

The authors show that central nervous system myelin lacking proteolipid protein (PLP) induces mitochondrial dysfunction, including altered motility, degeneration, and ectopic smooth endoplasmic reticulum interactions, leading to axonal structural defects and degeneration. Mutated PLP occurs in hereditary spastic paraplegia, and these cellular effects provide potential insight into the pathology of the disease.
Abstract
Hereditary spastic paraplegia (HSP) is a neurological syndrome characterized by degeneration of central nervous system (CNS) axons. Mutated HSP proteins include myelin proteolipid protein (PLP) and axon-enriched proteins involved in mitochondrial function, smooth endoplasmic reticulum (SER) structure, and microtubule (MT) stability/function. We characterized axonal mitochondria, SER, and MTs in rodent optic nerves where PLP is replaced by the peripheral nerve myelin protein, P0 (P0-CNS mice). Mitochondrial pathology and degeneration were prominent in juxtaparanodal axoplasm at 1 mo of age. In wild-type (WT) optic nerve axons, 25% of mitochondria–SER associations occurred on extensions of the mitochondrial outer membrane. Mitochondria–SER associations were reduced by 86% in 1-mo-old P0-CNS juxtaparanodal axoplasm. 1-mo-old P0-CNS optic nerves were more sensitive to oxygen-glucose deprivation and contained less adenosine triphosphate (ATP) than WT nerves. MT pathology and paranodal axonal ovoids were prominent at 6 mo. These data support juxtaparanodal mitochondrial degeneration, reduced mitochondria–SER associations, and reduced ATP production as causes of axonal ovoid formation and axonal degeneration.
Introduction
Myelin insulates nerve fibers and promotes rapid propagation of action potentials. In addition to its role in nerve conduction, myelin facilitates the long-term survival of axons (Nave and Trapp, 2008). Loss or disruption of this myelin-trophic support results in axonal degeneration, which is the major cause of permanent neurological disability in inherited and acquired diseases of myelin (Trapp and Nave, 2008). Experimental manipulations of myelin gene expression have established that individual myelin proteins play essential roles in providing axonal trophic support and that these trophic functions are independent of any role these proteins play in myelin formation. Null mutations of the myelin-associated glycoprotein (MAG; Yin et al., 1998), proteolipid protein (PLP; Griffiths et al., 1998), and 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP; Lappe-Siefke et al., 2003) cause axonal degeneration. MAG and CNP deletions have little, if any, effect on central nervous system (CNS) myelination, whereas the PLP-null CNS is hypomyelinated. Axonal pathologies that precede axonal degeneration include altered axonal transport and axonal ovoid formation in PLP- and CNP-null mice (Griffiths et al., 1998; Lappe-Siefke et al., 2003) and reduced neurofilament phosphorylation and smaller axonal diameters in MAG-null mice (Yin et al., 1998). These pathologies are most prominent in paranodal regions of the myelin internode, where myelin–axonal communication is most likely to occur.
Hereditary spastic paraplegia (HSP) is a diverse group of genetic disorders with a common clinical feature of lower limb spasticity caused by degeneration of long CNS axons. Over 70 HSP gene loci have been identified, and 22 HSP-causing genes have been cloned (Blackstone, 2012; Novarino et al., 2014). Whole exome sequencing and protein network analysis identified 18 additional gene candidates for HSP (Novarino et al., 2014). Proteins encoded by most of these HSP genes are enriched in axons, where they participate in mitochondrial function (DiMauro et al., 2013), fast axonal transport (Saugier-Veber et al., 1994; Edgar et al., 2004), ER morphogenesis/function (Rismanchi et al., 2008; Montenegro et al., 2012), purine metabolism (Akizu et al., 2013), and membrane trafficking (Wright et al., 2011). Four HSP-associated genes (PLP, fatty acid 2-hydroxylase, connexin 47, and MAG) are enriched in myelinating oligodendrocytes (Novarino et al., 2014). PLP is the most abundant protein in CNS myelin. Deletions and point mutations of the PLP gene cause HSP2 (Saugier-Veber et al., 1994; Starling et al., 2002; Edgar et al., 2004; Inoue, 2005). Mice null for PLP develop a late-onset (>12 mo) axonal degeneration characterized by altered axonal transport and axonal ovoids that initially accumulate in juxtaparanodal axoplasm (Griffiths et al., 1998). We generated a mutant mouse model (referred to as P0-CNS mice) in which PLP is replaced with P0 protein, the major structural protein of peripheral nervous system myelin. P0-CNS and PLP-null optic nerves display similar axonal pathology (Griffiths et al., 1998; Edgar et al., 2004; Yin et al., 2006). Compared with PLP-null mice, P0-CNS mice have shorter myelin internodes, more paranodal axoplasm, increased axonal pathology, increased neurological disability, and less than half the life span of PLP-null mice (Yin et al., 2006). P0-CNS mice, therefore, develop an accelerated form of HSP2, where axonal structural pathology is prominent by 6 mo of age. Although experimentally induced P0-CNS mice display many of the pathological and clinical features of HSP, there is no naturally occurring mammalian counterpart. Because of the rapid progression of axonal pathology, P0-CNS mice provide a more convenient HSP animal model than the original PLP-null mice and were used in this study.
Although little is known about the mechanisms underlying myelin-induced axon ovoid formation, an imbalance between axonal energy supply and demand has been postulated to cause axonal degeneration in primary diseases of myelin (Trapp and Stys, 2009; Campbell and Mahad, 2012). Because of their length, axons must depend on their local environment for energy substrates. Up to 98% of the axonal surface is covered by myelin. Therefore, the oligodendrocyte is a likely source of axonal ATP substrates. Recent studies support the concept that oligodendrocytes transfer ATP substrates to axons via monocarboxylate transporter 1 (MCT1), which is enriched in paranodal regions of the myelin internode (Lee et al., 2012; Morrison et al., 2013; Saab et al., 2013, 2016). In the present study, we find that mitochondrial dysfunction/degeneration and reduced axonal ATP production are prominent features of axonal pathology in P0-CNS mice. Because P0-CNS mice have clinical and pathological phenotypes similar to those caused by mutations in proteins enriched in or associated with axonal mitochondria and smooth ER (SER), our experiments suggest that reduced mitochondrial function in juxtaparanodal axoplasm may be a convergent mechanism of axonal degeneration in many forms of HSP and possibly other neurodegenerative diseases.
Results
Axonal mitochondria in P0-CNS axons
We used serial blockface scanning 3D EM to reconstruct segments of 1-mo-old wild-type (WT; Fig. 1, A–C) and P0-CNS (Fig. 1, D–F) optic nerve axons at nanometer resolution. We reconstructed axonal mitochondria (Fig. 1, C and F; individual mitochondria are displayed as different colors) and compared their numbers and sizes in juxtaparanodal axoplasm (mitochondria within 15 µm of nodes) and internodal axoplasm (located >15 µm from the node). Compared with WT axons, there was a significant increase (approximately twofold) in the number of juxtaparanodal mitochondria in P0-CNS axons (Fig. 1 G). The number of internodal mitochondria was similar in P0-CNS and WT axons (Fig. 1 G). Individual mitochondria in both juxtaparanodal and internodal P0-CNS axoplasm, however, were significantly shorter (Fig. 1 H) and slightly thicker (Fig. 1 I) when compared with WT mitochondria. Compared with WT axons, the volume of individual mitochondria in P0-CNS fibers was decreased by ∼50% in juxtaparanodal axoplasm (Fig. 1 J). Because of increased mitochondrial numbers, however, total mitochondrial volume was significantly increased in P0-CNS juxtaparanodal axoplasm when compared with mitochondrial volume in WT juxtaparanodal axoplasm (Fig. 1 K).
Figure 1.

Mitochondria in 1-mo-old WT and P0-CNS optic nerve axons. (A–F) 3D EM reconstructions of 1-mo-old WT (A–C) and P0-CNS (D–F) optic nerve segments showing 3D organization of fibers (A and D), a single-slice view of a myelinated axon (B and E), and 3D reconstructions of mitochondria in single axons (C and F; individual mitochondria are shown as different colors). Bars, 1 µm. m, mitochondria. (G) Mitochondrial numbers were increased in P0-CNS juxtaparanodal axoplasm and were similar in WT and P0-CNS internodal axoplasm. (H and I) Compared with mitochondria in WT axons, individual mitochondria in P0-CNS axons were significantly shorter (H) and slightly thicker (I). (J) The volumes of individual mitochondria were decreased in P0-CNS juxtaparanodal axoplasm and internodal axoplasm. (K) Total mitochondrial volumes were increased in P0-CNS juxtaparanodal axoplasm and were similar in WT and P0-CNS internodal cytoplasm. Jxpn, juxtaparanodal; Ax, axon; P, paranode; N, node. (G–K) Bar, median; box, 25–75%; whiskers show range; **, P < 0.01; ***, P < 0.001; WT versus P0-CNS, Wilcoxon signed-rank test with Bonferroni’s correction for multiple comparisons. Data were pooled for mitochondria (H–J, n = 44, 115, 210, and 198) and axons (G and K, n = 19, 22, 37, and 40) from three animals for analysis.
