

Abstract
Elevated lipogenesis is a common characteristic of cancer and metabolic diseases. Sterol regulatory element-binding proteins (SREBPs), a family of membrane-bound transcription factors controlling the expression of genes important for the synthesis of cholesterol, fatty acids and phospholipids, are frequently upregulated in these diseases. In the process of SREBP nuclear translocation, SREBP-cleavage activating protein (SCAP) plays a central role in the trafficking of SREBP from the endoplasmic reticulum (ER) to the Golgi and in subsequent proteolysis activation. Recently, we uncovered that glucose-mediated N-glycosylation of SCAP is a prerequisite condition for the exit of SCAP/SREBP from the ER and movement to the Golgi. N-glycosylation stabilizes SCAP and directs SCAP/SREBP trafficking. Here, we describe a protocol for the isolation of membrane fractions in human cells and for the preparation of the samples for the detection of SCAP N-glycosylation and total protein by using western blot. We further provide a method to monitor SCAP trafficking by using confocal microscopy. This protocol is appropriate for the investigation of SCAP N-glycosylation and trafficking in mammalian cells.
Keywords: Cellular Biology, Issue 117, SCAP, N-glycosylation, SREBPs, endoplasmic reticulum, membrane protein, lipid synthesis
Introduction
Deregulation of lipid metabolism is a common characteristic of cancer and metabolic diseases1-6. In these processes, sterol regulatory element-binding proteins (SREBPs), a family of transcription factors, play a critical role in controlling the expression of genes important for the uptake and synthesis of cholesterol, fatty acids, and phospholipids7-10.
SREBPs, including SREBP-1a, SREBP-1c, and SREBP-2, are synthesized as inactive precursors that bind to the endoplasmic reticulum (ER) membrane by virtue of two transmembrane domains7. The N-terminus of SREBPs contains a DNA-binding domain for the transactivation of target genes11. The C-terminus of SREBP precursors binds to SREBP cleavage-activating protein (SCAP)12,13, a polytopic membrane protein that plays a pivotal role in the regulation of SREBP stability and activation14-16.
The implementation of transcriptional function requires the translocation of the SCAP/SREBP complex from the ER to the Golgi, where two proteases sequentially cleave SREBP and release its N-terminal fragment, which then enters into the nucleus for the transactivation of lipogenic genes7. In these processes, the levels of sterols in the ER membrane control the exit of the SCAP/SREBP complex from the ER7,17. Under high sterol conditions, sterol binds to SCAP or ER-resident insulin-induced gene protein-1 (Insig-1) or -2 (Insig-2), enhancing the association of SCAP with Insigs that retain the SCAP/SREBP complex in the ER18-20. When sterol levels decrease, SCAP dissociates with Insigs. This leads to a SCAP conformational change, which allows SCAP interaction with common coat proteins (COP)II complex. The complex mediates the incorporation of the SCAP/SREBP complex into the budding vesicles and directs its transport from the ER to the Golgi21,22. Upon translocation to the Golgi, SREBPs are sequentially cleaved by Site-1 and Site-2 proteases, leading to the release of the N-terminus7,23-29.
SCAP protein carries three N-linked oligosaccharides at asparagine (N) positions N263, N590, and N64115. We recently revealed that glucose-mediated N-glycosylation of SCAP in these sites is a prerequisite condition for SCAP/SREBP trafficking from the ER to the Golgi under low sterol conditions30-32. Loss of SCAP glycosylation via mutation of all three asparagine to glutamine (NNN to QQQ) disables the trafficking of the SCAP/SREBP complex and results in the instability of SCAP protein and reduction of SREBP activation31. Our recent data also demonstrate that SREBP-1 is highly activated in glioblastoma and regulated by SCAP N-glycosylation4,31,33,34. Targeting SCAP/SREBP-1 signaling is emerging as a new strategy to treat malignancies and metabolic syndromes1,3,35-38. Therefore, it is important to develop an effective method to analyze SCAP protein and N-glycosylation levels and monitor its trafficking in human cells and patient tissues.
The luminal region of SCAP protein (amino acids 540-707) in the ER contains two N-glycosylation sites (N590 and N641) that are protected from proteolysis when intact membranes are treated with trypsin15. This luminal fragment has a molecular weight of ~ 30 kDa that is small enough to allow the resolution of individual glycosylated variants of SCAP by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)15,31. Here, we provide a method to detect N-glycosylation of SCAP and the total protein in human cells. This protocol is derived from the method described in the publications from the Brown and Goldstein laboratory15 and our recent publication31. The protocol can be used in the study of SCAP protein from mammalian cells.
