

Abstract
The development and use of small-molecule inhibitors of the adherence of P. gingivalis to oral streptococci represents a potential therapy for the treatment of periodontal disease as these organisms work in tandem to colonize the oral cavity. Earlier work from these laboratories demonstrated that a small synthetic peptide was an effective inhibitor of the interaction between P. gingivalis and S. gordonii and that a small-molecule peptidomimetic would provide a more stable, less expensive and more effective inhibitor. An array of 2-(azidomethyl)- and 2-(azidophenyl)-4,5-diaryloxazoles having a full range of hydrophobic groups were prepared and reacted with substituted arylacetylenes to afford the corresponding ‘click’ products. The title compounds were evaluated for their ability to inhibit P. gingivalis’ adherence to oral streptococci and several were found to be inhibitory in the range of (IC50) 5.3–67μM.
Keywords: Azides, Biofilms, Click Chemistry, Oxazoles, Peptidomimetics
Graphical Abstract
Inhibition of the interaction of streptococcal antigen I/II and P. gingivalis Mfa proteins by 1,2,3-triazole-based peptidomimetics.
1. Introduction
Periodontal disease is caused by a consortium of anaerobic bacteria including Porphyromonas gingivalis, Tannerella forsythsis and Treponema denticola1. These organisms have been designated as the “red complex” and are strongly associated with chronic adult periodontitis. Current methods to treat periodontitis involves scaling and root planing and in more severe cases, surgery may be required to reduce pocket depth. In general, therapeutic approaches are lacking that specifically target periodontal pathogens like P. gingivalis and furthermore, treatments that prevent or limit the re-colonization of the oral cavity by P. gingivalis after treatment procedures are not available. Although the primary niche for P. gingivalis is in a mixed community of bacterial species in the subgingival pocket, upon initial entry into the oral cavity it must first colonize supragingival plaque2. Our previous results suggest that the interaction of P. gingivalis with oral streptococci is important for this early colonization event3,4. Thus, this interaction represents an ideal point for therapeutic intervention to control colonization (or re-colonization) of oral tissues by P. gingivalis. Adherence of P. gingivalis to streptococci is species specific and is driven by a protein-protein interaction that occurs between the minor fimbrial antigen (Mfa) of P. gingivalis and the antigen I/II (Ag I/II) polypeptide of streptococci5–7. This interaction induces a response in P. gingivalis that may be involved in its adaptation to biofilm growth in the oral cavity8. Daep et al. also identified a discrete domain in Ag I/II protein that mediates its interaction with Mfa and showed that this region resembles the eukaryotic nuclear receptor (NR) box protein-protein interaction domain5,6. Furthermore, variation in the sequence and structure of the NR box in different members of the Ag I/II family of proteins accounted for the species selectivity of P. gingivalis adherence to oral streptococci5. We also showed that the NR box is comprised of two functional peptide motifs, VXXLL and NITVK, and that a synthetic peptide encompassing both motifs functioned as a potent inhibitor of P. gingivalis adherence to streptococci and significantly reduced P. gingivalis virulence in vivo7. Together, these results suggest that P. gingivalis colonization of the oral cavity can be controlled by preventing its initial association with streptococci and that inhibitors of the Mfa-AgI/II interaction represent potential therapeutic agents to control periodontal disease. However, the use of peptides as therapeutic agents has limitations arising from the relatively high cost of peptide synthesis and their susceptibility to degradation by proteases expressed by oral organisms, including P. gingivalis itself. To overcome these limitations, we sought to produce a potent and stable inhibitor that is not based on a peptide backbone but mimics the natural peptide substrate recognized by Mfa. Since the streptococcal NR box-like sequence contains two functional motifs, we envisioned that the design and study of small-molecule, non-peptide based inhibitors of the Mfa/AgI/II interaction encompassed the employment of two synthetic small-molecule scaffolds joined together via the “click” reaction9,10. Within the expansive area of nitrogen/oxygen heterocycles, we have identified the 2,4,5-trisubstituted oxazole framework as a starting point for the NITVK-associated inhibitors of Mfa/AgI/II interaction11–13 and show here that these compounds potently block P. gingivalis adherence to streptococci in vitro when clicked with substituted arylalkynes. As click chemistry involves the classical cycloaddition reaction between an alkyl or aryl azide and an alkyl or aryl alkyne, the oxazoles were functionalized with the azide moiety and an array of arylalkynyl click partners containing hydrophobic groups were chosen for both evaluating the click reactions and exploring and evaluating the minimal structural requirements for the VXXLL mimic. The newly-formed 1,2,3-triazole resulting from the click reaction may function as a polar, slightly basic backbone of the complete peptidomimetic and depending on the spacer group between the triazole and the oxazole, may affect the topology of the entire molecule. It may also be noted that triazole rings can function as mild hydrogen bond acceptors as well as structures with π-stacking potential.14
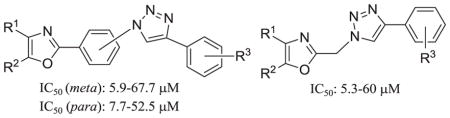
2. 1 Design of the Target Compounds
