

Abstract
Lytic polysaccharide monooxygenases (LPMOs) represent a recent addition to the carbohydrate‐active enzymes and are classified as auxiliary activity (AA) families 9, 10, 11, and 13. LPMOs are crucial for effective degradation of recalcitrant polysaccharides like cellulose or chitin. These enzymes are copper‐dependent and utilize a redox mechanism to cleave glycosidic bonds that is dependent on molecular oxygen and an external electron donor. The electrons can be provided by various sources, such as chemical compounds (e.g., ascorbate) or by enzymes (e.g., cellobiose dehydrogenases, CDHs, from fungi). Here, we demonstrate that a fungal CDH from Myriococcum thermophilum (MtCDH), can act as an electron donor for bacterial family AA10 LPMOs. We show that employing an enzyme as electron donor is advantageous since this enables a kinetically controlled supply of electrons to the LPMO. The rate of chitin oxidation by CBP21 was equal to that of cosubstrate (lactose) oxidation by MtCDH, verifying the usage of two electrons in the LPMO catalytic mechanism. Furthermore, since lactose oxidation correlates directly with the rate of LPMO catalysis, a method for indirect determination of LPMO activity is implicated. Finally, the one electron reduction of the CBP21 active site copper by MtCDH was determined to be substantially faster than chitin oxidation by the LPMO. Overall, MtCDH seems to be a universal electron donor for both bacterial and fungal LPMOs, indicating that their electron transfer mechanisms are similar.
Keywords: lytic polysaccharide monooxygenase, cellobiose dehydrogenase, electron transfer, electron donor, hydrogen peroxide, chitin, cellulose, enzyme kinetics
Abbreviations
- AA
auxiliary activity
- CAZy
carbohydrate‐active enzyme database
- CAZymes
carbohydrate‐active enzymes
- CDH
cellobiose dehydrogenase
- CYT
cytochrome
- DH
dehydrogenase
- DP
degree of polymerization
- FAD
flavin adenine dinucleotide
- LPMO
lytic polysaccharide monooxygenase
- ox
oxidized
Introduction
Lytic polysaccharide monooxygenases (LPMOs) are copper‐dependent enzymes that employ an oxidative mechanism to cleave the glycosidic bonds of polysaccharides.1, 2, 3, 4, 5 The main substrates for LPMOs are insoluble polysaccharides such as chitin or cellulose,1, 2, 3 but LPMO activity has also been demonstrated for xyloglucan,6 xylan7 soluble cello‐oligosaccharides,8 and starch.9, 10 LPMOs are currently classified as family 9, 10, 11, and 13 of the auxiliary activities (AAs) in the CAZy database.11 So far, AA9, AA11, and AA13‐type LPMOs (LPMO9s, LPMO11s, and LPMO13s) have only been identified in fungi, whereas AA10‐type LPMOs (LPMO10s) have been found in eukaryotes, prokaryotes, and viruses. The active site of these enzymes contains a solvent exposed copper‐ion that is coordinated by two conserved histidines in a histidine brace.3, 12, 13 The role of the copper ion is to activate a dioxygen molecule that leads to hydroxylation of either the C1 or C4 carbon of the substrate.4, 14, 15 The hydroxylation event yields an unstable hemiketal intermediate that results in spontaneous cleavage of the glycosidic bond through an elimination reaction.4, 16 For catalysis by LPMOs to occur, the copper ion must be reduced by an external electron donor prior to the activation of dioxygen. It is known from laboratory experiments that functional electron donors include small‐molecule reductants like ascorbic acid1 or gallic acid,3 lignin present in plant cell walls,17, 18, 19 certain redox‐active proteins, such as cellobiose dehydrogenase (CDH)4, 20, 21 and phenolic compounds that undergo redox cycling by glucose‐methanol‐choline oxidoreductases.22 CDH has only been found in fungi. Several studies have reported that LPMOs and CDHs are cotranscribed and coexpressed during fungal growth on plant cell wall material,23, 24, 25 which has led to the notion that CDH may be the primary natural electron donor for (fungal) LPMOs.26
CDHs are two‐domain proteins comprising a flavin adenine dinucleotide (FAD)‐binding dehydrogenase (DH) domain coupled to a heme‐binding cytochrome (CYT) domain.27, 28, 29 CDH oxidizes disaccharides or oligosaccharides to their corresponding aldonic acids. In this process, the two electrons obtained from the substrate are stored in the DH domain by reduction of the FAD. By internal electron transfer, one electron can be transferred from the DH domain to the CYT domain (by reduction of the heme group). CDHs can thereby perform two‐electron reduction reactions (via the DH domain) or one‐electron reductions (via the CYT domain). CDHs are capable of efficient transfer of electrons to both small chemical compounds and proteins.30, 31, 32 In the absence of a good electron acceptor, O2 can also be reduced to yield H2O2 (or in some in cases).33 A simplified scheme illustrating the probable CDH‐LPMO system is shown in Figure 1.
Figure 1.
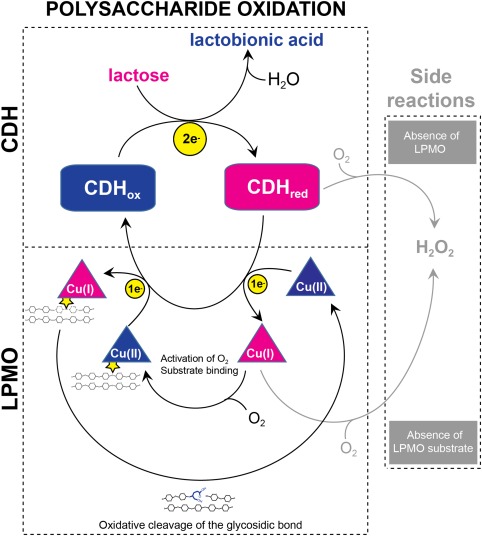
Lytic polysaccharide oxidation by the CDH‐LPMO system. The LPMO is illustrated by a triangular cartoon, CDH in a square cartoon with rounded corners. Electrons are shown by yellow circles. Enzymes are colored blue in their oxidized form and pink in their reduced form. The LPMO substrate is indicated by two tethered chains representing a polysaccharide crystal.