Protocol
1. Detection of Endogenous SCAP Protein in Human Cells
- Cell Culture and Treatment
- Seed ~ 1 × 106 U87 cells in a 10 cm dish with Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and incubate the cells at 37 °C and 5% CO2 for 24 hr before treatment.
- Wash cells once with phosphate-buffered saline (PBS), then switch cells to fresh DMEM medium with or without glucose (5 mM) for 12 hr.
- Preparation of Cell Membrane Fractions and Nuclear Extracts
- Wash cells once with PBS, scrape into 1 ml PBS, and centrifuge at 1,000 x g for 5 min at 4 °C.
- Resuspend cells in an ice-cold buffer containing 10 mM HEPES-KOH (pH 7.6), 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium ethylenediaminetetraacetic acid (EDTA), 1 mM sodium ethylene glycol tetraacetic acid (EGTA), 250 mM sucrose, and a mixture of protease inhibitors (5 µg/ml pepstatin A, 10 µg/ml leupeptin, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), and 25 µg/ml N-acetyl-Leu-Leu-Norleu-al (ALLN)) for 30 min on ice.
- Pass cell extracts through a 22 G x 1 1/2 needle 30 times and centrifuge at 890 x g at 4 °C for 5 min to isolate nuclei.
- Use the supernatant for the separation of membrane fractions (step 1.2.7). Resuspend the nuclear pellet in 0.1 ml of buffer C (20 mM HEPES/KOH pH 7.6, 0.42 M NaCl, 2.5% (v/v) glycerol, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA and a mixture of protease inhibitors (5 µg/ml pepstatin A, 10 µg/ml leupeptin, 0.5 mM PMSF, 1 mM DTT, and 25 µg/ml ALLN)).
- Rotate the suspension at 4 °C for 60 min and centrifuge at 20,000 x g in a microcentrifuge for 20 min at 4 °C. The supernatant is designated as "nuclear extracts".
- Heat the nuclear extracts at 100 °C for 10 min with 5x loading buffer (312.5 mM Tris-HCl pH 6.8, 10% SDS, 50% glycerol, 12.5% β-mercaptoethanol, and 0.05% bromophenol blue) before subjecting to SDS-PAGE.
- Centrifuge the supernatant from the original 890 x g spin at 20,000 x g for 20 min at 4 °C. For subsequent western blot analysis, dissolve the pellet in 0.1 ml of SDS lysis buffer (10 mM Tris-HCl pH 6.8, 100 mM NaCl, 1% (v/v) sodium dodecyl sulfate (SDS), 1 mM sodium EDTA, and 1 mM sodium EGTA). This is designated "membrane fraction". NOTE: We use 20,000 x g for 20 min to pellet membranes. 100,000 x g for 10 - 20 min has been used in the papers from the Brown and Goldstein laboratory15.
- Incubate the membrane fraction at 37 °C for 30 min and determine the protein concentration by Bradford protein assay. Add 1 µl 100x bromophenol blue solution before subjecting the samples to SDS-PAGE.
- Load 25 - 50 µg of total membrane proteins on a 10% SDS-PAGE gel39. Run the gel at 80 V for 15 min, then change to 130 V and run for another 115 min. Transfer at 140 mA for 130 min39.
- For Western blotting analysis, use anti-SCAP antibody to detect the total SCAP protein. Use protein disulfide isomerase (PDI), an ER-resident protein40, as an internal control. Use anti-SREBP-1 antibody to detect the N-terminal band of SREBP-1. Use Lamin A as an internal control for nuclear extracts31.
2. Analysis of SCAP N-glycosylation in Human Cells
- Cell Culture, Transfection, and Treatment
- For membrane analysis, seed ~1 × 106 HEK293T cells in a 10 cm dish with DMEM supplemented with 5% FBS and incubate the cells at 37°C and 5% CO2 for 24 hr before transfection.
- Dilute 4 - 8 µg GFP-SCAP plasmid (a gift from Dr. Peter Espenshade)22 in 1 ml reduced serum medium and mix gently.
