

Abstract
As stated by the prevailing amyloid cascade hypothesis, Alzheimer's disease (AD) is caused by the aggregation and cerebral deposition of long amyloid‐β peptide (Aβ) species, which are released from a C‐terminal amyloid precursor protein fragment by γ‐secretase. Mutations in its catalytic subunit presenilin‐1 (PS1) increase the Aβ42 to Aβ40 ratio and are the major cause of familial AD (FAD). An opposing hypothesis states that loss of essential presenilin functions underlies the disease. A major argument for this hypothesis is the observation that the nearly inactive PS1 L435F mutant, paradoxically, causes FAD. We now show that the very little Aβ generated by PS1 L435F consists primarily of Aβ43, a highly amyloidogenic species which was overlooked in previous studies of this mutant. We further demonstrate that the generation of Aβ43 is not due to a trans‐dominant effect of this mutant on WT presenilin. Furthermore, we found Aβ43‐containing plaques in brains of patients with this mutation. The aberrant generation of Aβ43 by this particular mutant provides a direct objection against the presenilin hypothesis.
Keywords: Alzheimer's disease, amyloid‐β peptide 43, γ‐secretase, neurodegeneration, presenilin
Subject Categories: Neuroscience
Introduction
According to the widely believed amyloid cascade hypothesis, Alzheimer's disease (AD) is triggered by the pathogenic accumulation of long amyloid‐β peptide (Aβ) species such as Aβ42 in the brain of affected patients. Aβ is derived from sequential processing of the β‐amyloid precursor protein (APP) by β‐ and γ‐secretase (Lichtenthaler et al, 2011). The latter cleavage starts at the ε‐site within the APP transmembrane domain and following a series of carboxy‐terminal trimming steps ultimately releases two major Aβ forms, Aβ40 as principal product as well as smaller amounts of Aβ42 (Lichtenthaler et al, 2011). Due to its higher hydrophobicity, the slightly longer Aβ42 peptide is more prone to aggregation than Aβ40, a property, which likely underlies its neurotoxicity (Haass & Selkoe, 2007). The amyloid cascade hypothesis is strongly supported by genetic evidence from familial AD (FAD) cases showing that most mutations in APP as well as in presenilin‐1 (PS1) and its homologue presenilin‐2 (PS2), which constitute the catalytic subunit of γ‐secretase (Lichtenthaler et al, 2011), cause an increased ratio of Aβ42 to Aβ40 (Scheuner et al, 1996), i.e. that FAD mutations are associated with substrate and protease. Moreover, a mutation in immediate vicinity to the β‐secretase cleavage site in APP has recently been identified in the Icelandic population that protects against AD (Jonsson et al, 2012), while a double mutation at this site occurring in a Swedish family (swAPP) results in an exceptionally strong increase of Aβ generation (Citron et al, 1992).
Since overproduction of Aβ42 in APP transgenic mice failed to induce neurodegeneration despite severe amyloidosis (Irizarry et al, 1997) and because many PS1 and PS2 FAD mutations strongly impair processing of γ‐secretase substrates such as Notch (Weggen & Beher, 2012), an alternative hypothesis for the disease mechanism of AD, the “presenilin hypothesis”, has been suggested, which states that AD is caused by a loss of essential presenilin functions (Shen & Kelleher, 2007). This has the important implication that therapeutic inhibition of γ‐secretase to lower Aβ will not be beneficial in AD. Rather, it will be important to maintain normal presenilin function. The presenilin hypothesis was initially based on the observation that conditional PS knockout mice display classical features of neurodegeneration that also occur in AD (Saura et al, 2004). It received further support when a remarkable FAD mutation, PS1 L435F, was identified (Heilig et al, 2010). Biochemical analyses using cell culture models and recently also knockin mice revealed that this FAD mutant did not support presenilin endoproteolysis, an autocatalytic process to activate γ‐secretase (Lichtenthaler et al, 2011), and nearly completely abrogated γ‐secretase activity toward its substrates APP and Notch1 (Heilig et al, 2010, 2013; Xia et al, 2015). Thus, PS1 L435F represents a highly unusual PS1 FAD mutation that supports the concept that loss of presenilin function constitutes an essential trigger of early onset dementia, probably being more important than an elevated ratio of Aβ42/Aβ40 (Shen & Kelleher, 2007).
Results and Discussion
In a previous screen for mutations defective in presenilin autoproteolysis, two PS1 loss‐of‐function mutations, R278I, a known FAD mutation (Godbolt et al, 2004), and L435H, a synthetic mutation of L435, which severely reduced the processing of APP and Notch1 were described (Nakaya et al, 2005). Intriguingly, mass spectrometry (MS) analysis revealed that both PS1 R278I and L435H gave rise to abnormally high, nearly exclusive generation of Aβ43 (Nakaya et al, 2005), a finding that has been recapitulated in vivo for PS1 R278I knockin mouse models (Saito et al, 2011). Although Aβ43 is secreted only in very minor amounts under physiological conditions (Page et al, 2008; Saito et al, 2011), it is found in Aβ plaques in sporadic and FAD brain (Iizuka et al, 1995; Parvathy et al, 2001; Welander et al, 2009; Saito et al, 2011; Sandebring et al, 2013). Like Aβ42, it is highly neurotoxic, although the extent of toxicity compared to Aβ42 varies among assays (Saito et al, 2011; Burnouf et al, 2015; Meng et al, 2015). Aβ43 is deposited earlier than Aβ42 in mouse models of AD indicating that Aβ43 is a potent and probably also a primary nucleation factor of Aβ aggregates in vivo (Saito et al, 2011; Zou et al, 2013). Taken together, this indicated that, similarly to the PS1 R278I or the L435H mutants, also PS1 L435F might secrete Aβ43, a property that was not investigated in previous studies (Heilig et al, 2010, 2013; Xia et al, 2015).