Our initial approach toward the design of the peptidomimetics was focused on the installation of three dimensional hydrophobic moieties that will mimic both the VXXLL and NITVK motifs of the helical peptide. The 2,4,5-trisubstituted oxazole scaffold was chosen as the peptidomimetic model of the NITVK motif. The oxazole ring comprises a central torus whereby two aromatic rings, each located at the 4 and 5 positions of the heterocycle, bear hydrophobic residues (Figure 1). The positions of the hydrophobic residues may be placed ortho, meta or para on each ring at the 4- or 5-position of the oxazole. The hydrophobic residues may take the form of halogens, alkoxy groups or alkyl groups. The planarity of the substituted oxazole ring excludes any stereochemical considerations thereby simplifying the initial design of the inhibitory NITVK scaffolds. The NITVK scaffolds were designed with the azide function of the click coupling partner positioned with at least a one-carbon spacer or a phenyl-ring spacer between the azide moiety and the oxazole torus with the azidoalkyl or azidoaryl group located at the 2-position of the oxazole. The azides take the form of three general groups which vary at the 2-position of the 4,5-diaryloxazole. The Group I azide precursors 10-13 possess aryl groups situated at the 4- and 5- positions of the oxazole ring. Both aryl groups are substituted at the para positions with fluorine, chlorine, bromine or methoxy groups and have the azidomethyl reacting group at the 2-position of the oxazole. Similarly, the Group II azide precursors 18-21 possess substituted aryl groups situated at the 4- and 5-positions of the oxazole and bear fluorine, chlorine, bromine and methoxy groups at the para positions. In contrast to the Group I candidates, compounds 18-21 possess a meta-azidophenyl group at the 2-position of the oxazole. The Group III targets 26-29 have the same para aryl substituent pattern at oxazole positions 4 and 5, but employ a para-azidophenyl reacting group at position 2 of the oxazole. So the structural and substituent differences of the Group I, II and III azides offer a great breath of molecular diversity for the NITVK mimic. Our initial design of the VXXLL-mimic required an alkynyl reacting group and encompassed a scaffold of lesser structural complexity, namely a carbocyclic aromatic ring bearing hydrophobic groups (Table 1).15 While, previous workers have identified di- and trialkylaminotriazines as VXXLL mimics in research involving the nuclear receptor boxes,16,17 we first opted for the albeit simpler and planar aromatic analogue bearing the characteristic symmetrically-opposed hydrophobic groups. The alkylaminotriazine scaffold (Table 1) possesses non-polar hydrophobic N-alkyl groups which mimic the one valine and two leucine groups in the helical peptide. Given the requirement of the alkynyl reacting group at position 1 of the aryl ring, the only requirement remaining was two hydrophobic substituents, each at positions 3 and 5. For the hydrophobic substituents, we chose the halogens (F, Cl, Br), methoxy (OCH3) and trifluoromethyl (CF3) groups, all of which were situated on aromatic rings. In terms of hydrophobicity trends, F>Cl>Br with the methoxy group being more polar and less hydrophobic than bromine and the trifluoromethyl group being more hydrophobic than a single fluorine. Given that the naphthalene core has been implicated in mimicking the double leucine motif in earlier studies,16 our selection included an alkynylnaphthalene bearing a hydrophobic methoxy group (Table 1). It should be noted that our initial selections of simple aryl-substituted VXXLL mimics constituted only aryl rings which were singly-substituted with hydrophobic groups. The singly-substituted aryl components are functionalized with a terminal acetylene moiety to facilitate the 3+2 cycloaddition or “click” reaction with the azide-functionalized NITVK mimics.18–20 Consequently, lead optimization may occur through adjustment of the hydrophobic groups on both the mono-aryl and diaryloxazole scaffolds as well as the topology and size of the alkyl-triazole or aryl-triazole linker.
Figure 1.

General azide/alkyne scaffold construction
Table 1.
Aryl Alkynyl Click Partners 30-401

|
Arranged in order of increasing molecular weight
See Experimental for Preparation
2.2 Synthesis
Our syntheses of the Group I compounds begins with the preparation of 2-(azidomethyl)-4,5-diaryloxazoles 10-13 from the corresponding unsubstituted or substituted benzoins (Scheme 1). Benzoin or the substituted benzoins 1-4 are esterified with chloroacetyl chloride in the presence of pyridine to give the aryl-substituted chloroacetyl esters 6-9 (43–85%). The chloroacetyl (benzoin) esters 6-9 are then cyclized to the corresponding 2-chloromethyloxazoles with ammonium acetate in acetic acid followed by immediate treatment with sodium azide in N,N-dimethylformamide (DMF) to provide the Group I azidomethyloxazoles 10-13. The progress of the cyclization reactions were monitored by thin-layer chromatography, normally take 1–3 hours and afforded the product chloromethyloxazoles in 71–80% yield. The air-stable and non-hygroscopic products (10-13) obtained from the room-temperature azide displacement reaction are purified using gravity-column silica gel chromatography with mixtures of hexanes/ethyl acetate as the mobile phase. Our synthesis of the Group II 3-azidophenyl precursors 18-21 begins with the esterification of benzoin or substituted benzoins 2-5 with 3-azidobenzoyl chloride/4-dimethylaminopyridine (48–93%, Scheme 2). The resulting azidobenzoyl esters 14-17 were cyclized to the corresponding 2-(3-azidophenyl) oxazoles 18-21 using ammonium acetate in acetic acid (115 °C/3 h/44–63%). The synthesis of the Group