- Add 8 - 16 µl DNA transfection reagent by pipetting directly into the 1 ml reduced serum medium containing plasmids and mix gently.
- Incubate the transfection reagent/plasmid complex for 25 min at room temperature.
- Add transfection complex to the cells with fresh medium and continue to culture for 24 hr at 5% CO2 and 37 °C.
- Wash once with PBS and treat the cells for 12 hr with or without glucose (5 mM) in the presence or absence of tunicamycin (1 µg/ml), an N-glycosylation inhibitor.
Prepare cell membrane pellets as described in steps 1.2.1, 1.2.2, 1.2.3, and 1.2.7.
- Detection of SCAP N-glycosylation
- Trypsin Proteolysis
- Resuspend the membrane pellets from cells overexpressing GFP-SCAP in 114 µl of buffer containing 10 mM HEPES·KOH (pH 7.4), 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, and 100 mM NaCl.
- Incubate 57 µl aliquots of membrane proteins in the absence or presence of 1 µl of trypsin (1 µg/µl), in a total volume of 58 µl, for 30 min at 30 °C.
- Stop reactions by the addition of 2 µl (400 units) of soybean trypsin inhibitor.
- Deglycosylation by Peptide-N-Glycosidase F (PNGase F)
- For subsequent treatment with PNGase F, add 10 µl of solution containing 3.5% (wt/vol) SDS and 7% (vol/vol) 2-mercaptoethanol to each sample.
- After heating at 100 °C for 10 min, sequentially add 7 µl of 0.5 M sodium phosphate (pH 7.5), 7 µl of 10% (v/v) Nonidet P-40 with 12× protease inhibitors, and 2 µl (0.5 U/µl) of PNGase F.
- After incubation at 37 °C for 3 hr, stop reactions by the addition of 5x loading buffer (312.5 mM Tris-HCl pH 6.8, 10% SDS, 50% glycerol, 12.5% β-mercaptoethanol, and 0.05% bromophenol blue). Heat the mixtures at 100 °C for 10 min and subject to SDS-PAGE. Load 25-50 µg of total membrane proteins on a 10% SDS-PAGE gel39. Run the gel at 80 V for 15 min, then change to 130 V for another 115 min. Transfer at 140 mA for 130 min.
- For the Western blot, use 10 µg/ml anti-SCAP (9D5) antibody to detect N-glycosylation and total SCAP protein.
3. Detection of SCAP Trafficking from ER to the Golgi by Using GFP-SCAP in Human Cells
Seed 2.5 × 105 U87 cells in a 60 mm dish with DMEM supplemented with 5% FBS and incubate the cells at 37 °C and 5% CO2 for 24 hr before transfection.
Dilute 4 µg GFP-SCAP plasmid in 0.5 ml reduced serum medium and mix.
Add 8 µl DNA transfection reagent by pipetting directly into 0.5 ml reduced serum medium containing plasmids and mix gently.
Incubate the transfection reagent/plasmid complex for 25 min at room temperature.
Add transfection complex to the cells with fresh medium and continue to culture for 24 hr at 5% CO2 and 37 °C.
Reseed 5 - 10 × 104 cells into a 6-well plate containing a glass cover slip (22 mm × 22 mm) and continue to culture at 37 °C and 5% CO2 for 24 hr.
Wash once with PBS and treat the cells for 12 hr with or without glucose (5 mM) in the presence or absence of tunicamycin (1 µg/ml).
Wash cells once with PBS and fix in 4% formaldehyde for 10 min.
Wash cells three times with PBS, invert the coverslips, mount on slides together with antifade reagent, and seal the coverslips using nail polish.
Check the GFP-SCAP trafficking using a 63X oil immersion objective in a laser scanning confocal microscope41.
Representative Results
Figure 1 shows the detection of endogenous SCAP protein and SREBP-1 nuclear form in human glioblastoma U87 cells in response to glucose stimulation by using western blot. The membrane protein SCAP is detected in the "membrane fraction". The nucleus form (N-terminal) of SREBP-1 is detected in the "nuclear extracts". The level of SCAP protein (PDI serves as an internal control of ER membrane proteins) and SREBP-1 nuclear form are markedly enhanced by glucose stimulation. These results demonstrate that the protocol is able to detect the endogenous SCAP protein in human cells.
