of P2X7R103. Additional studies in models with endogenously arising tumors would further confirm the relevance of ICD as well as the contribution of DAMPs to this process.
Fig 2. Contribution of DAMPs to tumor rejection via ICD.
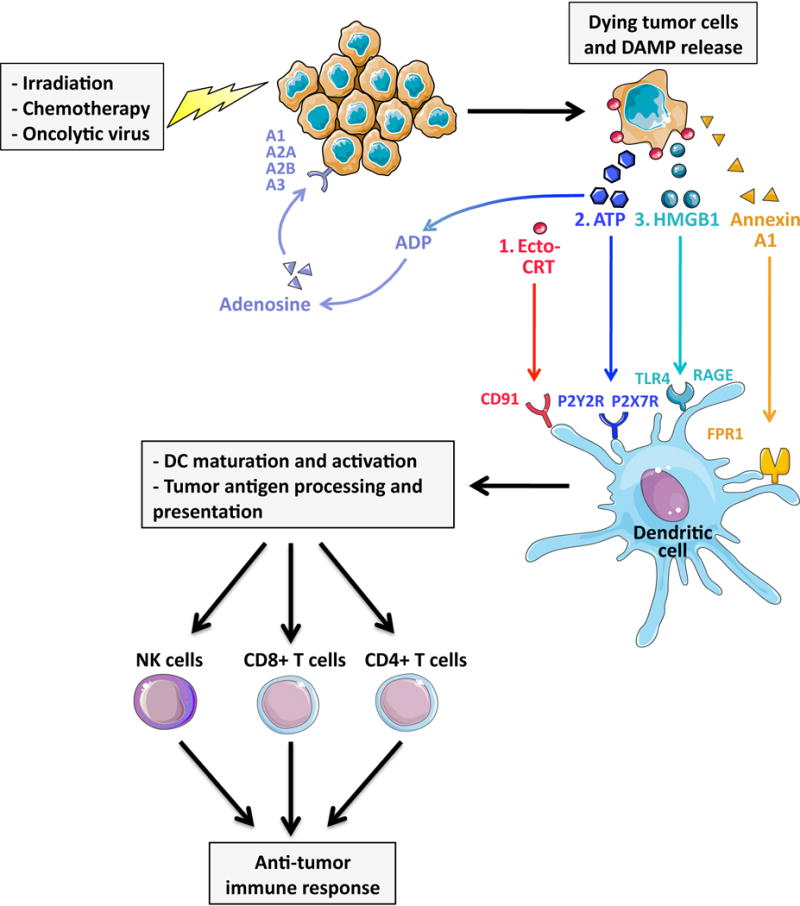
Immunogenic cell death (ICD), induced by various anti-cancer therapies strongly relies on the activation of DAMP signaling pathways. Following exposure irradiation, treatment with select chemotherapeutic agents and or infections with oncolytic viruses, tumor cells release DAMPs in the following order: 1. pre-apoptotic exposure of the ER chaperone calreticulin on the cell surface (ecto-CRT); 2. early apoptotic secretion of ATP; 3. post-apoptotic release of HMGB1. These DAMPs engage their respective receptors including CD91, P2X7R, P2Y2R, RAGE and TLR4 on the surface of dendritic cells (DC), triggering DC engulfment of dying cells, tumor antigen processing and presentation. In addition, Annexin A1, via its receptor FPR1, is required to bring DC into close proximity to dying tumor cells. DC maturation and activation ultimately foster potent anti-tumor responses via recruitment and activation of CD4+ and CD8+ T cells and natural killer (NK) cells.
Calreticulin
CRT is one of the best-characterized mediators of ICD. CRT translocation triggered by chemotherapy and UVC irradiation relies on PERK-mediated eIF2α phosphorylation followed by capase-8 mediated BAP31-dependent activation of BAX/BAK proteins99. However, eIF2α phosphorylation and caspase-8 are dispensable for photodynamic therapy (PDT)-induced ecto-CRT exposure6. Ecto-CRT functions as a pro-phagocytic signal for DC5 and promotes the IL-6 and TNF-mediated priming of the T helper 17 (Th17) cells through scavenger receptor CD91107. The presence of ecto-CRT has also been demonstrated in immune cells, including DC, where it was found to interact with NY-ESO-1, a tumor associated antigen with distinctively strong immunogenicity108, confirming its role in linking tumor and host immune response. Of note, the recombinant N-terminal fragment of CRT was sufficient to stimulate B cells and macrophage activation109, arguing for a potent immunostimulatory role of the soluble form of CRT. Interestingly, resistance to anti-cancer vaccination induced by ICD was associated with a defect in ecto-CRT exposure resulting from low endogenous CRT protein levels and rescued by exogenous reconstitution of ecto-CRT110. In the same study, CRT expression was a predictive biomarker of anticancer therapy response in cancer patients and a potential regulator of phagocytosis in tumors in ICD clinical settings110. Although necessary, CRT translocation is not sufficient to elicit an anti-tumor response, which relies on additional signaling pathways involved in antigen processing and presentation and immune cells polarization.