To investigate this possibility, we stably transfected HEK293 cells expressing swAPP (HEK293/sw) with cDNA encoding the PS1 L435F mutant or, as controls, PS1 WT and the catalytically inactive PS1 D385A mutant. As shown in Fig 1A, the constructs were robustly expressed in pooled clones and underwent normal γ‐secretase complex formation as demonstrated by the replacement of endogenous PS2 and the maturation of nicastrin (NCT). We confirmed the previously reported loss‐of‐function phenotype for PS1 L435F showing virtually absence of presenilin endoproteolysis (Fig 1A), accumulation of uncleaved APP C‐terminal fragments (CTFs) comparable to the catalytic inactive PS1 D385A mutation (Fig 1B) and nearly no Aβ total production (Fig 1C). Next, we investigated the Aβ profiles generated by these mutants. Tris‐Bicine‐Urea SDS‐PAGE analysis, which allows electrophoretic separation of individual Aβ species (Wiltfang et al, 1997), showed a band for PS1 L435F that migrated faster than that of Aβ42 for PS1 WT and at a comparable height of the Aβ43 band observed for the PS1 L166P mutant that was included in this analysis as a reference FAD mutant that besides high amounts of Aβ42 also generates Aβ43 (Moehlmann et al, 2002; Page et al, 2008) (Fig 1D). These data strongly suggests that PS1 L435F secretes Aβ43 and in even higher amounts than Aβ42 (Fig 1D). To further confirm these data, single‐cell clones stably overexpressing PS1 WT or PS1 L435F (Fig 1E) were used next for a quantitative analysis of Aβ species using a previously described highly sensitive Aβ43‐specific ELISA (Saito et al, 2011). As shown in Fig 1F, Aβ43 was identified in the PS1 L435F mutant as prominent and more abundant species than Aβ42. Tris‐Bicine‐Urea SDS‐PAGE analysis of the Aβ species co‐migrated with standard peptides (Fig 1G) as well as MS analysis (Fig 1H) confirmed this result. Finally, consistent with the strong loss of function in APP processing, generation of the APP intracellular domain (AICD) was nearly abolished by the mutant (Fig EV1A). Thus, the loss‐of‐function phenotype of PS1 L435F is associated with an abnormal relative overproduction of Aβ43.
Figure 1. The PS1 L435F FAD mutant strongly impairs APP processing while generating Aβ43.

- PS1, PS2, and NCT were analyzed in cell lysates by immunoblotting using antibodies PS1N (PS1), BI‐HF5C (PS2), and N1660 (NCT), respectively.
- Full‐length APP and APP CTFs were analyzed by immunoblotting using antibody 6687.
- Conditioned media were analyzed for secreted APPs by immunoblotting using antibody 22C11 and for total Aβ by combined immunoprecipitation/immunoblotting using antibodies 3552/2D8.
- Conditioned media were analyzed for individual Aβ species on Tris‐Bicine‐Urea SDS‐PAGE by combined immunoprecipitation/immunoblotting of Aβ using antibodies 3552/2D8. Pooled clones of HEK293/sw cells stably transfected with PS1 L166P were used as reference. Note that more sample was loaded for the PS1 L435F mutant to facilitate analysis.
- PS1 expression levels were analyzed in cell lysates of HEK293/sw cells stably expressing PS1 WT or PS1 L435F by immunoblotting using antibodies PS1NT and 5E12, respectively.
- Conditioned media were analyzed by ELISA specific for Aβ40, Aβ42, and Aβ43. Data represent mean ± s.e.m. (n = 6). Absolute levels and Aβ ratios are shown.
- ecreted Aβ was analyzed as in (D). To verify individual Aβ species, Aβ standards were co‐migrated.
- Total Aβ in conditioned media was analyzed by MALDI‐TOF MS following immunoprecipitation with antibody 4G8. Observed (Aβ42, 4513.6; Aβ43, 4615.3) and predicted molecular masses (Aβ42, 4514.1; Aβ43, 4615.2) were in good agreement.
Figure EV1. The PS1 L435F FAD mutant strongly impairs AICD generation.

- Levels of full‐length APP, APP CTFs, and AICD were analyzed in cell lysates of single‐cell clones of HEK293/sw cells stably expressing PS1 WT or PS1 L435F by immunoblotting using antibody Y188. S.E., short exposure; L.E., long exposure.
- Levels of endogenous full‐length APP, APP CTFs, and AICD were analyzed in cell lysates of PS1/2−/− MEF cells stably transduced with PS1 WT or PS1 L435F by immunoblotting using antibody Y188 as in (A). S.E., short exposure; L.E., long exposure.