Figure 2 shows the analysis of SCAP N-glycosylation and total GFP-SCAP protein levels in response to glucose stimulation and tunicamycin treatment in HEK293T cells expressing GFP-labeled hamster SCAP. Figure 2A shows the thematic structure of SCAP protein across the ER membrane carrying three N-glycosylation sites. The fragment of SCAP containing a.a 540 - 707, located in the 6th luminal loop in the ER, is trypsin-resistant15 and is used for analysis of N-glycosylation on the sites N590 and N641. As shown in Figure 2B (upper panel), after trypsin treatment, the higher weight bands (containing a.a 540 - 707) indicate SCAP protein containing one or two N-linked oligosaccharides (N590 and N641) in the presence of glucose and absence of PNGase or tunicamycin (detected by IgG-9D5 antibody). In the presence of PNGase F, N-linked oligosaccharides were removed from the SCAP protein, causing the fragment (a.a 540 - 707) to move faster in SDS-PAGE. Consistently, blocking the N-glycosylation initiation process by tunicamycin completely abolished SCAP N-glycosylation, indicated by the appearance of a lower band of SCAP fragment (Figure 2B, upper panel). Moreover, total GFP-SCAP protein (detected by an antibody against GFP) was reduced in the absence of glucose or via tunicamycin treatment (Figure 2B, lower panel). These results demonstrate that the protocol is suitable for the analysis of SCAP N-glycosylation.
Figure 3 shows that GFP-SCAP trafficking from the ER to the Golgi in response to glucose stimulation was suppressed by tunicamycin treatment in U87 cells. As shown in Figure 3A, confocal microscopy images show that GFP-SCAP protein resides in the ER in the absence of glucose, as shown by its co-localization with PDI, an ER-resident protein (red). In contrast, in the presence of glucose, GFP-SCAP moves to the Golgi, shown by the co-localization with the Golgi protein marker Giantin (red) (Figure 3B). Moreover, tunicamycin treatment suppressed glucose-induced trafficking of GFP-SCAP from the ER to the Golgi (Figure 3A and B).
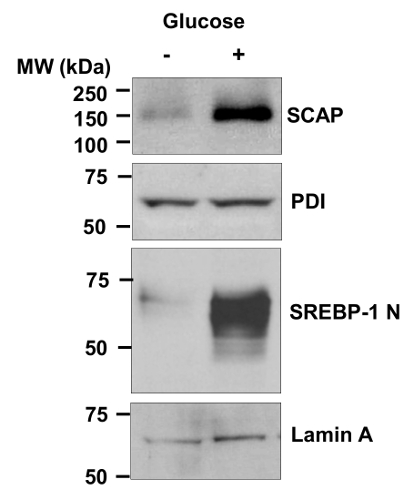 Figure 1.Detection of Endogenous SCAP Protein and SREBP-1 Nuclear Form in U87 Cells. Western blot analysis of membrane and nuclear extracts from human primary glioblastoma U87 cells cultured in serum-free media with or without glucose (5 mM) for 12 hr. Anti-SCAP antibody against human SCAP a.a 450 - 500 was used to detect the endogenous SCAP in human cells. The active form (N-terminal) of SREBP-1 is detected in the "nuclear extracts" using antibody against the a.a 301 - 407 fragment of human SREBP-1. This figure has been modified with permission from 31. Please click here to view a larger version of this figure.
Figure 1.Detection of Endogenous SCAP Protein and SREBP-1 Nuclear Form in U87 Cells. Western blot analysis of membrane and nuclear extracts from human primary glioblastoma U87 cells cultured in serum-free media with or without glucose (5 mM) for 12 hr. Anti-SCAP antibody against human SCAP a.a 450 - 500 was used to detect the endogenous SCAP in human cells. The active form (N-terminal) of SREBP-1 is detected in the "nuclear extracts" using antibody against the a.a 301 - 407 fragment of human SREBP-1. This figure has been modified with permission from 31. Please click here to view a larger version of this figure.
