

Abstract
Cholangiocarcinoma is a highly lethal cancer with limited therapeutic options. Recent genomic analysis of cholangiocarcinoma has revealed the presence of fibroblast growth factor receptor 2 (FGFR2) fusion proteins in up to 13% of intrahepatic cholangiocarcinoma (iCCA). FGFR fusions have been identified as a novel oncogenic and druggable target in a number of cancers. In this study, we established a novel cholangiocarcinoma patient derived xenograft (PDX) mouse model bearing an FGFR2-CCDC6 fusion protein from a metastatic lung nodule of an iCCA patient. Using this PDX model, we confirmed the ability of the FGFR inhibitors, ponatinib, dovitinib and BGJ398, to modulate FGFR signaling, inhibit cell proliferation and induce cell apoptosis in cholangiocarcinoma tumors harboring FGFR2 fusions. In addition, BGJ398 appeared to be superior in potency to ponatinib and dovitinib in this model. Our findings provide a strong rationale for the investigation of FGFR inhibitors, particularly BGJ398, as a therapeutic option for cholangiocarcinoma patients harboring FGFR2 fusions.
Keywords: Intrahepatic Cholangiocarcinoma, FGFR2 fusion, Ponatinib, Dovitinib, BGJ398
1. Introduction
Cholangiocarcinoma is a lethal biliary tract cancer with suboptimal treatment outcomes. The overall incidence of cholangiocarcinoma has been increasing in Western countries [1; 2; 3; 4; 5]. Survival of patients with cholangiocarcinoma is poor, with a five-year survival rate of less than 10%, due to the typical advanced stage at presentation and limited treatment options [6; 7]. Although surgical treatments are potentially curative for selected patients, most patients are not eligible for surgery. Additionally, current standard combination chemotherapy with gemcitabine and cisplatin does not achieve long-term survival [8]. There is therefore a pressing need to develop novel therapies against this cancer.
The fibroblast growth factor receptor (FGFR) pathway has been linked to tissue-organ development, maturation, and homeostasis via the regulation of cell proliferation, invasion, and angiogenesis [9]. Aberrant FGFR pathway activity can enable malignant transformation and promote tumor progression. Constitutively active FGFR signaling can result from gain-of-function mutations, gene amplifications, and chromosomal translocations. Recent studies have revealed gene fusions of FGFR2 with multiple partners in intrahepatic cholangiocarcinoma (iCCA) as well as in other cancers [10; 11].
In recent studies using Next Generation Sequencing, FGFR2 fusions have been identified in 13% to 50% of iCCA patients [12; 13; 14; 15; 16; 17]. FGFR2 fusions involving FGFR2-BICC1 and FGFR2-AHCYL1 were identified in 13.6% of patients with iCCA [13]. This study also showed that FGFR2-fusions occur almost exclusively in the intrahepatic subtype of cholangiocarcinoma, with almost no occurrences in perihilar cholangiocarcinoma or distal cholangiocarcinoma. These observations have also been confirmed by studies using an FGFR2 break-apart Fluorescent In Situ Hybridization (FISH) assay, in which FGFR2 fusions were observed in 13% (12 of 96) of iCCA but not in any perihilar or distal cholangiocarcinomas [15]. FGFR2 fusion-positive cholangiocarcinomas have distinct pathologic features, including an intraductal or tubular anastomosing growth pattern, and absence of stem-like markers. Tumors harboring FGFR fusions have demonstrated enhanced sensitivity to FGFR inhibitors, suggesting that cholangiocarcinoma patients with FGFR2 fusions may benefit from targeted FGFR2 kinase inhibition [13; 17].
Preliminary evidence of antitumor activity was observed in an iCCA patient with a FGFR2-MGEA5 fusion treated with ponatinib and similarly in an FGFR2-TACC3 fusion-positive iCCA patient treated with pazopanib and subsequently ponatinib [14]. Based on these data and supporting preclinical studies, a number of clinical trials prospectively investigating the activity of FGFR small molecule inhibitors in iCCAs bearing FGFR2 fusions have been initiated [11; 18]. Therefore, the identification of FGFR2 fusions may warrant a novel molecular classification of iCCA and suggests a new therapeutic opportunity for iCCAs driven by these fusions.