Because axonal pathology is most prominent in distal juxtaparanodes of adult P0-CNS axons (Yin et al., 2006), we compared mitochondrial numbers and axonal diameters in distal and proximal juxtaparanodal axoplasm in 1-mo-old WT (Fig. 2 A) and P0-CNS optic nerve fibers (Fig. 2 B). Mitochondrial numbers (Fig. 2 C) and axonal diameters (Fig. 2 D) were significantly increased in P0-CNS distal juxtaparanodal axoplasm when compared with WT distal juxtaparanodal axoplasm. Similar, but less dramatic, changes in mitochondrial numbers were present in proximal juxtaparanodal axoplasm (Fig. 2 C). The increase in distal juxtaparanodal axon diameter correlated with the positioning of mitochondria beneath the axolemma (Fig. 2 B). In 1-mo-old P0-CNS axons, mitochondria did not accumulate beneath Schmidt-Lanterman incisures (SLIs; Fig. S1, A and B), which form in P0-CNS internodes (Yin et al., 2008), nor were they accompanied by accumulations of vesicles and dense bodies, which were present in 6-mo-old P0-CNS mice (Fig. S1 C). We next asked whether mitochondria in 1-mo-old P0-CNS axons displayed ultrastructural changes associated with dysfunction and/or degeneration and whether these changes were more prominent in juxtaparanodal axoplasm than in internodal axoplasm. Mitochondria were binned into four groups: elongated with normal cristae (Fig. 2 E), round with normal cristae (Fig. 2 F), round with reduced/abnormal cristae (Fig. 2 G), and round with few or no cristae (Fig. 2 H). There was little difference in the distributions of mitochondrial morphologies in the internodal and juxtaparanodal axoplasm of WT myelinated fibers or in the internodal axoplasm of P0-CNS myelinated fibers (Fig. 2 I). Elongated mitochondria comprised ∼70% of the mitochondria in both internodal (69%) and juxtaparanodal (73%) regions of WT axoplasm. 16% were round with normal cristae, and 11% were round with reduced or absent cristae. In 1-mo-old P0-CNS juxtaparanodal axoplasm, only 22% of mitochondria were elongated, 44% were round with normal cristae, and 34% were round with reduced or absent cristae (Fig. 2 I). These data establish that mitochondrial pathology is a prominent feature in juxtaparanodal axoplasm from 1-mo-old P0-CNS myelinated fibers. We also considered whether microtubule (MT) loss in juxtaparanodal axoplasm may have accounted for mitochondrial accumulation in 1-mo-old P0-CNS axons. WT juxtaparanodal/nodal axoplasm (Fig. 2 J) and the core of the P0-CNS juxtaparanodal/nodal axoplasm (Fig. 2 K) contained similar MT orientations (Fig. 2, J and K, green lines) and MT densities (Fig. 2 L). Thus, disassembly of MTs does not appear to be responsible for mitochondrial accumulation and displacement in 1-mo-old P0-CNS juxtaparanodal axoplasm.
Figure 2.

Mitochondrial pathology and MT distribution in juxtaparanodal regions of 1-mo-old P0-CNS optic nerve axons. (A) An electron micrograph shows a mitochondrion in WT juxtaparnodal axoplasm. (B) Mitochondria accumulated in P0-CNS juxtaparanodal axoplasm and were positioned beneath the axolemma. (C and D) Mitochondrial numbers (C) and axonal diameters (D) were similar in WT proximal and distal juxtaparanodal axoplasm (A). Compared with WT axons, mitochondrial numbers (C) were increased in P0-CNS juxtaparanodal axoplasm, and this resulted in increased axonal diameters (D). These increases were greater in distal compared with proximal juxtaparanodal axoplasm (B–D). (E–H) Mitochondrial morphology was binned into elongated with normal cristae (E), rounded with normal cristae (F), rounded with reduced cristae (G), and round with sparse or no cristae (H). (I) Most WT and internodal P0-CNS mitochondria were elongated, but most P0-CNS juxtaparanodal mitochondria were rounded with increased frequency of disrupted cristae. (J and K) MTs were traced in WT (J, green overlay) and P0-CNS juxtaparanodal axoplasm (K, green overlay). Bars, 0.5 µm. (L) MT density was similar in WT and the core of P0-CNS juxtaparanodal axoplasm. MTs were rarely detected in the swollen axoplasm that contained displaced mitochondria (B and K). m, mitochondria. (C, D, and L) Mean ± SEM of axonal values from three animals; *, P < 0.05; **, P < 0.02; t test.
Mitochondrial outer membrane extensions and SER associations are altered in P0-CNS axons
Mitochondria and SER interact morphologically and functionally (Hayashi et al., 2009). To date, knowledge of the 3D distribution of SER in WT nerves is limited, and studies have not examined whether axonal SER or mitochondria–SER associations are altered in myelin diseases. To investigate the integrity of the SER network and its physical association with mitochondria, we traced SER and mitochondria in serial 3D EM images from 1-mo-old WT (Fig. 3, A–C) and P0-CNS (Fig. 3, D–F) myelinated optic nerve axons. Axonal SER cisternae formed a continuous and highly branched network of tubular structures (10–30 nm wide) in WT myelinated axons (Fig. 3 B). The overall distribution/density of SER was similar in WT (Fig. 3 B) and P0-CNS (Fig. 3 E) axons, with ∼3–4 µm of SER length per micrometer cubed of axoplasm (Fig. 3 G). The SER in P0-CNS axons, however, was often fragmented and was not as continuous as WT axonal SER (Fig. 3 E). Blunt ends of SER tubules were increased 2.3-fold in the juxtaparanodal axoplasm of P0-CNS fibers (Fig. 3 H).
Figure 3.

Mitochondria–SER associations and mitochondrial outer membrane extensions in 1-mo-old WT and P0-CNS optic nerve axons. (A–F) Images of 1-mo-old WT (A–C) and P0-CNS (D–F) optic nerves showing a single-slice view of myelinated axons by EM (A and D), 3D projections of SER (B and E), and individual mitochondria (C and F, multiple colors). m, mitochondria. (G and H) SER length relative to axonal volume was similar in WT and P0-CNS juxtaparanodal and internodal regions. (H) However, the number of SER termini per micrometer cubed of the axon was 2.5-fold greater in P0-CNS juxtaparanodal regions. (I and J) Extensions of the mitochondrial outer membranes occurred at the longitudinal ends of WT mitochondria (I, arrows). The SER closely apposed WT mitochondria and outer membrane extensions in single EM slices (I, arrowheads), and this association was fully delineated by 3D reconstruction of segmented SER (J, green) and mitochondria (J, red). (K and L) Mitochondria in P0-CNS juxtaparanodal axoplasm rarely extended outer membranes, and in 3D reconstructions (L), the surrounding SER was fragmented. (M) The percentage of mitochondria with outer membrane extensions was significantly reduced in P0-CNS juxtaparanodal and internodal axoplasm. (N) SER associations with mitochondria were reduced in P0-CNS juxtaparanodal axoplasm (P < 0.002). When present, outer membrane extensions in P0-CNS juxtaparanodal axoplasm were significantly shorter than outer membrane extensions in WT mitochondria or in P0-CNS internodal mitochondria (includes mitochondria with lengths of zero). (O) Total SER associated with mitochondria was greater in WT than in P0-CNS axons. (P and Q) SER associated with the mitochondrial body (P) and outer membrane extensions (Q) was extensive in WT axoplasm but was reduced in P0-CNS juxtaparanodal mitochondria (P) and was rare on extensions (Q). (R) Optic nerve axons contained elongated mitochondria with outer membrane extensions that increased mitochondrial length by two thirds and provided an interface for SER interaction (a). SER–mitochondrial outer membrane extensions (a) had a higher surface area to volume ratio than the main mitochondrion (b). In P0-CNS, increased proportions of rounded mitochondria with shorter or absent extensions (c) and SER fragmentation (e) reduced SER associations. (G, H, and M) Mean ± SEM; t test; means of three animals (1,528 mitochondria). (N–Q) Box plots: bar, median; box, 25–75%; whiskers, 97.5–2.5%. Wilcoxon signed-rank test with Bonferroni’s correction; mitochondria (n = 28, 42, 63, and 62) pooled from three animals. *, P < 0.05; **, P < 0.01; ***, P < 0.005. Bars: (A–F) 1 µm; (I–L) 0.5 µm.
We next compared the relationship between SER and mitochondria in WT (Fig. 3, I and J) and P0-CNS (Fig. 3, K and L) axons and measured the length of SER that apposed the mitochondrial outer membrane by <50 nm (Fig. 3, M–Q). In addition to SER–outer membrane associations on the main body of the mitochondrion, 3D reconstructions identified lateral extensions of the mitochondrial outer membrane (Fig. 3 I, arrows). These extensions had a luminal diameter of ∼120 nm (Fig. 3 I), and mitochondrial inner membranes were rarely present. Approximately 70% of the mitochondria in 1-mo-old WT optic nerves had mitochondrial outer membrane extensions (Fig. 3, I and J), which increased total mitochondrial length by ∼66% (Fig. S2 A). The median mitochondrial length was ∼1.7 µm (Fig. 1 H), whereas the median length of mitochondrial outer membrane extensions was ∼1.2 µm (Fig. 3 N). Three quarters of elongated mitochondria had extensions, as did half of the round mitochondria with normal cristae (Table S1). Approximately 10% of round mitochondria with reduced or absent cristae had extensions. Compared with WT axons, there was a 47% reduction in the percentage of P0-CNS internodal mitochondria with outer membrane extensions (Fig. 3 M) and a 69% decrease in the number of juxtaparanodal mitochondria with extensions (Fig. 3 M). Mitochondrial outer membrane extensions were significantly shorter in P0-CNS juxtaparanodal axoplasm (Fig. 3 N).