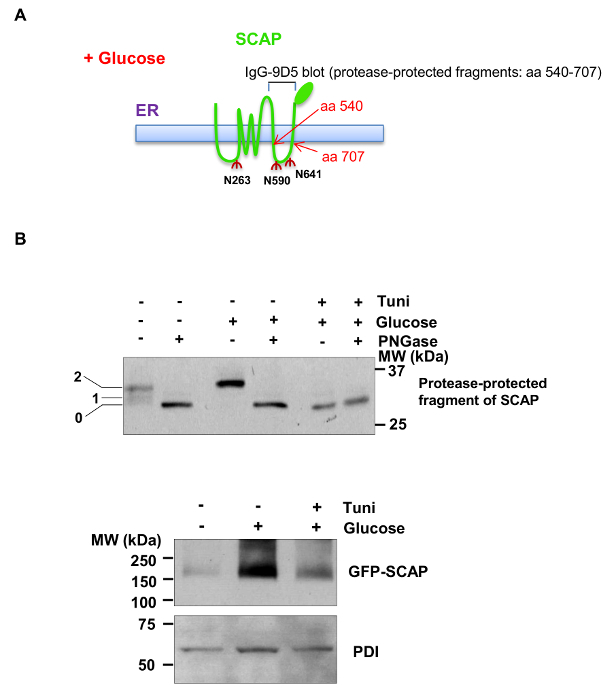 Figure 2.Western Blot Analysis of SCAPN-glycosylation and GFP-SCAP Protein Levels. A) Schematic diagram showing the SCAP structure spanning cross the ER membrane. There are three asparagine (N) residues in SCAP, all of which reside in the ER lumen and are modified by N-linked oligosaccharides. The fragment of SCAP containing amino acids 540-707 is resistant to trypsin digestion. It contains two N-linked oligosaccharide modification sties, N590 and N641, which can be detected by IgG-9D5 antibody against the a.a 540 - 707 of hamster SCAP by western blot. B) Western blot analysis of SCAP N-glycosylation (upper: with trypsin treatment) or GFP-SCAP protein levels (lower: no trypsin treatment) from HEK293T cells transiently transfected with GFP-SCAP for 24 hr and then cultured in serum-free media with or without glucose (5 mM) in the presence or absence of tunicamycin (1 µg/ml), a N-glycosylation inhibitor, for 12 hr. The numbers on the left side of the blot indicate the number of N-glycosylation sites on SCAP protein (N590 and N641 sites) (upper, detected by IgG-9D5 antibody). Lower panel for GFP-SCAP was detected by an antibody against GFP protein. This figure has been modified with permission from 31. Please click here to view a larger version of this figure.
Figure 2.Western Blot Analysis of SCAPN-glycosylation and GFP-SCAP Protein Levels. A) Schematic diagram showing the SCAP structure spanning cross the ER membrane. There are three asparagine (N) residues in SCAP, all of which reside in the ER lumen and are modified by N-linked oligosaccharides. The fragment of SCAP containing amino acids 540-707 is resistant to trypsin digestion. It contains two N-linked oligosaccharide modification sties, N590 and N641, which can be detected by IgG-9D5 antibody against the a.a 540 - 707 of hamster SCAP by western blot. B) Western blot analysis of SCAP N-glycosylation (upper: with trypsin treatment) or GFP-SCAP protein levels (lower: no trypsin treatment) from HEK293T cells transiently transfected with GFP-SCAP for 24 hr and then cultured in serum-free media with or without glucose (5 mM) in the presence or absence of tunicamycin (1 µg/ml), a N-glycosylation inhibitor, for 12 hr. The numbers on the left side of the blot indicate the number of N-glycosylation sites on SCAP protein (N590 and N641 sites) (upper, detected by IgG-9D5 antibody). Lower panel for GFP-SCAP was detected by an antibody against GFP protein. This figure has been modified with permission from 31. Please click here to view a larger version of this figure.
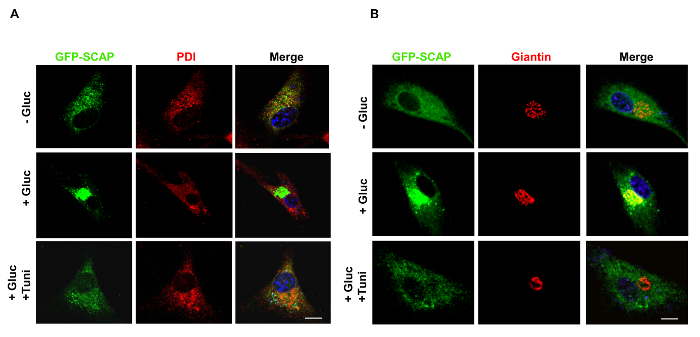 Figure 3. Detection of SCAP Trafficking from the ER to the Golgi. A, B) Confocal microscopy images show that GFP-SCAP resides in the ER or moves to the Golgi in the absence or presence of glucose (5 mM) with/without tunicamycin (1 µg/ml) treatment in U87 cells for 12 hr. PDI shows ER staining (red) (A). Giantin, the Golgi marker (red) (B). DAPI (blue) stains the cell nucleus. The scale bars represent 10 µm. Figure 3A is new data and has not been published before. Figure 3B has been modified with permission from 31. Please click here to view a larger version of this figure.