ATP and adenosine
Extracellular ATP is a critical effector in ICD103. Similarly to CRT translocation, ATP release seems to be dependent on cell death stimulus and modalities. PTD-induced, pre-apoptotic release of ATP relies on the overlapping classical secretory pathway as well as PERK-regulated secretory and PI3 kinase-dependent extracellular trafficking pathways and is independent of BAX/BAK. Early apoptotic secretion of ATP has been shown to be pannexin 1 hemichannels-dependent following UVC irradiation111 and autophagy-dependent in dying cells undergoing chemotherapy (anthracyclines and oxaliplatin)6, 100. Once released from the dying cancer cells, ATP displays a dual effect, both acting as a chemotaxis inducer and activator of the inflammasome pathway depending on its extracellular concentration112. ATP released by apoptotic cells has been shown to promote P2Y2R dependent-phagocytic clearance113 and P2X7R-dependent activation of the NLRP3 inflammasome by chemotherapeutic agents. The subsequent activation of caspase-1 then leads to the secretion of IL-1β and polarization of IFNγ-producing CD8+ T cells100, 103. ATP from dying cancer cells attracts myeloid cells to the site of cell death104, and ATP receptors P2Y2R and P2X7R stimulate the recruitment of DC and inflammatory cells into the tumor stroma as well as the maturation of T cells into the cytotoxic CD8+ phenotype103, 104, 114. P2X7R, which has a low affinity for ATP, is critical to support antitumor immune response and restrict tumor progression specially via its expression on host hematopoietic cells114. Ivermectin, an anti-parasitic drug, has been shown to display anti-tumor properties in vivo, due to its ability to stimulate P2X4R/P2X7R/Pannexin-1 signaling and to promote a novel form of cancer cell death involving a combination of apoptosis and highly inflammatory regulated necrosis, consistent with pyroptosis115. Noteworthy, adenosine, resulting from ATP hydrolysis by the ectoenzymes CD73 and CD39 exerts effect that are often opposite to those of ATP (see details in previous section), such as the promotion of an immunosuppressive environment via A2A receptors76, 116, 117 as well as chemotaxis and metastasis via A2B receptors79. However, adenosine can also induce tumor cell death, via A1, A2A, A2B and A3 receptors19,118, thereby additionally contributing to limit tumor growth (Fig 2). Therefore, the kinetics of ATP release and conversion to adenosine, as well as the large number of receptors for ATP and adenosine need to be taken into consideration when targeting this system for ICD induction.
Annexin A1/FPR1
Formyl peptide receptor 1 (FPR1) is a PRR that – besides recognizing N-formylated peptides from bacteria – also interacts with several DAMPs including annexin A1. Recent studies have shown that a single nucleotide polymorphism, which suppresses FPR1 signaling, was associated with reduced survival in patients receiving adjuvant chemotherapy for breast or colorectal cancer105. In experimental models, FPR1 expression on the host and expression of FPR1 ligand annexin A1 on tumor cells were required for chemotherapy-induced reduction of tumor growth. Likewise, treatment with FPR1 antagonist cyclosporine H also abolished anti-cancer effects of chemotherapy105. Mechanistically, FPR1 was dispensable for the recruitment of DC to the tumor bed, but was required for DC to come into close proximity to dying cancer cells, take up tumor-associated antigens and cross-present them to T cells105.