- Cell‐free generation of AICD from recombinant APP C100‐His6 substrate by CHAPSO‐solubilized γ‐secretase of stably transduced PS1/2−/− MEF cells was analyzed by immunoblotting using Penta‐His (AICD total) and anti‐AICD neo‐epitope (AICD Val50) antibodies, respectively.
Source data are available online for this figure.
To explain the apparent paradox that PS1 L435F causes amyloid plaque deposition and AD in mutation carriers despite a near complete functional loss, it was suggested that the mutant protein inhibits and alters the catalytic activity of PS1 WT by a trans‐dominant effect to stimulate generation of Aβ42 by PS1 WT (Heilig et al, 2013). To exclude the possibility that Aβ43 generation resulted from residual PS1 WT activity trans‐dominantly disturbed by PS1 L435F, PS1/2−/− double‐knockout mouse embryonic fibroblast (MEF) cells stably transduced with PS1 L435F, i.e. with the mutant as sole presenilin present in the cell, were analyzed next. As expected, the PS1 L435F mutant was hardly endoproteolysed, caused a strong loss of function of γ‐secretase activity as judged from the accumulation of endogenous mouse APP CTFs similar to that in the parental PS1/2−/− MEFs (Fig 2A) and almost completely blocked AICD formation (Fig EV1B and C). As shown by Tris‐Bicine‐Urea SDS‐PAGE analysis, Aβ43 became the major Aβ species secreted by the mutant upon transient overexpression of human APPsw‐6myc (Kretner et al, 2013) (Fig 2B)—being generated in even higher relative amounts than in the HEK293/sw single‐cell clone expressing the mutant (Fig 1G). Additional quantitative analysis by ELISA further confirmed this result and showed again that Aβ43 was the predominant Aβ species by the mutant when it was expressed in PS1/2−/− MEF cells (Fig 2C). Since these data demonstrate that the generation of Aβ43 by PS1 L435F is an intrinsic property of this mutant and not due to a trans‐dominant effect on a dimeric PS1 WT/L435F complex, we thus finally revisited the human PS1 L435F FAD cases and asked whether Aβ43 was deposited in the brain of the affected two siblings analyzed previously (Heilig et al, 2010). Sequencing of genomic DNA isolated from frozen frontal cortex confirmed the presence of the PS1 L435F mutation in these cases and its absence in a control FAD case with a different PS1 mutation (Fig EV2). As shown in Figs 3 and EV3, substantial Aβ43 deposition was found in frontal cortex and hippocampus of both PS1 L435F cases. The staining intensity of Aβ43‐positive plaques was much higher in the PS1 L435F mutation cases compared to the FAD case with a different PS1 mutation and an additional sporadic AD (SAD) case reaching the staining intensity levels of Aβ42, which is considered to be the major compound of Aβ plaques. Consistent with the previous report (Heilig et al, 2010), cotton wool plaques were observed in the two PS1 L435F mutation cases (Fig EV4A and B) that were frequently associated with neuritic pathology (Fig EV4C and D).
Figure 2. The PS1 L435F FAD mutant generates Aβ43 as the predominant long Aβ species independent of PS1 WT .
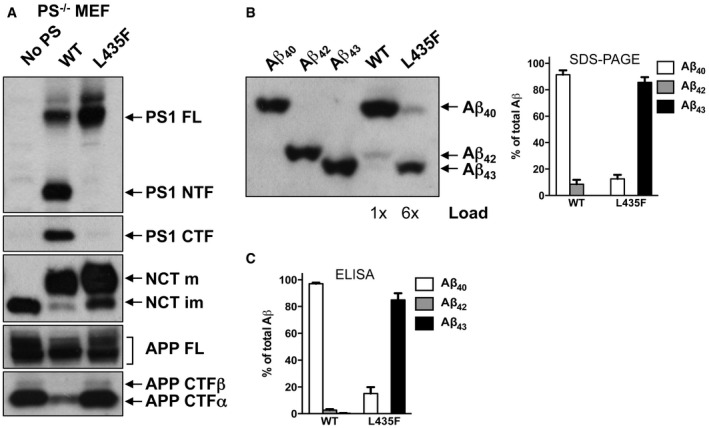
- PS1/2−/− MEF cells stably transduced with PS1 WT or PS1 L435F were analyzed for PS1 expression and APP processing by immunoblotting as in Fig 1A. Antibody 5E12 was used for the detection of the PS1 CTF.
- Stably transduced PS1/2−/− MEF cells of A were transiently transfected with APPsw‐6myc and conditioned media were analyzed for Aβ species by Tris‐Bicine‐Urea SDS‐PAGE. Individual Aβ species were verified by co‐migration with Aβ standards (left panel) and quantified (right panel). Data represent mean ± s.e.m. of n = 3 independently performed transfections. Note that more sample was loaded for the PS1 L435F mutant to facilitate immunoblot analysis.
- Secreted Aβ from cells in B was quantified by ELISA. Data represent mean ± s.e.m. of n = 5 (PS1 WT) or 6 (PS1 L435F) independently performed transfections.
Source data are available online for this figure.
Figure EV2. DNA sequence analysis of PS1 exon 12.