In WT axons, the total SER on mitochondria was slightly greater for juxtaparanodal mitochondria (2.4-µm median length) than for internodal mitochondria (1.7-µm median length; Fig. 3 O). In WT juxtaparanodal axoplasm, 1.8 µm of SER (median length) was associated with the body of the mitochondria (Fig. 3 P), and 0.6 µm was associated with mitochondrial outer membrane extensions (Fig. 3 Q). Total SER associated with mitochondria was significantly reduced in both P0-CNS internodal (1.0-µm median value) and juxtaparanodal (0.5-µm median value) axoplasm (Fig. 3 O). In P0-CNS juxtaparanodal axoplasm, SER associated with mitochondria was reduced by 86% (Fig. 3 O). SER associated with the mitochondrial body (0.6 µm) was reduced by 66% (Fig. 3 Q and Fig. S2 B), and SER associated with mitochondrial outer membrane extensions were reduced by almost 100% (Fig. 3 P and Fig. S2, C and D). As shown schematically in Fig. 3 R (a), extensions on mitochondria provide an increased interface for mitochondrial outer membrane–SER associations. The smaller luminal diameter of the extensions may also provide a higher surface area to volume ratio for molecular exchange than on the mitochondrial body (Fig. 3 R, b). Reductions in mitochondrial length and outer membrane extension size in rounded mitochondria (Fig. 3 R, c and d) decrease mitochondria–SER associations in P0-CNS axons. Fragmentation of the SER (Fig. 3 R, e) further reduces mitochondria–SER associations and disrupts SER continuity.
Axonal function and ATP production
To investigate whether reduced ATP production is associated with mitochondrial degeneration and reduced mitochondria–SER associations in P0-CNS optic nerves, we compared 1-mo-old WT and P0-CNS nerves. The drop in ATP levels (Fig. 4 B, orange) was further validated in experiments where optic nerves were exposed to a metabolic challenge caused by oxygen-glucose deprivation (OGD; Fig. 4 A). Optic nerve function was monitored quantitatively as the area under the supramaximal compound action potential (CAP). The CAP area is proportional to the total number of excited axons and represents a convenient and reliable means of monitoring optic nerve axon function (Cummins et al., 1979; Stys et al., 1991). When challenged with 30 min of OGD, P0-CNS optic nerves (Fig. 4 A, orange circles) show an accelerated loss of function (Fig. 4, A and E; 20 min for P0-CNS and 30 min for control, P = 0.008) compared with control nerves (Fig. 4 A, green circles). There was also a statistically significant reduction (P = 0.03) in the percentage of P0-CNS axons recovering from 30 min of OGD (Fig. 4 A, inset). The rapid loss of axon function and the reduction in nerve function recovery are most likely caused by decreased ATP production, as previously shown (Baltan et al., 2008; Baltan, 2009), and not by glutamate excitotoxicity because 30 min of OGD leads to minimal glutamate accumulation (Tekkök et al., 2007).
Figure 4.
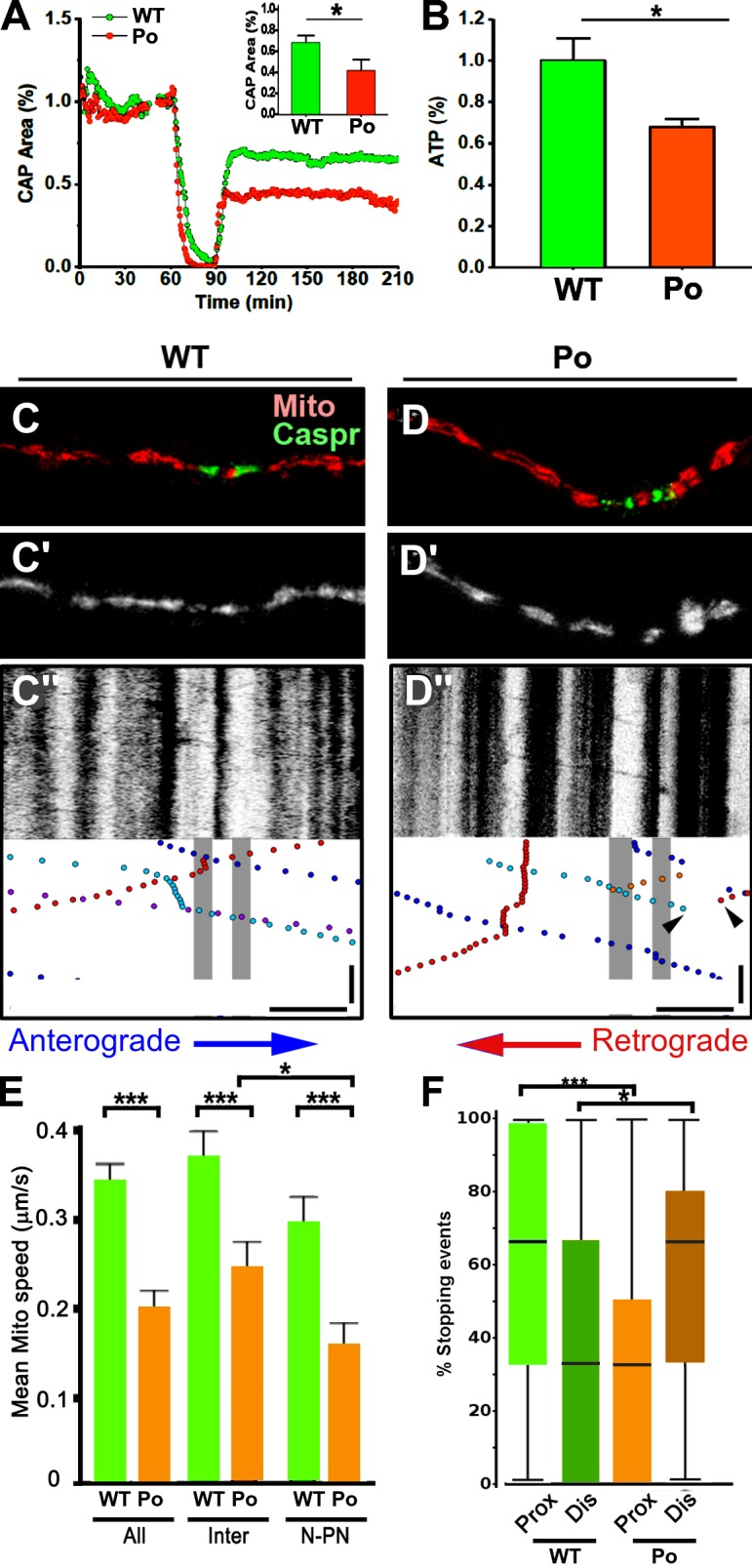
Increased vulnerability to energy deprivation and altered mitochondrial transport in optic nerves from P0-CNS mice. (A) Recordings of CAPs in 1-mo-old P0-CNS mice (orange circles, n = 4) showed a rapid loss of CAPs and reduced recovery after 30 min of OGD compared with age-matched controls (green circles, n = 6). The inset bar graph summarizes recovery at the end of 2 h after OGD exposure. *, P = 0.026. (B) ATP levels were reduced in optic nerves from 1-mo-old P0-CNS mice compared with controls. ATP levels were corrected to protein and normalized to WT controls. *, P = 0.015. (C–F) In organotypic slice cultures, mitochondrial transport was altered in myelinated P0-CNS axons. (C and D) Node locations were identified after time-lapse imaging by immunostaining for Caspr (green) and registered to axonal Mito-Dendra2 labeling (red). Live time-lapse images of mitochondria (C′ and D′) are converted to kymographs (C″ and D″), where cessation of mitochondrial transport is frequently observed in P0-CNS axons (P0; D″, arrowheads). Bars: (horizontal) 10 µm; (vertical on kymograph) 100 s. (E) The mean speed of motile mitochondria was significantly reduced in P0-CNS axons compared with WT axons over the entire axon segment studied (All) and within internodal (Inter) and nodal–paranodal axoplasm (N-PN). The mean mitochondrial speed in P0-CNS axons was significantly reduced in nodal–paranodal axoplasm compared with internodal axoplasm. Bars and lines are mean + SEM. n = 27, 28, 23, 27, 22, and 24. (F) Mitochondrial stopping events were quantified in proximal (Prox) and distal (Dis) regions of the nodes. Stopping events were more common in the proximal juxtaparanode in WT axons and in the distal juxtaparanodal axoplasm in P0-CNS axons. Bars with boxes and lines are median, interquartile range, and maximum/minimum. *, P < 0.05; ***, P < 0.001; t test; n = 32 and 23 (F) axons from three WT and three P0-CNS explants.