HMGB1
Although it was initially thought that HMGB1 release primarily occurred following necrosis, several studies also pointed out that HMGB1 release could also be associated with apoptosis, specifically during secondary necrosis. Post-apoptotic release of HMGB1, as triggered by radiotherapy or chemotherapy (anthracyclines), enhances DC-mediated antigen presentation via TLR423. In this context, HMGB1 acts as a critical mediator of the TLR4-dependent processing of exogenous tumor antigens by DC but does not promote DC migration and maturation which primarily involves the HMGB1-RAGE axis119. HMGB1 stimulates DC maturation through RAGE and leads to T-helper 1 polarization120. Interestingly, RAGE-mediated HMGB1 endocytosis has recently been described as a trigger of pyroptosis in macrophages121, which could represent a potential feedback mechanism promoting anti-tumor immunity during ICD. Although chemotherapy-induced HMGB1-TLR4 axis signaling has mainly been involved in eliciting anti-tumor responses, HMGB1 has also been showed to foster an immunosuppressive and pro-tumor environment (see previous section; Fig. 1, Table 1), which could negatively impact the outcome of anti-cancer therapies. The opposites roles of HMGB1 in tumorigenesis and ICD following anti-cancer therapies might be attributable to its redox status that defines its molecular interaction and activities122, 123, and plays a key role in cell fate regulation124, 125. Indeed, reducible HMGB1 induces Rage/Beclin1-dependent autophagy in cancer cells, promoting tumor resistance to chemotherapeutic agents or radiotherapy, whereas oxidized HMGB1 enhances the cytotoxicity of these chemotherapeutics and triggers apoptosis mediated by the caspase-9/-3 intrinsic pathway124. Of note, the extracellular milieu, known to be oxidative under physiologic conditions, is altered and highly variable under pathologic conditions, such as cancer, as evidenced by in vitro culture of different cancer cell lines126. In vivo, the tumor microenvironment tends to be pro-oxidative which would diminish the pro-inflammatory properties of HMGB1 via oxidation-mediated inactivation122, 123, 127.
Finally, loss of membrane integrity occurring during primary or secondary necrosis leads to the release of uric acid, DNA, ATP, and N-formyl peptides that can participate and reinforce the anti-tumor response. As such, DNA released after chemotherapy-induced cell death70 can efficiently stimulate an antigen-specific anti-tumor immune response, and uric acid enhances tumor immune rejection97.
4. Tumor-specific DAMPs versus DAMPs from non-tumor cells following cytostatic therapy
The most profound release of DAMPs occurs in response to cytostatic therapies. However, these therapies are by no means tumor-specific and induce cell death in rapidly proliferating cells in many organs including bone marrow, gastrointestinal tract and hair follicles. Hence, it is likely that a significant proportion of DAMPs are released from non-tumor compartments in response to cytostatic therapies. Additional studies are required to determine whether DAMPs from healthy cells in these compartments modulate inflammatory and immune responses including ICD in the setting of cytostatic therapies.
Another key point that has not been sufficient addressed is the potential role of yet unknown tumor cell-specific DAMPs. The majority of studies on the role of DAMPs in tumors have been based on DAMPs discovered in models of non-malignant disease. Due to the profound aberrations of a wide range of pathways in tumor cells, tumor-specific DAMPs could be generated by a number of mechanisms, including posttranslational alterations and alterations in the secretion of mediators that are typically retained within healthy cells. In this regard, tumor-specific DAMPs could also be a cargo of exosomes, whose secretion is commonly upregulated in tumor cells.
5. Exploiting DAMPs for tumor prevention or anti-tumor therapy
It is conceivable that DAMPs can be targeted in different stages of carcinogenesis. Considering that constant cell death favors tumor development in organs such as the liver, one could envision blocking DAMP signaling as tumor-preventative strategy. On the other hand, the recently established key role of DAMPs in the immune system’s response to tumors, in particular in the setting of anti-tumor therapies, suggests that activating DAMPs signaling pathways might be exploited for anti-tumor therapies, analogous to PRR-mediated activation of anti-tumor pathways by “Coley’s toxin”. However, this view is likely to be simplistic as PRR-induced immunostimulatory signals may not only fail in chronic setting but even get “hijacked” by the tumor, resulting in tumor-promoting inflammation rather than an efficient anti-tumor response in the long-run. For this reason, it appears more promising to activate DAMP signaling pathways in conjunction with cystostatic therapies as well as therapies that prevent immune exhaustion such as checkpoint inhibitors. This might ensure continuous activation of anti-tumor responses and ICD, and might be particular appealing in settings when tumor mass is small and release of DAMPs minimal, e.g. as a type of adjuvant therapy after curative resection. In this regard, a recent study has identified FPR1 as determinant of chemotherapy-induced immunity and survival in adjuvant setting. Although these data are promising and may open new possibilities for treatment, we still need a better understanding of the role of specific DAMPs and their cellular targets as these responses are likely to be mediated by multiple DAMPs in complex signaling networks. It is likely that contribution of DAMPs and their receptors to carcinogenesis and ICD are organ-, tumor- and context-specific, and that key DAMPs in this setting are yet-to-be identified molecules or DAMPs with specific post-translational modifications. Moreover, it is conceivable that immuno-suppressive effects of chemotherapy may dominate in some organs as suggested by studies that demonstrated chemotherapy-induced plasma cell recruitment and subsequent plasma cell-mediated inhibition of chemotherapeutic efficacy128. Finally, it is also likely that DAMPs shape the tumor microenvironment which in turn provides essential support for the tumor129. Hence, targeting select DAMPs that contribute to a tumor-promoting microenvironment may be beneficial. In summary, further understanding of the diverse roles of DAMPs in cancer is required before DAMPs can be exploited for therapeutic strategies, and it is likely that these therapeutic approaches might incorporate both activation and inhibition of DAMP signaling pathways.