A heterozygous C to T missense mutation at residue 435 changing leucine to phenylalanine is confirmed in two FAD cases (#1 and #2), but not found in a third control FAD case (#3).
Figure 3. Deposition of Aβ isoforms in the frontal cortex of AD cases with and without PS1 L435F mutation.

Immunohistochemical detection of Aβ40 (left column), Aβ42 (medium column), and Aβ43 (right column) in consecutive frontal cortex paraffin sections of two FAD cases with PS1 L435F mutation (cases #1 and #2, 1st and 2nd rows), another FAD case with different PS1 mutation (case #3, 3rd row), and one SAD case (case #4, fourth row). In both PS1 L435F mutation cases, the numerous Aβ plaques mainly contain Aβ42 and Aβ43 but less Aβ40. This is in contrast to Aβ plaques of the control FAD and SAD cases, in which solely Aβ42 predominates and Aβ43 is sparse, even levels of Aβ40 seem to be lower than in the PS1 L435F cases. Note that in both PS1 L435F cases, several plaques are larger than those seen in cases #3 and #4, representing cotton wool plaques. Scale bar = 500 µm. Magnification is identical in all pictures.
Figure EV3. Deposition of Aβ isoforms in the hippocampus of AD cases with and without PS1 L435F mutation.
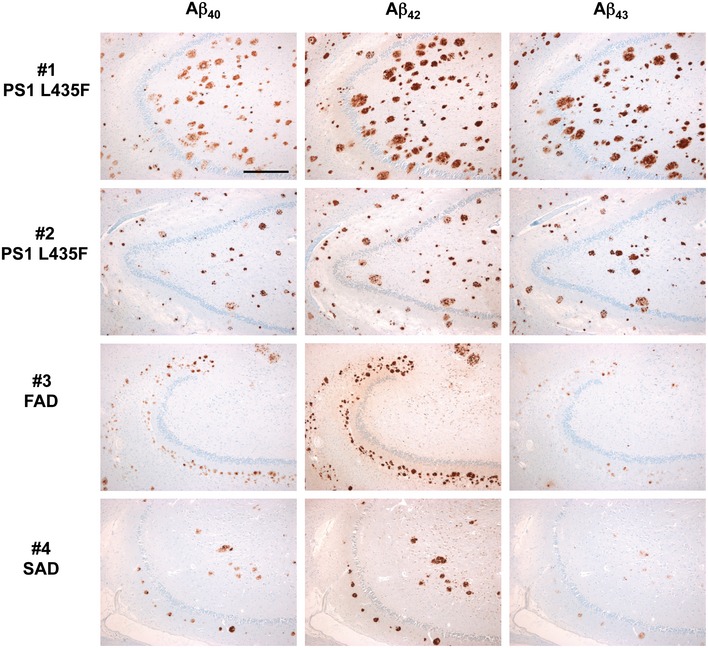
Immunohistochemical detection of Aβ40 (left column), Aβ42 (medium column), and Aβ43 (right column) in consecutive hippocampus paraffin sections (hilus region) of two FAD cases with PS1 L435F mutation (cases #1 and #2, 1st and 2nd rows), one FAD case with different PS1 mutation (case #3, 3rd row) and one sporadic SAD case (case #4, fourth row). Identical to findings in the frontal cortex (Fig 3), plaques in both PS1 L435F cases contain abundant Aβ43 in contrast to plaques of cases without that mutation (cases #3 and #4). Aβ42 levels are similar in plaques of all cases, whereas Aβ40 levels seem to be slightly higher in plaques of PS1 L435F cases. Note that the plaque size in PS1 L435F cases (in particular in case #1) is much larger than in cases #3 and #4; these large plaques represent cotton wool plaques. Scale bar = 500 µm. Magnification is identical in all pictures.
Figure EV4. Cotton wool plaques in the frontal cortex of PS1 L435F mutation case #1.
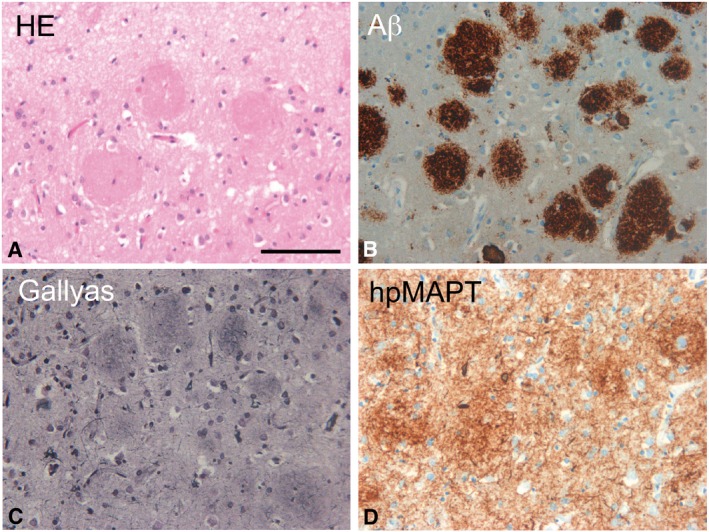
- In conventional hematoxylin–eosin (HE) stains, cotton wool plaques impress as round pink‐colored structures in the neuropil. Scale bar = 100 µm. Magnification is identical in all pictures.
