

Abstract
Synthetic biology (or chemical biology) is a growing field to which the chemical synthesis of proteins, particularly enzymes, makes a fundamental contribution. However, the chemical synthesis of catalytically active proteins (enzymes) remains poorly documented because it is difficult to obtain enough material for biochemical experiments. We chose calstabin, a 107‐amino‐acid proline isomerase, as a model. We synthesized the enzyme using the native chemical ligation approach and obtained several tens of milligrams. The polypeptide was refolded properly, and we characterized its biophysical properties, measured its catalytic activity, and then crystallized it in order to obtain its tridimensional structure after X‐ray diffraction. The refolded enzyme was compared to the recombinant, wild‐type enzyme. In addition, as a first step of validating the whole process, we incorporated exotic amino acids into the N‐terminus. Surprisingly, none of the changes altered the catalytic activities of the corresponding mutants. Using this body of techniques, avenues are now open to further obtain enzymes modified with exotic amino acids in a way that is only barely accessible by molecular biology, obtaining detailed information on the structure‐function relationship of enzymes reachable by complete chemical synthesis.
Keywords: synthetic biology, calstabin, total synthesis, catalytic activity, crystallography
Short abstract
PDB Code(s): 5HKG
Introduction
Since the development of peptide solid‐phase synthesis,1 peptide chemistry has gained more and more recognition and successes.2, 3 Peptide chemistry was reborn in many areas of modern pharmacology and helped develop penetrating peptides,4 as well as new tools of natural origin and the possibility of synthesizing new pharmacological agents. Peptides are the source of immense diversity when composed of the 20 natural amino acids, and even greater when the many available exotic amino acids are concerned.5 The idea of possible complementation between the targets (essentially proteins) with such small peptides made of the same basic building blocks led many laboratories to build peptide libraries (e.g.,6) as a potent source of diversity in the search of inhibitors or other types of interacting compounds, at least as starting points for medicinal chemistry programs.
Peptides, such as insulin, Neuropeptide Y, erythropoietin, various growth factors and interleukins, could nowadays be obtained by chemical synthesis, but reaching proteins (particularly catalytically active enzymes) by complete chemical synthesis remains rare. Though the demand for artificial (or synthetic) proteins is great, only thirteen such enzymes has been obtained since the first completely synthetic enzymes of Hirschmann et al.7 and Gutte and Merrifield8 (see Table 1 for details) Until the rise of chemical ligation, synthesis of long peptides potentially with catalytic activity, was a very difficult exercise likely limited by overall yields.19 Even if each step has a yield of 99%, after 100 steps only 1% of the starting material would remain. Nevertheless, several dozens of publications have reported the successful synthesis of long peptides (∼100 amino acids). These peptides were sometimes quality‐controlled only on the basis of their biological activities rather than on their biophysical and physical characteristics. It seems that one of the reasons for this fact is sometimes the amount of final material that is obtained that is too small to permit further characterization (see also the notion of scalability by Chalker20).
Table 1.
Reported Synthetic Enzymes
| Enzyme | (Reference year) | Size of the sequence | Catalytic activity type | Synthesis strategya |
|---|---|---|---|---|
| Ribonuclease S | 7 | 104 aab | RNase S catalytic activity | SPPSa |
| Ribonuclease A | 8 | 124 aa | RNase A catalytic activity | SPPS |
| HIV1 protease | 9 | 99 aac | Protease catalytic activity | SPPS |
| 4‐Oxalocrotonate Tautomerase | 10 | 62 aa | Tautomerase activity | SPPS |
| Phospholipase A2 | 11 | 124 aa | Phospholipase activity | NCLa |
| Cytochrome b562 | 12 | 106 aad | Electron transfer capacity | NCL |
| H‐Ras | 13 | 166 aa | GTPasee | |
| Lysozyme | 14 | 130 aa | Lysozyme‐mediated degradation | NCL |
| HTLV protease | 15 | 126 aab | protease catalytic activity | NCL |
| DapA | 16 | 312 aaf | 4‐hydroxy‐tetrahydrodipicolinate synthase | NCL |
| (ΔN59) Sortase A | 17 | 142 aag | Ligase catalytic activity | |
| Barnase | 18 | 113 aa | Rnase activity | NCL |
| Calstabin | present work | 107 aa | Proline isomerase | NCL |
SPPS stands for Solid Phase Peptide Synthesis and NCL stands for Native Chemical Ligation. In all cases the SPPS was used to synthesize the fragments in the NCL approach, while SPPS was used exclusively before the rising of NCL.
The S47C mutant was produced.
All cysteines were replaced by α‐amino‐n‐butyric acid.
In the form of a SeMet7 enzyme.
The measured activity was GTP/GDP exchange, not GTPase activity.
Including a 10‐aa N‐terminal His‐tag linked to a 10‐aa thrombin cleavage site, an alanine to cysteine mutation (A77).
All methionines were replaced by norleucines.
In our company, we entered the peptide chemistry domain a long time ago through various approaches (see e.g.,21, 22, 23), and only recently did we decide to explore the possibility of producing, refolding, characterizing, and even crystallizing synthetic proteins. The goal behind this program was and still is to master those technologies in our laboratory, in order to explore several hard‐to‐produce enzymes, to modify Biologics to render them more stable, as well as to launch some structure/mutations relationship studies on target enzymes implicated in key physio‐pathological pathways.20 Among these enzymes that we are currently studying, we chose to present here the results on calstabin, a peptidyl‐prolyl isomerase also known as FK506 binding protein (FKBP) that belongs to the general family of protein chaperones known as immunophilins.24 The many roles of calstabin in physiological pathways make it an interesting target for pharmacology; it also has possible implications in Tau oligomerization,25, 26 heartbeat,27 and skeletal muscle fatigue.28 From the pharmacological point of view, many laboratories have searched and found small molecule ligands with regulating properties for this particular enzyme (for review see Liu et al.29). In other words, calstabin has been well described and characterized by its pharmacology or by binding.
In the present study, the complete synthesis of calstabin on the tens of milligrams scale, its purification, characterization, refolding, and peptidyl‐proline isomerase activities, and the crystallization of wild‐type calstabin and mutants are described. The results show the capacity to produce enzyme of a fair size (between 100 and 200 amino acids), opening the way for larger molecules.
Results
Calstabin isoforms
Calstabin exists in several isoforms that have been cloned in the past.30 However, little is known about these isoforms. The shorter cloned and synthetic isoforms lack any proline isomerase activity (F. Cogé, M. Bacchi, and J.A. Boutin, in preparation). We took advantage of the synthesis strategy to have access to the main isoforms. Therefore, despite a general description of our strategy, the shorter versions of this enzyme were not used in the present study. Nevertheless, their characteristics in terms of purity and mass are presented in Supporting Information Figures S1–S3. We obtained four isoforms that we discovered or were reported previously30, 31 by standard cloning, expression, and purification. The full process of refolding and catalytic activity measurement was performed on the four isoforms. Although the refolding was successful, no catalytic activity was recorded for the three shorter forms. Therefore, we concentrated our comparative efforts on the native form, which spans 1–107 in the protein sequence. It was important for us to have the native recombinant enzyme in the same experiments compared to the synthetic forms, comprising the same sequence, in order to check any step of activity and/or characterization (including refolding) between two proteins from different origins.
Polypeptide synthesis
The synthetic polypeptides were obtained according to classical Merrifield solid phase synthesis1 using two‐fragment ligation.1 9, 32 Thanks to the SEA resin and extensive descriptions in the literature, together with past expertise in our laboratories, the various polypeptides were obtained with fair yield and purity. Figures 1, 2, 3 recapitulate the analytical data for the two peptides and the full length protein in terms of yield for the final purification and mass spectrometric analyses. Notably, the yield pertains to after the two polypeptides were obtained and ligated together, not the starting points, when yield is often 99% per amino acid step. Producing several tens of milligrams was mandatory to our present program, as the amount of material necessary for further characterization is often large. For future enzymatic projects in this area such as systematic ab initio screening or systematic co‐crystallization in the frame of a discovery program, at least 10 times more material will be needed (see Chalker20 for further discussion on scalability). At this stage it is important to realize that the products obtained by chemistry were at least as pure as the material from recombinant origin used in the same contexts, and in any case more than 95% pure, although microheterogeneity can be seen especially from the protein 1 (native calstabin) spectrum. The mass spectrometry analyses clearly showed that a single peptide species was present in the preparation using the standard, reverse‐phase step of purification followed by lyophilization. Figures 1, 2, 3 (peptide 1, protein 3, and protein 1, respectively, see Table 2) show that each polypeptide was obtained as a single peak with a molecular weight corresponding to the sequence (Table 3). The structure of the recombinant calstabin clearly shows that this enzyme possesses 2 cysteines that are not involved in an S–S bridge, this leads to two free cysteines (and their corresponding SH) that can impair a clear analysis of the purity of the final product.
Figure 1.
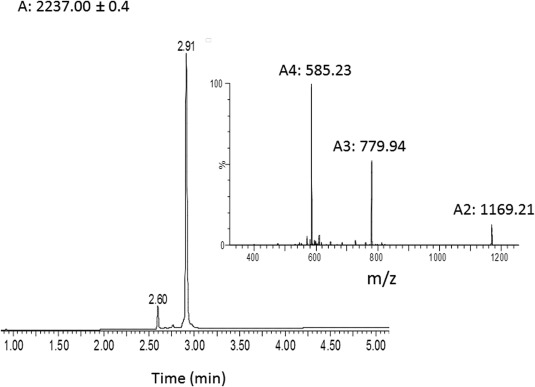
Characterization of purified peptide 1. Analytical HPLC profile (λ = 214 nm) and ESI mass spectra of the chemically synthesized calstabin (1–21) (average mass: 2334.7 Da, observed: 2337.00 ± 0.4 Da). These data were obtained with the purified synthetic peptide segment (135 mg, 23% yield for 0.25 mmol).