References
- 1.Dvorak HF. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 2.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and Its Role in Rsv-Mediated Tumor Formation. Science. 1985;230:676–678. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 3.Hockel M, Dornhofer N. The Hydra Phenomenon of Cancer: Why Tumors Recur Locally after Microscopically Complete Resection. Cancer Res. 2005;65:2997–3002. doi: 10.1158/0008-5472.CAN-04-3868. [DOI] [PubMed] [Google Scholar]
- 4.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 6.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A Novel Pathway Combining Calreticulin Exposure and Atp Secretion in Immunogenic Cancer Cell Death. EMBO J. 2012;31:1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and Damps in Cancer Therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 8.Chen GY, Nunez G. Sterile Inflammation: Sensing and Reacting to Damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, et al. The Grateful Dead: Damage-Associated Molecular Pattern Molecules and Reduction/Oxidation Regulate Immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 10.Kono H, Rock KL. How Dying Cells Alert the Immune System to Danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic Cell Death in Cancer Therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 13.Balkwill F, Mantovani A. Inflammation and Cancer: Back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Neriah Y, Karin M. Inflammation Meets Cancer, with Nf-Kappab as the Matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 15.Wynn TA, Ramalingam TR. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasparakis M, Vandenabeele P. Necroptosis and Its Role in Inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 17.Kreuzaler P, Watson CJ. Killing a Cancer: What Are the Alternatives? Nat Rev Cancer. 2012;12:411–424. doi: 10.1038/nrc3264. [DOI] [PubMed] [Google Scholar]
- 18.Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased Level of Extracellular Atp at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS One. 2008;3:e2599. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, Inflammation and Cancer: A Leading Role for Adenosine. Nat Rev Cancer. 2013;13:842–857. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- 20.Srikrishna G, Freeze HH. Endogenous Damage-Associated Molecular Pattern Molecules at the Crossroads of Inflammation and Cancer. Neoplasia. 2009;11:615–628. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. Hmgb1 and Rage in Inflammation and Cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 22.Bresnick AR, Weber DJ, Zimmer DB. S100 Proteins in Cancer. Nat Rev Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-Like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 24.Guo ZS, Naik A, O’Malley ME, Popovic P, Demarco R, Hu Y, et al. The Enhanced Tumor Selectivity of an Oncolytic Vaccinia Lacking the Host Range and Antiapoptosis Genes Spi-1 and Spi-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 25.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a Signaling Platform That Assembles in Response to Genotoxic Stress and Loss of Iaps. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 26.Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many Stimuli Pull the Necrotic Trigger, an Overview. Cell Death Differ. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of Lrp on the Phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 28.Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular Mechanisms of Atp Secretion During Immunogenic Cell Death. Cell Death Differ. 2014;21:79–91. doi: 10.1038/cdd.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antonioli L, Pacher P, Vizi ES, Hasko G. Cd39 and Cd73 in Immunity and Inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic Cells Hyperacetylate Chromatin Protein Hmgb1 to Redirect It Towards Secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, et al. Hypoxia Induced Hmgb1 and Mitochondrial DNA Interactions Mediate Tumor Growth in Hepatocellular Carcinoma through Toll-Like Receptor 9. J Hepatol. 2015;63:114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, et al. Combined Evaluation of Lc3b Puncta and Hmgb1 Expression Predicts Residual Risk of Relapse after Adjuvant Chemotherapy in Breast Cancer. Autophagy. 2015;11:1878–1890. doi: 10.1080/15548627.2015.1082022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, et al. Cancer Cell Secretion of the Damp Protein Hmgb1 Supports Progression in Malignant Mesothelioma. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 Proteins. Curr Mol Med. 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a Key Pathway Required for the Sterile Inflammatory Response Triggered by Dying Cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 36.