Mitochondrial transport in P0-CNS axons
The accumulation of organelles in axonal ovoids in PLP-null and P0-CNS axons has been associated with impaired axonal transport (Griffiths et al., 1998; Edgar et al., 2004; Yin et al., 2006). We compared axonal mitochondrial distribution and transport in myelinated organotypic slice cultures from WT and P0-CNS cerebella (Fig. 4, C–F) as described previously (Ohno et al., 2011). Time-lapse images of fluorescently tagged Mito-Dendra2–positive axonal mitochondria were recorded in myelinated Purkinje cell axons, and kymographs of stationary and motile mitochondria were generated (Fig. 4, C’, C”, D’, and D”). After live imaging, cultures were fixed, and nodal regions of the imaged axons were delineated by immunostaining for Caspr and registered with live imaging of the same fiber (Fig. 4, C and D). Large stationary mitochondria did not translocate in either the anterograde or retrograde directions (Fig. 4, C” and D”, vertical stripes). Compared with WT axons, the mean transport speed of small motile mitochondria in P0-CNS axons was reduced by 40% over the total length of the axon (Fig. 4 E, All), by 38% in internodal axoplasm (Fig. 4 E, Inter), and by 50% in nodal–juxtaparanodal axoplasm (Fig. 4 E, N-PN). Motile mitochondria preferentially stopped in the proximal juxtaparanodal region in WT axons (Fig. 4 F). In contrast, motile mitochondria preferentially slowed or stopped in the distal juxtaparanodal region in P0-CNS axons (Fig. 4 F). These results demonstrate that mitochondrial transport in myelinated P0-CNS axons is impaired and preferentially affected in distal juxtaparanodal regions.
MT organization and stability in P0-CNS optic nerve axons
MTs are the substrate for fast axonal transport of mitochondria (Hollenbeck and Saxton, 2005). The EM data described in Fig. 2 (J–L) support normal MT orientations and densities in 1-mo-old P0-CNS optic nerves. To investigate whether MT organization was affected in distal paranodal axoplasm of older P0-CNS axons, we examined 6-mo-old WT and P0-CNS axons with high-voltage EM tomography. MTs in WT nodal and paranodal axoplasm were parallel to each other and the longitudinal orientation of the axon (Fig. 5 A). In contrast, paranodal MTs in the P0-CNS optic nerve axon (Fig. 5 B) had significantly shorter lengths (620 nm vs. 1,856 nm in WT) and greater curvature, including some that turned 180°. Thus, MT lengths in P0-CNS optic nerve axons were reduced threefold compared with control nerves (Fig. 5 C). Reduced MT stability appears to play a role in paranodal axonal pathology in 6-mo-old P0-CNS optic nerves. MTs can exist as stable polymers of α/β-tubulins with long assembly/disassembly half-lives, or they can rapidly cycle between de- and repolymerization states. The insoluble/soluble acetylated α-tubulin ratio, which is considered to be an indication of MT stability (Schulze et al., 1987; Baas and Black, 1990), was similar (∼0.8) in 1- and 6-mo-old WT and 1-mo-old P0-CNS optic nerves. In contrast, this ratio was significantly decreased in 6-mo-old P0-CNS optic nerves (Fig. 5 D). These data indicate a decrease in MT stability in 6-mo-old P0-CNS optic nerve axons.
Figure 5.

MT organization and stability are altered in mature P0-CNS optic nerve axons. (A) Axonal MTs in 3D EM tomograms of 6-mo-old WT optic nerves are straight and oriented parallel to the longitudinal axis of the axon. (B) MTs in 6-mo-old P0-CNS optic nerves are disorganized. (C) MT lengths in P0-CNS optic nerve axons are decreased threefold compared with MT lengths in WT axons (P < 0.00000001). n = 46 and 566 MTs (WT and P0-CNS). (D) The soluble/insoluble tubulin ratio, an indicator of MT stability, declines with age in P0-CNS mice. n = 3, 3, 6, and 6 preps (1 mo WT, 1 mo P0-CNS, 6 mo WT, and 6 mo P0-CNS). (E–G) Quantification and localization of phosphorylated tau epitopes in 6-mo-old WT and P0-CNS optic nerve samples. (E and F) The phospo-tau epitopes, Ser199–202, Ser396, and Ser404, were decreased two- to threefold in P0-CNS samples (pooled samples analyzed by Kinexus; see Materials and methods). (F) Single pooled samples are from 10 WT and 20 P0-CNS nerves. (G) Double immunofluorescent labeling for the axonal marker Tuj1 (tubulin-βIII; green) and either the phospho-tau epitope Ser396 (red) or Ser199/202 tau (red) localized these phospho-tau epitopes in axonal ovoids. (H–J) Quantification and localization of GSK3-β, GSK3-β phospho-Ser9, and CDK5 in 6-mo-old WT and P0-CNS optic nerves. (H and I) GSK3-β was increased and GSK3-β phospho-Ser9 was decreased in P0-CNS optic nerves. CDK5 levels were also increased in P0-CNS optic nerves. (C, D, and I) Bars and lines are mean + SEM. ***, P < 0.001; t test. n = 3 and 3 preps (WT and P0-CNS). (J) Staining of 6-mo-old P0-CNS optic nerve sections with neurofilament (NF; green), Tuj1 (tubulin-βIII; green), GSK3-β (red), and CDK5 (red). Expression of GSK3-β (red) and CDK5 (red) was enriched in ovoids. GSK3-β phospho-Ser9 (red) was not detected in ovoids. (G and J) Bars, 5 µm.
Tau phosphorylation is increased in P0-CNS axons
Axonal MT stability is also mediated by the MT-binding protein Tau. Tau stimulates MT polymerization, promotes MT stabilization, and modulates binding of kinesin and dynein to MTs (Hasegawa, 2006). Tau–MT binding is inhibited by phosphorylation within and adjacent to multiple MT-binding domains (Bramblett et al., 1993). We compared the levels of phosphorylation at four tau residues in optic nerve homogenates from 6-mo-old WT and P0-CNS mice (Fig. 5 E) and detected a two- to threefold increase in the phosphorylation of Ser396 and Ser199/Ser202 in P0-CNS samples (Fig. 5 F). Phospho-Ser396 tau flanks MT-binding repeats and modulates MT binding (Bramblett et al., 1993; Johnson and Stoothoff, 2004; Schneider and Mandelkow, 2008). Whole optic nerve biochemical analysis may include other sources of tau, including oligodendrocytes (Anderson et al., 2008, 2010). We colocalized phospho-Ser396 and phospho-Ser202 with the axonal markers tubulin-βIII or neurofilament and confirmed that they were enriched in axonal swellings in P0-CNS optic nerves (Fig. 5 G). Thus, tau hyperphosphorylation may contribute to MT instability in paranodal axonal swellings.
Multiple kinases can phosphorylate tau (Ferrer et al., 2005; Schneider and Mandelkow, 2008), including the β subunit of glycogen synthase 3 (GSK3) and CDK5. In Western blots, the GSK3 isoform (GSK3-β) and phosphorylation of GSK3-β at Ser9 were similar in 1-mo-old WT and P0-CNS optic nerves. In contrast, levels of GSK3-β subunits were significantly increased in 6-mo-old P0-CNS optic nerves, whereas GSK3-β phospho-Ser9 levels were significantly reduced (Fig. 5, H and I). Because GSK3 phosphorylation inactivates GSK3 kinase activity, elevated levels of GSK3 and reduced GSK3 phosphorylation in P0-CNS optic nerves are consistent with increased GSK3 activity and tau phosphorylation. CDK5 was also increased in 6-mo-old P0-CNS optic nerves (Fig. 5, H and I). GSK3-β and CDK5 were enriched in axon ovoids in P0-CNS optic nerves (Fig. 5 J). GSK3-β and GSK3-β phospho-Ser9 staining were faint in normal-appearing axons and present in glial processes. GSK3-β phospho-Ser9 staining was found in glial processes but not in P0-CNS axonal ovoids (Fig. 5 J), which supports increased GSK3-β kinase activity.
Discussion
Our results implicate mitochondrial degeneration and decreased mitochondria–SER associations as primary causes of axonal degeneration in a rodent model of inherited myelin disease. Decreased ATP levels, reduced mitochondrial cristae, and decreased recovery from OGD associate decreased axonal energy metabolism with these mitochondrial pathologies. Reduced mitochondria–SER associations implicate altered Ca2+ homeostasis as an additional contributor to juxtaparanodal axonal pathology in 1-mo-old P0-CNS optic nerves. Collectively, our results support the concept that oligodendrocytes sustain axonal viability by providing ATP substrates to the paranodal axoplasm and that reduced energy production is the likely cause of axonal degeneration in P0-CNS mice. Although the molecular mechanism responsible for this metabolic coupling remains to be established, it is likely to involve multiple oligodendrocyte/myelin proteins, including those that cause HSP (PLP, MAG, connexin 47, and fatty acid 2-hydroxylase).