Figure 2.
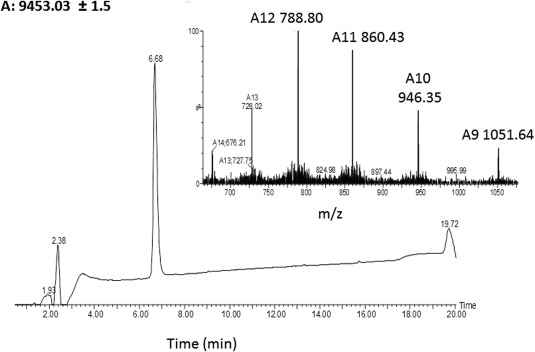
Characterization of purified protein 3. Analytical HPLC profile (λ = 214 nm) and electrospray ionization mass spectrometry of the chemically synthesized calstabin (22–107) (average mass: 9451.8 Da, observed: 9453.03 ± 1.50 Da). These data were obtained with the purified synthetic peptide segment (120 mg, 2.5% yield for 0.5 mmol).
Figure 3.
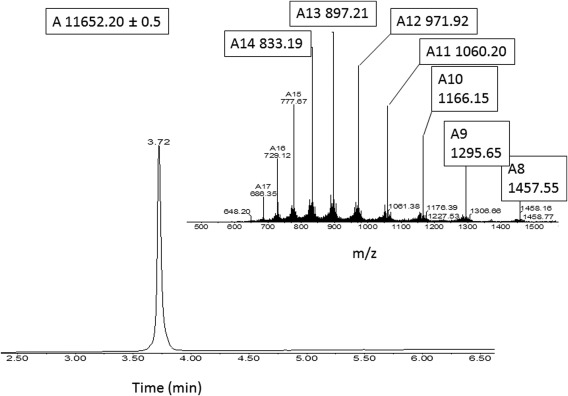
Characterization of purified protein 1. Analytical HPLC profile (λ = 214 nm) and electrospray ionization mass spectrometry of the chemically synthesized calstabin (1–107) (average mass: 11651.4 Da, observed: 11652.20 ± 0.05 Da). These data were obtained with the purified synthetic protein.
Table 2.
Summary of the Various Sequences Used in the Study
| Name | Sequence | Illustration | |
|---|---|---|---|
| Protein 1 NM_004116 | Natural sequence | (M)GVEIETISPGDGRTFPKKGQTCVVHYTGMLQNGKKFDSSRDRNKPFKFRIGKQEVIKGFEEGAAQMSLGQRAKLTCTPDVAYGATGHPGVIPPNATLIFDVELLNLE | Figure 3 |
| Protein 2 (Rat, 1994) | Natural sequence | (M)GVEIETISPGDGRTFPKKGQTCVVHYTGMLQNGKKFDSSRDRNKPFKFRIGKQEVIKGFEEGAAQLGPLSPLPICPHPC | |
| Peptide 1 | Calstabin‐SEAoff (1‐21) | GVEIETISPGDGRTFPKKGQT‐SEAoff | Figure 1 |
| Peptide 2 | Calstabin (N‐ε‐2,4‐dinitrophenyl‐l‐lysine)‐SEAoff (1‐21) | GVEIETISPGDGRTFPK‐K(Dnp)‐GQT‐SEAoff | Supporting Information Figure S4 |
| Protein 3 | Cys‐calstabin (22‐107) | CVVHYTGMLQNGKKFDSSRDRNKPFKFIGKQEVIKGFEEGAAQMSLGQRAKLTCTPDVAYGATGHPGVIPPNATLIFDVELLNLE | Figure 2 |
| Peptide 3 | Calstabin (N‐ω‐methyl‐l‐arginine)‐SEAoff (1‐21) | GVEIETISPGDG‐R(Me)‐TFPKKGQT‐SEAoff | Supporting Information Figure S5 |
| Protein 4 | Calstabin (N‐ε‐2,4‐dinitrophenyl‐l‐lysine) | GVEIETISPGDGRTFPK‐K(Dnp)‐GQTCVVHYTGMLQNGKKFDSSRDRNKPFKFRI‐GKQEVIKGFEEGAAQMSLGQRAKLTCTPDVAYGATGHPGVIPPNATLIFDVELLNLE | Supporting Information Figure S7 |
| Protein 5 | Calstabin (N‐ω‐methyl‐l‐arginine) | GVEIETISPGDG‐R(Me)‐TFPKKGQTCVVHYTGMLQNGKKFDSSRDRNKPFKFR‐IGKQEVIKGFEEGAAQMSLGQRAKLTCTPDVAYGATGHPGVIPPNATLIFDVELLNLE | Supporting Information Figure S8 |
| Protein 6 | TAMRA‐labeled calstabin | TAMRA‐GVEIETISPGDGRTFPKKGQTCVVHYTGMLQNGKKFDSSRDRNKPFKFR‐IGQEVIKGFEEGAAQMSLGQRAKLTCTPDVAYGATGHPGVIPPNATLIFDVELLNLE‐OH | Supporting Information Figure S9 |
| Peptide 4 | TAMRA‐labeled‐calstabin‐ SEAoff (1‐21) | TAMRA‐GVEIETISPGDGRTFPKKGQT‐SEAoff | Supporting Information Figure S6 |
| Peptide 5 | Natural sequence (1‐79) | GVEIETISPGDGRTFPKKGQTCVVHYTGMLQNGKKFDSSRDRNKPFKFRIGKQEVIKGFEEGAAQLGPLSPLPICPHPC | Supporting Information Figure S1 |
| Peptide 6 | Natural sequence (1‐56) | GVEIETISPGDGRTFPKKGQTCVVHYTASLVGMIKEAWKANSFPLLPARAIHGASF | Supporting Information Figure S2 |
| Peptide 7 | Natural sequence (1‐31) | GVEIETISPGDGRTFPKKGQTCVVHYTGEVE | Supporting Information Figure S3 |
Table 3.
Summary of the Yields Obtained for the Calstabins Synthesized in the Present Study
| Protein | Peptide A | Peptide B | Final product (mg) | Final yield (%) | Figure |
|---|---|---|---|---|---|
| Native calstabin (Protein 1) | Calstabin‐SEAoff (1–21) 60 mg | Cys‐Calstabin (22–107) 40 mg | 29.3 | 40 | 3 |
| Calstabin (N‐ε‐2,4‐dinitrophenyl‐l‐lysine) (Protein 4) | Calstabin (K(Dnp))‐SEAoff (1–21) 16.7 mg | Cys‐calstabin (22–107) 12 mg | 6.5 | 31 | S7 |
| Calstabin (N‐ω‐methyl‐l‐arginine) (Protein 5) | Calstabin (R(Me)) SEAoff (1–21) 16.7 mg | Cys‐calstabin (22–107) 11.3 mg | 8 | 39 | S8 |
| TAMRA‐labeled calstabin (Protein 6) | TAMRA calstabin SEAoff (1–21) 3.4 mg | Cys‐calstabin (22–107) 14 mg | 6.9 | 39 | S9 |
Folding of synthetic calstabin
From the synthetic process, we anticipated that no activity could be recorded from the crude material, even after lyophilization. Therefore, the synthetic calstabin lyophilizate was refolded using the Pierce protein refolding kit. The homogeneity of the refolded enzymes was studied by analytical size exclusion chromatography (SEC). For all tested conditions, the native (synthetic) protein (protein 2, Table 2) was obtained as two species: P1 and P2 (see Fig. 4). Because of the apparent concentrations of both species, there was no difference in the sequence of the two species present in the preparation, as the analysis of the pure material could not have missed nearly 50% of species other than the one recorded in the mass spectrogram (Fig. 3). Furthermore, the recombinant protein in the same experimental conditions led to a single peak coeluting with P2 (data not shown). Therefore, P2 is the active, fully refolded protein and was taken as the reference for refolding optimization. We screened different conditions for denaturation and refolding. For the denaturation process, different denaturing agents were tested, including 6M guanidine, SDS, and TFA 0.1%, each in the presence of 5 mM DTT or 5 mM TCEP, and DMSO. We also tested different concentrations of the protein during denaturation (0.1, 0.5, and 1 mg mL−1). For the refolding process, two methods were tested: successive dialysis against a non‐denaturing buffer (15 different buffers were screened according to the Pierce refolding kit, vide supra) or rapid dilution (factor 100) of the protein solubilized at 10 mg mL−1 in DMSO. We made a couple of attempts to provide the chemically synthesized protein with 1% recombinant protein to help the refolding. As the ratio of P2/P1 remained the same, we considered this attempt a failure. Out of all these conditions, only the concentration of the protein during denaturation affected the percentage of P1 versus P2. This optimization led to an improvement of the overall repartition between inactive P1 and active P2 from 30% to 50%.
Figure 4.
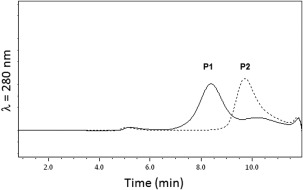
Analytical size exclusion chromatography of the folded synthetic calstabin. The partially folded, chemically synthesized calstabin (1–107) was submitted to gel filtration on a Superdex 75 5/150 GL equilibrated with 50 mM Tris/HCl, 300 mM NaCl (pH 7.5) buffer filtered (0.1 µM) at a flow rate of 0.2 mL min−1. The protein elution was monitored at 280 nm.