III 2(4-azidophenyl)-4,5-diaryloxazoles paralled that of the Group II compounds (Scheme 3). Benzoins 2-5 were esterified with 4-azidobenzoyl chloride/DMAP which afforded the 4-azido benzoyl esters 22-25 (72–93%). The esters were then cyclized to the oxazoles 26-29 with ammonium acetate in acetic acid (47–69%). The chromatographic conditions for purification of Group II and Group III azides were similar to that used for the Group I compounds. Both the Group I, Group II and Group II azides were reacted with arylalkynes (30-40, Table 1) using a system composed of aqueous tetrahydrofuran (THF), copper sulfate and sodium ascorbate over a reaction period of sixteen hours at room temperature (Tables 2, 3 and 4). After workup of the reaction mixtures, the product triazoles (41-90, Tables 2, 3 and 4) were purified by gravity-column chromatography on silica gel using hexanes/ethyl acetate or chloroform/methanol as the mobile phase. The pure triazoles were isolated as crystalline compounds whose composition and structures were confirmed by NMR and HRMS (See Experimental). The product yields for the click reactions of azidomethyl oxazoles 10-13 and arylalkyne reactants 30-40 are detailed in Table 2 (Compounds 41-66). The product yields for the click reactions of the meta-substituted azidophenyloxazoles 20-21 and arylalkyne reactants selected from 30-40 are detailed in Table 3 (Compounds 67-80) and the product yields for the click reactions of the para-substituted azidophenyloxazoles 26-27 and arylalkyne reactants selected from 30-40 are detailed in Table 4 (Compounds 81-90). The regiochemistry of the dipolar cycloaddition ‘click’ reaction involves products in which the substitution could be 1,4- or 1,5- on the triazole ring depending on the reaction conditions. All of the click products described herein (Tables 2, 3 and 4) were formed as a result of copper (I) catalysis which gives exclusively the 1,4- or “anti”-substituted 1,2,3-triazole.18 While the acetylenes 30-39 were used to explore both the reactivity demand of the click reaction and evaluate the hydrophobic nature of the substituent groups which mimic the VXXLL motif, we sought a more specifically-designed scaffold which utilized hydrophobic groups attached to a more hydrophilic scaffold. An interesting scaffold which would accommodate both a charge and a wide range of hydrophobic groups is acetylenic triazine 40 (Table 1). Using the 1,3,5-triazine as the central torus, a wide range of hydrophobic groups may be accommodated, and in the case of 40, the isopentyl (3-methylbutyl) groups were chosen for the synthesis (Scheme 4). Lithiation of ethynyltrimethylsilane at 0°C followed by addition of the organolithium reagent to commercially-available cyanuric chloride gives the mono-(trimethylsilylethynyl) dichlorotriazine 91. Direct addition of excess isopentylamine gave the acetylenic diaminotriazine 92 (51%) which was purified by column chromatography. Desilylation of 92 with tetra-n-butylammonium fluoride (TBAF) in THF afforded the ethynyl diaminotriazine coupling partner 40 (84%, Scheme 4).
Scheme 1.

Synthesis of Group I Azide Pecursors, 2-(Azido-methyl)-4,5-Diphenyloxazoles 10-13. Reagents/Conditions: (a) chloroacetyl chloride/DMAP/CH2Cl2/5 °C to rt/16 h; (b) NH4OAc/HOAc/115 °C/3 h; (c) NaN3/DMF/rt/3 h.
Scheme 2.

Synthesis of Group II Azide Precursors, 2-(3-Azidophenyl)-4,5-Diphenyloxazoles 18-21. Reagents/Conditions: (a) DMAP/CH2Cl2/5 °C to rt/16 h; (b) NH4OAc/HOAc/115 °C/3 h.
Scheme 3.

Synthesis of Group III Azide Precursors, 2-(4-Azidophenyl)-4,5-Diphenyloxazoles 26-29. Reagents/Conditions: (a) DMAP/CH2Cl2/5 °C to rt/16 h; (b) NH4OAc/HOAc/115 °C/3 h.
Table 2.
Activities of 2-(triazolylmethyl)-4,5-Diphenyloxazole Click Products 41-66.
| ||||
|---|---|---|---|---|
| Compound (Yield%)1 | IC50 (μM) | S. gordonii growth inhibition (%) | P. gingivalis growth inhibition (%) | Concentration (μM) |
| 41, R1, R2=Ph, R3=3-Pyridyl (99) | >60 | |||
| 42, R1, R2=Ph, R3=2-Methylphenyl (90.5) | >56.3 | |||
| 43, R1, R2=Ph, R3=3-Fluorophenyl (99) | >60 | |||
| 44, R1, R2=Ph, R3=4-Fluorophenyl (98) | >60 | |||
| 45, R1, R2=Ph, R3=2-Methoxyphenyl (86) | >60 | |||
| 46, R1, R2=Ph, R3=2-(Trifluoromethyl)phenyl (89) | >60 | |||
| 47, R1, R2=Ph, R3=4-(n-Pentyl)phenyl (94) | 35.5 | 6.6 | −14.5 | 60 |
| 48, R1, R2=Ph, R3=6-Methoxynaphthalene-2-yl (99) | >60 | |||
| 49, R1, R2=Ph, R3=4-(Phenoxy)phenyl (86) | >60 | |||
| 50, R1, R2=Ph, R3=3,5-di-(Trifluoromethyl)phenyl (97) | >60 | |||
| 51, R1, R2=Ph, R3=6-(N2,N4-disopentyl-1,3,5-triazine)-2,4-diamine (90) | 36.9 | 6.8 | 72.8 | 60 |
| 52, R1, R2=4-Fluorophenyl, R3=3-Pyridyl (66) | 59.8 | |||
| 53, R1, R2=4-Fluorophenyl, R3=3-Fluorophenyl (72) | >60 | |||
| 54, R1, R2=4-Fluorophenyl, R3=2-Methoxyphenyl (92) | >60 | |||
| 55, R1, R2=4-Fluorophenyl, R3=2-(Trifluoromethyl)phenyl (98) | >60 | |||
| 56, R1, R2=4-Fluorophenyl, R3=6-Methoxynaphthalene-2-yl(97) | 29.3 | 0.2 | −3.3 | 60 |
| 57, R1, R2=4-Chlorophenyl, R3=3-Pyridyl (99) | >60 | |||
| 58, R1, R2=4-Chlorophenyl, R3=3-Fluorophenyl (89) | >60 | |||
| 59, R1, R2=4-Chlorophenyl, R3=2-Methoxyphenyl (93) | 56.8 | 6.6 | −19.6 | 60 |
| 60, R1, R2=4-Chlorophenyl, R3=2-(Trifluoromethyl)phenyl (89) | 56.7 | |||
| 61, R1, R2=4-Chlorophenyl, R3=6-Methoxynaphthalene-2-yl (95) | 5.3 | −3.8 | 0.3 | 60 |
| 62, R1, R2=4-Bromophenyl, R3=3-Pyridyl (85) | 50.9 | |||
| 63, R1, R2=4-Bromophenyl, R3=3-Fluorophenyl (95) | >60 | |||
| 64, R1, R2=4-Bromophenyl, R3=2-Methoxyphenyl (96) | 43.0 | 11.1 | −12.4 | 60 |
| 65, R1, R2=4-Bromophenyl, R3= 2-(Trifluoromethyl)phenyl (96) | >60 | |||
| 66, R1, R2=4-Bromophenyl, R3=6-Methoxynaphthalene-2-yl (92) | 33.4 | −8.6 | −9.9 | 60 |
Yields are for isolated pure compounds
Table 3.