Figure 3. Detection of SCAP Trafficking from the ER to the Golgi. A, B) Confocal microscopy images show that GFP-SCAP resides in the ER or moves to the Golgi in the absence or presence of glucose (5 mM) with/without tunicamycin (1 µg/ml) treatment in U87 cells for 12 hr. PDI shows ER staining (red) (A). Giantin, the Golgi marker (red) (B). DAPI (blue) stains the cell nucleus. The scale bars represent 10 µm. Figure 3A is new data and has not been published before. Figure 3B has been modified with permission from 31. Please click here to view a larger version of this figure.
Discussion
In this study, we describe a protocol for the isolation of membrane fractions in human cells and for the preparation of the samples for the detection of SCAP N-glycosylation and total protein by using western blot. We further provide a method to monitor SCAP trafficking by using GFP-labeling and confocal microscopy. The method is specifically used to analyze membrane protein and is an important tool to investigate SCAP N-glycosylation and trafficking.
Compared with the method using various lectins to recognize different N-glycan chains and identify N-glycosylated proteins42, our method described here uses PNGase F to remove protein-linked N-glycan and detects the migration shift to identify the glycosylation of SCAP protein fragment a.a 540-707. The limitation of the lectin method is dependent upon the identification of the appropriate type of lectin to recognize the specific N-glycan. Our PNGase F cleavage method provides a simple procedure to analyze SCAP N-glycosylation. Furthermore, the method can also be applied to analyze SCAP N-glycosylation and protein level in human patients or animal tissues after homogenization2. Our study provides a new tool to analyze the signature of lipid metabolism in cancer and metabolic diseases.
In this study, the plasmid of GFP-labeled SCAP used for analysis of N-glycosylation and trafficking in human cells is hamster-origin. SCAP antibody (IgG-9D5) that is raised against the amino acids 540-707 fragment of SCAP protein can only detect the protein and N-glycosylation of the hamster SCAP, such as in the Chinese hamster ovarian (CHO) cell line15. The antibody against amino acids 450 - 500 of human SCAP is able to detect the endogenous SCAP protein in human cells, as shown in Figure 1. To detect SCAP N-glycosylation in human cells or tissues, we need to develop a specific antibody against the amino acids 540 - 707 fragment of human SCAP. In addition, identification of a specific lectin to recognize SCAP-bound N-glycan is an alternative way to analyze human SCAP N-glycosylation42. Moreover, defining the composition and structure of each N-glycan chain binding in SCAP (N263, N590 and N641) will facilitate our understanding of the underlying mechanism of SCAP trafficking from the ER to the Golgi.
Furthermore, to obtain the best results for the detection of SCAP N-glycosylation, we adjusted some reaction parameters according to our experimental conditions, which are modified from the methods described in the publications from the Brown and Goldstein Laboratory15. To successfully detect SCAP protein and its N-glycosylation, the quality of the membrane fractions and nuclear extracts are very important. We directly determined the protein concentration after the membrane fractions incubated at 37 °C, avoiding putting the sample back on ice. The method described here can be widely applied to different fields of research, including cancer, cardiovascular disease, diabetes, and obesity.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to Drs. Mike S. Brown and Joseph L. Goldstein for their agreement to publish this method according to the methods described in their publications. We appreciate Dr. Peter Espenshade for sharing the GFP-SCAP plasmid. This work was supported by NIH grants NS072838 and NS079701 to D.G., American Cancer Society Research Scholar Grant RSG-14-228-01-CSM to D.G., and OSUCCC Pelotonia Postdoctoral Fellowship to C.C. We also appreciate the support from the Ohio State Neuroscience Core (P30 NS038526) and OSUCCC Translational Therapeutic Program seed grant and start-up funds to D.G.