SEC‐MALS analysis of the sample did not agree with a mixture of monomeric form (P2) and dimeric form (P1). However, the different elution times suggested a higher hydrodynamic radius for P1 compared to P2. To characterize the two species, refolded material was subjected to preparative SEC, which led to a good separation. After isolating the two peaks, we analyzed the proteins separately with MALS/RI‐coupled analytical chromatography (Fig. 5). P2 was homogeneous and pure on SDS‐PAGE, whereas P1 was mainly homogeneous with traces of a smaller species. SDS‐PAGE also showed minute impurities at lower molecular weights that could account for 2–3% of the total purity of the preparation. Furthermore, it is also possible that micro‐heterogeneity (as can be observed from the mass spectrum of the Fig. 3) was responsible of the lack of folding/activity of P1. The experiment described on Figure 6 was therefore conducted, in order to better appreciate the composition of P2. Two ion populations can be observed: population A with a measured mass of 11,649 ± 2 Da (monomer) and a minor population B with a measured mass of 23,298 ± 2 Da (dimer). Their charge state distributions are compatible with the retention of a compact native‐like conformation in the gaz‐phase.
Figure 5.
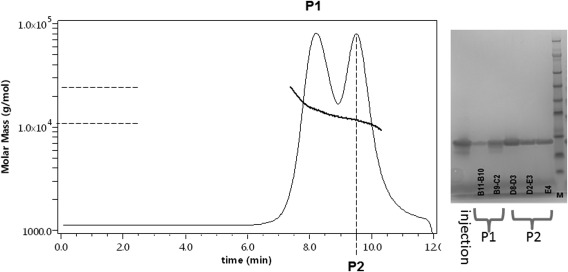
Analytical size‐exclusion chromatogram and SDS‐PAGE analysis. After separation by gel filtration, samples of P1 and of P2 were analyzed separately using a Superdex 75 5/150 GL equilibrated with 50 mM Tris/HCl, 300 mM NaCl (pH 7.5) buffer coupled to a multi‐angle light scattering detector and an RI detector. The differential refractive index was recorded as a function of time. P1 is represented by a solid line and P2 is represented by a dashed line. Samples of the eluates were analyzed by SDS‐PAGE. 1: sample before size exclusion chromatography, P1: first chromatographic peak, P2: second chromatographic peak. The right lane was filled with protein markers.
Figure 6.
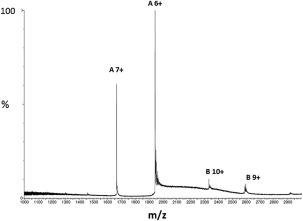
Nano‐ESI of synthetic calstabin, fraction P2 in native conditions. Nano‐ESI spectra of synthetic calstabin (P2) in native conditions (200 mM ammonium acetate pH 7.4).
Secondary structure of refolded materials P1 and P2
To study the secondary structures of these two species, we recorded their far‐UV CD spectra and that of one of the recombinant proteins under the same conditions (Fig. 7). Recombinant and isolated synthetic calstabin (P2) had very similar far‐UV CD spectra with a well‐defined minimum at 213 nm and maximum at 201 nm. This spectral identity supports conservation of the overall secondary structure after refolding the chemically synthesized calstabin in solution. This type of conclusion was reached on the basis of similar experiments on the phospholipase A2, also chemically synthesized and compared, as in here, to its recombinant counterpart.11 Both spectra were in agreement with a secondary structure composed mainly of beta‐sheets, which is in agreement with the published structure of calstabin (PDB reference: 1C9H). Interestingly, the isolated synthetic calstabin (P1) had a far‐UV CD spectrum of a poorly to non‐folded protein as previously suggested by the SEC‐MALS analysis. This finding strongly suggests that part of the synthetic protein was not refolded and, therefore, behaved differently than the recombinant or synthetic P2 species. In addition, uncharacterized synthetic proteins, particularly those adjudged on the basis of their biological activity, may be composed of two or more species, all with the correct molecular weight and the same retention time under analytical chromatographic conditions, but one folded, the other not.
Figure 7.
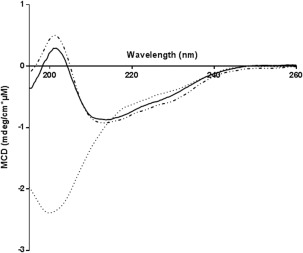
CD spectra of the chemically synthesized calstabins after separation by gel filtration compared to recombinant calstabin. P1‐synthetic calstabin (dotted line), P2‐synthetic calstabin (dashed dotted line), and recombinant calstabin (solid line). Spectra were recorded at 20°C in 20 mM potassium phosphate buffer (pH 7.7).
Thermal stability of refolded synthetic calstabin (P2)
We compared the stability of the refolded synthetic protein (P2) to the stability of the recombinant protein by measuring their melting temperatures (T m) via CD (Fig. 8). The thermal denaturation curve fit a single sigmoid, which is consistent33 with a cooperative two‐state thermal denaturation mechanism as expected from the literature.34 The fit of the thermal denaturation curve gave us access to the T m of the proteins and showed that the synthetic and recombinant calstabin have the same thermal stabilities (52.2°C ± 0.4°C and 52.4°C ± 0.07°C, respectively, at 50 µM in 20 mM potassium phosphate buffer, pH 7.7). Therefore, the proteins are not only similar in sequence, as expected, but also have probably identical secondary structures.
Figure 8.
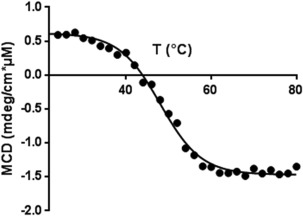
Thermal denaturation curve of synthetic calstabin. Molar CD of P2 at 202 nm in 20 mM potassium phosphate buffer (pH 7.7) during thermal denaturation at 1°C min−1. The solid line is the theoretical curve fitted to the experimental data points.
Structural study
To further prove the lack of differences between the synthetic and natural enzymes, we crystallized and determined the X‐ray crystal structure of the synthetic calstabin in complex with a binder: rapamycin, with a resolution of about 1.5 Å (Table 4). The overall structures of the synthetic and recombinant calstabin are very similar. Superposition of synthetic and recombinant calstabin [Fig. 9(B)] highlighted that they are isomorphous, with an average root mean square deviation (rmsd) value calculated onto all 107 Cα atoms of 0.263 Å (see statistics in Table IV). Small conformation changes are localized in solvent‐exposed regions belonging to loops and not involved in ligand binding. Five β‐strands define one side of the molecule whereas a small α−helix (58–65) spans three of these strands: (3–8), (72–77), (99–109) on the other side. Residues belonging to the other three β‐strands (22–31), (35–39), (46–50), the helix and two long loops (51–57), (80–98) form a hydrophobic binding pocket that harbors rapamycin. Hydrogen bonds that rapamycin establishes with residues D37, Q53, I56, Y82 are conserved both in the recombinant and synthetic calstabin structures [Fig. 8(C)]. The structure of the synthetic enzyme (corresponding to the native sequence in complex with rapamycin) was deposited into the protein data bank (PDB) under access code 5HKG. To the best of our knowledge, there is less than a handful of synthetic crystallized enzymes deposited in the PDB, for example, the human lysozyme code 1IWT,14 ribonuclease A, code: 2NUI 35 and many cocrystals of the HIV protease, see for instance code 3HLO.36
Table 4.
Data Collection and Refinement Statistics of Chemically Obtained Calstabin 2 Crystal
| Data collection | |
|---|---|
| Space group | P212121 |
| a, b, c (Å) | 45.2 48.61 53.36 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 0.97857 |
| Molecules per ASU | 1 |
| Resolution (Å) | 50–1.50 (1.59–1.50) |
| No. reflections | |
| Total | 99,026 (16,092) |
| Unique | 19,433 (1891) |
| Completeness (%) | 99.93 (99.27) |
| Redundancy | 5.14 (5.25) |
| Mean I/sigma (I) | 23.06 (11.92) |
| R sym (%) | 4.5 (14.1) |
| Refinement | |
| Resolution (Å) | 10–1.5 (1.55–1.50) |
| R free/R factor (%) | 19.46 (23.91)/17.31 (17.90) |
| Root mean square deviation: | |
| Bond distances (Å) | 0.014 |
| Bond angles (°) | 1.93 |
| Number of atoms | |
| Macromolecule | 846 |
| Drug | 84 |
| Water | 147 |
| Ramachandran plot | |
| Most favored regions (%) | 97 |
| Additionally allowed (%) | 3 |
| Outliers (%) | 0 |
Values in parentheses are for the highest resolution shell.
Figure 9.
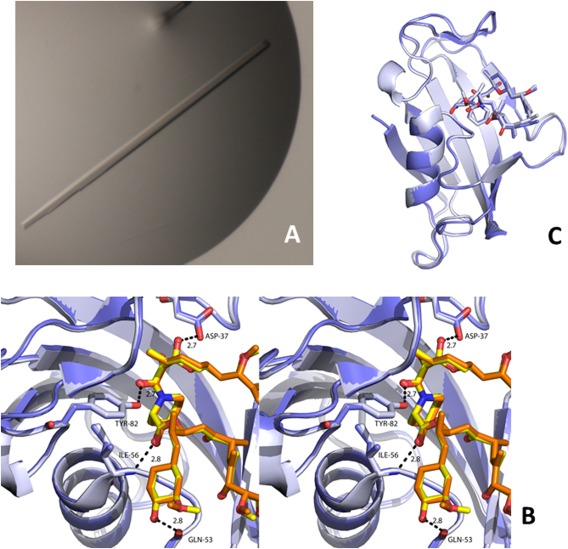
Visualization of the synthetic calstabin crystal. A. Picture of a crystal of the chemically synthesized, wild‐type sequence, calstabin in complex with rapamycin. Microphotograph of a crystal of the chemically synthesized calstabin. B. Stereo diagram of the binding pocket of the calstabin‐rapamycin complex. The recombinant and synthetic complexes are shown in light blue (calstabin)/orange (rapamycin) and slate (calstabin)/yellow (rapamycin), respectively. O atoms and N atoms are shown in red and blue, respectively, in both the structures. Hydrogen bonds are shown as dashed black lines. C. Superposition of the complexes of rapamicyn with the recombinant (grey) and synthetic Calstabin (violet). (Protein and ligand are represented in cartoon and stick mode, respectively). Protein coordinates were deposited under the PDB ID: 5HKG).Structural identity between the native recombinant calstabin and chemically synthesized calstabine. Reconstruction of X‐ray diffraction maps led to identical structures for the synthetic and recombinant calstabin proteins. The coordinates were deposited in the Protein Data Bank under the entry 5HKG.