Activities of 2-(3-triazolylphenyl)-4,5-Diphenyloxazole Click Products 67-80
| ||||
|---|---|---|---|---|
| Compound (Yield%)1 | IC50 (μM) | S. gordonii growth inhibition (%) | P. gingivalis growth inhibition (%) | Concentration (μM) |
| 67, R1, R2=4-Methoxyphenyl, R3=3-Pyridyl (89) | 57.7 | |||
| 68, R1, R2=4-Methoxyphenyl, R3=3-Fluorophenyl (97) | 5.9 | −9.1 | −2.6 | 40 |
| 69, R1, R2=4-Methoxyphenyl, R3=4-Fluorophenyl (86) | 53.0 | −22.1 | −14.6 | 40 |
| 70, R1, R2=4-Methoxyphenyl, R3=2-Methoxyphenyl (83) | 15.4 | 0.8 | −6.2 | 40 |
| 71, R1, R2=4-Methoxyphenyl, R3=2-(Trifluoromethyl)phenyl (95) | 27.0 | −9.7 | −1.6 | 40 |
| 72, R1, R2=4-Methoxyphenyl, R3=4-(n-Pentyl)phenyl (99) | >60 | |||
| 73, R1, R2=4-Methoxyphenyl, R3=6-Methoxynaphthalene-2-yl (77) | 13.0 | −1.3 | 3.0 | 20 |
| 74, R1, R2=4-Methoxyphenyl, R3=4-Phenoxyphenyl (89) | >60 | |||
| 75, R1, R2=4-Methoxyphenyl, R3=3,5-di-(Trifluoromethyl)phenyl (95) | 47.4 | |||
| 76, R1, R2=4-Bromophenyl, R3=3-Pyridyl (72) | 31.0 | |||
| 77, R1, R2=4-Bromophenyl, R3=3-Fluorophenyl (85) | 5.0 | −5.7 | −4.3 | 5 |
| 78, R1, R2=4-Bromophenyl, R3=2-Methoxyphenyl (71) | 15.0 | 10.5 | 4.3 | 40 |
| 79, R1, R2=4-Bromophenyl, R3=2-(Trifluoromethyl)phenyl (90) | 15.2 | 2.0 | −0.6 | 40 |
| 80, R1, R2=4-Bromophenyl, R3=6-Methoxynaphthalene-2-yl (91) | 20.3 | |||
Yields are for isolated pure compounds.
Table 4.
Activities of 2-(4-triazolylphenyl)-4,5-Diphenyloxazole Click Products 81-90

| ||||
|---|---|---|---|---|
| Compound (Yield%)1 | IC50 (μM) | S. gordonii growth inhibition (%) | P. gingivalis growth inhibition (%) | Concentration (μM) |
| 81, R1, R2=4-Fluorophenyl, R3=3-Pyridyl (78) | 25.6 | |||
| 82, R1, R2=4-Fluorophenyl, R3=3-Fluorophenyl (92) | 51.5 | |||
| 83, R1, R2=4-Fluorophenyl, R3=2-Methoxyphenyl (81) | 7.7 | −7.8 | −0.5 | 20 |
| 84, R1, R2=4-Fluorophenyl, R3=2-(Trifluoromethyl)phenyl (83) | 12.8 | −11.0 | 0.1 | 40 |
| 85, R1, R2=4-Fluorophenyl, R3=6-Methoxynaphthalene-2-yl (83) | 52.2 | |||
| 86, R1, R2=4-Chlororophenyl, R3=3-Pyridyl (100) | 40.9 | |||
| 87, R1, R2=4-Chlorophenyl, R3=3-Fluorophenyl (33) | 15.7 | 8.7 | 11.6 | 40 |
| 88, R1, R2=4-Chlorophenyl, R3=2-Methoxyphenyl (100) | 37.3 | |||
| 89, R1, R2=4-Chlorophenyl, R3=2-(Trifluoromethyl)phenyl (94) | 25.1 | |||
| 90, R1, R2=4-Chlorophenyl, R3=6-Methoxynaphthalene-2-yl (74) | 32.4 | |||
Yields are for isolated pure compounds.
Scheme 4.

Synthesis of Acetylenic Triazine Precursor 40. Reagents/Conditions: (a) Ethynyltrimethylsilane/n-Bu-Li/THF/0 °C/1 h; (b) isopentylamine/48 h/0 °C to rt; (c) TBAF/THF/rt.