Loss of PLP function causes axonal degeneration in both mouse and human. We consider loss of PLP function as the underlying cause of axonal pathology in P0-CNS optic nerves. The accelerated rate of axonal degeneration in P0-CNS mice (compared with PLP-null mice) is the result of an increase in juxtaparanodal regions caused by shorter myelin internodes (Yin et al., 2006). Mice with both PLP and P0 have normal life spans and no axonal pathology (Yin et al., 2006), so it is unlikely that P0 expression in CNS myelin directly impacts axonal viability. Expression of P0 in the absence of PLP, however, does induce the formation of SLIs in optic nerve myelin internodes (Yin et al., 2008). SLIs, which are small cytoplasmic channels that connect outer and inner margins of the myelin sheath, are prominent features of mammalian peripheral nervous system myelin but are normally absent from CNS myelin internodes. In adult P0-CNS optic nerve axons, axoplasm beneath SLIs can contain occasional accumulations of mitochondria (Yin et al., 2008). Mitochondria did not accumulate in axoplasm beneath SLIs in 1-mo-old P0-CNS optic nerves (Fig. S1, A and B). Although SLIs may contribute to axonal pathology in adult P0-CNS optic nerves, they do not appear to impact mitochondrial viability in 1-mo-old P0-CNS mice. Because SLIs are not a feature of PLP-null optic nerves, they can contribute to, but are not essential for, CNS axonal pathology.
The pathological hallmark of P0-CNS myelinated axons is accumulation of axonal organelles at distal juxtaparanodes. Our data suggest that this late-onset axonopathy (6–9 mo) is a consequence of reduced ATP production and of mitochondrial degeneration that is apparent at 1 mo of age. MT pathology prominent at 6 mo (decreased length, decreased stability, altered orientation, and increased phosphorylated tau) was not present at 1 mo of age. Accumulation of small vesicles, which is a prominent feature of 6-mo-old P0-CNS juxtaparanodal axoplasm (Fig. S1 C), was not present in 1-mo-old P0-CNS juxtaparanodal axoplasm (Figs. 1 E, 2 B, and 3, D, I, and K). Vesicular fast axonal transport is fueled by the glycolytic enzyme GAPDH, which binds to and travels with axonal vesicles in a huntingtin-dependent fashion (Zala et al., 2013). The dissociation of vesicular transport from mitochondrial respiration assures fast vesicular transport in axoplasm devoid of mitochondria. In contrast, nodal function depends on mitochondrial respiration and oligodendrocyte-derived ATP substrates. The juxtaparanodal enrichment of mitochondrial pathology and ovoid formation in HSP reflects the high-energy demand of saltatory conduction and the select vulnerability of juxtaparanodal mitochondria to mutations in myelin genes.
Stationary mitochondria were increased in 1-mo-old P0-CNS paranodal axoplasm (Fig. 4). Two events could provide a possible mechanism for stationary mitochondrial generation in juxtaparanodal axoplasm. First, juxtaparanodal axonal mitochondria could undergo fission and increase the mitochondrial number. Second, increased stopping of transported motile mitochondria at distal juxtaparanodes (as demonstrated by our time-lapse imaging experiments; Fig. 4) and their fusion with mitochondria could increase mitochondrial volume. As previously demonstrated for demyelinated axons (Kiryu-Seo et al., 2010; Ohno et al., 2011, 2014), P0-CNS axons may increase mitochondrial volume in juxtaparanodal axoplasm as a response to increased energy demands caused by mitochondrial degeneration and reduced ATP production. Degenerating mitochondria are displaced to peripheral juxtaparanodal axoplasm and increase axonal diameter (Fig. 2). When neurofilaments accumulate in axons as the result of ββ′-iminodipropionitrile toxicity, they are also displaced to the peripheral axoplasm (Griffin et al., 1978). Peripheral displacement of altered axonal organelles appears to be a mechanism used by axons to maintain a central core of normal axoplasm.
Recent studies support the concept that MCT1, which is enriched in myelin paranodes, releases lactate that is internalized into paranodal axoplasm and used by mitochondria for oxidative metabolism and ATP biosynthesis (Fünfschilling et al., 2012; Lee et al., 2012; Morrison et al., 2013, 2015; Saab et al., 2013; Hirrlinger and Nave, 2014). Attenuation of MCT1 levels in vivo and in vitro results in axonal degeneration (Lee et al., 2012). Pharmacological inhibition of MCT1 in organotypic slice cultures also causes axonal degeneration, which can be rescued by adding free lactate to the culture medium (Lee et al., 2012). Because lactate transfer cannot be directly measured, sorting primary and secondary effects of lactate deficiency in the environment of mitochondrial degeneration will require manipulation of MCT1 function in oligodendrocytes.
Mitochondrial dysfunction and increased intraaxonal Ca2+ are considered to be convergent mechanisms of axonal degeneration in chronic neurodegenerative diseases (Coleman, 2005). The ability to reconstruct mitochondria and SER in WT and P0-CNS axons at nanometer resolution allowed us to identify and expand previously described extensions of the mitochondrial outer membrane (Spacek and Lieberman, 1980). Approximately 70% of WT optic nerve axonal mitochondria had extensions of their outer membrane, which increased mitochondrial length and mitochondria–SER associations by 66%. Sites of close opposition between mitochondria and SER are referred to as the mitochondria-associated ER membrane (MAM; Hayashi et al., 2009). Although the molecular composition of the MAM is not fully characterized, the SER MAM contains several Ca2+-binding chaperones (Hayashi et al., 2009). It is possible that mitochondrial outer membrane extensions have a unique molecular signature that reflects specialized or enhanced MAM functions. In support of a specialized function, not all CNS axons have mitochondrial outer membrane extensions. Originally described in CNS axons (Spacek and Lieberman, 1980), they are abundant in optic nerves and rare in the corpus callosum and cerebellar axons (Ohno et al., 2011, 2014). They are not common in rodent liver (Ohno et al., 2014) but are a prominent feature of mitochondria in Sertoli cells (Rashed, 2010). Mitochondria in 1-mo-old P0-CNS paranodal axoplasm rarely extended their outer membranes, and the SER was fragmented. The 86% reduction in mitochondria–SER associations in P0-CNS axons may alter Ca2+ buffering and play a pivotal role in juxtaparanodal axonal dysfunction.
Paranodal axonal swellings are a pathological hallmark of HSPs caused by protein mutations that are enriched in myelin and axonal organelles (mitochondria, MTs, and SER; Blackstone, 2012). Axon ovoids are also a common feature in peripheral nerves from individuals with Charcot-Marie-Tooth disease. Charcot-Marie-Tooth is caused by mutations in neuronal or Schwann cell genes and is considered to be the peripheral nerve equivalent of HSP (Timmerman et al., 2013). Correlations between mutated genes and protein network analysis have also identified significant overlap between HSP genes and genes implicated in amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease (Novarino et al., 2014). The major objective of this study was to elucidate the mechanism responsible for axonal degeneration in P0-CNS mice. Mitochondrial degeneration, reduced mitochondria–SER associations, and reduced ATP production are prominent features of axonal degeneration in P0-CNS axons and may drive aspects of axonal degeneration in a variety of CNS diseases.
Materials and methods
Animals
We generated transgenic mice that expressed mouse P0 cDNA (Lemke and Axel, 1985) ligated to a 9.1-kb region of the mouse myelin basic protein promoter. This line has equal amounts of P0 and PLP in CNS myelin (PLP/P0-CNS mice; Yin et al., 2006). By crossing PLP-null mice with PLP/P0-CNS mice, we obtained mice that were null for PLP and homozygous for the P0 transgene (P0-CNS). These mice have been described in detail previously (Yin et al., 2006) and consist of B6129F1 (PLP null), FVB (PLP/P0-CNS mice), and B6129F1/FVB (P0-CNS) backgrounds. Because the PLP gene is X linked, our experiments only included male P0-CNS mice. WT male mice were used as controls and were of the same genetic background as P0-CNS mice.
3D EM reconstruction using serial blockface scanning EM
Tissue preparation and imaging were performed as previously described (Ohno et al., 2011) using en block staining methods (Deerinck et al., 2010). In brief, P0-CNS and WT mice at 1 and 6 mo of age were perfused with 0.1-M sodium cacodylate buffer containing 2.5% glutaraldehyde (Electron Microscopy Sciences) and 4% paraformaldehyde. Optic nerves were dissected, cut into 5-mm segments, postfixed in 0.1% tannic acid in buffer, washed, and then stained successively with osmium ferricyanide, tetracarbohydrazide, aqueous osmium tetroxide, saturated aqueous uranyl acetate, and Walton’s lead aspartate stains, as described previously (Deerinck et al., 2010). Tissues were then dehydrated in graded ethanols and embedded in Procure 812 embedding resin at 60°C for 48 h.
Nerve segments were mounted on aluminum pins, trimmed, surrounded with silver paste, and examined in a Sigma VP scanning electron microscope (ZEISS) fitted with a 3View in-chamber ultramicrotome (Gatan) and a low-kilovolt backscattered electron detector (Gatan). Longitudinally oriented axons were imaged covering areas ∼80 µm wide and 80–400 µm in length. Images were collected at 1.8–2.5 kV, depending on tissue contrast, and up to 500 slices were cut at a thickness of 40–100 nm. Imaging was conducted using a 30-µm aperture in high-current mode and imaged at room temperature (21°C at a chamber vacuum of 10−6 mbar and working distance of 5.7 mm).