Catalytic activity of synthetic calstabin
The peptidyl‐prolyl cis‐trans isomerase (PPIase) activities of the synthetic and recombinant wild‐type calstabin were measured by the protease‐coupled assay described by Harrison and Stein37 (Fig. 10). This convenient assay takes advantage of the fact that chymotrypsin hydrolyzes peptide Xxx‐Xaa‐Pro‐Phe‐pNA when the Xaa‐Pro bond is in its cis conformation, but not when it is in its trans conformation. Therefore, the authors designed an assay in which the chymotrypsin is given a peptide substrate in its trans conformation. In the presence of calstabin, it will be transformed into its cis conformer and become a substrate for chymotrypsin. The para‐nitroaniline is then released and is measurable. The synthetic calstabin had a similar catalytic activity as the recombinant enzyme (Table 5), with undistinguishable affinity and catalytic parameters.
Figure 10.
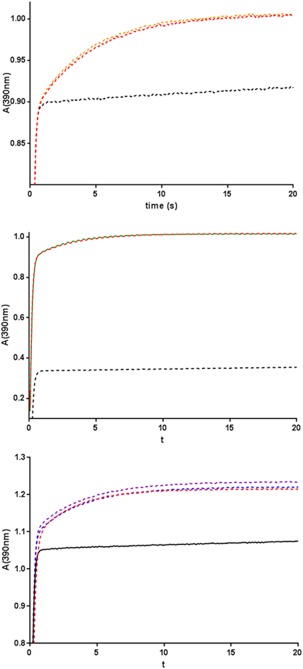
PPIase activities of the synthetic and recombinant calstabins. Absorbance of the p‐Na group released after digestion of the peptide Nsuc‐Ala‐Leu‐Pro‐Phe‐pNa in the presence of a large excess of chymotrypsin without calstabin (black line) or with synthetic calstabin (0.52 µM, orange line), TAMRA‐calstabin (0.7 µM, green line), K18(Nε‐(2,4‐dinitrophenyl)‐l‐lysine) calstabin (0.52 µM, blue line), R13 (N‐ω‐methyl‐l‐arginine) calstabin (0.52 µM, purple line), or recombinant calstabin (control; same concentration as synthetic proteins, red line). 2 The abbreviations used are: MALS, multi‐angle light scattering; pNa: para‐nitroaniline; PPIase: peptidyl‐prolyl cis‐trans isomerase; RI: refractive index; rmsd: root mean square deviation; SEC: size‐exclusion chromatography; SDS‐PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; TAMRA: 5‐carboxytetramethylrhodamine; T m: melting temperature.
Table 5.
Specific Activity (k iso) and k cat/K m Value of Refolded Synthetic Calstabin Versus Recombinant Calstabin
| k iso [s−1] (×10−2)a | k cat/K m [mM−1 s−1]b | |
|---|---|---|
| Recombinant calstabin (protein 1) | 23.3 ± 0.06 | 442 ± 29 |
| Synthetic calstabin (protein 1) | 23.8 ± 0.1 | 438 ± 11 |
| Chymotrypsin only | 1.03 |
Recorded at 0.52 µM for calstabin concentration.
Calculated for enzyme concentrations between 0.4 and 1 µM.
Modification of the native sequence
As a first step toward systematic incorporation of exotic amino acids one at a time in precisely defined and unique locations along the sequence, we chose the easy way of modifying calstabin, in the small N‐terminal fragment (from amino acid 1 to amino acid 21). The mutants were prepared with altered peptides as summarized in Table 2. Peptides 2, 3, and 4 were ligated with protein 3, creating proteins 4, 5, and 6, respectively. These peptides and proteins were analyzed and the results presented in Supporting Information Figures S4–S9. The corresponding proteins were refolded in the same way as the wild‐type calstabin and tested for catalytic activity. We achieved the refolding of a TAMRA‐tagged synthetic calstabin, R13(N‐ω‐methyl‐l‐arginine), and K18(Nε‐(2,4‐dinitrophenyl)‐l‐lysine) (data not shown). All of the synthetic calstabins had similar catalytic activity, whether they were native or slightly modified by exotic amino acids. Interestingly, after isolation of the well‐folded proteins by SEC, they had recovered 100% of the catalytic activity of the recombinant enzyme (Fig. 10, Table 6).
Table 6.
k cat/K m Value of Synthetic Native and Refolded Mutant Calstabin Versus Recombinant Calstabin
| Enzyme | k cat/K m (mM−1 s−1) |
|---|---|
| Recombinant calstabin (protein 1) | 438 ± 17 |
| Synthetic calstabin (protein 1) | 438 ± 17 |
| R13 (N‐ω‐methyl‐l‐arginine) calstabin (protein 5) | 470 ± 41 |
| K18 (Nε‐(2,4‐dinitrophenyl)‐l‐lysine) calstabin (protein 4) | 508 ± 18 |
| TAMRA‐calstabin (protein 6) | 494 ± 37 |
Calculate + d for enzyme concentrations between 0.4 and 1 µM.
Discussion
A recent review by Engelhard38 reminds us that the chemical synthesis of proteins has been around for many years. The author stressed that the progress made in this area has been slow but benchmarked by the solid‐phase synthesis discovery by Merrifield,1 the chemical ligation discovery by Kent's group,1 9 and its further development by Muir et al.39 It is interesting that, despite these available tools, there are still “only” 13 reported enzymes that have been synthesized chemically (see Table 1 for details). The first complete synthesis of an enzyme, probably ribonuclease A,40 was a tour‐de‐force. It is clear that, even at that time (1969), beyond pure novelty, this technology permitted to anticipate in a distant future, the possibility to master mutagenesis via solid‐phase chemical modifications in the protein sequence created by inserting natural amino acids, while directed mutagenesis by molecular biology was still in its infancy. The second paper dealing with enzyme synthesis was from Stephen Kent's group.9 The HIV‐1 protease was chosen as the target of this synthesis because the d‐protease is obtained by total synthesis, the only option for having access to such an unnatural enzyme. This completed the historical observations of Pasteur and Fischer concerning the natural structure of peptide backbones and their (only) influence on the secondary, ternary, and quaternary structures related to the recognition of their substrates, which can be no other than chiral (d‐peptides serving as a substrate for the d‐protease instead of l‐peptides). Of note, this seminal work led to considerable progress in the discovery of HIV protease inhibitors including further cocrystallizations of those inhibitors with the synthetic enzyme, a unique example in the literature (see Kent41 for review). Later, the same group synthesized the 106‐amino‐acid cytochrome b562 and the 130‐amino‐acid lysozyme, and even more recently the 142‐amino‐acid sortase AdN59.12 The closest relative of the present article is certainly the work on ras by Becker et al.13 In that paper, the 166‐amino‐acid synthetic enzyme was refolded and tested for its capacity to exchange GTP and GDP. Completion of the refolding was not demonstrated, as the authors probably thought that 100% of the polypeptide refolded uniformly rather than 50% as in our case. Each time the goal has been the same, demonstrating that the length of the sequence was less of a problem despite low yields, leading to minute amounts of enzyme at the end of the process, until finally one group synthesized the 312‐amino‐acid 4‐hydroxy‐tetrahydrodipicolinate synthase (DapA).16 This work stresses, at least to a certain extent, the lack of practical boundaries whenever protein synthesis is concerned.
Wu and Schultz42 stated that the need for chemical proteins is immense because of the “generation of therapeutic proteins with enhanced pharmacology,” “use of p‐nitrophenylalanine mutants to break immunological self‐tolerance,” “use of photocaged amino acids to photoactivate enzymatic activity,” “use of redox amino acids as mechanistic probes of electron transfer in enzymes,” and “α‐hydroxyl acids for protein purification with ‘traceless’ affinity tags.” All of these reasons to explore this domain further are still valid. Introducing diversity into proteins (and particularly, but not exclusively in enzymes) will permit exploration of the intimate mechanisms of action of catalysis or ameliorate the performance and specificity of enzymes. There are two ways to reach such diversity, which Schultz called “expanding the genetic code”; one is the beautiful manipulation of microorganisms in which one can force specific t‐RNAs to be acylated with unnatural (or exotic) amino acids43, 44 and to encode a protein in which an amino acid has been introduced at a particular location. By considering the large number of publications generated by the Schultz group and others on the basis of these technologies, it seems that an alternative approach would not be necessary. Unfortunately, due to the tremendous amount of molecular biology and cellular biology work needed to get these approaches up and running, alternative routes are welcome. Another possible route to obtain exotic amino acid‐modified proteins would be the use of chemical synthesis of peptides ultimately linked together by chemical ligation.32 Ligation is an excellent solution for obtaining proteins. Another limitation of solid‐phase peptide synthesis is that the polypeptide grows longer, increasing the insolubility and leading to objects that are very difficult to handle. The use of chemical ligation obviously permits the use of shorter fragments and results in higher final yields.45, 46
Though long peptides (not to mention proteins or enzymes) have been synthesized for quite a long time, they are quite often poorly biophysically characterized. Beside its purity, as measured by liquid chromatography coupled to mass spectrometry, the purity of a peptide is too often adjudged by its biological activity, such as binding to its receptor or its activity as a growth factor, regardless of any other considerations, regardless to the possible lack of appropriate folding, as in the present example of at least 50% of the pure preparation. Of course, there are many examples of such peptides characterized in minute details, but many others are not.