- Immunohistochemistry for Aβ (4G8 antibody) even visualizes plaques that are not visible in HE stains. Note that Aβ in cotton wool plaques is densely packed and that cotton wool plaques are large with diameters between 50 and 100 µm.
- A Gallyas silver stain identifies most cotton wool plaques as neuritic plaques. However, the number of dystrophic neurites (black threads) crossing these plaques is low.
- In immunohistochemical stains for hyperphosphorylated microtubuli‐associated protein tau (hp‐MAPT) (AT‐8 antibody), a dense network of neuropil threads is seen. Agglomerations of threads represent neuritic cotton wool plaques.
Taken together, although the PS1 L435F mutant leads to a near complete loss‐of‐function phenotype with only very little residual catalytic activity, we demonstrate that the mutant secretes substantial amounts of Aβ43 in cell culture models, higher than that of Aβ42. In line with these data, our neuropathological analysis showed that Aβ43 is deposited in high amounts in the brain of the mutation carriers. As evident from the nearly exclusive Aβ43 secretion by PS1 L435F in the absence of PS1 WT, the generation of pathogenic Aβ species can, however, not result from PS1 WT activity disturbed by the heterozygous expression of PS1 L435F as recently proposed (Heilig et al, 2013). Overall, the PS1 L435F is very similar to the PS1 R278I FAD mutation, for which a strong loss‐of‐function phenotype is associated with nearly exclusive secretion of Aβ43 (Nakaya et al, 2005; Saito et al, 2011). Homozygosity of PS1 R278I is embryonic lethal in mice; however, heterozygous mice (PS1 WT/R278I) are viable and develop plaque pathology associated with memory impairment when crossed with APP transgenic mice (Saito et al, 2011). Thus, analogous to PS1 R278I, the PS1 L435F mutation might induce FAD by Aβ43 production rather than by a loss of γ‐secretase function as proposed previously. Indeed, considering that an increase of the Aβ42/Aβ40 ratio is sufficient to accelerate formation and stabilization of toxic oligomeric Aβ species even when the absolute Aβ concentration is very low (Kuperstein et al, 2010), it becomes clear that severe loss‐of‐function mutations that almost completely abolish total Aβ generation could cause FAD when they preferentially cause generation of Aβ43 rather than Aβ42 and Aβ40. Our results thus add to other criticisms raised against the presenilin hypothesis. For example, not all PS1 FAD mutations are associated with reduced γ‐secretase activity (Weggen & Beher, 2012) and mutations in PS1, NCT, and PEN‐2 that fully abolish γ‐secretase function are associated with familial acne inversa, but not with dementia (Wang et al, 2010). The presenilin hypothesis can also not sufficiently explain APP FAD mutations and is inconsistent with a protective APP mutation in the Icelandic population, which reduces the risk for AD by reducing total Aβ levels (Jonsson et al, 2012). In conclusion, our data suggest that the essential and primary trigger of FAD in mutations like PS1 L435F is the secretion of Aβ43 arguing against the presenilin hypothesis.
Materials and Methods
Antibodies
Antibodies to PS1 N‐terminal (PS1N, Capell et al, 1997, immunoblot (IB) 1:1,000) and PS2 C‐terminal fragments (BI.HF5c, Steiner et al, 1999, IB: 1:2,000), respectively, as well as antibodies to the C‐terminus of APP (6687, Steiner et al, 2000, IB 1:1,000), and to total Aβ (3552, Yamasaki et al, 2006, immunoprecipitation (IP) 1:500–1:7,500 and 2D8, Shirotani et al, 2007, IB 3 μg/ml), have been described previously. Neoepitope‐specific antibody to Val50 of AICD (IB 5 μg/ml) was a gift from Eli Lilly and Company and has been described before (Chavez‐Gutierrez et al, 2012). End‐specific antibodies to Aβ40, Aβ42, and Aβ43 characterized previously (Saito et al, 2011) were obtained from IBL (JP18580, JP18582, and JP 18583, immunohistochemistry (IHC) 2 μg/ml). Antibodies NT1 to the PS1 NTF (Covance SIG‐39194, IB 1:2,000), N1660 to NCT (Sigma N1660, IB 1:1,000), 22C11 to secreted soluble APP (Merck Millipore MAB348, IB 1:5,000), 4G8 to Aβ (Covance SIG‐39220, IP 1:500–1:2,500; and BioLegend 800701, IHC 1:500), Y188 to the APP C‐terminus (Abcam ab32136, IB 1:4,000), Penta‐His (Qiagen 34460, IB 1:2,000), and AT‐8 to hyperphosphorylated microtubule‐associated protein tau (Thermo Scientific MN1020, IHC 1:200) were obtained from the indicated companies. Rat monoclonal antibody 5E12 (IB 2.5 μg/ml) of IgG2a subclass was raised against residues 313‐333 (SKYNAESTERESQDTVAENDD) of human PS1.
Cell lines
Pooled as well as single‐cell clones of HEK293 cells stably co‐expressing Swedish mutant APP with WT and mutant PS1 constructs were generated and cultured as described (Steiner et al, 2000; Page et al, 2008). Culture, transfection, and viral transduction of PS1/2−/− double‐knockout MEF cells have been described before (Kuhn et al, 2010; Kretner et al, 2013).