References
- Ru P, Williams TM, Chakravarti A, Guo D. Tumor metabolism of malignant gliomas. Cancers (Basel) 2013;5:1469–1484. doi: 10.3390/cancers5041469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YA, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell metabolism. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Bell EH, Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncology. 2013;2:289–299. doi: 10.2217/cns.13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009. pp. 12932–12937. [DOI] [PMC free article] [PubMed]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Guo D, Cloughesy TF, Radu CG, Mischel PS. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle. 2010;9:211–212. doi: 10.4161/cc.9.2.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ. Expanding Roles for SREBP in Metabolism. Cell metabolism. 2012;16:414–419. doi: 10.1016/j.cmet.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Zhang SC. Coordination of lipid metabolism in membrane biogenesis. Annual review of cell and developmental biology. 2009;25:539–566. doi: 10.1146/annurev.cellbio.24.110707.175344. [DOI] [PubMed] [Google Scholar]
- Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends in endocrinology and metabolism: TEM. 2012;23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R, et al. Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1) The Journal of biological chemistry. 1994;269:17267–17273. [PubMed] [Google Scholar]
- Sakai J, Nohturfft A, Goldstein JL, Brown MS. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. The Journal of biological chemistry. 1998;273:5785–5793. doi: 10.1074/jbc.273.10.5785. [DOI] [PubMed] [Google Scholar]
- Sakai J, et al. Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. The Journal of biological chemistry. 1997;272:20213–20221. doi: 10.1074/jbc.272.32.20213. [DOI] [PubMed] [Google Scholar]
- Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87:415–426. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, Brown MS, Goldstein JL. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. The Journal of biological chemistry. 1998;273:17243–17250. doi: 10.1074/jbc.273.27.17243. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, Hua X, Brown MS, Goldstein JL. Recurrent G-to-A substitution in a single codon of SREBP cleavage-activating protein causes sterol resistance in three mutant Chinese hamster ovary cell lines. Proc Natl Acad Sci U S A. 1996;93:13709–13714. doi: 10.1073/pnas.93.24.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- Yang T, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Xia ZP, Adams CM, Rawson RB. Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci U S A. 2002;99:16672–16677. doi: 10.1073/pnas.262669399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11694–11699. doi: 10.1073/pnas.182412799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315–323. doi: 10.1016/s0092-8674(00)00037-4. [DOI] [PubMed] [Google Scholar]
- Sakai J, et al. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RB, DeBose-Boyd R, Goldstein JL, Brown MS. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J Biol Chem. 1999;274:28549–28556. doi: 10.1074/jbc.274.40.28549. [DOI] [PubMed] [Google Scholar]
- Zelenski NG, Rawson RB, Brown MS, Goldstein JL. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J Biol Chem. 1999;274:21973–21980. doi: 10.1074/jbc.274.31.21973. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade PJ, Cheng D, Goldstein JL, Brown MS. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. The Journal of biological chemistry. 1999;274:22795–22804. doi: 10.1074/jbc.274.32.22795. [DOI] [PubMed] [Google Scholar]
- Guo D. SCAP links glucose to lipid metabolism in cancer cells. Mol Cell Oncol. 2016;3 doi: 10.1080/23723556.2015.1132120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell. 2015;28:569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ. Sugar Makes Fat by Talking to SCAP. Cancer Cell. 2015;28:548–549. doi: 10.1016/j.ccell.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. 2009;2:82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014;20:2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EH, Guo D. Biomarkers for malignant gliomas. Malignant Gliomas, Radiation Medicine Rounds. 2012;3:339–357. [Google Scholar]
- Geng F, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res. 2016. [DOI] [PMC free article] [PubMed]
- Ru P, et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. 2016. [DOI] [PMC free article] [PubMed]
- Green MR, Sambrook J. Molecular Cloning: A Laboratory Manual (Fourth Edition) 2012.
- Uehara T, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Miyashita T. Confocal microscopy for intracellular co-localization of proteins. Methods Mol Biol. 2015;1278:515–526. doi: 10.1007/978-1-4939-2425-7_34. [DOI] [PubMed] [Google Scholar]
- Cao J, Guo S, Arai K, Lo EH, Ning M. Studying extracellular signaling utilizing a glycoproteomic approach: lectin blot surveys, a first and important step. Methods Mol Biol. 2013;1013:227–233. doi: 10.1007/978-1-62703-426-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]