In the present study, we chose to synthesize the 107‐amino‐acid calstabin using a two‐fragment ligation process. The final protein, as well as its two initial fragments, was more than 95% pure in LC‐MS analysis. A refolding process was performed after a series of trials and errors, followed by purification onto a gel filtration column. The chromatogram clearly showed two species in the calstabin preparation. We separated out the two species, one being folded (P2) the other not (P1), despite our renewed efforts. Regarding the nonrefolded part of the sample, calstabin seems to not be able to form dimeric or multimeric species. It will be important to improve the yield of refolding in order to use all of the advantages of the chemistry while expanding its application to enzymes, and more generally, proteins. Interestingly, even if P1 could not be refolded well, it does not lead to aggregation. The impurities on SDS‐PAGE could be explained by traces of some deletion, though they are invisible in the mass spectrum of the final product. Oxidation of residues, such as methionine or cysteine, could also explain the inability of P1 to refold properly. Mass spectrometric analysis of P1 has not provided more information because of a broad signal, probably caused by hard ionization of the species or the heterogeneity of the sample. To the contrary, the minute examination of the mass spectrum of P2 leads to fine signal, translating the purity of the sample.
To benefit from the fact that the synthesized enzyme could incorporate exotic amino acids, we decided to produce several mutants with amino acids placed in easy to reach locations (e.g., in the rather short peptide 1, see Table 2). Here, we limited ourselves to the description of the N‐terminal variants obtained from full chemical synthesis. With the ligation approach it was “easy” to synthesize a peptide (protein 3, spanning amino acids 22–107) and to ligate it to the various variants of the N‐terminal region, such as peptide 1 (the natural sequence), peptide 2 (the variant in which an analogue of lysine was introduced at position 13), peptide 3 (in which an arginine analogue was inserted at position 18 instead of an arginine), and peptide 4 (in which the fluorescent probe was introduced at the N‐terminal glycine). The results demonstrate that the chemical synthesis of calstabin and its refolding result in a fully structured enzyme and that the modification of the N‐terminal fragment by the exotic amino acids does not impair the refolding of the enzyme nor change its catalytic activity. Chemical synthesis is a way to introduce a fluorescent group into an enzyme without tryptophan. This protocol could also be used to introduce some non‐natural amino acids, creating a protein that could be grafted onto a surface or covalently reacted with another enzyme or chemical.
Interestingly, alternative splicing of calstabin (as in isoforms 1 and 2) led to two proteins of 107 and 79 amino acids, as well as other shorter versions. These isoforms are present in biological samples but lack any proline isomerization activity, strongly suggesting that calstabin has roles other than its chaperone‐like capacity.
It is probably the low yield that prevented progress in this area of chemical synthesis of enzymes. By producing milligrams of protein, the use of the polypeptides was naturally limited and a systematic approach difficult to obtain, mainly because of the final cost of such a project. We thought that the main interests in the complete synthesis of enzymes resided in the new capacity to better understand a structure‐function relationship (see discussions in Craik et al.,3 Young and Schultz47 and Wright et al.48). In other words, how would the modification of a given amino acid at a single position change the features of an enzyme in terms of characteristics, catalytic activity, and crystal structure? Of course, at that stage, the use of natural amino acids would be useless, as the production of recombinant proteins would lead to the same protein with much less effort. It would then be of major interest to change amino acids to their exotic analogues, going beyond the D‐unnatural amino acids for the synthetic/exotic ones.16, 49 Incidentally, more than a thousand alternative amino acids are commercially available. The recent literature shows the systematic introduction of the chemical ligation approach, and important progress in terms of yields.50
One step further in protein science would be the availability of techniques leading to the production of greater amounts of synthetic proteins. For instance, it is not possible, in this context, to work on the basis of 30 µg obtained after the complete synthesis of one of the F‐ATPase subunits.51 Currently, yield is the main obstacle to a generalized use of synthetic proteins as an alternative to other techniques leading to the incorporation of exotic amino acids in enzymes.44 The use of the chemical approach to modify enzymes with unnatural (or exotic) amino acids will open up new avenues in understanding the intimate mode of action of enzyme catalysis and ways to produce enzymes with alternative, unnatural specificities.
Materials and Methods
Materials
All reagents were purchased from Sigma‐Aldrich (Saint Louis, MO) unless otherwise indicated. Superdex 75 columns were purchased from GE Healthcare (Orsay, France). Peptide Nsuc‐Ala‐Leu‐Pro‐Phe‐pNa was purchased from Bachem (Bubendorf, Switzerland). Absorbance and UV–visible spectra were recorded on a Nanodrop 2000c Thermoscientific. Analytical chromatography was performed on a HPLC Agilent 1260 Affinity, a MALS DAWN 8+ (Wyatt Technology, CA), and a RI Optilab T‐rEX (Wyatt Technology, CA). Preparative chromatography was performed with a FPLC AKTA Explorer (GE Healthcare, Orsay, France). All columns were purchased from GE Healthcare. CD experiments were performed with a Jasco J‐815 CD Spectrophotometer (JASCO UK, Great Dunmow, Essex, England). Stopped Flow experiments were performed with a SFM 300 (Bio‐Logic, Claix, France) and PMS 250 for UV measurement (Bio‐Logic, Claix, France). Dialysis was performed with Slide‐A‐Lyzer dialysis cassettes of 2 kDa MWCO (Life Technologies, Thiais, France). Ethyl (hydroxyimino) cyanoacetate (Oxyma), N,N′‐diisopropyl‐carbodiimide (DIC), and N‐Fmoc‐protected amino acids were obtained from Iris Biotech GmbH (Marktredwitz, Germany). Side‐chain protecting groups used for the amino acids were Fmoc‐Arg(Pbf)‐OH, Fmoc‐Asn(Trt)‐OH, Fmoc‐Asp(OtBu)‐OH, Fmoc‐Cys(Trt)‐OH, Fmoc‐Gln(Trt)‐OH, Fmoc‐Glu‐(OtBu)‐OH, Fmoc‐His(Trt)‐OH, Fmoc‐Lys(Boc)‐OH, Fmoc‐Thr(tBu)‐OH, and Fmoc‐Tyr(tBu)‐OH. Fmoc‐Asp(OtBu)‐(Dmb) Gly‐OH, Fmoc‐Asp(OtBu)‐Ser(psiMe,Mepro)‐OH, Fmoc‐Arg(Me,Pbf)‐OH, Fmoc‐Lys(Dnp)‐OH, Fmoc‐Lys(Boc)‐OH and 5(6)‐carboxy‐tetramethyl‐rhodamine (TAMRA) were purchased from Novabiochem (Merck‐Milipore, Darmstadt, Germany) and Iris Biotech (Marktredwitz, Germany). Bis(2‐sulfanylethyl)‐aminotritylpoly‐styrene (SEA PS) and pre‐loaded Wang resin were obtained from X'prochem (Lille, France) and Iris Biotech (Marktredwitz, Germany), respectively. Triisopropylsilane (TIS), 2,4,6‐trinitrobenzenesulfonic acid solution 1% (w/v) in DMF (TNBS), tris(2‐carboxy‐ethyl)phosphine hydrochloride (TCEP), guanidine hydrochloride, and 4‐mercap‐tophenyl acetic acid (MPAA) were purchased from Sigma–Aldrich (St Louis, MO). All reagents were purchased at the purest grade available. Peptide synthesis grade N,N‐dimethyl‐formamide (DMF), dichloromethane (CH2Cl2, DCM), diethylether (Et2O), acetonitrile (CH3CN), N,N‐diisopropylethylamine (DIEA), acetic anhydride (Ac2O), piperidine, and trifluoroacetic acid (TFA) were purchased from Sigma–Aldrich (St Louis, MO) or Iris Biotech GmbH (Marktredwitz, Germany). All buffers were prepared with MilliQ water (Millipore, Darmstadt, Germany). The pH of buffer solutions was adjusted using NaOH or HCl.
Analysis and purification
Reactions were monitored by analytical UPLC‐MS (UPLC Acquity HClass coupled to a SQ Mass Detector 2 [Waters Corp, Guyancourt, France)] on a reverse phase column [BEH C18, 150 × 2.1 mm2, Waters Corp, (Guyancourt, France)], at 60°C using an appropriate gradient of increasing buffer B concentration in buffer A (flow rate 0.6 mL min−1, buffer A = 0.05% formic acid in H2O; buffer B = 0.05% formic acid in CH3CN). The column eluate was monitored by UV at 214 nm. The mass spectra were acquired on a quadrupole detector [SQ Mass Detector 2 (Waters Corp, Guyancourt, France)] equipped with an electrospray source: flow rate, 5 µL min−1; ionization mode, ES+; m/z range, 300–2000; source voltage, capillary voltage 3 kV and cone voltage 30 V; source temperature, desolvation temperature 500°C; source gas flow, desolvation 900 L h−1; tune resolution, source temperature 150°C; extractor, 3 V; RF lens, 2.5 V. Calculated masses were based on the average isotope composition. The purification of crude peptides was performed with a preparative reverse phase HPLC system (Waters Delta Prep 4000) using a preparative C18 reverse phase column (10 µm, 120 Å, 50 × 300 mm2, (Vydac Denali, ISS, Dartford, UK) and appropriate gradient of increasing concentration of buffer B in buffer A (flow rate 80 mL min−1). The fractions containing the purified target peptide were identified by UV measurement (Waters 2489 UV/Visible detector) at 214 nm and selected fractions combined and lyophilized.