Since the discovery of CDHs, their in vivo role has been uncertain. The widespread occurrence of these enzymes has generated several hypotheses on their functions, especially linked to their ability to produce H2O2. Hydrogen peroxide can act as an antimicrobial agent34, 35 or together with Fe(II), create hydroxyl radicals, one of the strongest oxidizing agents in aqueous systems,36 in a Fenton‐reaction. It has been suggested that such hydroxyl radicals can degrade or modify cellulose, indicating a role of CDHs in unspecific plant cell wall degradation.37, 38, 39 The discovery of LPMOs and the notion that these enzymes are good electron acceptors for one‐electron transfer from the CYT domain of CDH26 has shed new light on these issues. Notably, reduced LPMOs are also able to produce H2O2 in the absence of a substrate.8, 40
The specificity of the electron transfer reaction between CDHs and LPMOs has hitherto not been investigated in detail, one key question being if there is any specificity at all or if an enzyme such as CDH can reduce any LPMO. In this study, we report on the ability of a fungal CDH to activate bacterial LPMO10s, including an analysis of the rate of electron transfer between the proteins. We show that both chitin and cellulose‐active LPMO10s can use CDH as a source of electrons and we compare the functionality of the LPMO10‐CDH interaction with previously studied LPMO9‐CDH interactions. Importantly, so far, the LPMO literature is almost devoid of kinetic data, which is likely due to the difficulty of obtaining linear progress curves, which again is likely due to the use of unstable small molecule reductants. We show here that careful experimental design based on using CDH/lactose for the generation of reducing equivalents gives superior control of the reaction kinetics. In fact, we show that the rate of an LPMO reaction can be measured by simply monitoring product formation by CDH.
Results
Electron transfer from MtCDH to CBP21
Stopped‐flow experiments with reduced MtCDH and copper‐saturated CBP21‐Cu(II) showed fast electron transfer between the proteins. During the interaction between the enzymes, a monophasic reaction was observed and the electron transfer rates were obtained from a single exponential fit. The electron transfer rate increased proportionally with the concentration of CBP21 (Fig. 2), which indicates a fast, bimolecular reaction. At 50 µM CBP21, the observed electron transfer rate reached 32 s−1. The observed re‐oxidation rate of the heme b cofactor by oxygen (in an air saturated buffer) in absence of CBP21 was 0.013 s−1, which is very low. The observed FAD reoxidation rate of the oxidative half‐reaction was k obs = 3.2 min−1 (data not shown).
Figure 2.
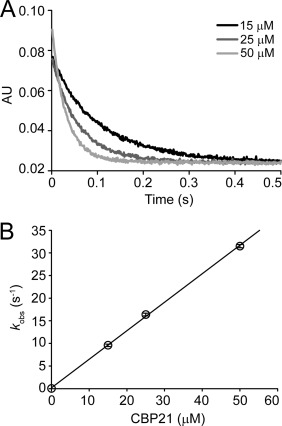
Reaction of reduced MtCDH‐heme with CBP21‐Cu(II). A: Oxidation of 5 µM MtCDH by CBP21 (15, 25, and 50 µM) was followed in a stopped‐flow spectrometer at 563 nm. Observed electron transfer rates are plotted in (B). Partially reduced MtCDH was obtained by reduction with cellobiose, in 50 mM sodium phosphate buffer, pH 6.0. Error bars show the standard deviation of three replicates. Concentrations are those after mixing.
Activation of bacterial LPMOs by MtCDH
Qualitative enzyme activity assays showed that MtCDH can act as an electron donor for CBP21 and ScLPMO10C from Streptomyces coelicolor, which target chitin and cellulose as substrates, respectively (Fig. 3). Analysis of reaction products by UPLC, HPAEC‐pulsed amperometric detector (PAD) and by MALDI‐TOF MS showed that the products were aldonic acids giving double sodium adducts that are characteristic for the presence of a carboxylic group1, 41; MS results not shown.
Figure 3.
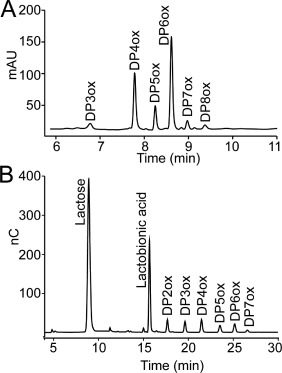
Product profiles from LPMO‐CDH reactions. Chromatographic analysis of reaction products arising from incubation of 1.5 µM MtCDH and 3.0 mM lactose with (A) 1.0 µM CBP21 and 10 mg/mL β‐chitin or (B) 1.0 µM ScLPMO10C and 10 mg/mL Avicel for 4 h. Both reactions were buffered in 25 mM Bis‐Tris pH 6.0. Peaks are labeled as follows: DP, degree of polymerization; ox, oxidized at C1 (aldonic acids). In‐house made standards were used to verify product identities and product distributions were also verified by MALDI‐TOF MS (not shown). Some (small) peaks in both chromatograms were not possible to identify (unlabeled peaks in the chromatograms) and most likely represent background noise.
Dose response experiments
In order to determine a suitable lactose concentration for the MtCDH‐CBP21 experiments, the effect of lactose concentrations ranging from 0.5 to 10 mM was investigated. The quantity of LPMO‐generated products (which were all converted to chitobionic acid by chitobiase treatment) increased with increasing lactose concentrations, plateauing at 3.0 mM [Fig. 4(A)]. Using 3.0 mM lactose as substrate for MtCDH, the effect of varying the concentration of MtCDH was investigated. These experiments showed that faster initial rates (i.e., higher product levels after 4 h) were obtained by increasing the MtCDH concentration up to 3 µM, whereas the yield after 24 h showed a maximum at 1.5 µM MtCDH [Fig. 4(B)]. Thus, increasing the MtCDH concentration seems to be beneficial for the short term activity of CBP21, but reduces LPMO activity during longer incubation times. For comparison, the dose‐response relationship between CBP21 and ascorbate as electron donor was also investigated (Fig. 5). Increasing concentrations of ascorbate resulted in both higher initial rates of chitin oxidation and final yields of oxidized products, except for the highest ascorbate concentration (10 mM), which resulted in a progress curve with an initial phase similar to that of 5 mM ascorbate, but had a lower final yield. It is notable that the progress curves with ascorbate showed linearity during the first 1.5 h of the reaction only, after which LPMO activity declined substantially. This is different when using MtCDH, as shown below.
Figure 4.
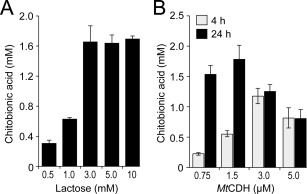
Dose‐response experiments. Accumulation of oxidized products was measured after 4 h (gray bars, panel B only) and 24 h (black bars). A: Degradation of 10 mg/mL β‐chitin by 1.0 µM CBP21, 1.5 µM MtCDH at varying concentrations of lactose. B: Degradation of 10 mg/mL β‐chitin by 1.0 µM CBP21 in the presence of 3.0 mM lactose and varying concentrations of MtCDH. The standard deviations for all experiments are shown by error bars (n = 3). All reactions (panels A and B) were buffered in 25 mM Bis‐Tris, pH 6.0.
Figure 5.
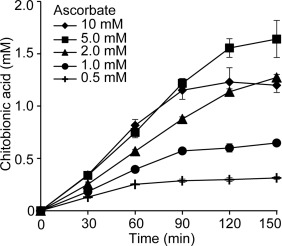
CBP21 activity at varying concentrations of ascorbic acid. Time course reactions were monitored for the degradation of 10 mg/mL β‐chitin by 1.0 µM CBP21 in the presence of ascorbate concentrations ranging from 0.5 to 10 mM as indicated in the graph inset. Standard deviations are shown by error bars (n = 3). All reactions were conducted in 50 mM Bis‐Tris, pH 6.0.