To assess the functional activity of the click products, compounds 41-90 were examined for inhibition of P. gingivalis adherence to S. gordonii using an established biofilm model system8 as described in the Experimental. A representative dose response series of inhibition of P. gingivalis adherence is shown in Figure 2. For each compound tested, an IC50 value for adherence inhibition was approximated after quantifying the ratio of green to red fluorescence and these values are shown in Tables 2, 3, and 4. Compounds were then classified into three groups; strong inhibitors of adherence where IC50 was <10μM (compounds 61, 68, 77 and 83), moderate inhibitors of adherence where IC50 was <20μM (compounds 70, 73, 78, 79, 84 and 87) and the remainder were weak inhibitors of adherence (IC50 >20μM). In surveying the structural relationships among the most active compounds listed above, click compound 61, bearing the triazolylmethyl “linker” is derived from the Group I 2-(azidomethyl)oxazoles. The Group II oxazoles give rise to compounds 68, 70, 73, 77, 78 and 79 and possess a meta-triazolyl linker (Table 3). In contrast, the Group III oxazoles are precursors for compounds 83, 84 and 87 which have a para-triazolyl linker (Table 4). Of all the most active compounds mentioned above, save for 68, 70 and 73, every compound bears the full range of halogen substituents at both para-positions of the 4,5-diphenyloxazole.21,22 The most active compounds derived from Group III (83, 84, 87) bear para-fluorines on the phenyl rings of the NITVK-mimic and render that section of the molecule the most hydrophobic.23 Compounds 61 and 83 bear arylmethoxy groups while 68 and 77 bear meta-substituted fluorines derived from the VXXLL mimic. As a group, the most active compounds (68, 70, 73, 77, 78 and 79) were derived from the meta-substituted azidophenyl precursors whereby the click reaction gave the meta-substituted triazole linker between the oxazole scaffold and VXXLL mimic. For example, 73 possesses the combination of two 4,5-para-methoxy groups and the highly hydrophobic naphthalene ring which allows the optimal interaction within the VXXLL binding motif. Within the same group, 68 and 70 bear the para-methoxy combination and with meta-fluoro (68) and ortho-methoxy (70) VXXLL mimics. In terms of molecular shape, the click compounds derived from the Group II azides have a characteristic “L” shape which is imparted by the meta-juxtaposition of the 2-oxazole group and the newly-formed triazole group on the phenyl ‘spacer’ ring. The shape adopted by the para- or 1,4-alignment of the 2-oxazole group and triazole groups on the phenyl ‘spacer’ ring is more linear by comparison. In contrast, compound 61 exhibits a more flexible topology whereby the methylene group or spacer conjoining the oxazole and triazole rings allows for several degrees of rotation and multiple binding modes to the helical peptide. Linear or L-shaped scaffolds are common in the design of small-molecule inhibitors, especially when combined with the multi-pronged, hydrophobic functionality found in all three groups of the inhibitors described herein.24 To confirm that the reduction of P. gingivalis levels resulted from inhibition of its adherence to S. gordonii rather than a general toxic effect of the compounds, active compounds from all groups were tested for inhibition of P. gingivalis and S. gordonii planktonic growth at the concentration listed in Tables 2, 3, and 4. As shown, few of the compounds had a significant effect on planktonic growth, suggesting that they are not functioning as antibiotics but rather as inhibitors of P. gingivalis adherence. In addition, our preliminary results suggest that the compounds have little cytotoxic activity against a gingival epithelial cell line (not shown).
Figure 2.

Dose dependent inhibition of P. gingivalis adherence to S. gordonii by compound 78. Two species biofilms comprising S. gordonii (red) and P. gingivalis (green) were formed as described in the Experimental in the absence (control) and presence of 5 – 40uM of compound 78.
4. Conclusions
An array of small-molecule peptidomimetics which carry the core interacting mimics of the biofilm-inhibitory BAR peptide have been prepared by click reactions in moderate to high yield. Using a simple two-species biofilm model, several synthetic targets proved to inhibit the adherence of P. gingivalis to oral streptococci by interfering with the interaction of Mfa-AgI/II. While this model does not represent the complexity of the oral biofilm in vivo, it does reflect an initial interaction that may be essential for P. gingivalis colonization of the oral cavitiy. Therefore, our results suggest that these compounds represent potential targeted therapeutics to limit P. gingivalis colonization. Both the azide and the alkynyl reacting components contain moderate to strongly-acting hydrophobic groups such as halogens and alkoxy groups which are situated on aromatic rings. These compounds are more compact, possess increased stability and can be synthesized and purified at a much lower expense than the polypeptides they mimic. The key adjustment parameters are the inclusion of suitable hydrophobic groups on both the NITVK and VXXLL mimics. The inhibitory results indicated that the interactions were predominantly through hydrophobic forces and that suitable substitutions in the aryl rings could increase those interactions as part of the optimization process. As such, there is a strong indication that affinity of the small-molecule scaffolds increases with both molecular weight and hydrophobicity and even includes the aromatic nature of the parent structures.25 Given the relatively high numbers of halogens in the active compounds, one cannot exclude the possibility of secondary interactions which involve electrostatic influences such as halogen bonding.26,27 The lead candidates in the Mfa/AgI/II inhibitor evaluation exemplify a number of features common to many protein-protein inhibitor interaction problems. The most active candidates and their development involve leads that have characteristic shapes or topologies, significant hydrophobicity and the conjunction of several low to high affinity pharmacophores.28 The optimization process involving many of the compounds described in this communication is ongoing and will further reveal many of the properties and interactions responsible for inhibitory activity. As newer compounds are involving in this Mfa/AgI/II ‘click’ study, they will be the subject of future communications from these laboratories.
5. Experimental
5.1 Chemistry
5.1.1 General Methods
Unless otherwise specified, all solvents and reagents were ACS grade and were used as supplied. Alkynes 30-39 were commercially available and were used as supplied. All melting points (mp) were determined using a Thomas-Hoover apparatus. Infrared spectra were recorded on a Perkin-Elmer Spectrum Version 10.02.00 instrument and absorptions are reported as reciprocal centimeters (cm−1). Proton (1H) and carbon (13C) nmr spectra were recorded on a Varian INOVA instrument (400MHz, 100MHz for 13C) or a Bruker instrument at (500 MHz, 125MHz for 13C). High resolution mass spectra (HRMS) were performed using electrospray ionization (ESI). All air and moisture-sensitive reactions were run in oven-dried glassware under an atmosphere of dry nitrogen. Gravity-column chromatography was performed on Silica Gel 60 (E. Merck, 7734, 70-230 mesh). Thin-layer chromatography (TLC) was performed with glass-backed plates (E. Merck, 5715, Silica Gel 60 F254 2.5mm thickness) and visualized using 2% anisaldehyde in ethanol, 2.5% phosphomolybdic acid in ethanol or 10% sulfuric acid in ethanol.