Protein synthesis strategies
Calstabin, a small 12 kDa enzyme composed of 107 amino acids, does not require cofactors or metal ions for folding or catalytic activity. The protein has three shorter isoforms (31, 56, and 79 amino acids in length). Because of the shorter lengths, these calstabins were synthesized using step‐by‐step SPPS methodology. For the longer isoforms and derivatives, we used the native chemical ligation (NCL) method and assembled smaller fragments synthesized by solid‐phase peptide synthesis (SPPS). All of the isoforms share the same N‐terminal sequence, which contains a cysteine in position 21 that can be used as the ligation site during NCL. In addition to the four isoforms, we also synthesized other non‐native derivatives based on the full‐length calstabin. All calstabin proteins and derivatives were synthesized using the same cysteine ligation site, except the 31 and 56‐amino‐acid isoforms, which were synthesized step‐by‐step.
Synthesis of shorter calstabin isoforms
Peptide elongation was performed on Fmoc‐Glu(OtBu)‐Wang resin (Tentagel, 0.25 mmol g−1) or Fmoc‐Phe‐Wang resin (Tentagel, 0.25 mmol g−1) and Fmoc‐Cys(Trt)‐Wang resin (Tentagel, 0.25 mmol g−1) using the standard Fmoc/tert‐butyl chemistry described above. During synthesis, the pseudo proline building block Fmoc‐Asp(OtBu)‐Ser(psiMe,Mepro)‐OH was used for the sequence‐specific introduction of Asp‐Ser residues. After complete synthesis of the peptide fragment, deprotection and cleavage were performed with TFA/TIS/EDT/H2O (94/1/2.5/2.5 by vol; 10 mL for 0.1 mmol of resin) for 3 h.
Preparation of synthetic calstabin
Peptide elongation was performed using standard Fmoc/tert‐butyl chemistry on an automated peptide synthesizer (SymphonyX Synthesizer, Protein Technologies, Tucson, AZ) on a 0.5 mmol scale. Double couplings were performed using a fivefold molar excess of each Fmoc‐l‐amino acid, fivefold molar excess of Oxyma, and fivefold molar excess of DIC. A capping step was performed after each coupling with Ac2O in DMF. The various sequences of the proteins are summarized in Table 2. More details are given about the main proteins synthesized in Table 3, with some details on the yields and amounts obtained.
Synthesis of SEA peptide fragment (general procedure)
Peptide elongation was performed on Fmoc‐Thr(OtBu)‐SEA PS resin (0.5 mmol, 0.077 mmol g−1) using the standard Fmoc/tert‐butyl chemistry described above. During synthesis, the building block Fmoc‐Asp(OtBu)‐(Dmb)Gly‐OH was used for the sequence‐specific introduction of Asp‐Gly residues. Typical procedures for the synthesis and purification of SEAoff peptide segments were described previously.52, 53, 54 Briefly, peptide fragment deprotection and cleavage were performed with TFA/TIS/H2O/EDT (94/1/2.5/2.5 by vol; 30 mL for 0.25 mmol of resin) for 3 h.
Synthesis of Cys peptide fragment (general procedure)
Peptide elongation was performed manually on Fmoc‐Glu(OtBu)‐Wang resin (Tentagel, 0.5 mmol, 0.25 mmol g−1) using the standard Fmoc/tert‐butyl chemistry described above. During synthesis, the pseudo proline building block Fmoc‐Asp(OtBu)‐Ser(psiMe,Mepro)‐OH was used for the sequence‐specific introduction of Asp‐Ser residues. After complete synthesis of the peptide fragment, deprotection and cleavage were performed with TFA/phenol/H2O/thioanisole/EDT/DMS/NH4I (81/5/3/5/2.5/2/1.5 by vol; 30 ml for 0.25 mmol of resin) for 3 h.
Ligation (general procedure)
MPAA (34 mg, 0.2 mmol) and tris(2‐carboxyethyl)phosphine hydrochloride (TCEP) (58 mg, 0.2 mmol) were dissolved in 6 M guanidine–HCl, 0.1 M pH 7.2 sodium phosphate buffer (1 mL). NaOH (6 M) was then added to adjust the pH to 7. SEAoff‐peptide A and Cys‐peptide segment B (2.7 eq) were dissolved together in the above solution (final peptide concentration 3 mM). The solution was stirred for 60 h at room temperature under argon atmosphere and the progress of the ligation monitored by LC‐MS. After 60 h, the reaction mixture was diluted with water (3 mL), acidified with 10% aqueous TFA (200 μL), and extracted with diethylether to remove the excess MPAA. TCEP (36.5 mg) was added to the aqueous phase and agitated for 20 min. The crude product was directly purified by reversed‐phase HPLC using a linear water (buffer A: H2O 0.1% TFA)‐acetonitrile (buffer B: ACN 0.1% TFA) gradient (0–20% of buffer B in 1 min then 20 to 80% of buffer B in 121 min) to obtain the purified synthetic protein (see Table 3).
Production and purification of recombinant calstabin
A 100 mL of 2YT medium [1.6% (w/v) Tryptone; 1% (w/v) yeast extract; 0.5% (w/v) NaCl] supplemented with 100 μg mL−1 ampicillin was inoculated in a 1‐L Erlenmeyer flask with an aliquot of a glycerol stock of the Escherichia coli strain BL21(DE3)/hFKBP12.6‐GST pGEX6P3 at 37°C for 16 h under agitation (200 rpm). A total of 5 L of 2YT medium supplemented with 100 μg mL−1 ampicillin was inoculated with this preculture in a 7‐L bioreactor at 37°C, aerated at 1 v.v.m, and under agitation at 300 rpm. The production was induced by the addition of 1 mM IPTG at an OD600 of 1 for 21 h at 23°C. The cells were harvested by centrifugation at 6000g for 10 min at 4°C. The cell pellet was frozen at −20°C. A pellet equivalent to 5 L of production was resuspended in 500 mL of lysis buffer (10 mM imidazole, 300 mM sucrose, 1 mM DTT, 10 μM pepstatin A, 8.5 μM bestatin, 1 μM E64, 1 mM PMSF adjusted to pH 7.4). The suspension was homogenized and the cells sonicated three times for 1 min with pulses of 0.5‐in. at 100% power. The soluble fraction was recovered by centrifugation at 16,000g for 1 h at 4°C. Purification procedures were performed at 4°C. A total of 500 mL of intracellular soluble fraction was loaded onto 50 mL of glutathione agarose resin equilibrated in buffer A (Buffer A: 10 mM imidazole, 300 mM sucrose, 1 mM DTT pH 7.4; Buffer B: 10 mM imidazole, 300 mM sucrose, 1 mM DTT, 10 mM glutathione pH 7.4). The lysate was fixed for 16 h, washed with buffer A for 5 min, and then eluted three times with buffer B for 25 min each. Nearly 240 mg of GST‐calstabin was used for the PreScission cleavage. This quantity was contained in elutions 1 and 2. Then, 170 mL of 2× cleavage buffer (100 mM Tris‐HCl pH 7.5, 300 mM NaCl, 2 mM DTT, 2 mM EDTA) was added to 170 mL of GST elution (elutions 1 and 2). A total of 2400 U of PreScission protease was added. The mix was incubated at 4°C under agitation for 16 h. The cleaved calstabin was extensively dialyzed with a Spectra/Por Membrane (MWCCO of 3.5 kDa) at least three times for 3 h against 2 L of buffer C (10 mM imidazole, 300 mM sucrose, 1 mM DTT, 10% glycerol, pH 7.4). The purification procedures were performed at 4°C using an AKTA explorer. The cleaved calstabin was loaded on a 33 mL glutathione agarose column previously equilibrated with buffer A. The column was washed with 100 ml of buffer A and then eluted with 10 × 10 mL of buffer B (10 mM imidazole, 300 mM sucrose, 1 mM DTT, 10 mM glutathione, pH 7.4). Flow through and first wash fractions containing calstabin were concentrated three times until 0.5 mg mL−1 using an AMICON system with a 5 kDa cut off membrane. Five milligrams of total protein were injected into a S75 26/60 prep‐grade 320 ml column and eluted with 1 column volume of buffer A (PBS, 1 mM DTT). The purified calstabin was concentrated until near 1 mg mL−1 using an AMICON system with a 5 kDa cut off membrane. Then, calstabin was dialyzed with a Spectra/Por Membrane (MWCCO of 3.5 kDa) twice for 3 h against 2 × 2 L of 1X PBS.
Molecular biology
Gene sequencing and MS analysis were in agreement with the sequence GPLGSMGVEI ETISPGDGRT FPKKGQTCVV HYTGMLQNGK KFDSSRDRNK PFKFRIGKQE VIKGFEEGAA QMSLGQRAKL TCTPDVAYGA TGHPGVIPPN ATLIFDVELL NLE. The GPLGS fragment, which is the only difference from the sequence of synthetic calstabin, comes from the GST tag.
Protein refolding
The lyophilized peptide was solubilized at 0.5 mg/ml in a buffer containing 100 mM Tris‐HCl, 6M guanidinium hydrochloride, and 5 mM DTT (pH 8) and incubated at 37°C for 30 min. At that stage, numerous tests were performed using the Pierce® refolding kit (ThermoFisher Scientific, Illkirch‐Graffenstaden, France), but only the following condition led to an active synthetic enzyme. Therefore, the protein was refolded by dialysis 2 × 2 h at 4°C against a buffer containing 50 mM Tris‐HCl and 20 mM NaCl (pH 7.5). A final extensive dialysis was performed against an analysis buffer. For refolding by dilution, the lyophilized peptide was solubilized at 8 mg/ml in DMSO and diluted drop by drop in a Tris buffer (50 mM NaCl, 20 mM KCl, 0.8 mM, pH 7.7) + 5 mM DTT in order to obtain 1% DMSO. After 4 h at 4°C, the samples were concentrated to a final concentration of ∼0.5 mg mL−1.
Preparative size‐exclusion chromatography
For preparative SEC, samples were concentrated to 2.5 mg mL−1 using an Amicon Ultra‐4 concentrator with an MWCO of 2000 Da and centrifuged at 20,300g for 20 min. Proteins were injected on a Superdex 75 10/300 GL (GE Healthcare, Orsay, France) equilibrated with 50 mM Tris/HCl and 300 mM NaCl (pH 7.5) buffer at 0.8 mL min−1. The protein elution was monitored at 280 nm.