LPMO kinetics
Using the optimum conditions obtained from the dose‐response experiments (Fig. 4) the reaction kinetics of CBP21 with the MtCDH/lactose system as electron‐donor was monitored [Fig. 6(A)]. The product formation curve was essentially linear and enzyme activity remained stable for up to 10 h. The linear part of the curve has a slope of 2.2 µM min−1 indicating an apparent rate of 2.2 min−1 for CBP21 (note that in this experiment only solubilized oxidized products are monitored; see below for further details). An identical experiment where MtCDH + lactose was substituted by 1.0 mM ascorbate, showed a bi‐phasic product formation curve, where the rate of product formation dropped substantially after ∼1.5 h of incubation [Fig. 6(A)], as was also observed in the CBP21‐ascorbate dose‐response experiment depicted in Figure 5.
Figure 6.
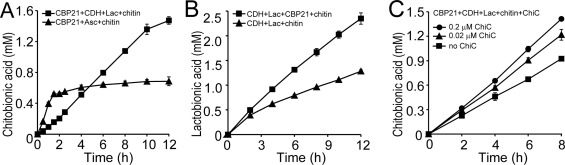
Degradation of β‐chitin by CBP21 and oxidation of lactose by MtCDH. A: Time course analysis of degradation of 10 mg/mL β‐chitin (equivalent to 24.6 mM chitobiose) by 1.0 µM CBP21 using either 1.5 µM MtCDH and 3.0 mM lactose (squares) or 1.0 mM ascorbate (triangles) as reducing agents. B: Oxidation of lactose by 1.5 µM MtCDH in the presence (squares) or absence (triangles) of 1.0 µM CBP21. C: Degradation of β‐chitin by 1.0 µM CBP21 in the presence of 1.5 µM MtCDH, 3.0 mM lactose and 0, 0.02, or 0.2 µM ChiC. All reactions contained 10 mg/mL β‐chitin. The standard deviations for all experiments are shown by error bars (n = 3). All reactions (panels A, B, and C) were buffered in 25 mM Bis‐Tris, pH 6.0.
The influence of CBP21 on the activity of MtCDH was quantified by monitoring lactose oxidation in the presence of chitin and in the presence or absence of the LPMO [Fig. 6(B)]. In the presence of CBP21 a linear progress curve was observed (slope = 3.23 µM min−1), whereas the CBP21 deficient reaction shows an initial burst, followed by linear progress (slope = 1.67 µM min−1).
Soluble products formed by CBP21 represent only a part of the LPMO activity and in order to quantify the total amount of oxidized products formed during catalysis, an endo‐chitinase (ChiC from S. marcescens) was added to CBP21/MtCDH/lactose/chitin reaction mixtures. In the presence of 0.02 or 0.2 µM ChiC, the formation of oxidized products over time was still linear [Fig. 6(C); i.e., chitinase activity does not reduce substrate availability]. The chitinase containing reactions yielded 1.3 times (0.02 µM ChiC) and 1.5 times (0.2 µM ChiC) more oxidized products compared with reactions only containing CBP21 and MtCDH/lactose [Fig. 6(C)]. Therefore, the actual product formation rate for MtCDH/lactose fueled CBP21 is ∼3.3 µM min−1, corresponding to an apparent rate constant of 3.3 min−1. It is noteworthy that the apparent rates of chitin oxidation (3.3 µM min−1) and lactose oxidation (3.23 µM min−1) are very similar. The reduction in product formation rate after 10 h is likely due to substrate depletion, since at that point, that is, at 1.4 mM oxidized products, ∼6% of the disaccharides in the substrate is oxidized, which is similar to previously observed maximum values.1
Generation of H2O2 by MtCDH and CBP21
The generation of H2O2 in enzyme reactions containing MtCDH and/or CBP21 was quantified at various time points during a 90‐min incubation period (Fig. 7). Reactions containing MtCDH and lactose showed production of H2O2 that increased over time to ∼150 µM after 90 min. CBP21 alone did not generate H2O2 [Fig. 7(B)] but, expectedly, generated H2O2 when also ascorbate was added to the reaction [Fig. 7(A)]. When CBP21 and its substrate, β‐chitin, were both present in the reaction containing either MtCDH + lactose or ascorbate as electron donors, formation of H2O2 was very low and decreased over time. In fact, for the reaction with MtCDH + lactose as electron donor, H2O2 could only be detected immediately after mixing the reaction mixture constituents (∼10 µM) and was below the detection limit of the assay in subsequent measurements. A similar trend was observed for ascorbate, but the initial level of H2O2 was slightly higher (∼20 µM) and the decrease was slower, reaching ∼5 µM after 90 min. In a control reaction, it was shown that the presence of chitohexaose, which is a soluble chitin fragment that is not cleaved by CPB21, had no effect on H2O2‐production by the CDH‐lactose‐CBP21 system [Fig. 7(B)]. Taken together, these observations show that reducing equivalents are channeled towards oxidative polysaccharide cleavage rather than direct oxygen reduction and that the presence of LPMO substrate prevents the formation of H2O2 by both CDH and the LPMO.
Figure 7.
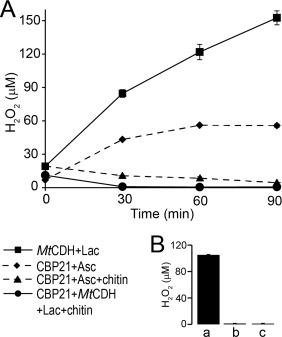
Analysis of H2O2 generated in reactions containing CBP21 and MtCDH. A: Time course analysis of H2O2 generated in reactions containing 1.0 µM CBP21 and 1.0 mM ascorbate (diamonds), 1.0 µM CBP21, 1.0 mM ascorbate, and 10 mg/mL β‐chitin (triangles), 1.5 µM MtCDH and 3.0 mM lactose (squares) and 1.5 µM MtCDH, 3.0 mM lactose, 1.0 µM CBP21, and 10 mg/mL β‐chitin (circles). B: Control experiments showing generation of H2O2 in reactions containing 0.9 mM (GlcNAc)6, 1.0 µM CBP21, 1.5 µM MtCDH, and 3.0 mM lactose (a), 1.0 µM CBP21 in buffer (b) and 1.5 µM MtCDH in buffer (c). Reactions were analyzed after 90 min incubation at 40°C and 1000 rpm. All reactions (panels A and B) were buffered in 25 mM Bis‐Tris, pH 6.0. Standard deviations for all experiments are shown by error bars (n = 3).