Protein analysis
PS1 and PS2, NCT, full‐length APP, C‐terminal APP fragments, secreted APPs and Aβ were analyzed as described (see Kretner et al, 2013 and references therein). Individual Aβ species were quantified by a previously characterized ELISA (Saito et al, 2011) using end‐specific C‐terminal antibodies to Aβ40, Aβ42, and Aβ43 obtained from IBL according to the instructions of the supplier. MS analysis of secreted Aβ species immunoprecipitated with antibody 4G8 was performed as described previously (Page et al, 2008) except that samples were analyzed on a 4800 MALDI‐TOF/TOF Analyzer (Applied Biosystems/MDS SCIEX). To analyze individual Aβ species by immunoblotting, Tris‐Bicine‐Urea SDS‐PAGE was used (Wiltfang et al, 1997). To improve the separation of longer Aβ species, we used a 12% stacking gel containing 4M urea and an 8% separation gel containing 8M urea. Quantitation of bands from immunoblots was done using the LAS‐4000 image reader (Fujifilm Life Science) and Multi‐Gauge V3.0 software for analysis.
Cell‐free γ‐secretase assay
Membrane fractions of PS1/2−/− MEF cells stably transduced with PS1 WT or PS1 L435F were prepared as described (Sastre et al, 2001) and solubilized with 1% CHAPSO. Following a clarifying spin (100,000 × g, 30 min, 4°C), γ‐secretase activity was assessed using recombinant 1.4 μM C100‐His6 substrate (Edbauer et al, 2003) in assay buffer (150 mM sodium citrate pH 6.4, 0.5 mg/ml phosphatidylcholine, 10 mM DTT, 0.1 mg/ml BSA, 0.25% CHAPSO, 1 × PI) in the presence or absence of 0.5 μM L‐685,458 (Merck Millipore). Samples were separated by SDS–PAGE on 10‐20% Tris‐Tricine gels (Invitrogen), and AICD generation was analyzed by immunoblotting using antibodies Penta‐His or anti‐AICD Val50.
Patient samples
Autopsy tissue samples and paraffin sections were provided by the Massachusetts Alzheimer's Disease Research Center. All human autopsy tissue was collected in accordance with the protocol approved by the Institutional Review Board of the Massachusetts General Hospital in accordance with the Ethical Principles and Guidelines for the Protection of Human Subjects of Research (the “Belmont Report”) and the requirements of the Health Insurance Portability and Accountability Act (HIPAA) of 1996, as well as applicable regulations. Consent for research use of tissues was obtained from the next of kin of the deceased at the time of death and prior to performance of the autopsy. All cases are listed in Table EV1.
Mutation analysis
DNA was extracted from frozen tissue samples of all FAD cases using Maxwell System (Promega GmbH, Mannheim, Germany) and quantified by NanoPhotometer® P‐Class (Implen GmbH, München, Germany). Two primers (forward primer, 5′‐TTGCCTGAAAATGCTTTCATAATTAT‐3′; reverse primer, 5′‐GGAATGCTAATTGGTCCATAAAAG‐3′) were designed to amplify a 199‐bp product flanking the hot spot mutation L435F in the exon 12 of PS1. DNA was amplified by PCR using the Multiplex PCR Kit (Qiagen, Hilden, Germany) following manufacturer's instructions. PCRs were performed in a total volume of 25 μl with 20–30 ng genomic DNA. The PCR mixture was performed with an initial denaturation for 15 min at 95°C, cycled 38 times (94°C for 30 s, 54°C for 60 s, and 72°C for 120 s) and 72°C for 10 min for a final extension. Visualization of the PCR product was performed by gel electrophoresis. PCR product was purified with the DNA Clean & Concentrator Kit (Zymo Research Europe GmbH, Freiburg, Germany) and directly sequenced on an ABI 3130 Genetic Analyser (Applied Biosystem, CA) using the same primers as above.
Histological stains
Hematoxylin–eosin stains and Gallyas silver stains were performed according to standard protocols.
Immunohistochemistry
Deparaffinized sections were pretreated with 90% formic acid for 5 min before incubation with rabbit polyclonal antibodies specific to Aβ40, Aβ42, or Aβ43 or with mouse monoclonal antibody 4G8 recognizing all Aβ isoforms. For the detection of hyperphosphorylated microtubuli‐associated protein tau, sections were microwaved before incubation with mouse monoclonal antibody AT‐8. Immunohistochemistry was performed with a Ventana BenchMark using the i‐view DAB detection kit.