Images were reconstructed and registered using ImageJ/FIJI software (National Institutes of Health). Axons, SER, and mitochondria were segmented in each slice by a combination of signal intensity–based thresholding and manual segmentation using ImageJ with FIJI plugins or Reconstruct software (SynapseWeb; Fiala, 2005) and rendered using Blender software or Amira (v. 5.5.0; FEI Visualization Sciences Group), as described previously (Ohno et al., 2011). Volumes were calculated as the sum of areas in each slice multiplied by the slice thickness. Lengths were measured as 3D distances using polyline tools. Axonal and mitochondrial mean diameters were calculated using length and volume and assuming a cylindrical geometry. SER associated with the mitochondrial surface was measured as the length of SER within 50 nm of the mitochondrial surface. SER termini (SER fragmentation) were counted manually on projection images of a 5-µm-long segment of axon and expressed as termini per SER volume. Reconstructed single-slice views were assembled from multiple regions of interest obtained along a user-defined path through the stack (x, y, and z coordinates), e.g., along an axon centerline.
MT stability assay and Western blots
WT and P0-CNS optic nerves from 1- and 6-mo-old mice were homogenized and lysed in an MT stabilization buffer containing 100 mM Pipes, pH 6.9, 5 mM MgCl2, 1 mM EGTA, 30% glycerol, 0.1% Nonidet P-40, 0.1% Triton X-100, 0.1% Tween 20, 0.1% β-mercaptoethanol, 0.001% antifoam, 1 mM dithiothrietol, 1 mM GTP, 10 mM ATP, 10 µM Taxol, and 10 µl protease inhibitor cocktail (Cytoskeleton, Inc.). After centrifugation at 100,000 g for 30 min, supernatants containing soluble tubulin were separated from pellets containing polymerized tubulin. The pellets were dissolved in double-distilled deionized water for Western blotting with acetylated α-tubulin antibodies (mouse monoclonal; Sigma-Aldrich). Data are expressed as the insoluble/soluble acetylated α-tubulin ratio.
Phospho-site–specific kinase screening was performed by Kinexus Bioinformatics Corp. using antibody microarrays. Pooled samples from 20 (1 mo old) and 10 (6 mo old) optic nerves from WT and P0-CNS mice were collected and submitted to Kinexus for antibody microarray (KAM-1.3) testing using 500 Pan-Ab and 300 Phospho-Ab, which included phosphorylation states of tau and kinases. Analysis was undertaken using the KPSS 9.1 program by Kinexus.
Western blotting was performed on WT and P0-CNS optic nerves homogenized in 10 volumes of Tris-buffered saline containing 1 mM EDTA, 1 mM PMSF, and protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentrations were determined by protein assays (Bio-Rad Laboratories). Proteins were separated on 4–12% NuPAGE MES gels (Thermo Fisher Scientific) and then transferred onto polyvinylidene fluoride membranes. Primary antibodies for Western blots included GSK3-αβ (mouse monoclonal; Enzo Life Sciences), 3β-pSer9 (rabbit polyclonal; Cell Signaling Technology), and Cdk5 (mouse monoclonal; EMD Millipore), with GAPDH as a loading control.
Immunocytochemistry
6-mo-old WT and P0-CNS mice were perfused with 4% paraformaldehyde. Optic nerves were removed, fixed overnight, and then cut at a thickness of 10 µm in a cryostat (Leica Biosystems). Sections were double immunostained for axons with Neurofilament-H (rabbit polyclonal; AbD Serotec) or tubulin-βIII (TUJ1; mouse monoclonal; Covance) and GSK3-αβ (mouse monoclonal), GSK3β-pSer9 (rabbit polyclonal), tau–phospho Ser396, tau–phospho Ser202 (rabbit polyclonal; Abcam), or Cdk5 (mouse monoclonal). Secondary antibodies were Alexa Fluor 488 or Alexa Fluor 594 conjugated (Invitrogen). Confocal images were collected using an SP5 confocal microscope (Leica Biosystems) using a 63× 1.4 NA oil immersion lens and acquiring images at room temperature using 488-nm and 561-nm laser lines and 1,024 × 1,024 pixel formats. Step sizes of 0.5 µm were used for z series acquisition. Images were reconstructed using ImageJ/FIJI software.
High-voltage transmission electron microscope tomography
WT and P0-CNS mice were perfused with 4% paraformaldehyde and 2.5% glutaraldehyde. Semi-thin serial sections (0.5 µm) of optic nerves were cut, placed on formvar-coated slotted grids, and stained with 1% uranyl acetate and 1% lead citrate. 3D reconstructions of axonal contents were collected as previously described (Perkins et al., 2008). In brief, for each reconstruction, a series of images at regular tilt increments was collected with a 4000EX intermediate-voltage electron microscope (JEOL USA) operated at 400 kV. Tilt series were recorded on film at ×20K at 2° angles from −60° to 60°. Negatives were digitized to produce images with 1.41 nm/pixel. The IMOD package (Kremer et al., 1996) was used for rough alignment, with fine alignment and reconstruction performed using the TxBR package (Lawrence et al., 2006). Volume segmentation was performed by manual tracing using Xvoxtrace (Perkins et al., 1997), and reconstructions were visualized using Analyze (Mayo Foundation), Synu (Hessler et al., 1996), and Amira (Mercury/TGS) software.
Preparation of optic nerves, recording techniques, and OGD
These procedures were performed as previously described (Baltan et al., 2010, 2011, 2013). In brief, mouse optic nerves were obtained from 1-mo-old P0-CNS and WT mice and placed in a perfusion chamber superfused with artificial cerebrospinal fluid (ACSF) and continuously aerated by a humidified gas mixture of 95% O2/5% CO2. All experiments were performed at 37°C. Suction electrodes back-filled with ACSF were used for stimulation (Isostim 520; WPI) and for recording the CAP. The recording electrode was connected to an amplifier (Axoclamp 900A; Molecular Devices), and the signal was amplified 50 times, filtered at 30 kHz, and acquired at 20–30 kHz. Axon pClamp 10 software (Molecular Devices) was used for data acquisition. Stimulus pulse (50-µs duration) strength was adjusted to evoke the maximum CAP and then increased another 25% for supramaximal stimulation. The supramaximal CAP was elicited every 30 s. OGD stress was induced by switching to glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. OGD was applied for 30 min, ACSF and O2 were restored, and CAPs were recorded for up to 5 h after the end of OGD. Optic nerve function was monitored quantitatively as the area under the supramaximal CAP. Original data were normalized by setting the mean of initial baseline values (measured over 15 min) to a value of 1.0. Results from several nerves were pooled, averaged, and plotted against time. All data are presented as the mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t tests or analyses of variance (ANOVAs). ANOVAs were followed by Bonferroni’s post-hoc test (Prism 4; GraphPad Software). The n values indicate the number of optic nerves.
ATP measurements
ATP extraction was performed as previously described (Khan, 2003) and modified for optic nerve as described in previous publications (Baltan et al., 2010, 2011). In brief, nerves were diced and placed in 75 µl of 10% perchloric acid (HClO4), sonicated, and centrifuged at 4,500 rpm for 10 min at 4°C. The supernatant was neutralized with 30 µl of 2.5 M potassium hydroxide (KOH) and centrifuged at 14,000 rpm for 10 min at 4°C. Total cellular ATP was measured using a bioluminescence assay kit (11 699 709 001; Roche). The absorbance was measured at 560 nm. The blank values were subtracted from the raw data, and ATP concentrations were calculated from a log–log plot of the standard curve and were normalized to protein concentration. The values are expressed as micromoles of ATP per milligram of protein. Protein content was quantified in one nerve, and ATP levels were measured in the other.
Myelinating organotypic cerebellar slice cultures
Tissue preparation and imaging were performed as previously described (Ohno et al., 2011). Myelinating organotypic cultures were generated from 150–200-µm-thick slices of P8-10 cerebellum from WT and P0-CNS mice (Dusart et al., 1997). The slices were cultured on Millicell organotypic culture inserts (EMD Millipore) in 6-well plates containing 50% minimal essential medium, 25% horse serum, 25% HBSS, 6 mg/ml d-glucose, 1× Glutamax (Invitrogen), and penicillin/streptomycin. 2 h after plating, the lentiviral vectors were injected into the Purkinje cell layer using a glass microinjection needle (Kasri et al., 2008).
Lentivirus with mitochondria-targeted Dendra2 was prepared using a pLenti6/V5 directional TOPO cloning kit (Invitrogen) according to the manufacturer’s protocol with slight modifications (Hirai, 2008). In brief, 293FT cells were transfected with pLentiV5/6 lentiviral vector including Mito-Dendra2 (Takara Bio Inc.) and packaging plasmids. The medium was changed 4 h after the transfection. Viral supernatants were collected 24 h after the transfection, filtered with 0.22 µm polyvinylidene fluoride membrane (EMD Millipore), and centrifuged at 100,000 g for 2 h. The viral pellet was suspended in a 1/100 volume of PBS.