5.1.2. 2-Oxo-1,2-diphenylethyl-2-chloroacetates (6-9)
The acyloins 1-5 (1.3–2 mmol, 1.0 equiv) were dissolved in dichloromethane (40 mL) at room temperature followed by the addition of 4-dimethylaminopyridine (2–3 mmol, 1.5 equiv) under nitrogen. The resultant solution was cooled (0–5°C) and chloroacetyl chloride (1.7–2.6 mmol, 1.3 equiv) was added by syringe. The reaction mixture was warmed gradually to room temperature and then stirred (16 h) under nitrogen. The reaction mixture was then washed with water (30 mL), 5% aqueous HCl (30 mL), and 5% aqueous sodium bicarbonate (30 mL). The organic layer was separated, dried over anhydrous sodium sulfate, filtered and concentrated. The crude residues was purified by gravity-column chromatography (hexane/ethyl acetate, 9:1) to provide the pure chloroacetyl esters 6-9.
5.1.3. 2-(Azidomethyl)-4,5-diphenyloxazoles (10-13)
The chloro-acetyl esters 6-9 (1.1–1.7 mmol, 1.0 equiv) were dissolved in glacial acetic acid (50 mL) and to the clear solution was added ammonium acetate (17–27 mmol, 15 equiv). The reaction mixture was then heated at 115°C (oil bath, 3 h) under a nitrogen atmosphere. Upon completion of the reaction as indicated by TLC (hexane/ethyl acetate, 9:1), the mixture was allowed to cool to room temperature. The reaction mixture was then poured into cold water (40 mL), slowly neutralized with saturated aqueous sodium bicarbonate and then extracted with dichloromethane (3 × 25 mL). The organic extracts were combined, dried over anhydrous sodium sulfate and concentrated to obtain the crude residues which were purified by gravity-column chromatography (hexane/ethyl acetate, 9:1) to afford the intermediate chloromethyloxazoles which were used directly in the next step. Each intermediate chloromethyl oxazole (0.6–0.9 mmol, 1.0 equiv) was dissolved in DMF (10 mL) at room temperature. To the clear solution was added sodium azide (0.6–1.0 mmol, 1.1 equiv) and the mixture was stirred at room temperature (3 h) under a nitrogen atmosphere. The reaction progress was monitored by TLC (hexane/ethyl acetate, 9:1) and when completed the DMF was removed under high vacuum. The residual crude oil was then purified by gravity-column chromatography (hexane/ethyl acetate, 9:1) to obtain the corresponding 2-azidomethyl-(4,5)-diphenyloxazoles 10-13.
5.1.4. General Procedure for the Synthesis of the Substituted 2-Oxo-1,2-Diphenylethyl-3-Azidobenzoates 14-17 and the 2-Oxo-1,2-Diphenylethyl-4-Azidobenzoates 22-25
The substituted benzoin 2-5 (1.3–2.0 mmol, 1.0 equiv.) was dissolved in dichloromethane (50 mL) at room temperature. The resulting clear solution was cooled (0–5°C) followed by addition of DMAP (2.0–3.0 mmol, 1.5 equiv.). 3- or 4-azidobenzoyl chloride (prepared by treating the corresponding carboxylic acids (1.7–2.6 mmol, 1.3 equiv.) with thionyl chloride), dissolved in dichloromethane (20mL) was added dropwise to the cooled solution while stirring under nitrogen. After the reaction was complete, the reaction mixture was washed with 5% aqueous HCl (2 × 50 mL) followed by 5% aqueous sodium bicarbonate. The organic layer was separated, and dried over anhydrous sodium sulfate and filtered. Concentration of the filtrate gave a crude residue which was chromatographed on silica gel (hexane/ethylacetate, 9:1) to afford the pure 3-azidobenzoyl esters 14-17 or 4-azidobenzoyl esters 22-25.
5.1.5. General Procedure for the Synthesis of 2-(3-azidophenyl) 4,5-diphenyloxazoles (18-21) or 2-(4-azidophenyl)-4,5-diphenyl-oxazoles oxazoles (26-29)
The 3-azidobenzoyl esters 14-17 or the 4-azidobenzoyl esters 22-25 (0.4–0.5 mmol, 1.0 equiv) were dissolved in glacial acetic acid (50 mL) at room temperature. To the clear solution was added ammonium acetate (6–8 mmol, 15 equiv) under a nitrogen atmosphere and the resulting solution was heated at 115 °C (oil bath, 3h). After completion of the reaction as indicated by TLC (hexanes/ethyl acetate, 9:1), the reaction mixture was allowed to cool to room temperature, poured into cold water (30 mL) and slowly neutralized with saturated aqueous sodium bicarbonate. The mixture was then extracted with dichloromethane (3 × 25 mL) and the organic extracts were combined and dried over anhydrous sodium sulfate. Removal of the drying agent and concentration gave the corresponding crude 2-(3-azidophenyl)-4,5-disubstituted oxazoles 18-21 or the 2-(4-azidophenyl0-4,5-diphenyloxazoles 26-29. Both series of compounds were further purified by gravity-column chromatography on silica gel (hexane/ethyl acetate, 9:1).
5.1.6. N2,N4-Diisopentyl-6-(2-(trimethylsilyl)ethynyl)-1,3,5-triazine-2,4-diamine 92
To a solution of ethynyltrimethylsilane (0.53g, 0.76mL, 5.4mmol) in dry THF (5.0mL) was added n-butyllithium (1.6M solution in hexane, 3.37mL, 5.4mmol) by syringe at 0°C under argon while stirring. After one hour, the resultant solution of lithioethynyltrimethylsilane was canulated dropwise onto a solution of cyanuric chloride (1.0g, 5.4mmol) in dry THF (5.0mL) while stirring. The addition of the lithiated acetylene resulted in a viscous red-brown suspension. Stirring was continued (2h/0°C), then isopentylamine (1.89g, 2.51mL, 10.8mmol) was added dropwise by syringe at 0°C. The reaction mixture was allowed to come to room temperature and stirring was continued (48h). Column chromatography of the crude mixture on silica gel provided pure triazine 91 (0.96g, 51.1%).