Discussion
CDHs are universal electron donors for LPMOs. CDHs have been shown to act as an electron donor for several fungal family AA9 LPMOs (LPMO9s4, 20). In the present study, we show that MtCDH can efficiently transfer electrons from lactose to two bacterial LPMOs that only share 23% sequence identity, target different substrates and share less than 15% sequence identity with their fungal counterparts (Figs. 2 and 3). Observed electron transfer rates up to 32 s−1 were reached, meaning that electron transfer from CDH to the bacterial LPMOs is as efficient as transfer to LPMO9s26 or to other protein electron acceptors like cyt c.42 This indicates that CDHs can act as a general electron donor for LPMOs, independent of LPMO origin and seemingly independent of LPMO sequence. It has been suggested that LPMO9s share a conserved region located distantly from the active site, which may have evolved to interact with the CDH CYT domain for electron transfer.43, 44 This region is not conserved in the bacterial LPMO10s. A conserved site for potential protein‐protein interactions was neither found on the surface of the CYT domain of CDH.26 All in all, available data indicate that electron transfer between CDH and LPMOs does not depend on protein‐protein interactions and conserved docking sites. Electron transfer is more likely to only involve the actual sites of oxidation and reduction, which, for LPMOs, is the copper site and its conserved histidine brace.
Putative protein electron donors in bacterial LPMO‐containing chitinolytic/cellulolytic enzyme systems have not yet been identified, but the redox active protein “cbp2D” from Cellvibrio japonicus has been shown to be crucial for degradation of crystalline cellulose by the bacterium and has been proposed to represent a bacterial counterpart to CDH.45 Indeed, the present data add support to the emerging notion that LPMOs may receive electrons from many sources.22 The combination of a chitin‐active LPMO (CBP21) with CDH offered unprecedented possibilities for in‐depth studies of the CDH‐LPMO interplay, since CDH does not act on the LPMO substrate (which is chitin, not cellulose).
Use of MtCDH provides linear kinetics for CBP21
Despite intense research on LPMOs in recent years, kinetic data are scarce. It has been difficult to obtain linear progress curves, which is likely due to the common use of small molecule electron donors such as ascorbate, reduced glutathinone, and l‐cysteine. Such reducing agents are prone to autooxidation, which not only depletes the concentration of reductant, but also generates reactive oxygen species that can affect the stability of the proteins in the reaction. Indeed, a recent study reported that H2O2 generated by futile LPMO activity reduced the activity of the glycoside hydrolases present in the enzyme cocktail.46 We show here that, when using an optimal LPMO:CDH ratio, the progress curves are linear until substrate depletion comes into play [Fig. 6(A)]. Catalysis by LPMOs is slow (in the “per minute” range), whereas reduction of the LPMO active site copper can be substantially faster (in the “second range”; Fig. 2). With this in mind, it is not surprising that the data in Figures 4 and 5 show that dosing of electrons is important. When feeding electrons too fast to the LPMOs, side reactions are likely to occur, leading to generation of H2O2 (either by the LPMO itself or by CDH lacking sufficient electron acceptors) and possibly the generation of reactive oxygen species. While an initial faster product formation rate is obtained, LPMO activity ceases more rapidly, which not only may preclude kinetic analysis but which also is disadvantageous in an applied setting. Comparison of Figure 4 (CDH/lactose) with Figure 5 (ascorbic acid) clearly shows that, when dosing the CDH optimally, CDH comprises a stable electron‐donating system for the LPMO, in contrast to unstable ascorbic acid [Fig. 6(A)]. Notably, the CDH/lactose system allows tuning of LPMO activity in chitin degradation reactions by simply regulating the concentration of lactose.
It is likely that ascorbate is depleted at the time point where the slope of the progress curves becomes drastically reduced [Figs. 4 and 6(A)]. Interestingly, CBP21 activity does not completely cease after this time point [Fig. 6(A)], which means that CBP21 obtains electrons from elsewhere, possibly from the chitin itself. Indeed, the original discovery that CBP21 boosts chitin degradation by chitinases was based on experiments that did not involve an externally added electron donor.47
The electron donor is rate limiting for CBP21
While the data in Figures 4 and 5 show that too high electron supply may be detrimental for overall process efficiency, they also clearly show that the availability of electrons is a rate limiting factor in the reaction. Looking only at initial CPB21 rates, there are clear dose‐response effects. For ascorbate, it seems that 5 mM is the optimal concentration, yielding an apparent initial rate of 13 min−1 (0.22 s−1) for CBP21 (Fig. 5). Interestingly, this is in the same range as the observed rates for a cellulose‐active fungal LPMO using the highly efficient light‐induced electron transfer system.48 The increase to 10 mM ascorbate gives a similar initial rate, but a lower yield, indicating either enzyme inactivation or O2 depletion through ascorbate autooxidation (O2 depletion being less likely due to the rigorous shaking of the reaction mixture). The same trend (faster initial rate, but lower final yield) is observed when increasing the concentration of MtCDH. MtCDH is able to generate substantial amounts of H2O2 in the absence of an electron acceptor (Fig. 7) and one could thus expect production of H2O2 if there is a shortage of electron acceptor. It should be noted that H2O2 generated by either ascorbate or MtCDH likely is the downstream product of other reactive oxygen species generated, like , which is more reactive than H2O2 and which may damage the enzymes.
Oxidation rates of lactose and chitin are equal
Under the conditions used here MtCDH oxidized lactose at a steady‐state turnover rate of ∼1.1 min−1. This is in agreement with the observed FAD re‐oxidation rate of the oxidative half‐reaction (k obs = 3.2 min−1), which represents the theoretical upper limit of CDH's oxygen reactivity. Figure 2 shows that CBP21 is a much better electron acceptor for MtCDH than oxygen with observed rates in the order of tenths per second. Consequently, in the presence of an LPMO and its substrate, lactose oxidation by MtCDH did not lead to the formation of H2O2 (Fig. 7) indicating that MtCDH transfers all its electrons to CBP21 and that CBP21 becomes re‐oxidized by acting on its polysaccharide substrate. The slow steady‐state turnover rate of chitin oxidation, compared to the fast electron transfer rate observed from MtCDH to CBP21 in solution, indicates that the transfer of the first electron is not the rate limiting step of the reaction. Furthermore, to perform successful catalysis, CBP21 must bind to the insoluble substrate, meaning that successful catalysis by the LPMO also depends on the substrate binding equilibrium (CBP21 has a binding dissociation constant of ∼1 µM 47). Moreover, the monooxygenase reaction requires two electrons, thus requiring interaction with two reduced MtCDH molecules (or the same molecule twice) to complete the catalytic cycle. The mechanism of LPMO reduction by MtCDH is not known, but is thought to occur in part through direct reduction of the active site copper by MtCDH's CYT domain.26 While it is highly likely that transfer of the first electron [i.e. reduction of Cu(II) to Cu(I)] occurs in solution, the nature of the second electron transfer step is not clear. Several scenarios are thinkable, the discussion of which is beyond the scope of this article. All these scenarios have possible rate‐limiting steps, which could, for example, relate to the electron transfer itself via another route than via the copper19, 43 or to partial dissociation of the LPMO from the substrate, if such dissociation would be needed to channel the second electron via the copper.