Purkinje cell axons with Dendra2-positive mitochondria were imaged by confocal microscopy as reported previously (Ohno et al., 2011). Glass-bottom dishes with culture medium and pieces of Millicell membrane with attached slices were transferred into a microincubator (PDMI-2; Medical System) attached to an inverted laser-scanning confocal microscope (TCS SP; Leica Biosystems). The slices were illuminated with 488-nm light through a 40× oil immersion objective (NA 1.21; Leica Biosystems). Images of mitochondria were collected at 1,024 × 1,024 pixel resolution every 6 s. Imaging at 6-s intervals did not induce mitochondrial fragmentation, acute mitochondrial swelling, or cessation of axonal transport.
Kymographs of mitochondria in time-lapse images were produced using ImageJ as described previously (Miller and Sheetz, 2004). Motile mitochondria were identified as moving diagonal lines in kymographs. Mean velocity of motile mitochondria was measured by dividing total displacement distance by total time in juxtaparanodal (<15 µm from nodes) and internodal (15–40 µm from nodes) axoplasm. After time-lapse imaging, slices were fixed with buffered 4% paraformaldehyde for 6 h and immunostained for Caspr to establish the location of the paranodal/nodal axoplasm (Ohno et al., 2011). The immunostained images were coregistered with kymographs, and stopping events were quantified in proximal and distal juxtaparanodal regions (∼2.5–17.5 µm from the center of nodes).
Statistical analysis
Mitochondrial size, shape, and volume data were tested for normal distributions using the Kolmogorov-Smirnoff test (α = 0.05). Where normally distributed, groups were compared using ANOVAs and two-tailed Student’s t tests with Bonferroni’s correction for multiple comparisons where appropriate. Kruskal-Wallis comparisons and Wilcoxon signed-rank tests (two-tailed with Bonferroni corrections to α levels for multiple comparisons) were used for nonparametric data. Mitochondrial pathology categorical data were compared between groups using Fisher’s exact test. Statistical testing was performed using R or SigmaStat software.
Online supplemental material
Fig. S1 shows electron micrographs of optic nerve axons from 1- and 6-mo-old P0-CNS mice. Fig. S2 shows how mitochondrial outer membrane extensions increase mitochondria–SER associations in optic nerve axons from 1-mo-old mice. Table S1 shows the percentage of mitochondria with normal and pathological morphologies (Fig. 2, E–I) bearing outer membrane extensions.
Supplementary Material
Acknowledgments
The authors would like to thank Peiman Lahsaei for assistance with electron tomography, Emily Benson, Graham Gobieski, and Huan Zhong for assistance with tracing, and Dr. Christopher Nelson for editorial assistance.
This work was partially supported by National Institutes of Health grants 5R01NS038186 (to B.D. Trapp), 5R01AG033720 (to S. Baltan), 5P41RR004050 (to M.H. Ellisman), and P41GM103412-24 (to M.H. Ellisman). N. Ohno was supported by a postdoctoral fellowship from the National Multiple Sclerosis Society (grant FG1834).
The authors declare no competing financial interests.
Footnotes
Abbreviations used:
- ACSF
- artificial cerebrospinal fluid
- ANOVA
- analysis of variance
- CAP
- compound action potential
- CNP
- 2′,3′-cyclic nucleotide 3′-phosphodiesterase
- CNS
- central nervous system
- HSP
- hereditary spastic paraplegia
- MAG
- myelin-associated glycoprotein
- MAM
- mitochondria-associated ER membrane
- MT
- microtubule
- OGD
- oxygen-glucose deprivation
- PLP
- proteolipid protein
- SER
- smooth ER
- SLI
- Schmidt-Lanterman incisure
- WT
- wild type
References
- Akizu N., Cantagrel V., Schroth J., Cai N., Vaux K., McCloskey D., Naviaux R.K., Van Vleet J., Fenstermaker A.G., Silhavy J.L., et al. . 2013. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell. 154:505–517. 10.1016/j.cell.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J.M., Hampton D.W., Patani R., Pryce G., Crowther R.A., Reynolds R., Franklin R.J., Giovannoni G., Compston D.A., Baker D., et al. . 2008. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain. 131:1736–1748. 10.1093/brain/awn119 [DOI] [PubMed] [Google Scholar]
- Anderson J.M., Patani R., Reynolds R., Nicholas R., Compston A., Spillantini M.G., and Chandran S.. 2010. Abnormal tau phosphorylation in primary progressive multiple sclerosis. Acta Neuropathol. 119:591–600. 10.1007/s00401-010-0671-4 [DOI] [PubMed] [Google Scholar]
- Baas P.W., and Black M.M.. 1990. Individual microtubules in the axon consist of domains that differ in both composition and stability. J. Cell Biol. 111:495–509. 10.1083/jcb.111.2.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S. 2009. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 15:126–133. 10.1177/1073858408324788 [DOI] [PubMed] [Google Scholar]
- Baltan S., Besancon E.F., Mbow B., Ye Z., Hamner M.A., and Ransom B.R.. 2008. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J. Neurosci. 28:1479–1489. 10.1523/JNEUROSCI.5137-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S., Inman D.M., Danilov C.A., Morrison R.S., Calkins D.J., and Horner P.J.. 2010. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 30:5644–5652. 10.1523/JNEUROSCI.5956-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S., Murphy S.P., Danilov C.A., Bachleda A., and Morrison R.S.. 2011. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci. 31:3990–3999. 10.1523/JNEUROSCI.5379-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S., Morrison R.S., and Murphy S.P.. 2013. Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics. 10:798–807. 10.1007/s13311-013-0201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C. 2012. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35:25–47. 10.1146/annurev-neuro-062111-150400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramblett G.T., Goedert M., Jakes R., Merrick S.E., Trojanowski J.Q., and Lee V.M.. 1993. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 10:1089–1099. 10.1016/0896-6273(93)90057-X [DOI] [PubMed] [Google Scholar]
- Campbell G.R., and Mahad D.J.. 2012. Mitochondrial changes associated with demyelination: consequences for axonal integrity. Mitochondrion. 12:173–179. 10.1016/j.mito.2011.03.007 [DOI] [PubMed] [Google Scholar]
- Coleman M. 2005. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 6:889–898. 10.1038/nrn1788 [DOI] [PubMed] [Google Scholar]
- Cummins K.L., Perkel D.H., and Dorfman L.J.. 1979. Nerve fiber conduction-velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr. Clin. Neurophysiol. 46:634–646. 10.1016/0013-4694(79)90101-9 [DOI] [PubMed] [Google Scholar]
- Deerinck T.J., Bushong E.A., Lev-Ram V., Shu X., Tsien R.Y., and Ellisman M.H.. 2010. Enhancing serial block-face scanning electron microscopy to enable high resolution 3-D nanohistology of cells and tissues. Microsc. Microanal. 16S2:1138–1139. 10.1017/S1431927610055170 [DOI] [Google Scholar]
- DiMauro S., Schon E.A., Carelli V., and Hirano M.. 2013. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 9:429–444. 10.1038/nrneurol.2013.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusart I., Airaksinen M.S., and Sotelo C.. 1997. Purkinje cell survival and axonal regeneration are age dependent: an in vitro study. J. Neurosci. 17:3710–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar J.M., McLaughlin M., Yool D., Zhang S.C., Fowler J.H., Montague P., Barrie J.A., McCulloch M.C., Duncan I.D., Garbern J., et al. . 2004. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 166:121–131. 10.1083/jcb.200312012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I., Gomez-Isla T., Puig B., Freixes M., Ribé E., Dalfó E., and Avila J.. 2005. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2:3–18. 10.2174/1567205052772713 [DOI] [PubMed] [Google Scholar]
- Fiala J.C. 2005. Reconstruct: a free editor for serial section microscopy. J. Microsc. 218:52–61. 10.1111/j.1365-2818.2005.01466.x [DOI] [PubMed] [Google Scholar]
- Fünfschilling U., Supplie L.M., Mahad D., Boretius S., Saab A.S., Edgar J., Brinkmann B.G., Kassmann C.M., Tzvetanova I.D., Möbius W., et al. . 2012. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J.W., Hoffman P.N., Clark A.W., Carroll P.T., and Price D.L.. 1978. Slow axonal transport of neurofilament proteins: impairment of beta,beta’-iminodipropionitrile administration. Science. 202:633–635. 10.1126/science.81524 [DOI] [PubMed] [Google Scholar]
- Griffiths I., Klugmann M., Anderson T., Yool D., Thomson C., Schwab M.H., Schneider A., Zimmermann F., McCulloch M., Nadon N., and Nave K.A.. 1998. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 280:1610–1613. 10.1126/science.280.5369.1610 [DOI] [PubMed] [Google Scholar]
- Hasegawa M. 2006. Biochemistry and molecular biology of tauopathies. Neuropathology. 26:484–490. 10.1111/j.1440-1789.2006.00666.x [DOI] [PubMed] [Google Scholar]
- Hayashi T., Rizzuto R., Hajnoczky G., and Su T.P.. 2009. MAM: more than just a housekeeper. Trends Cell Biol. 19:81–88. 10.1016/j.tcb.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler D., Young S.J., and Ellisman M.H.. 1996. A flexible environment for the visualization of three-dimensional biological structures. J. Struct. Biol. 116:113–119. 10.1006/jsbi.1996.0019 [DOI] [PubMed] [Google Scholar]