5.1.7. 6-Ethynyl-N2,N4-diisopentyl-1,3,5-triazine-2,4-diamine 40
To a prechilled (0°C) solution of N2,N4-diisopentyl-6-((trimethylsilyl)ethynyl)-1,3,5-triazine-2,4-diamine (500 mg, 1.4 mmol) in dry THF (25 mL) was slowly added tetra-n-butylammonium fluoride (TBAF, 0.72 mL, 0.72 mmol) under a nitrogen atmosphere. The resulting solution was stirred at 5–10°C (1 h). After the reaction was complete as confirmed by TLC, cold water (25 mL) was added to the reaction mixture followed by extraction with dichloromethane (2 × 25 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated to obtain crude residue. The residue was purified by column chromatography (hexane/EtOAc, 7/3→hexane/EtOAc, 3/7) to give pure 40 (331mg, 84%) as light yellow solid: Rf. 0.48 (CHCl3/MeOH, 9:1).
5.1.8. General Procedure for the Synthesis of Click Compounds (41-90)
To a solution of Group I (41-66), Group II (20-21) or Group III (26-27) azides (0.1–0.18 mmol) in anhydrous THF (1.5 mL) were added alkynes 30-40 (0.11–0.2 mmol) and solid copper sulfate (0.1–0.18 mmol) at room temperature. To this mixture was then added a solution of sodium ascorbate (0.5–0.09 mmol) in water (1 mL). The resulting reaction mixture was stirred at room temperature (16 h). The progress of reaction, as indicated by formation of the less-mobile product, was monitored by TLC. After completion of reaction as indicated by TLC, the THF was removed under vacuum and the residue was partitioned between dichloromethane (15 mL) and water (10 mL). The organic layer was separated, dried over anhydrous sodium sulfate and filtered. Concentration of the filtrate afforded the usually solid crude residues which was purified by column chromatography (hexanes/EtOAc, 7.5:2.5; or chloroform/methanol, 9:1). to give the corresponding pure triazoles 41-90 (See Supporting Information).
5.2 Biology
5.2.1. Bacterial Strains and Culture Conditions
P. gingivalis ATCC33277 was grown in reduced trypticase soy broth (Difco) supplemented with 0.5 percent yeast extract, 1 μg/mL menadione, and 5 μg/mL hemin. Twenty five milliliters of media were reduced for 24 hours under anaerobic conditions by equilibrating in an atmosphere consisting of 10% CO2, 10% H2, and 80% N2. Following equilibration, P. gingivalis was inoculated in the media and grown for 48 hours at 37 °C under anaerobic conditions. S. gordonii DL-1 (1) was cultured aerobically without shaking in brain heart infusion (BHI) broth supplemented with 1 percent yeast extract for 16 hours at 37 °C. For some compounds that inhibited P. gingivalis adherence to S. gordonii, we determined their affect the planktonic growth of P. gingivalis and S. gordonii. Broth cultures were grown as described above and compared to cultures grown in the absence of the compound. The concentration of each compound used in the growth inhibition experiments is shown in Tables 2, 3 and 4. Cell density of each culture was determined by measuring the optical density at 600nm (O.D.600nm) after 24 hrs of growth. The percent growth inhibition/stimulation was calculated using the following equation: [(O.D.600nm of the control/O.D.600nm of the culture grown in the presence of compound) − 1] × 100.
5.2.2. Biofilm Model for in vitro Analysis of P. gingivalis Adherence to S. gordonii
To prepare bacterial cells for biofilm culture, 10 ml of an overnight S. gordonii culture was centrifuged at 5600 rpm for 5 min and the cell pellet was suspended in 1 mL sterile PBS. Subsequently, 20 μL of 5 mg/ml hexidium iodide (Molecular Probes) was added to the cell suspension and incubated for 15 min with gentle shaking at room temperature in the dark. The labeled cells were centrifuged as described above, washed with phosphate buffered saline (10 mM Na2HPO4, 18 mM KH2PO4, 1.37 M NaCl, and 2.7 mM KCl, pH 7.2; [PBS]) and the cell pellet was suspended in PBS at a final O.D.600nm of 0.8. Similarly, 10 mL of a 48 hr culture of P. gingivalis was centrifuged at 5600 rpm for 15 min, the cell pellet was suspended in 1mL PBS and 20 μl of 5(6)-carboxyfluorescein N-hydroxysuccinimide ester (4 mg/ml, Molecular Probes, Inc.) was added. After incubation for 30 min with gentle shaking at room temperature in the dark, the suspension was centrifuged and washed as described above and suspended in PBS at a final O.D.600nm of 0.4. For biofilm cultures, 1mL of labeled S. gordonii cells was added to each well of a 12-well microtiter plate (Greiner Bio-one) containing a circular coverslip (Fisher brand) and incubated in an anaerobic chamber with rotary shaking for 24 hrs at 37 °C. Unbound cells were removed by aspiration and 1 ml of labeled P. gingivalis cells containing the desired concentration of test compound was added and incubated under anaerobic conditions for 22 hrs at 37 °C. Test compounds were dissolved in dimethylsulfoxide (DMSO) to generate 1000X stock solutions and were routinely tested over a final concentration range of 0 – 60 μM. 1 μL of the appropriate stock solution was added to each 1 mL aliquot of labeled P. gingivalis cells and incubated for 30 minutes at room temperature prior to adding the suspension to the microtiter plate wells. For control biofilms, 1 μL of DMSO was added to 1 mL of labeled P. gingivalis and incubated as described above.