Electrophoresis
SDS‐PAGE was performed using Nu‐PAGE 4–12% in MES buffer (Invitrogen). Samples were boiled for 5 min at 95°C with LDS sample buffer (Invitrogen) and 50 mM DTT. Migration was done under 200 V for 40 min.
Analytical size‐exclusion chromatography with MALS
For SEC, samples were concentrated to at least at 0.3 mg mL−1 using an Amicon Ultra‐4 concentrator with an MWCO of 2000 Da and centrifuged at 20,300g for 20 min. Proteins were analyzed on a Superdex 75 5/150 GL column (GE Healthcare, Orsay, France) equilibrated with 50 mM Tris/HCl and 300 mM NaCl (pH 7.5) buffer filtered (0.1 µM) at a flow rate of 0.2 mL min−1. Protein elution was monitored at 280 nm. HPLC was equipped with a multi‐angle light scattering detector (DAWN 8+ Wyatt Technology, Santa Barbara, CA) and an RI detector (Optilab T‐rEX Wyatt Technology, Santa Barbara, CA).
Native mass spectrometry
Nano‐ESI mass spectra were acquired on a time‐of‐flight mass spectrometer (LCT Premier XE, Waters, Milford, MA) upgraded with the Non‐Covalent Enhancement kit and fitted with an automated chip‐based NanoESI system (Nanomate 200, Advion Biosciences, Ithaca, NY). Standard nanospray parameters were used (chip voltage 1500 V, gas pressure 0.3 psi). Before native ESI‐MS measurements, synthetic calstabin sample (Fraction 2) was desalted in 200 mM ammonium acetate (pH 7.4) by five successive concentration/dilution buffer‐exchange steps with Amicon Ultra centrifugal filter (3 kDa cut‐off, Pierce, Rockford, IL). Synthetic calstabin was diluted to 10 µM. The mass spectrometer was carefully tuned with gentle desolvation parameters to preserve protein folding during the ionization/desorption process. The cone, ion guide 1 and aperture 1 voltages were optimized to 50 V, 50 V, and 10 V, respectively. The pressure in the source region was 1.9 mbar. Ions were detected with a multichannel plate (MCP) detector set at 2200 V. Native mass spectra were recorded in the positive V mode on the mass range m/z 500–5000 after calibration with 2 µM horse heart myoglobin dissolved in acetonitrile/water/formic acid (50:50:1, v/v). Data were acquired and processed using MassLynx v4.1 (Waters, Milford, MA). Spectra acquired for 1–2 min were combined and smoothed before centering.
Circular dichroism
CD measurements were performed after extensive dialysis against a 20 mM potassium phosphate buffer (pH 7.7). For secondary structure analysis at 20°C, spectra were acquired at 20°C with a 0.1 cm path at 50 nm min−1 scanning speed. For denaturation analysis, spectra were acquired between 20 and 80°C with a 0.1 cm path and an increase of 1°C min−1.
Peptidyl‐prolyl cis–trans isomerase (PPIase) catalytic activity
The PPIase activity of calstabin37, 55, 56 was measured in a spectrophotometric assay. The protease‐coupled assay relied on chymotrypsin's inability to cleave peptide substrates with a cis‐Pro at the P2 position, such as in the peptide Suc‐Ala‐Ala‐Pro‐Phe‐pNA. The progress of the reaction was followed with an increase of absorbance at 390 nm corresponding to free pNa (ɛ390nm = 13,400 M−1 cm−1). The reaction occurred at 10°C with a stopped flow for 120 s. Chymotrypsin was prepared at 12 mg mL−1 in 50 mM Hepes and 100 mM NaCl (pH 8) buffer. The stock solution of substrate (Nsuc‐Ala‐Leu‐Pro‐Phe‐pNa) was 10 mM in DMSO. The peptide was then diluted in 50 mM Hepes and 100 mM NaCl (pH 8) buffer at a final concentration of 200 µM. Calstabin was four times more concentrated by the addition of working buffer. For each measure, 50 µL of chymotrypsin, 50 µL of enzyme, and 100 µL of substrate was expulsed at 10 mL s−1. There were six shots for each condition. Harrison and Stein37 showed that, in the presence of excess chymotrypsin, the progress curves for the production of p‐nitroaniline were in first‐order in substrate with a rate constant k iso corresponding to the specific activity of the PPIase after the burst of the initially present trans‐substrate. The experimental data could be fitted with two exponentials, the first one corresponding to the reaction of the initially present trans‐substrate and the second to the kinetics of the PPIase activity, giving us the specific activity of the enzyme depending on its concentration. To avoid dependence on the concentration of this parameter, we can calculate the k cat/K m value using the formula k cat/K m = (k iso– k chimo)/[PPIase], where k chimo corresponds to the parameters calculated without the enzyme and to the natural isomerization of proline in water.
Protein crystal
The synthetic calsatbin (residues 1–107) was solubilized at a final concentration of 66 μM in 50 mM HCl‐Tris and 150 mM NaCl (pH 8). The enzyme (at 66 µM) was mixed with rapamycin at a final concentration of 369 µM in the presence of 6.4% MeOH and then incubated for 1 h on ice. The enzyme complex was subsequently concentrated at 492 µM. The sample was centrifuged at 11,000g for 15 min before crystallization. Hanging drops of synthetic calstabin were set up by mixing equimolar ratio of the complex and the reservoir containing 16% PEG 8K, 10% glycerol, 500 mM KCl, and 50 mM Hepes (pH 7). First crystals appeared after 2 days at 17°C by vapor diffusion and were optimized using the micro‐seeding technique. The optimized crystals were long rods [Fig. 9(A)] that were cryo‐cooled in a final solution containing 18% PEG 8K, 10% glycerol, 500 mM KCl, and 50 mM Hepes (pH 7).
X‐ray diffraction
X‐ray data sets were collected in the Proxima 1 beamline station at the Synchrotron SOLEIL (Saclay, France). Diffraction data were indexed and scaled with XDS.57 Molecular replacement solution was obtained with Phaser, CCP4 suite58 using recombinant calstabin bound to rapamycin (PDB entry ID: 1C9H) as search model. Model building and refinement were carried out with the Coot59 and Buster programs.60 Data collection and refinement statistics are summarized in Table 4. Figures were made using PyMOL software (Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC).
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.
Supporting Information Figure 8.
Supporting Information Figure 9.
Acknowledgments
The authors thank GTP Technology for the production of recombinant calstabin and San Francisco Edit for the revision of the manuscript. The authors thank the Synchrotron SOLEIL Program Committee for allowing data collection under project number 99150097.
References
- 1. Merrifield RB (1965) Automated synthesis of peptides. Science 150:178–185. [DOI] [PubMed] [Google Scholar]
- 2. Nestor JJ, Jr (2009) The medicinal chemistry of peptides. Curr Med Chem 16:4399–4418. [DOI] [PubMed] [Google Scholar]
- 3. Craik DJ, Fairlie DP, Liras S, Price D (2013) The future of peptide‐based drugs. Chem Biol Drug Des 81:136–147. [DOI] [PubMed] [Google Scholar]
- 4. Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES (2012) Cell‐penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des 80:639–646. [DOI] [PubMed] [Google Scholar]
- 5. Boutin JA, Fauchere AL (1996) Combinatorial peptide synthesis: statistical evaluation of peptide distribution. Trends Pharmacol Sci 17:8–12. [DOI] [PubMed] [Google Scholar]
- 6. Marasco D, Perretta G, Sabatella M, Ruvo M (2008) Past and future perspectives of synthetic peptide libraries. Curr Protein Pept Sci 9:447–467. [DOI] [PubMed] [Google Scholar]
- 7. Hirschmann R, Nutt RF, Veber DF, Vitali RA, Varga SL, Jacob TA, Holly FW, Denkewalter RG (1969) Studies on the total synthesis of an enzyme. V. The preparation of enzymatically active material. J Am Chem Soc 91:507–508. [DOI] [PubMed] [Google Scholar]
- 8. Gutte B, Merrifield RB (1971) The synthesis of ribonuclease A. J Biol Chem 246:1922–1941. [PubMed] [Google Scholar]
- 9. Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SB (1989) Conserved folding in retroviral proteases: crystal structure of a synthetic HIV‐1 protease. Science 245:616–621. [DOI] [PubMed] [Google Scholar]
- 10. Fitzgerald MC, Chernushevich I, Standing KG, Kent SBH, Whitman CP (1995) Total chemical synthesis and catalytic properties of the enzyme enantiomers L‐ and D‐4‐oxalocrotonate tautomerase. J Am Chem Soc 117:11075–11080. [Google Scholar]
- 11. Hackeng TM, Mounier CM, Bon C, Dawson PE, Griffin JH, Kent SB (1997) Total chemical synthesis of enzymatically active human type II secretory phospholipase A2. Proc Natl Acad Sci USA 94:7845–7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Low DW, Hill MG, Carrasco MR, Kent SB, Botti P Total synthesis of cytochrome b562 by native chemical ligation using a removable auxiliary. Proc Natl Acad Sci USA 98:6554–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Becker CF, Hunter CL, Seidel R, Kent SB, Goody RS, Engelhard M (2003) Total chemical synthesis of a functional interacting protein pair: the protooncogene H‐Ras and the Ras‐binding domain of its effector c‐Raf1. Proc Natl Acad Sci USA 100:5075–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durek T, Torbeev VY, Kent SB (2007) Convergent chemical synthesis and high‐resolution X‐ray structure of human lysozyme. Proc Natl Acad Sci USA 104:4846–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li C, Li X, Lu W (2010) Total chemical synthesis of human T‐cell leukemia virus type 1 protease via native chemical ligation. Biopolymers 94:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weinstock MT, Jacobsen MT, Kay MS (2014) Synthesis and folding of a mirror‐image enzyme reveals ambidextrous chaperone activity. Proc Natl Acad Sci USA 111:11679–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng FK, Zhang L, Wang YT, Schneewind O, Kent SB (2014) Total chemical synthesis of the enzyme sortase A(DeltaN59) with full catalytic activity. Angew Chem Int Ed Engl 53:4662–4666. [DOI] [PubMed] [Google Scholar]