Author contributions
BK, JT, CH, and HS conceived and designed experiments. BK and JT performed biochemical experiments. AF performed mass spectrometry analysis. TA performed and evaluated histological and immunohistochemical stains and documented their results. JM and AG were responsible for DNA sequence analysis. PK and SFL generated stably transduced MEF cells. EK generated monoclonal antibody 5E12. BK, JT, AF, CH, TA, and HS analyzed data and interpreted results. HS supervised the project and wrote the paper with contributions from BK, JT, JM, and TA.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
The etiology of Alzheimer's disease (AD) is currently explained by two opposing hypotheses. The prevailing and widely accepted amyloid cascade hypothesis states that abnormal accumulation of longer amyloid‐β peptide (Aβ) species, such as Aβ42 triggers the disease. An alternative hypothesis, the presenilin hypothesis, states that loss of essential presenilin functions causes the disease and that aberrant Aβ generation is a secondary process. As proteolytic subunit of γ‐secretase, presenilin not only generates various Aβ species differing in their C‐termini from the amyloid precursor protein (APP) but also cleaves many other crucial substrates such as Notch‐1. As a consequence, maintaining rather than inhibiting presenilin function should be beneficial in AD treatment strategies. Thus, clarifying the relevance of the presenilin hypothesis has obvious implications for AD drug development.
Results
A major argument for the presenilin hypothesis has been the phenotype of the presenilin‐1 (PS1) L435F mutant. This mutant causes a severe loss of function with nearly undetectable Aβ generation, but intriguingly causes familial AD (FAD). To explain this paradox, a trans‐dominant effect of the mutant on PS1 WT was proposed. We now show that, strikingly, the PS1 L435F mutant causes a robust generation of Aβ43, a species, which was not investigated in previous analyses of this mutant. We further show that PS1 L435F also produces Aβ43 in the absence of WT presenilin excluding a dominant‐negative mechanism of this mutant. Finally, unlike in controls, neuropathological analysis of the PS1 L435F FAD cases revealed a robust deposition of Aβ43 in postmortem brain tissues.
Impact
The finding that the PS1 L435F mutant preferentially generates the previously overlooked highly amyloidogenic Aβ43 by its intrinsic residual activity supports the concept of the amyloid cascade hypothesis. Accordingly, loss of essential presenilin functions is unlikely to be the primary trigger of AD. Thus, disease‐modifying approaches for AD aiming at selectively targeting the generation and/or clearance of amyloidogenic Aβ species should remain a major focus to develop therapeutic interventions to AD.
For more information
http://www.alzforum.org/news/research-news/mutant-presenilin-knock-mice-mimic-knockouts-stir-old-debate: A controversial discussion whether loss of presenilin function is at the core of AD after it was reported that PS1 L435F knockin mice display cognitive decline and neurodegeneration (Xia et al, 2015).
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Figure EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgements
We are grateful to Bart De Strooper, Ralph A. Nixon, Alison Goate, and Paul Szekeres for reagents; Matthew Frosch and Brad Hyman for providing paraffin sections and frozen brain tissue samples; and Alice Sülzen, Brigitte Kraft, and Michael Ruiter for technical assistance. We also thank Axel Imhof for giving access to the mass spectrometer of the Protein Analysis Unit of the Ludwig‐Maximilians‐University Munich and Ignasi Forné, Pierre Schilcher, and Andreas Schmidt for technical help with MS analysis. This work was supported by the Friedrich‐Baur‐Stiftung (HS), the Deutsche Forschungsgemeinschaft (FOR2290) (HS and SFL), the Breuer Foundation Alzheimer Award (SFL), an advanced grant of the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant Agreement No. 321366‐Amyloid (CH) and the general legacy of Mrs. Ammer (to the LMU/chair of CH).
EMBO Mol Med (2016) 8: 458–465
References
- Burnouf S, Gorsky MK, Dols J, Gronke S, Partridge L (2015) Aβ43 is neurotoxic and primes aggregation of Aβ40 in vivo. Acta Neuropathol 130: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Saffrich R, Olivo JC, Meyn L, Walter J, Grunberg J, Mathews P, Nixon R, Dotti C, Haass C (1997) Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J Neurochem 69: 2432–2440 [DOI] [PubMed] [Google Scholar]
- Chavez‐Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H et al (2012) The mechanism of γ‐secretase dysfunction in familial Alzheimer disease. EMBO J 31: 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo‐Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the β‐amyloid precursor protein in familial Alzheimer's disease increases β‐protein production. Nature 360: 672–674 [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of γ‐secretase activity. Nat Cell Biol 5: 486–488 [DOI] [PubMed] [Google Scholar]
- Godbolt AK, Beck JA, Collinge J, Garrard P, Warren JD, Fox NC, Rossor MN (2004) A presenilin 1 R278I mutation presenting with language impairment. Neurology 63: 1702–1704 [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β‐peptide. Nat Rev Mol Cell Biol 8: 101–112 [DOI] [PubMed] [Google Scholar]
- Heilig EA, Xia W, Shen J, Kelleher RJ 3rd (2010) A presenilin‐1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of γ‐secretase activity. J Biol Chem 285: 22350–22359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ 3rd (2013) Trans‐dominant negative effects of pathogenic PSEN1 mutations on γ‐secretase activity and Aβ production. J Neurosci 33: 11606–11617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka T, Shoji M, Harigaya Y, Kawarabayashi T, Watanabe M, Kanai M, Hirai S (1995) Amyloid β‐protein ending at Thr43 is a minor component of some diffuse plaques in the Alzheimer's disease brain, but is not found in cerebrovascular amyloid. Brain Res 702: 275–278 [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT (1997) Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J et al (2012) A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 488: 96–99 [DOI] [PubMed] [Google Scholar]