Interestingly, our experimental approach, with quantification of chitobionic acid (product resulting from CBP21 catalysis), lactobionic acid (product resulting from MtCDH catalysis), and reduction of O2 to H2O2, allowed us to monitor the total flow of electrons in the reaction system. Thus, we were able to show that under conditions where the carbohydrate substrates were not limiting and where the CBP21:MtCDH ratio used yielded a linear progress curve, chitin and lactose were oxidized at an identical speed amounting to ∼3 µM min−1. Since CDH‐driven oxidation of one lactose molecule yields two electrons and the LPMO‐driven oxidation of chitin is thought to require two electrons, our results indicate that all electrons generated by MtCDH are consumed by CBP21 in the reaction. This is in agreement with the absence of H2O2 in reactions containing CBP21, MtCDH and the respective enzyme substrates (Fig. 7), that is, no futile O2 reduction takes place by either of the enzymes. These data thus support the proposed catalytic mechanism for LPMO mediated chitin oxidation.1, 44, 49 Interestingly, under the conditions employed here, measuring lactose oxidation, which is easier than measuring the total amount of oxidative cleavages in an insoluble substrate, provides a simple tool for quantitative monitoring of LPMO activity.
In conclusion, the present data provide the first detailed insight into the activation of a bacterial LPMO by a protein electron donor and show how linear kinetics may be obtained by using such a donor. Clearly, LPMOs are good electron acceptors and it is conceivable that all natural LPMO containing enzyme systems depend on a carefully balanced cascade of enzymatic redox reactions that are optimized for biomass conversion (or other, yet to be discovered LPMO functionalities44), while preventing nondesirable generation of reactive oxygen species.
Experimental Procedures
Protein expression and purification
CDH from the thermophilic ascomycetous fungus Myriococcum thermophilum (MtCDH) was expressed in Pichia pastoris and purified as previously reported.50 The production of MtCDH was performed at 4 L‐scale in a laboratory bioreactor (MBR, Switzerland) according to the Pichia Fermentation Process guidelines (Invitrogen). In short, the cultivation was initiated by adding 0.4 L of a preculture grown over night at 30°C and 120 rpm. Expression of recombinant protein was induced with methanol. The cultivation temperature was 30°C, the airflow rate was kept constant at 6 L min−1, and the stirrer speed was 800 rpm. Samples were taken regularly and checked for CDH activity. Purification of MtCDH was done by a two‐step chromatographic procedure (all equipment from GE Healthcare) using hydrophobic interaction chromatography (PHE‐Sepharose FF resin) and anion exchange chromatography (Source 15Q resin). The purest CDH fractions were pooled, concentrated using Amicon Ultra centrifugal filters (Millipore) with a molecular weight cut‐off of 10 kDa and sterile filtered (0.2 µm). The purity of MtCDH was confirmed by SDS‐PAGE.
Streptomyces coelicolor LPMO10C, ScLPMO10C (also known as CelS2), was expressed in E. coli as previously described.41 In brief, a fresh transformant containing the ScLPMO10C encoding plasmid was inoculated and grown in LB medium supplemented with ampicillin (100 µg/mL) at 37°C for ∼16 h without induction. The protein was harvested from the periplasmic space using a cold osmotic shock method.51 Purification was carried out by anion exchange chromatography using a 5 mL HiTrap DEAE FF column (GE Healthcare) in 50 mM Tris‐HCl pH 7.5 and the protein was eluted by applying a linear salt gradient (0–500 mM NaCl over 60 column volumes). Subsequently, the partially purified protein was loaded onto a HiLoad 16/60 Superdex 75 size exclusion chromatography column (GE Healthcare) operated with a running buffer consisting of 50 mM Tris‐HCl pH 7.5 and 200 mM NaCl. Fractions containing pure LPMO were pooled and concentrated using an Amicon Ultra centrifugal filter (Millipore) with a molecular weight cut‐off of 10 kDa, before the enzyme concentration was determined using the Bradford assay (Bio‐Rad).
Chitobiase from Serratia marcescens (SmGH20A) was expressed and purified as previously described by Loose et al.52 with minor changes. In short, BL21 star cells containing the pET30 Xa/LIC vector with the chb gene were grown in LB medium supplemented with 100 µg/mL kanamycin at 37°C to an OD600 = 0.5 after which protein production was induced by addition of IPTG to a final concentration of 0.3 mM, followed by incubation at 30°C for 5 h with shaking at 160 rpm. The culture was harvested and resuspended in lysis/binding buffer (20 mM Tris‐HCl pH 8.0, 5 mM imidazole). The cells were disrupted by incubating 30 min with 0.1 mg/mL lysozyme followed by sonication using a Vibra cell sonicator (Sonics) using 27% amplitude and a repeated cycle of 5 s on and 1 s off for a total duration of 3 min. The extract was loaded onto 5 mL Ni‐NTA Agarose resin (Protino, Macherey‐Nagel) using 20 mM Tris‐HCl, pH 8.0, 5 mM imidazole as running buffer. The protein was eluted with 20 mM Tris‐HCl, pH 8.0, 500 mM imidazole. The eluted chitobiase was concentrated and the imidazole was removed using an Amicon Ultra centrifugal filter (Millipore) with 10 kDa cut‐off. The enzyme concentration was determined using the Bradford assay (Bio‐Rad).
CBP21 from Serratia marcescens (SmLPMO10A) was expressed and purified as previously described by Vaaje‐Kolstad et al.47 In short, E. coli BL21 DE3 cells harboring a pRSETB vector containing the cbp21 gene were grown in TB‐medium supplemented with ampicillin (100 µg/mL) overnight in a Harbinger LEX bioreactor (Harbinger Biotech, Toronto, Canada) at 37°C. The cells were harvested by centrifugation and the periplasmic content was extracted using the cold osmotic shock method. The extract was adjusted to 20 mM Tris‐HCl, pH 8.0, 1.0 M (NH4)2SO4 and applied to 10 mL chitin beads (NEB) using a BioLogic chromatographic system from BioRad. After nonbound protein had passed through the column, CBP21 was eluted with 20 mM acetic acid. The protein was concentrated using an Amicon Ultra centrifugal filter with a 10 kDa cut‐off (Millipore) and the buffer was exchanged to 20 mM Tris‐HCl, pH 8.0. The protein concentration was determined using A280 and the theoretical extinction coefficient.
Chitinase 18C (ChiC) from S. marcescens was produced and purified as described previously by Vaaje‐Kolstad et al.47 In short, this was accomplished by expression of the enzyme in E. coli, followed by extraction of periplasmic proteins by cold osmotic shock and one‐step purification by standard ion exchange chromatography, using Q‐Sepharose Fast Flow at pH 9.4 and a 0–100 mM NaCl linear gradient for elution of the chitinase. The protein concentration was determined using A280 and the theoretical extinction coefficient.