- Hirai H. 2008. Progress in transduction of cerebellar Purkinje cells in vivo using viral vectors. Cerebellum. 7:273–278. 10.1007/s12311-008-0012-5 [DOI] [PubMed] [Google Scholar]
- Hirrlinger J., and Nave K.A.. 2014. Adapting brain metabolism to myelination and long-range signal transduction. Glia. 62:1749–1761. 10.1002/glia.22737 [DOI] [PubMed] [Google Scholar]
- Hollenbeck P.J., and Saxton W.M.. 2005. The axonal transport of mitochondria. J. Cell Sci. 118:5411–5419. 10.1242/jcs.02745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K. 2005. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 6:1–16. 10.1007/s10048-004-0207-y [DOI] [PubMed] [Google Scholar]
- Johnson G.V., and Stoothoff W.H.. 2004. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117:5721–5729. 10.1242/jcs.01558 [DOI] [PubMed] [Google Scholar]
- Kasri N.N., Govek E.E., and Van Aelst L.. 2008. Characterization of oligophrenin-1, a RhoGAP lost in patients affected with mental retardation: lentiviral injection in organotypic brain slice cultures. Methods Enzymol. 439:255–266. 10.1016/S0076-6879(07)00419-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H.A. 2003. Bioluminometric assay of ATP in mouse brain: Determinant factors for enhanced test sensitivity. J. Biosci. 28:379–382. 10.1007/BF02705114 [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S., Ohno N., Kidd G.J., Komuro H., and Trapp B.D.. 2010. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J. Neurosci. 30:6658–6666. 10.1523/JNEUROSCI.5265-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J.R., Mastronarde D.N., and McIntosh J.R.. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116:71–76. 10.1006/jsbi.1996.0013 [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C., Goebbels S., Gravel M., Nicksch E., Lee J., Braun P.E., Griffiths I.R., and Nave K.A.. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33:366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lawrence A., Bouwer J.C., Perkins G., and Ellisman M.H.. 2006. Transform-based backprojection for volume reconstruction of large format electron microscope tilt series. J. Struct. Biol. 154:144–167. 10.1016/j.jsb.2005.12.012 [DOI] [PubMed] [Google Scholar]
- Lee Y., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y., Tsingalia A., Jin L., Zhang P.W., et al. . 2012. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 487:443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G., and Axel R.. 1985. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 40:501–508. 10.1016/0092-8674(85)90198-9 [DOI] [PubMed] [Google Scholar]
- Miller K.E., and Sheetz M.P.. 2004. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117:2791–2804. 10.1242/jcs.01130 [DOI] [PubMed] [Google Scholar]
- Montenegro G., Rebelo A.P., Connell J., Allison R., Babalini C., D’Aloia M., Montieri P., Schüle R., Ishiura H., Price J., et al. . 2012. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 122:538–544. 10.1172/JCI60560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B.M., Lee Y., and Rothstein J.D.. 2013. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 23:644–651. 10.1016/j.tcb.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B.M., Tsingalia A., Vidensky S., Lee Y., Jin L., Farah M.H., Lengacher S., Magistretti P.J., Pellerin L., and Rothstein J.D.. 2015. Deficiency in monocarboxylate transporter 1 (MCT1) in mice delays regeneration of peripheral nerves following sciatic nerve crush. Exp. Neurol. 263:325–338. 10.1016/j.expneurol.2014.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave K.A., and Trapp B.D.. 2008. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31:535–561. 10.1146/annurev.neuro.30.051606.094309 [DOI] [PubMed] [Google Scholar]
- Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., Abdellateef M., Rosti B., Scott E., Mansour L., et al. . 2014. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 343:506–511. 10.1126/science.1247363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N., Kidd G.J., Mahad D., Kiryu-Seo S., Avishai A., Komuro H., and Trapp B.D.. 2011. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J. Neurosci. 31:7249–7258. 10.1523/JNEUROSCI.0095-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N., Chiang H., Mahad D.J., Kidd G.J., Liu L., Ransohoff R.M., Sheng Z.H., Komuro H., and Trapp B.D.. 2014. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc. Natl. Acad. Sci. USA. 111:9953–9958. 10.1073/pnas.1401155111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins G.A., Renken C.W., Song J.Y., Frey T.G., Young S.J., Lamont S., Martone M.E., Lindsey S., and Ellisman M.H.. 1997. Electron tomography of large, multicomponent biological structures. J. Struct. Biol. 120:219–227. 10.1006/jsbi.1997.3920 [DOI] [PubMed] [Google Scholar]
- Perkins G.A., Sosinsky G.E., Ghassemzadeh S., Perez A., Jones Y., and Ellisman M.H.. 2008. Electron tomographic analysis of cytoskeletal cross-bridges in the paranodal region of the node of Ranvier in peripheral nerves. J. Struct. Biol. 161:469–480. 10.1016/j.jsb.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashed R.-M.A. 2010. A possible interaction between the smooth endoplasmic reticulum and the mitochondria in Sertoli cells of adult rat testes. Egypt. J. Histol. 33:808–818. [Google Scholar]
- Rismanchi N., Soderblom C., Stadler J., Zhu P.P., and Blackstone C.. 2008. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 17:1591–1604. 10.1093/hmg/ddn046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab A.S., Tzvetanova I.D., and Nave K.A.. 2013. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 23:1065–1072. 10.1016/j.conb.2013.09.008 [DOI] [PubMed] [Google Scholar]
- Saab A.S., Tzvetavona I.D., Trevisiol A., Baltan S., Dibaj P., Kusch K., Möbius W., Goetze B., Jahn H.M., Huang W., et al. . 2016. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron. 91:119–132. 10.1016/j.neuron.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugier-Veber P., Munnich A., Bonneau D., Rozet J.M., Le Merrer M., Gil R., and Boespflug-Tanguy O.. 1994. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet. 6:257–262. 10.1038/ng0394-257 [DOI] [PubMed] [Google Scholar]
- Schneider A., and Mandelkow E.. 2008. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 5:443–457. 10.1016/j.nurt.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze E., Asai D.J., Bulinski J.C., and Kirschner M.. 1987. Posttranslational modification and microtubule stability. J. Cell Biol. 105:2167–2177. 10.1083/jcb.105.5.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spacek J., and Lieberman A.R.. 1980. Relationships between mitochondrial outer membranes and agranular reticulum in nervous tissue: ultrastructural observations and a new interpretation. J. Cell Sci. 46:129–147. [DOI] [PubMed] [Google Scholar]
- Starling A., Rocco P., Cambi F., Hobson G.M., Passos Bueno M.R., and Zatz M.. 2002. Further evidence for a fourth gene causing X-linked pure spastic paraplegia. Am. J. Med. Genet. 111:152–156. 10.1002/ajmg.10551 [DOI] [PubMed] [Google Scholar]
- Stys P.K., Ransom B.R., and Waxman S.G.. 1991. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 546:18–32. 10.1016/0006-8993(91)91154-S [DOI] [PubMed] [Google Scholar]
- Tekkök S.B., Ye Z., and Ransom B.R.. 2007. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J. Cereb. Blood Flow Metab. 27:1540–1552. 10.1038/sj.jcbfm.9600455 [DOI] [PubMed] [Google Scholar]
- Timmerman V., Clowes V.E., and Reid E.. 2013. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 246:14–25. 10.1016/j.expneurol.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Trapp B.D., and Nave K.A.. 2008. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31:247–269. 10.1146/annurev.neuro.30.051606.094313 [DOI] [PubMed] [Google Scholar]
- Trapp B.D., and Stys P.K.. 2009. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 8:280–291. 10.1016/S1474-4422(09)70043-2 [DOI] [PubMed] [Google Scholar]
- Wright M.H., Berlin I., and Nash P.D.. 2011. Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem. Biophys. 60:39–46. 10.1007/s12013-011-9181-9 [DOI] [PubMed] [Google Scholar]
- Yin X., Crawford T.O., Griffin J.W., Tu Ph., Lee V.M., Li C., Roder J., and Trapp B.D.. 1998. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J. Neurosci. 18:1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Baek R.C., Kirschner D.A., Peterson A., Fujii Y., Nave K.A., Macklin W.B., and Trapp B.D.. 2006. Evolution of a neuroprotective function of central nervous system myelin. J. Cell Biol. 172:469–478. 10.1083/jcb.200509174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Kidd G.J., Nave K.A., and Trapp B.D.. 2008. P0 protein is required for and can induce formation of Schmidt-Lantermann incisures in myelin internodes. J. Neurosci. 28:7068–7073. 10.1523/JNEUROSCI.0771-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D., Hinckelmann M.V., Yu H., Lyra da Cunha M.M., Liot G., Cordelières F.P., Marco S., and Saudou F.. 2013. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 152:479–491. 10.1016/j.cell.2012.12.029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.