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, et al. Hepatocyte Necrosis Induced by Oxidative Stress and Il-1 Alpha Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, et al. Inflammasome Activation and Il-1beta Target Il-1alpha for Secretion as Opposed to Surface Expression. Proc Natl Acad Sci U S A. 2011;108:18055–18060. doi: 10.1073/pnas.1109176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rider P, Carmi Y, Voronov E, Apte RN. Interleukin-1alpha. Semin Immunol. 2013;25:430–438. doi: 10.1016/j.smim.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Villarreal DO, Weiner DB. Interleukin 33: A Switch-Hitting Cytokine. Curr Opin Immunol. 2014;28:102–106. doi: 10.1016/j.coi.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/St2 Axis Promotes Breast Cancer Growth and Metastases by Facilitating Intratumoral Accumulation of Immunosuppressive and Innate Lymphoid Cells. Int J Cancer. 2014;134:1669–1682. doi: 10.1002/ijc.28481. [DOI] [PubMed] [Google Scholar]
- 41.Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. Il-33 Activates Tumor Stroma to Promote Intestinal Polyposis. Proc Natl Acad Sci U S A. 2015;112:E2487–2496. doi: 10.1073/pnas.1422445112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Zhu L, Lu X, Bian H, Wu X, Yang W, et al. Il-33/St2 Pathway Contributes to Metastasis of Human Colorectal Cancer. Biochem Biophys Res Commun. 2014;453:486–492. doi: 10.1016/j.bbrc.2014.09.106. [DOI] [PubMed] [Google Scholar]
- 43.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renehan AG, Zwahlen M, Egger M. Adiposity and Cancer Risk: New Mechanistic Insights from Epidemiology. Nat Rev Cancer. 2015;15:484–498. doi: 10.1038/nrc3967. [DOI] [PubMed] [Google Scholar]
- 45.Hecht SS. Cigarette Smoking and Lung Cancer: Chemical Mechanisms and Approaches to Prevention. Lancet Oncol. 2002;3:461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 46.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Qian BZ, Pollard JW. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Visser KE, Eichten A, Coussens LM. Paradoxical Roles of the Immune System During Cancer Development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 49.Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A Lymphotoxin-Driven Pathway to Hepatocellular Carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-Cell-Derived Lymphotoxin Promotes Castration-Resistant Prostate Cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic Lymphoid Structures Function as Microniches for Tumor Progenitor Cells in Hepatocellular Carcinoma. Nat Immunol. 2015;16:1235–1244. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng BQ, Jia CQ, Liu CT, Lu XF, Zhong N, Zhang ZL, et al. Serum High Mobility Group Box Chromosomal Protein 1 Is Associated with Clinicopathologic Features in Patients with Hepatocellular Carcinoma. Dig Liver Dis. 2008;40:446–452. doi: 10.1016/j.dld.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 53.Chung HW, Lee SG, Kim H, Hong DJ, Chung JB, Stroncek D, et al. Serum High Mobility Group Box-1 (Hmgb1) Is Closely Associated with the Clinical and Pathologic Features of Gastric Cancer. J Transl Med. 2009;7:38. doi: 10.1186/1479-5876-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, et al. Hmgb1 Inhibits Cell Death in Yeast and Mammalian Cells and Is Abundantly Expressed in Human Breast Carcinoma. FASEB J. 2003;17:1295–1297. doi: 10.1096/fj.02-0621fje. [DOI] [PubMed] [Google Scholar]
- 55.Choi YR, Kim H, Kang HJ, Kim NG, Kim JJ, Park KS, et al. Overexpression of High Mobility Group Box 1 in Gastrointestinal Stromal Tumors with Kit Mutation. Cancer Res. 2003;63:2188–2193. [PubMed] [Google Scholar]
- 56.Wild CA, Brandau S, Lotfi R, Mattheis S, Gu X, Lang S, et al. Hmgb1 Is Overexpressed in Tumor Cells and Promotes Activity of Regulatory T Cells in Patients with Head and Neck Cancer. Oral Oncol. 2012;48:409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 57.Wu D, Ding Y, Wang S, Zhang Q, Liu L. Increased Expression of High Mobility Group Box 1 (Hmgb1) Is Associated with Progression and Poor Prognosis in Human Nasopharyngeal Carcinoma. J Pathol. 2008;216:167–175. doi: 10.1002/path.2391. [DOI] [PubMed] [Google Scholar]
- 58.Meyer A, Staratschek-Jox A, Springwald A, Wenk H, Wolf J, Wickenhauser C, et al. Non-Hodgkin Lymphoma Expressing High Levels of the Danger-Signalling Protein Hmgb1. Leuk Lymphoma. 2008;49:1184–1189. doi: 10.1080/10428190802064909. [DOI] [PubMed] [Google Scholar]