Chitooligosaccharide oxidase (m‐ChitO) from Fusarium graminearum N‐terminally fused to maltose binding protein encoded by the pBAD‐MBP‐chitO expression vector was expressed as previously described53 with minor changes. The culture was harvested and the pellet was resuspended in 20 mM Tris‐HCl, pH 8.0, containing 10% glycerol. The cells were disrupted by sonication for 2.5 min (5 s on, 1 s off) at an amplitude of 30%. The crude extract was loaded on a 5 mL DEAE FF column (GE Healthcare) and m‐ChitO was eluted using a stepwise gradient from 15 mM NaCl to 250 mM NaCl in 20 mM Tris‐HCl, pH 8.0. Fractions containing m‐ChitO were pooled and concentrated using Amicon Ultra centrifugal filters (Millipore) with 10 kDa cut‐off. The concentrated protein sample was then subjected to a size exclusion chromatography step using a HiLoad 16/60 Superdex 75 size exclusion column (GE Healthcare), operated in 20 mM Tris‐HCl, pH 8.0. Fractions containing m‐ChitO that was at least 85% pure were pooled and concentrated. The enzyme concentration was determined using the Bradford assay (Bio‐Rad).
Cu(II) saturation and desalting of LPMOs
CBP21 and ScLPMO10C were saturated with Cu(II) according to the protocol described by Loose et al.52 Briefly, the LPMOs were saturated by incubating them with Cu(II)SO4 in a 1:3 molar ratio (enzyme:copper) at room temperature for 30 min. After saturation, excess Cu(II)SO4 was removed by passing the proteins through a PD MidiTrap G‐25 (GE Healthcare) desalting column using 25 mM Bis‐Tris, pH 6.0, as running buffer.
Stopped‐flow spectroscopy
Pre‐steady state kinetic studies measured the re‐oxidation of CDH's heme b cofactor by CPB21 and were performed with a SX‐20 stopped‐flow apparatus (Applied Photophysics, Leatherhead, UK) equipped with a flow cell with a path length of 10 mm and a diode array detector. Using the sequential mixing mode, 20 µM MtCDH and a 3‐fold molar excess of cellobiose were initially mixed (1:1) and held in an ageing loop until reoxidation of the FAD cofactor by ambient oxygen and hence full depletion of cellobiose was observed. After 95 s full reoxidation of the FAD cofactor was observed, while about ∼80% of the heme b was still reduced and was subsequently mixed (1:1) with the CPB21 solution. The reoxidation rate of the heme b cofactor was measured at 563 nm and was used to determine the electron transfer rate from MtCDH to CPB21 (k obs, s−1). The FAD re‐oxidation was measured at 449 nm and was used to determine the rate of the oxidative half‐reaction in the presence of oxygen (air saturated buffer = 250 µM). Observed traces were fitted to an exponential function using the Pro‐Data software suite (Applied Photophysics). All species were prepared in 50 mM sodium phosphate buffer, pH 6.0, and final concentrations of the enzymes in the measurement cell were 5.0 µM MtCDH and 15, 25, or 50 µM of CPB21. All measurements were carried out at 30°C in triplicates.
LPMO activity assays
Reactions containing 1.0 µM CBP21 or ScLPMO10C, with 10 mg/mL β‐chitin or Avicel, respectively, in the presence of 1.5 µM MtCDH and 3.0 mM lactose buffered in 25 or 50 mM BisTris pH 6.0 were incubated at 40°C in an Eppendorf Comfort Thermomixer with a temperature‐controlled lid, shaking at 1000 rpm. Samples were taken at various time points and immediately filtered using a 96‐well filter plate (Millipore) operated by a Millipore vacuum manifold to stop chitin oxidation. Samples used to monitor lactose oxidation over time were adjusted to 100 mM NaOH to stop MtCDH activity. For all CBP21 reactions, except reactions used to analyze the product profile, the soluble products were treated with 2.0 µM chitobiase for 2 h at 37°C to convert the chitooligosaccharides to chitobionic acid and GlcNAc. The resulting products were analyzed and quantified by UPLC as previously described by Loose et al.52 The analysis of products generated by ScLPMO10C is described below.
Product analysis by HPAEC‐PAD
Oxidized cello‐oligosaccharides generated by ScLPMO10C were analyzed by high performance anion exchange chromatography (HPAEC) using a Dionex Bio‐LC connected to a CarboPac PA1 column operated with a flow rate of 0.25 mL/min in 0.1 M NaOH (Eluent A) and a column temperature of 30°C. Products were separated as previously described54 using a stepwise gradient with increasing amount of eluent B (0.1 M NaOH and 1 M NaOAc) as follows: 0–10% B over 10 min, 10–30% B over 25 min, 30–100% B over 5 min, 100–0% B over 1 min, 0% B over 9 min. Oxidation of lactose over time by MtCDH was analyzed by separating lactose and lactobionic acid by HPAEC‐PAD, using a steeper gradient, as follows: 0–10% B over 10 min, 10–18% B over 10 min, 18–30% B over 1 min, 30–100% B over 1 min, 100–0% B over 0.1 min and 0% B over 13.9 min. Eluted oligosaccharides were monitored using a PAD and chromatograms were recorded using Chromeleon 7.0 software. In‐house made standards (see below) were run at regular intervals to allow quantification.
Product analysis by UPLC
Oxidized chitooligosaccharides (aldonic acids) were analyzed and quantified using an Aquity UPLC® BEH Amide 1.7 µm column run in HILIC (hydrophilic interaction) mode. To quantify chitobionic acid, a 2.1 × 50 mm column was used with the following gradient: 22% eluent A (15 mM Tris–HCl pH 8.0), 78% eluent B (100% acetonitrile): for 4 min, followed by a 1 min gradient to 62% B. The column was reconditioned by a 1 min gradient to initial conditions (22% A, 78% B) and additional running at these conditions for 1 min. To obtain a full product profile, a 2.1 × 150 mm column was used, applying the following gradient: 26% A and 74% B for 5 min, followed by a 2 min gradient to 62% B. These conditions were held for 1 min. The column was reconditioned by a 2 min gradient to 26% A and 74% B and additional running for 2 min. The flow rate was 0.4 mL/min and eluted chitooligosaccharides were monitored at 205 and 195 nm.
Production of chitobionic acid and lactobionic acid standards
Chitobionic acid standards were produced as previously described by Loose et al.52 In short, 2.0 mM chitobiose (95% pure, Megazyme) in 25 mM Bis‐Tris pH 6.0 were incubated with 0.1 mg/mL m‐ChitO over night at 22°C. The oxidized products were analyzed by UPLC. At least 97% of the chitobiose was oxidized.
Lactobionic acid standards were produced by incubating 3.0 mM lactose with 1.5 µM MtCDH in 25 mM Bis‐Tris pH 6.0. To speed up the reaction and obtain complete oxidation of all lactose added, 1.0 µM CBP21 and 10 mg/mL β‐chitin were added to the reaction (MtCDH will oxidize lactose substantially faster when an efficient electron acceptor like CBP21 is present in the reaction mixture). The samples were incubated in an Eppendorf Comfort Thermomixer with a temperature‐controlled lid, at 40°C and 1000 rpm. After 48–72 h samples were taken to assure complete oxidation of lactose to lactobionic acid. When full oxidation was reached, the sample was filtered (0.45 µm) and stored at −20°C until further use. Lactobionic acid could be base line separated from the oxidized chitooligosaccharides produced by CBP21 in the reaction using the HPAEC method described for analysis of oxidized cellooligosaccharides (see above).