5.2.3. Visualization of Biofilm Formation
To visualize P. gingivalis/S. gordonii biofilm formation, unbound P. gingivalis cells were removed by aspiration and coverslips were washed once with PBS. Biofilms were fixed by incubating the coverslips with 1 mL of 4% paraformaldehyde for 5 min followed by two washes with PBS. The coverslips were then removed, placed face down on a glass microscope slide containing a drop of anti-fade reagent (Life Technology) and sealed with nail polish. Visualization of biofilms was carried out by laser scanning confocal microscopy using a Leica SP8 confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL) using a 488 nm laser to detect labeled P. gingivalis and a 552 nm laser to detect S. gordonii. Z-plane scans of 25 μm in depth were collected at three randomly chosen frames on each coverslip using a z-step thickness of 0.7 μm. Background noise was minimized using software provided with the Leica SP8 and three dimensional reconstruction of the Z-plane scans and quantification of total green and red fluorescence was conducted using Volocity 6.3 Image analysis software (Perkin Elmer, Akron, Ohio). Data was expressed as the ratio of total green (P. gingivalis) to red (S. gordonii) fluorescence and the IC50 for each compound was defined as the concentration that reduced the ratio of green to red fluorescence by 50%. Experiments were carried out in triplicate for each concentration of test compound and three independent experiments were conducted for each compound. Graphpad InStat3 software was used for data analysis and statistical significance was defined as p < 0.05.
Supplementary Material
Acknowledgments
The measurement of high resolution mass spectra by the Texas A&M University Laboratory for Biological Mass Spectrometry, Dr. Vanessa Santiago and Dr. Bo Wang are acknowledged. Financial support from the NIH/NIDCR through grant 1RO1DE023206 is gratefully acknowledged.
Footnotes
Supplementary data (The 1H, 13C, FTIR) for Compounds 6-29, 40-90 and 92; and HRMS data for new compounds 7-9, 11-29, 40-90 and 92 along with experimental procedures associated with this article can be found, in the online version at http://dx.doi.org/.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr J Clin Periodontol. 1998;25:134. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 2.Wright CJ, Burns LH, Jack AA, Back CR, Dutton LC, Nobbs AH, Lamont RJ, Jenkinson HF. Mol Oral Micorbiol. 2013;28:83. doi: 10.1111/omi.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamont RJ, El-Sabaeny A, Park Y, Cook GS, Costerton JW, Demuth DR. Microbiol. 2002;148:1627. doi: 10.1099/00221287-148-6-1627. [DOI] [PubMed] [Google Scholar]
- 4.Park Y, Simionato MR, Sekiya K, Murakami Y, James D, Chen W, Hackett M, Demuth DR, Lamont RJ. Infect Immun. 2005;73:3983. doi: 10.1128/IAI.73.7.3983-3989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daep CA, James DM, Lamont RJ, Demuth DR. Infect Immun. 2006;74:5756. doi: 10.1128/IAI.00813-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daep CA, Lamont RJ, Demuth DR. Infect Immun. 2008;76:3272. doi: 10.1128/IAI.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daep CA, Novak EA, Lamont RJ, Demuth DR. Infect Immun. 2011;79:67. doi: 10.1128/IAI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda K, Tribble GT, Tucker CM, Anaya C, Shizukuishi S, Lewis JP, Demuth DR, Lamont RJ. Mol Microbiol. 2008;69:1153. doi: 10.1111/j.1365-2958.2008.06338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mocharla VP, Colasson B, Lee LV, Roper S, Sharpless KB, Wong CH, Kolb HC. Angew Chem Int Ed. 2005;44:116. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 10.Borman S. Chem and Eng News. 2002;80:29. [Google Scholar]
- 11.Patil PC, Luzzio FA, Demuth DR. Tetrahedron Lett. 2015;56:3039. doi: 10.1016/j.tetlet.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loner CM, Luzzio FA, Demuth DR. Tetrahedron Lett. 2012;53:5641. doi: 10.1016/j.tetlet.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demuth DR, Luzzio FA. US 9167820 B2 . Chem Abstr. 2013;158:243963.
- 14.Hua Y, Flood A. Chem Soc Rev. 2010;39:1262. doi: 10.1039/b818033b. [DOI] [PubMed] [Google Scholar]
- 15.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JR. ACS Chem Biol. 2008;5:282. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. J Med Chem. 2004;47:600. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 17.Davis JM, Tsou LK, Hamilton AD. Chem Soc Rev. 2007;36:326. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 18.Moses JE, Moorhouse AD. Chem Soc Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 19.Meldal M, Tornøe CW. Chem Rev. 2008;108:2952. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 20.Majumdar KC, Ray K. Synthesis. 2011:3767. [Google Scholar]
- 21.Liu F, Wang Li, Cong L, Wang L, Ou J, Wang M, Liu S. Molecules. 2012;17:2000. doi: 10.3390/molecules17022000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bissantz C, Kuhn B, Stahl M. J Med Chem. 2010;53:5061. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böhm H-J, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. Chem Bio Chem. 2004;5:637. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 24.Wilson CGM, Arkin MR. In: Small Molecule Inhibitors of Protein-Protein Interaction. Vassilev L, Fry D, editors. Springer; Heidelberg: 2011. p. 47. [Google Scholar]
- 25.Yin H, Lee G-I, Hamilton AD. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang E, editor. Wiley; New York: 2009. p. 293. [Google Scholar]
- 26.Priimagi A, Cavallo G, Metrangolo P, Resnati G. Acc Chem Res. 2013;46:2686. doi: 10.1021/ar400103r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auffinger, Hays P, Westof FA, Ho E, PS Proc Nat Acad Sci USA. 2004;101:16789. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrari S, Pellati F, Costi MP. In: Disruption of Protein-Protein Interfaces. Mangani S, editor. Springer-Verlag; Berlin: 2013. p. 34. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.