- Kretner B, Fukumori A, Kuhn PH, Perez‐Revuelta BI, Lichtenthaler SF, Haass C, Steiner H (2013) Important functional role of residue x of the presenilin GxGD protease active site motif for APP substrate cleavage specificity and substrate selectivity of γ‐secretase. J Neurochem 125: 144–156 [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF (2010) ADAM10 is the physiologically relevant, constitutive α‐secretase of the amyloid precursor protein in primary neurons. EMBO J 29: 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers‐Nolten I, Van Der Werf K, Subramaniam V et al (2010) Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J 29: 3408–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H (2011) Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J Neurochem 117: 779–796 [DOI] [PubMed] [Google Scholar]
- Meng P, Yoshida H, Tanji K, Matsumiya T, Xing F, Hayakari R, Wang L, Tsuruga K, Tanaka H, Mimura J et al (2015) Carnosic acid attenuates apoptosis induced by amyloid‐β 1‐42 or 1‐43 in SH‐SY5Y human neuroblastoma cells. Neurosci Res 94: 1–9 [DOI] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C et al (2002) Presenilin‐1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ42 production. Proc Natl Acad Sci USA 99: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya Y, Yamane T, Shiraishi H, Wang HQ, Matsubara E, Sato T, Dolios G, Wang R, De Strooper B, Shoji M et al (2005) Random mutagenesis of presenilin‐1 identifies novel mutants exclusively generating long amyloid β‐peptides. J Biol Chem 280: 19070–19077 [DOI] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Perez‐Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H et al (2008) Generation of Aβ38 and Aβ42 is independently and differentially affected by FAD‐associated presenilin 1 mutations and γ‐secretase modulation. J Biol Chem 283: 677–683 [DOI] [PubMed] [Google Scholar]
- Parvathy S, Davies P, Haroutunian V, Purohit DP, Davis KL, Mohs RC, Park H, Moran TM, Chan JY, Buxbaum JD (2001) Correlation between Aβx‐40‐, Aβx‐42‐, and Aβx‐43‐containing amyloid plaques and cognitive decline. Arch Neurol 58: 2025–2032 [DOI] [PubMed] [Google Scholar]
- Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J et al (2011) Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci 14: 1023–1032 [DOI] [PubMed] [Google Scholar]
- Sandebring A, Welander H, Winblad B, Graff C, Tjernberg LO (2013) The pathogenic Aβ43 is enriched in familial and sporadic Alzheimer disease. PLoS One 8: e55847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C (2001) Presenilin‐dependent γ‐secretase processing of β‐amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ 3rd, Kandel ER, Duff K et al (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron 42: 23–36 [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W et al (1996) Secreted amyloid β‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2: 864–870 [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ 3rd (2007) The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc Natl Acad Sci USA 104: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirotani K, Tomioka M, Kremmer E, Haass C, Steiner H (2007) Pathological activity of familial Alzheimer's disease‐associated mutant presenilin can be executed by six different γ‐secretase complexes. Neurobiol Dis 27: 102–107 [DOI] [PubMed] [Google Scholar]
- Steiner H, Romig H, Grim MG, Philipp U, Pesold B, Citron M, Baumeister R, Haass C (1999) The biological and pathological function of the presenilin‐1 ∆exon 9 mutation is independent of its defect to undergo proteolytic processing. J Biol Chem 274: 7615–7618 [DOI] [PubMed] [Google Scholar]
- Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim MG, Baumeister R et al (2000) Glycine 384 is required for presenilin‐1 function and is conserved in polytopic bacterial aspartyl proteases. Nat Cell Biol 2: 848–851 [DOI] [PubMed] [Google Scholar]
- Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T et al (2010) γ‐Secretase gene mutations in familial acne inversa. Science 330: 1065 [DOI] [PubMed] [Google Scholar]
- Weggen S, Beher D (2012) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal‐dominant Alzheimer's disease. Alzheimers Res Ther 4: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welander H, Franberg J, Graff C, Sundstrom E, Winblad B, Tjernberg LO (2009) Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J Neurochem 110: 697–706 [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Smirnov A, Schnierstein B, Kelemen G, Matthies U, Klafki HW, Staufenbiel M, Huther G, Ruther E, Kornhuber J (1997) Improved electrophoretic separation and immunoblotting of β‐amyloid (Aβ) peptides 1‐40, 1‐42, and 1‐43. Electrophoresis 18: 527–532 [DOI] [PubMed] [Google Scholar]
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ 3rd (2015) Presenilin‐1 knockin mice reveal loss‐of‐function mechanism for familial Alzheimer's disease. Neuron 85: 967–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A, Eimer S, Okochi M, Smialowska A, Kaether C, Baumeister R, Haass C, Steiner H (2006) The GxGD motif of presenilin contributes to catalytic function and substrate identification of γ‐secretase. J Neurosci 26: 3821–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Liu J, Watanabe A, Hiraga S, Liu S, Tanabe C, Maeda T, Terayama Y, Takahashi S, Michikawa M et al (2013) Aβ43 is the earliest‐depositing Aβ species in APP transgenic mouse brain and is converted to Aβ41 by two active domains of ACE. Am J Pathol 182: 2322–2331 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Source Data for Figure EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2