- 59.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The Hmgb1/Rage Axis Triggers Neutrophil-Mediated Injury Amplification Following Necrosis. J Clin Invest. 2015;125:539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A Novel Pathway of Hmgb1-Mediated Inflammatory Cell Recruitment That Requires Mac-1-Integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, et al. Rage Signaling Sustains Inflammation and Promotes Tumor Development. J Exp Med. 2008;205:275–285. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pusterla T, Nemeth J, Stein I, Wiechert L, Knigin D, Marhenke S, et al. Receptor for Advanced Glycation Endproducts (Rage) Is a Key Regulator of Oval Cell Activation and Inflammation-Associated Liver Carcinogenesis in Mice. Hepatology. 2013;58:363–373. doi: 10.1002/hep.26395. [DOI] [PubMed] [Google Scholar]
- 63.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of Rage-Amphoterin Signalling Suppresses Tumour Growth and Metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 64.He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue Damage-Associated “Danger Signals” Influence T-Cell Responses That Promote the Progression of Preneoplasia to Cancer. Cancer Res. 2013;73:629–639. doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 65.Ran S. The Role of Tlr4 in Chemotherapy-Driven Metastasis. Cancer Res. 2015;75:2405–2410. doi: 10.1158/0008-5472.CAN-14-3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou J, Chen X, Gilvary DL, Tejera MM, Eksioglu EA, Wei S, et al. Hmgb1 Induction of Clusterin Creates a Chemoresistant Niche in Human Prostate Tumor Cells. Sci Rep. 2015;5:15085. doi: 10.1038/srep15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo Y, Chihara Y, Fujimoto K, Sasahira T, Kuwada M, Fujiwara R, et al. High Mobility Group Box 1 Released from Necrotic Cells Enhances Regrowth and Metastasis of Cancer Cells That Have Survived Chemotherapy. Eur J Cancer. 2013;49:741–751. doi: 10.1016/j.ejca.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 68.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, et al. The Receptor for Advanced Glycation End Products (Rage) Sustains Autophagy and Limits Apoptosis, Promoting Pancreatic Tumor Cell Survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schlueter C, Weber H, Meyer B, Rogalla P, Roser K, Hauke S, et al. Angiogenetic Signaling through Hypoxia: Hmgb1: An Angiogenetic Switch Molecule. Am J Pathol. 2005;166:1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-Infiltrating Dcs Suppress Nucleic Acid-Mediated Innate Immune Responses through Interactions between the Receptor Tim-3 and the Alarmin Hmgb1. Nat Immunol. 2012;13:832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, et al. Expression of P2x7 Receptor Increases in Vivo Tumor Growth. Cancer Res. 2012;72:2957–2969. doi: 10.1158/0008-5472.CAN-11-1947. [DOI] [PubMed] [Google Scholar]
- 72.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin Activates the Inflammasome in Response to Toxins and Atp. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 73.Bianchi G, Vuerich M, Pellegatti P, Marimpietri D, Emionite L, Marigo I, et al. Atp/P2x7 Axis Modulates Myeloid-Derived Suppressor Cell Functions in Neuroblastoma Microenvironment. Cell death & disease. 2014;5:e1135. doi: 10.1038/cddis.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hofman P, Cherfils-Vicini J, Bazin M, Ilie M, Juhel T, Hebuterne X, et al. Genetic and Pharmacological Inactivation of the Purinergic P2rx7 Receptor Dampens Inflammation but Increases Tumor Incidence in a Mouse Model of Colitis-Associated Cancer. Cancer Res. 2015;75:835–845. doi: 10.1158/0008-5472.CAN-14-1778. [DOI] [PubMed] [Google Scholar]
- 75.Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2a Receptors Potently Suppresses the Metastasis of Cd73+ Tumors. Proc Natl Acad Sci U S A. 2013;110:14711–14716. doi: 10.1073/pnas.1308209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by Cd39 and Cd73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. Cd73 Has Distinct Roles in Nonhematopoietic and Hematopoietic Cells to Promote Tumor Growth in Mice. J Clin Invest. 2011;121:2371–2382. doi: 10.1172/JCI45559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. Cd73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc Natl Acad Sci U S A. 2013;110:11091–11096. doi: 10.1073/pnas.1222251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-Cd73 Antibody Therapy Inhibits Breast Tumor Growth and Metastasis. Proc Natl Acad Sci U S A. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100a8/A9 Activate Key Genes and Pathways in Colon Tumor Progression. Molecular cancer research : MCR. 2011;9:133–148. doi: 10.1158/1541-7786.MCR-10-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 Are Endogenous Activators of Toll-Like Receptor 4, Promoting Lethal, Endotoxin-Induced Shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 82.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-Mediated Upregulation of Chemoattractants and Recruitment of Myeloid Cells Predetermines Lung Metastasis. Nat Cell Biol. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 83.Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100a8-Serum Amyloid A3-Tlr4 Paracrine Cascade Establishes a Pre-Metastatic Phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 84.Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, et al. S100a8/A9 at Low Concentration Promotes Tumor Cell Growth Via Rage Ligation and Map Kinase-Dependent Pathway. J Leukoc Biol. 2008;83:1484–1492. doi: 10.1189/jlb.0607397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gupta S, Hussain T, MacLennan GT, Fu P, Patel J, Mukhtar H. Differential Expression of S100a2 and S100a4 During Progression of Human Prostate Adenocarcinoma. J Clin Oncol. 2003;21:106–112. doi: 10.1200/JCO.2003.03.024. [DOI] [PubMed] [Google Scholar]