Hydrogen peroxide assays
Hydrogen peroxide (H2O2) was quantified by using the Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (Molecular Probes) according to the instructions provided by the manufacturer. In short, the concentration of hydrogen peroxide was determined by mixing 5 µL of sample with 45 µL of 1 × reaction buffer (50 mM sodium phosphate, pH 7.4), followed by addition of 50 µL of the Amplex® Red working solution (100 µM Amplex® Red reagent and 0.2 U/mL horseradish peroxidase in 1 × reaction buffer) and incubation for 30 min at room temperature in 96‐well plates. The amount of the colorimetrically detectable product of the assay, resorufin, was quantified by measuring absorbance at 540 nm using a Multiskan FC spectrophotometer (Thermo Scientific). The standard curve, ranging from 0.5 to 17 µM, was made by diluting the H2O2 standard stock solution supplied with the kit in 1 × reaction buffer. Before addition of the Amplex® Red working solution, all standard samples were adjusted to 2.5 mM Bis‐Tris, pH 6.0, to generate conditions identical to those of the experimental samples.
Author Contributions
J.S.M.L. designed, performed and analyzed the experiments, and wrote the article. Z.F. designed, performed and analyzed the experiments, and contributed to writing the article. D.K. designed and supervised stopped‐flow experiments and wrote parts of the article. S.S. analyzed stopped‐flow experiments and drew graphs for the article. R.L. analyzed fast kinetic data and wrote parts of the article. V.G.H.E. proposed experiments, analyzed data, and wrote parts of the article. G.V‐K. initiated and supervised the project, proposed experiments, analyzed data, and wrote parts of the article.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Statement for Broader Audience: Lytic polysaccharide monooxygenases (LPMOs) are redox‐active enzymes that cleave the glycosidic bonds of recalcitrant polysaccharides. The present study shows that fungal cellobiose dehydrogenases can act as a universal electron donor for these enzymes and that appropriate dosing of electrons to the LPMO is important for stable activity. Finally, our results verify that two electrons are consumed by each LPMO reaction.
References
- 1. Vaaje‐Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330:219–222. [DOI] [PubMed] [Google Scholar]
- 2. Forsberg Z, Vaaje‐Kolstad G, Westereng B, Bunæs AC, Stenstrøm Y, MacKenzie A, Sørlie M, Horn SJ, Eijsink VGH (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci 20:1479–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JC, Johansen KS, Krogh KB, Jørgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter CP, Dupree P, Xu F, Davies GJ, Walton PH (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA 108:15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Phillips CM, Beeson WT, Cate JH, Marletta MA (2011) Cellobiose dehydrogenase and a copper‐dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa . ACS Chem Biol 6:1399–1406. [DOI] [PubMed] [Google Scholar]
- 5. Horn SJ, Vaaje‐Kolstad G, Westereng B, Eijsink VGH (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agger JW, Isaksen T, Várnai A, Vidal‐Melgosa S, Willats WG, Ludwig R, Horn SJ, Eijsink VGH, Westereng B (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc Natl Acad Sci USA 111:6287–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frommhagen M, Sforza S, Westphal AH, Visser J, Hinz SWA, Koetsier MJ, van Berkel WJH, Gruppen H, Kabel MA (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol Biofuels 8:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, Ludwig R, Haltrich D, Eijsink VGH, Horn SJ (2014) A C4‐oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello‐oligosaccharides. J Biol Chem 289:2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lo Leggio L, Simmons TJ, Poulsen JC, Frandsen KE, Hemsworth GR, Stringer MA, von Freiesleben P, Tovborg M, Johansen KS, De Maria L, Harris PV, Soong CL, Dupree P, Tryfona T, Lenfant N, Henrissat B, Davies GJ, Walton PH (2015) Structure and boosting activity of a starch‐degrading lytic polysaccharide monooxygenase. Nat Commun 6:5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA (2014) A family of starch‐active polysaccharide monooxygenases. Proc Natl Acad Sci USA 111:13822–13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aachmann FL, Sørlie M, Skjåk‐Bræk G, Eijsink VGH, Vaaje‐Kolstad G (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci USA 109:18779–18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hemsworth GR, Taylor EJ, Kim RQ, Gregory RC, Lewis SJ, Turkenburg JP, Parkin A, Davies GJ, Walton PH (2013) The copper active site of CBM33 polysaccharide oxygenases. J Am Chem Soc 135:6069–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper‐oxyl, oxygen‐rebound mechanism. Proc Natl Acad Sci USA 111:149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walton PH, Davies GJ (2016) On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr Opin Chem Biol 31:195–207. [DOI] [PubMed] [Google Scholar]
- 16. Beeson WT, Phillips CM, Cate JH, Marletta MA (2012) Oxidative cleavage of cellulose by fungal copper‐dependent polysaccharide monooxygenases. J Am Chem Soc 134:890–892. [DOI] [PubMed] [Google Scholar]
- 17. Dimarogona M, Topakas E, Christakopoulos P (2013) Recalcitrant polysaccharide degradation by novel oxidative biocatalysts. Appl Microbiol Biotechnol 97:8455–8465. [DOI] [PubMed] [Google Scholar]
- 18. Dimarogona M, Topakas E, Olsson L, Christakopoulos P (2012) Lignin boosts the cellulase performance of a GH‐61 enzyme from Sporotrichum thermophile . Bioresour Technol 110:480–487. [DOI] [PubMed] [Google Scholar]
- 19. Westereng B, Cannella D, Agger JW, Jørgensen H, Andersen ML, Eijsink VGH, Felby C (2015) Enzymatic cellulose oxidation is linked to lignin by long‐range electron transfer. Sci Rep 5:18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol 77:7007–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AK, Harreither W, Kittl R, Ludwig R (2012) Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl Environ Microbiol 78:6161–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kracher D, Scheiblbrandner S, Felice AK, Breslmayr E, Preims M, Ludwicka K, Haltrich D, Eijsink VGH, Ludwig R (2016) Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352:1098–1101. [DOI] [PubMed] [Google Scholar]