- 18. Mong SK, Vinogradov AA, Simon MD, Pentelute BL (2014) Rapid total synthesis of DARPin pE59 and barnase. Chembiochem 15:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dawson PE, Muir TW, Clark‐Lewis I, Kent SB (1994) Synthesis of proteins by native chemical ligation. Science 266:776–779. [DOI] [PubMed] [Google Scholar]
- 20. Chalker JM (2013) Prospects in the total synthesis of protein therapeutics. Chem Biol Drug Des 81:122–135. [DOI] [PubMed] [Google Scholar]
- 21. Boutin JA, Hennig P, Lambert PH, Bertin S, Petit L, Mahieu JP, Serkiz B, Volland JP, Fauchere JL (1996) Combinatorial peptide libraries: robotic synthesis and analysis by nuclear magnetic resonance, mass spectrometry, tandem mass spectrometry, and high‐performance capillary electrophoresis techniques. Anal Biochem 234:126–141. [DOI] [PubMed] [Google Scholar]
- 22. Boutin JA, Lambert PH, Bertin S, Volland JP, Fauchere JL (1999) Physico‐chemical and biological analysis of true combinatorial libraries. J Chromatogr B Biomed Sci Appl 725:17–37. [DOI] [PubMed] [Google Scholar]
- 23. Fauchere JL, Henlin JM, Boutin JA (1997) Peptide and nonpeptide lead discovery using robotically synthesized soluble libraries. Can J Physiol Pharmacol 75:683–689. [PubMed] [Google Scholar]
- 24. Sinars CR, Cheung‐Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J (2003) Structure of the large FK506‐binding protein FKBP51, an Hsp90‐binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci USA 100:868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blair LJ, Baker JD, Sabbagh JJ, Dickey CA (2015) The emerging role of peptidyl‐prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer's disease. J Neurochem 133:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hausch F (2015) FKBPs and their role in neuronal signaling. Biochim Biophys Acta 1850:2035–2040. [DOI] [PubMed] [Google Scholar]
- 27. Rullman E, Andersson DC, Melin M, Reiken S, Mancini DM, Marks AR, Lund LH, Gustafsson T (2013) Modifications of skeletal muscle ryanodine receptor type 1 and exercise intolerance in heart failure. J Heart Lung Transplant 32:925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reiken S, Gaburjakova M, Gaburjakova J, He Kl KL, Prieto A, Becker E, Yi Gh GH, Wang J, Burkhoff D, Marks AR (2001) beta‐adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104:2843–2848. [DOI] [PubMed] [Google Scholar]
- 29. Liu F, Wang YQ, Meng L, Gu M, Tan RY (2013) FK506‐binding protein 12 ligands: a patent review. Expert Opin Ther Pat 23:1435–1449. [DOI] [PubMed] [Google Scholar]
- 30. Arakawa H, Nagase H, Hayashi N, Fujiwara T, Ogawa M, Shin S, Nakamura Y (1994) Molecular cloning and expression of a novel human gene that is highly homologous to human FK506‐binding protein 12kDa (hFKBP‐12) and characterization of two alternatively spliced transcripts. Biochem Biophys Res Commun 200:836–843. [DOI] [PubMed] [Google Scholar]
- 31. Haddy A, Rusnak F (1994) Overexpression and characterization of a recombinant form of rat calcineurin A. Biochem Biophys Res Commun 200:1221–1229. [DOI] [PubMed] [Google Scholar]
- 32. Dawson PE, Kent SB (2000) Synthesis of native proteins by chemical ligation. Annu Rev Biochem 69:923–960. [DOI] [PubMed] [Google Scholar]
- 33. Stelea SD, Pancoska P, Benight AS, Keiderling TA (2001) Thermal unfolding of ribonuclease A in phosphate at neutral pH: deviations from the two‐state model. Protein Sci 10:970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Main ER, Fulton KF, Jackson SE (1999) Folding pathway of FKBP12 and characterisation of the transition state. J Mol Biol 291:429–444. [DOI] [PubMed] [Google Scholar]
- 35. Boerema DJ, Tereshko VA, Kent SB (2008) Total synthesis by modern chemical ligation methods and high resolution (1.1 A) X‐ray structure of ribonuclease A. Biopolymers 90:278–286. [DOI] [PubMed] [Google Scholar]
- 36. Torbeev VY, Kent SB (2007) Convergent chemical synthesis and crystal structure of a 203 amino acid “covalent dimer” HIV‐1 protease enzyme molecule. Angew Chem Int Ed Engl 46:1667–1670. [DOI] [PubMed] [Google Scholar]
- 37. Harrison RK, Stein RL (1990) Substrate specificities of the peptidyl prolyl cis–trans isomerase activities of cyclophilin and FK‐506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry 29:3813–3816. [DOI] [PubMed] [Google Scholar]
- 38. Engelhard M (2016) Quest for the chemical synthesis of proteins. J Pept Sci 22:246–251. [DOI] [PubMed] [Google Scholar]
- 39. Muir TW, Sondhi D, Cole PA (1998) Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci USA 95:6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gutte B, Merrifield RB (1969) The total synthesis of an enzyme with ribonuclease A activity. J Am Chem Soc 91:501–502. [DOI] [PubMed] [Google Scholar]
- 41. Kent S (2003) Total chemical synthesis of enzymes. J Pept Sci 9:574–593. [DOI] [PubMed] [Google Scholar]
- 42. Wu X, Schultz PG (2009) Synthesis at the interface of chemistry and biology. J Am Chem Soc 131:12497–12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun SB, Schultz PG, Kim CH (2014) Therapeutic applications of an expanded genetic code. Chembiochem 15:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiao H, Nasertorabi F, Choi SH, Han GW, Reed SA, Stevens RC, Schultz PG (2015) Exploring the potential impact of an expanded genetic code on protein function. Proc Natl Acad Sci USA 112:6961–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar KS, Bavikar SN, Spasser L, Moyal T, Ohayon S, Brik A (2011) Total chemical synthesis of a 304 amino acid K48‐linked tetraubiquitin protein. Angew Chem Int Ed Engl 50:6137–6141. [DOI] [PubMed] [Google Scholar]
- 46. Unverzagt C, Kajihara Y (2013) Chemical assembly of N‐glycoproteins: a refined toolbox to address a ubiquitous posttranslational modification. Chem Soc Rev 42:4408–4420. [DOI] [PubMed] [Google Scholar]
- 47. Young TS, Schultz PG (2010) Beyond the canonical 20 amino acids: expanding the genetic lexicon. J Biol Chem 285:11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wright TH, Vallee MR, Davis BG (2016) From chemical mutagenesis to post‐expression mutagenesis: a 50 Year odyssey. Angew Chem Int Ed Engl 55:5896–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milton RC, Milton SC, Kent SB (1992) Total chemical synthesis of a D‐enzyme: the enantiomers of HIV‐1 protease show reciprocal chiral substrate specificity [corrected]. Science 256:1445–1448. [DOI] [PubMed] [Google Scholar]
- 50. Seenaiah M, Jbara M, Mali SM, Brik A (2015) Convergent versus sequential protein synthesis: the case of ubiquitinated and glycosylated H2B. Angew Chem Int Ed 54:12374–12378. [DOI] [PubMed] [Google Scholar]
- 51. Wintermann F, Engelbrecht S (2013) Reconstitution of the catalytic core of F‐ATPase (alphabeta)3gamma from Escherichia coli using chemically synthesized subunit gamma. Angew Chem Int Ed Engl 52:1309–1313. [DOI] [PubMed] [Google Scholar]
- 52. Hou W, Zhang X, Li F, Liu CF (2011) Peptidyl N,N‐bis(2‐mercaptoethyl)‐amides as thioester precursors for native chemical ligation. Org Lett 13:386–389. [DOI] [PubMed] [Google Scholar]
- 53. Boll E, Drobecq H, Ollivier N, Blanpain A, Raibaut L, Desmet R, Vicogne J, Melnyk O (2015) One‐pot chemical synthesis of small ubiquitin‐like modifier protein‐peptide conjugates using bis(2‐sulfanylethyl)amido peptide latent thioester surrogates. Nat Protoc 10:269–292. [DOI] [PubMed] [Google Scholar]
- 54. Boll E, Ebran JP, Drobecq H, El‐Mahdi O, Raibaut L, Ollivier N, Melnyk O (2015) Access to large cyclic peptides by a one‐pot two‐peptide segment ligation/cyclization process. Org Lett 17:130–133. [DOI] [PubMed] [Google Scholar]
- 55. Kofron JL, Kuzmic P, Kishore V, Colon‐Bonilla E, Rich DH (1991) Determination of kinetic constants for peptidyl prolyl cis–trans isomerases by an improved spectrophotometric assay. Biochemistry 30:6127–6134. [DOI] [PubMed] [Google Scholar]
- 56. Harrison RK, Stein RL (1990) Mechanistic studies of peptidyl prolyl cis–trans isomerase: evidence for catalysis by distortion. Biochemistry 29:1684–1689. [DOI] [PubMed] [Google Scholar]
- 57. Kabsch W (2010) XDS. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2012) Exploiting structure similarity in refinement: automated NCS and target‐structure restraint in BUSTER. Acta Cryst D68:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.
Supporting Information Figure 8.
Supporting Information Figure 9.