- 86.Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, et al. Novel Effect of Antihelminthic Niclosamide on S100a4-Mediated Metastatic Progression in Colon Cancer. J Natl Cancer Inst. 2011;103:1018–1036. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 87.Bettum IJ, Vasiliauskaite K, Nygaard V, Clancy T, Pettersen SJ, Tenstad E, et al. Metastasis-Associated Protein S100a4 Induces a Network of Inflammatory Cytokines That Activate Stromal Cells to Acquire Pro-Tumorigenic Properties. Cancer letters. 2014;344:28–39. doi: 10.1016/j.canlet.2013.10.036. [DOI] [PubMed] [Google Scholar]
- 88.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of Dendritic Cell Differentiation and Accumulation of Myeloid-Derived Suppressor Cells in Cancer Is Regulated by S100a9 Protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. Rage, Carboxylated Glycans and S100a8/A9 Play Essential Roles in Colitis-Associated Carcinogenesis. Carcinogenesis. 2008;29:2035–2043. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vernon PJ, Loux TJ, Schapiro NE, Kang R, Muthuswamy R, Kalinski P, et al. The Receptor for Advanced Glycation End Products Promotes Pancreatic Carcinogenesis and Accumulation of Myeloid-Derived Suppressor Cells. J Immunol. 2013;190:1372–1379. doi: 10.4049/jimmunol.1201151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-Associated Uric Acid Crystals Activate the Nalp3 Inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 93.Kono H, Chen CJ, Ontiveros F, Rock KL. Uric Acid Promotes an Acute Inflammatory Response to Sterile Cell Death in Mice. J Clin Invest. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi Y, Evans JE, Rock KL. Molecular Identification of a Danger Signal That Alerts the Immune System to Dying Cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 95.Fini MA, Orchard-Webb D, Kosmider B, Amon JD, Kelland R, Shibao G, et al. Migratory Activity of Human Breast Cancer Cells Is Modulated by Differential Expression of Xanthine Oxidoreductase. J Cell Biochem. 2008;105:1008–1026. doi: 10.1002/jcb.21901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of Uric Acid to Cancer Risk, Recurrence, and Mortality. Clin Transl Med. 2012;1:16. doi: 10.1186/2001-1326-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu DE, Moore AM, Thomsen LL, Brindle KM. Uric Acid Promotes Tumor Immune Rejection. Cancer Res. 2004;64:5059–5062. doi: 10.1158/0008-5472.CAN-04-1586. [DOI] [PubMed] [Google Scholar]
- 98.Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, et al. Molecular Determinants of Immunogenic Cell Death Elicited by Anticancer Chemotherapy. Cancer Metastasis Rev. 2011;30:61–69. doi: 10.1007/s10555-011-9273-4. [DOI] [PubMed] [Google Scholar]
- 99.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of Pre-Apoptotic Calreticulin Exposure in Immunogenic Cell Death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 101.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The Interaction between Hmgb1 and Tlr4 Dictates the Outcome of Anticancer Chemotherapy and Radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 102.Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy Response of Spontaneous Mammary Tumors Is Independent of the Adaptive Immune System. Nat Med. 2012;18:344–346. doi: 10.1038/nm.2652. author reply 346. [DOI] [PubMed] [Google Scholar]
- 103.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the Nlrp3 Inflammasome in Dendritic Cells Induces Il-1beta-Dependent Adaptive Immunity against Tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 104.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 105.Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-Induced Antitumor Immunity Requires Formyl Peptide Receptor 1. Science. 2015;350:972–978. doi: 10.1126/science.aad0779. [DOI] [PubMed] [Google Scholar]
- 106.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 107.Pawaria S, Binder RJ. Cd91-Dependent Programming of T-Helper Cell Responses Following Heat Shock Protein Immunization. Nat Commun. 2011;2:521. doi: 10.1038/ncomms1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zeng G, Aldridge ME, Tian X, Seiler D, Zhang X, Jin Y, et al. Dendritic Cell Surface Calreticulin Is a Receptor for Ny-Eso-1: Direct Interactions between Tumor-Associated Antigen and the Innate Immune System. J Immunol. 2006;177:3582–3589. doi: 10.4049/jimmunol.177.6.3582. [DOI] [PubMed] [Google Scholar]