- 23. Yakovlev I, Vaaje‐Kolstad G, Hietala AM, Stefanczyk E, Solheim H, Fossdal CG (2012) Substrate‐specific transcription of the enigmatic GH61 family of the pathogenic white‐rot fungus Heterobasidion irregulare during growth on lignocellulose. Appl Microbiol Biotechnol 95:979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wymelenberg AV, Gaskell J, Mozuch M, Sabat G, Ralph J, Skyba O, Mansfield SD, Blanchette RA, Martinez D, Grigoriev I, Kersten PJ, Cullen D (2010) Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium . Appl Environ Microbiol 76:3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phillips CM, Iavarone AT, Marletta MA (2011) Quantitative proteomic approach for cellulose degradation by Neurospora crassa . J Proteome Res 10:4177–4185. [DOI] [PubMed] [Google Scholar]
- 26. Tan TC, Kracher D, Gandini R, Sygmund C, Kittl R, Haltrich D, Hallberg BM, Ludwig R, Divne C (2015) Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat Commun 6:7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cameron MD, Aust SD (2001) Cellobiose dehydrogenase‐an extracellular fungal flavocytochrome. Enzyme Microb Technol 28:129–138. [DOI] [PubMed] [Google Scholar]
- 28. Igarashi K, Momohara I, Nishino T, Samejima M (2002) Kinetics of inter‐domain electron transfer in flavocytochrome cellobiose dehydrogenase from the white‐rot fungus Phanerochaete chrysosporium . Biochem J 365:521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zamocky M, Ludwig R, Peterbauer C, Hallberg BM, Divne C, Nicholls P, Haltrich D (2006) Cellobiose dehydrogenase‐a flavocytochrome from wood‐degrading, phytopathogenic and saprotropic fungi. Curr Prot Pept Sci 7:255–280. [DOI] [PubMed] [Google Scholar]
- 30. Samejima M, Eriksson KEL (1992) A comparison of the catalytic properties of cellobiose ‐ quinone oxidoreductase and cellobiose oxidase from Phanerochaete chrysosporium . Eur J Biochem 207:103–107. [DOI] [PubMed] [Google Scholar]
- 31. Schou C, Christensen MH, Schülein M (1998) Characterization of a cellobiose dehydrogenase from Humicola insolens . Biochem J 330:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baminger U, Subramaniam SS, Renganathan V, Haltrich D (2001) Purification and characterization of cellobiose dehydrogenase from the plant pathogen Sclerotium (Athelia) rolfsii . Appl Environ Microbiol 67:1766–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mason MG, Nicholls P, Divne C, Hallberg BM, Henriksson G, Wilson MT (2003) The heme domain of cellobiose oxidoreductase: a one‐electron reducing system. Biochim Biophys Acta 1604:47–54. [DOI] [PubMed] [Google Scholar]
- 34. Thallinger B, Argirova M, Lesseva M, Ludwig R, Sygmund C, Schlick A, Nyanhongo GS, Guebitz GM (2014) Preventing microbial colonisation of catheters: Antimicrobial and antibiofilm activities of cellobiose dehydrogenase. Int J Antimicrob Agents 44:402–408. [DOI] [PubMed] [Google Scholar]
- 35. McDonnell G, Russell AD (1999) Antiseptics and disinfectants: activity, action, and resistance. Clin Microbiol Rev 12:147–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wood PM (1988) The potential diagram for oxygen at pH 7. Biochem J 253:287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kirk TK, Ibach R, Mozuch MD, Conner AH, Highley TL (1991) Characteristics of cotton cellulose depolymerized by a brown‐rot fungus, by acid, or by chemical oxidants. Holzforschung 45:239–244. [Google Scholar]
- 38. Hyde SM, Wood PM (1997) A mechanism for production of hydroxyl radicals by the brown‐rot fungus Coniophora puteana: Fe(III) reduction by cellobiose dehydrogenase and Fe(II) oxidation at a distance from the hyphae. Microbiology 143:259–266. [DOI] [PubMed] [Google Scholar]
- 39. Kremer SM, Wood PM (1992) Production of fenton reagent by cellobiose oxidase from cellulolytic cultures of Phanerochaete chrysosporium . Eur J Biochem 208:807–814. [DOI] [PubMed] [Google Scholar]
- 40. Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels 5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forsberg Z, Mackenzie AK, Sørlie M, Røhr ÅK, Helland R, Arvai AS, Vaaje‐Kolstad G, Eijsink VGH (2014) Structural and functional characterization of a conserved pair of bacterial cellulose‐oxidizing lytic polysaccharide monooxygenases. Proc Natl Acad Sci USA 111:8446–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rogers MS, Jones GD, Antonini G, Wilson MT, Brunori M (1994) Electron‐transfer from Phanerochaete chrysosporium cellobiose oxidase to equine cytochrome c and Pseudomonas aeruginosa cytochrome c‐551. Biochem J 298:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA (2015) Cellulose degradation by polysaccharide monooxygenases. Ann Rev Biochem 84:923–946. [DOI] [PubMed] [Google Scholar]
- 45. Gardner JG, Crouch L, Labourel A, Forsberg Z, Bukhman YV, Vaaje‐Kolstad G, Gilbert HJ, Keating DH (2014) Systems biology defines the biological significance of redox‐active proteins during cellulose degradation in an aerobic bacterium. Mol Microbiol 94:1121–1133. [DOI] [PubMed] [Google Scholar]
- 46. Scott BR, Huang HZ, Frickman J, Halvorsen R, Johansen KS (2015) Catalase improves saccharification of lignocellulose by reducing lytic polysaccharide monooxygenase‐associated enzyme inactivation. Biotechnol Lett 38:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vaaje‐Kolstad G, Houston DR, Riemen AH, Eijsink VGH, van Aalten DM (2005) Crystal structure and binding properties of the Serratia marcescens chitin‐binding protein CBP21. J Biol Chem 280:11313–11319. [DOI] [PubMed] [Google Scholar]
- 48. Cannella D, Möllers KB, Frigaard NU, Jensen PE, Bjerrum MJ, Johansen KS, Felby C (2016) Light‐driven oxidation of polysaccharides by photosynthetic pigments and a metalloenzyme. Nat Commun 7:11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hemsworth GR, Henrissat B, Davies GJ, Walton PH (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat Chem Biol 10:122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zamocky M, Schümann C, Sygmund C, O'Callaghan J, Dobson ADW, Ludwig R, Haltrich D, Peterbauer CK (2008) Cloning, sequence analysis and heterologous expression in Pichia pastoris of a gene encoding a thermostable cellobiose dehydrogenase from Myriococcum thermophilum . Protein Expr Purif 59:258–265. [DOI] [PubMed] [Google Scholar]
- 51. Manoil C, Beckwith J (1986) A genetic approach to analyzing membrane‐protein topology. Science 233:1403–1408. [DOI] [PubMed] [Google Scholar]
- 52. Loose JSM, Forsberg Z, Fraaije MW, Eijsink VGH, Vaaje‐Kolstad G (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett 588:3435–3440. [DOI] [PubMed] [Google Scholar]
- 53. Heuts DPHM, Winter RT, Damsma GE, Janssen DB, Fraaije MW (2008) The role of double covalent flavin binding in chito‐oligosaccharide oxidase from Fusarium graminearum . Biochem J 413:175–183. [DOI] [PubMed] [Google Scholar]
- 54. Westereng B, Agger JW, Horn SJ, Vaaje‐Kolstad G, Aachmann FL, Stenstrøm YH, Eijsink VGH (2013) Efficient separation of oxidized cello‐oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J Chromatogr A 1271:144–152. [DOI] [PubMed] [Google Scholar]