- 109.Hong C, Qiu X, Li Y, Huang Q, Zhong Z, Zhang Y, et al. Functional Analysis of Recombinant Calreticulin Fragment 39–272: Implications for Immunobiological Activities of Calreticulin in Health and Disease. J Immunol. 2010;185:4561–4569. doi: 10.4049/jimmunol.1000536. [DOI] [PubMed] [Google Scholar]
- 110.Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to Anticancer Vaccination Effect Is Controlled by a Cancer Cell-Autonomous Phenotype That Disrupts Immunogenic Phagocytic Removal. Oncotarget. 2015;6:26841–26860. doi: 10.18632/oncotarget.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 Channels Mediate ’Find-Me’ Signal Release and Membrane Permeability During Apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trautmann A. Extracellular Atp in the Immune System: More Than Just a “Danger Signal”. Sci Signal. 2009;2:pe6. doi: 10.1126/scisignal.256pe6. [DOI] [PubMed] [Google Scholar]
- 113.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adinolfi E, Capece M, Franceschini A, Falzoni S, Giuliani AL, Rotondo A, et al. Accelerated Tumor Progression in Mice Lacking the Atp Receptor P2x7. Cancer Res. 2015;75:635–644. doi: 10.1158/0008-5472.CAN-14-1259. [DOI] [PubMed] [Google Scholar]
- 115.Draganov D, Gopalakrishna-Pillai S, Chen YR, Zuckerman N, Moeller S, Wang C, et al. Modulation of P2x4/P2x7/Pannexin-1 Sensitivity to Extracellular Atp Via Ivermectin Induces a Non-Apoptotic and Inflammatory Form of Cancer Cell Death. Sci Rep. 2015;5:16222. doi: 10.1038/srep16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular Purine Metabolism and Signaling of Cd73-Derived Adenosine in Murine Treg and Teff Cells. Am J Physiol Cell Physiol. 2011;301:C530–539. doi: 10.1152/ajpcell.00385.2010. [DOI] [PubMed] [Google Scholar]
- 117.Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The Development and Immunosuppressive Functions of Cd4(+) Cd25(+) Foxp3(+) Regulatory T Cells Are under Influence of the Adenosine-A2a Adenosine Receptor Pathway. Front Immunol. 2012;3:190. doi: 10.3389/fimmu.2012.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daniele S, Zappelli E, Natali L, Martini C, Trincavelli ML. Modulation of A1 and A2b Adenosine Receptor Activity: A New Strategy to Sensitise Glioblastoma Stem Cells to Chemotherapy. Cell death & disease. 2014;5:e1539. doi: 10.1038/cddis.2014.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, et al. Release of High Mobility Group Box 1 by Dendritic Cells Controls T Cell Activation Via the Receptor for Advanced Glycation End Products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 120.Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, et al. High Mobility Group Box Protein 1: An Endogenous Signal for Dendritic Cell Maturation and Th1 Polarization. J Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 121.Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage Endocytosis of High-Mobility Group Box 1 Triggers Pyroptosis. Cell Death Differ. 2014;21:1229–1239. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually Exclusive Redox Forms of Hmgb1 Promote Cell Recruitment or Proinflammatory Cytokine Release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox Modification of Cysteine Residues Regulates the Cytokine Activity of High Mobility Group Box-1 (Hmgb1) Mol Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, et al. Hmgb1 Release and Redox Regulates Autophagy and Apoptosis in Cancer Cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, et al. Cytosolic Hmgb1 Controls the Cellular Autophagy/Apoptosis Checkpoint During Inflammation. J Clin Invest. 2015;125:1098–1110. doi: 10.1172/JCI76344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chaiswing L, Oberley TD. Extracellular/Microenvironmental Redox State. Antioxid Redox Signal. 2010;13:449–465. doi: 10.1089/ars.2009.3020. [DOI] [PubMed] [Google Scholar]
- 127.Policastro LL, Ibanez IL, Notcovich C, Duran HA, Podhajcer OL. The Tumor Microenvironment: Characterization, Redox Considerations, and Novel Approaches for Reactive Oxygen Species-Targeted Gene Therapy. Antioxid Redox Signal. 2013;19:854–895. doi: 10.1089/ars.2011.4367. [DOI] [PubMed] [Google Scholar]
- 128.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive Plasma Cells Impede T-Cell-Dependent Immunogenic Chemotherapy. Nature. 2015;521:94–98. doi: 10.1038/nature14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 130.Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic Regulation of the Expansion and Immunosuppressive Activity of Cd11b+Gr1+ Cells. J Immunol. 2011;187:6120–6129. doi: 10.4049/jimmunol.1101225. [DOI] [PMC free article] [PubMed] [Google Scholar]