We hypothesized that since a number of different FGFR2 fusion partners have been identified, there may be differential sensitivity of tumors bearing specific FGFR2 fusions to FGFR inhibitors. The optimal inhibitor for a particular tumor may therefore depend on the specific fusion partner as well as other molecular features of the tumor. In the present study, we developed a novel iCCA patient derived xenograft (PDX) mouse model, designated LIV31, that endogenously expresses an FGFR2-CCDC6 fusion protein and aimed to 1) investigate whether the FGFR inhibitor ponatinib enhances the antitumor effect of standard chemotherapy with gemcitabine and cisplatin, and 2) investigate the differential antitumor effects of the FGFR inhibitors ponatinib, dovitinib, and BGJ 398 in this novel iCCA mouse model. Finally, 3) since matrix metalloprotease (MMP) hyperactivity is a characteristic pathophysiologic event which promotes growth and metastasis of CCAs, we investigated the potential involvement of MMP expression in FGFR2 fusion signaling by assessing the effects of FGFR inhibitors on expression of MMP2, MMP3 and MMP9 in the LIV31 iCCA model.
2 Materials and Methods
2.1Reagents
Ponatinib (AP24534) was supplied by Ariad Pharmaceuticals (Cambridge, MA). NVP-BGJ398 and dovitinib (TKI-258) were purchased from Selleckchem (Houston, TX). Gemcitabine was from Sun Pharmaceutical (Cranbury, NJ). Cisplatin was from APP Pharmaceuticals (Schaumburg, IL). Primary antibodies against p-FGFR, p-AKT, AKT, p-ERK, ERK, p-FRS2, and cleaved caspase-3 were purchased from Cell Signaling (Danvers, MA); against MMP2, MMP3 and MMP9 from Abcam (Cambridge, MA); against β-actin from Sigma (St. Louis, MO); against total FGFR2 from Santa Cruz Biotechnology (Dallas, TX); against Ki-67 from Epitomics (Eugene, OR); against CD31 from Dako (Carpinteria, CA); and against FRS2 along with mouse/rabbit specific HRP/DAB (ABC) Detection IHC kit from Abcam Inc (Cambridge, UK). The In Situ Cell Death Detection Kit for TUNEL staining was from Roche (Indianapolis, IN). The RNAeasy mini Kit was from Qiagen (Valencia, CA)
2.2 Establishment of cholangiocarcinoma patient derived xenograft mouse model
Informed consent was obtained from the patient as part of a protocol approved by the Mayo Clinic Institutional Review Board. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the Mayo Clinic.
Freshly resected tissue of a lung metastasis from a patient with stage IV iCCA was chopped, mixed with an equal volume 0.1mL of Matrigel, and injected subcutaneously into the right flanks of 6-8 week old NOD/SCID/Il2rg null (NSG) mice. Tumors established in NSG mice were subsequently resected, minced in Matrigel, and injected into the flanks of nude mice.
2.3 FISH assay and G-band karyotyping
To characterize the chromosomal structure and confirm the presence of an FGFR2 gene rearrangement in the LIV31 tumor cells, karyotyping by G-banding, break-apart FISH and chromosomal painting were performed on LIV31 cells and formalin-fixed paraffin-embedded tumor tissue by an experienced FISH technologist as described previously [15].
Slides were prepared using cells cultured from the indicated specimens and were G-banded with Trypsin and Leishman's Stain. A total of 20 metaphases were obtained from at least two slides and analyzed in their entirety. Upon completion of analysis, karyograms were prepared from each representative clone observed.
The FGFR2 rearrangement was analyzed by break-apart FISH. Human bacterial artificial chromosomes (BACs) flanking the FGFR2 gene region were identified using the University of California Santa Cruz (UCSC) February 2009 Human Genome Assembly hg19. The 3’clones (RP11-878D21, CTD-2542P10, RP11-984I17, CTD-2291K12 and CTD-3237E5) were labeled by nick translation with Spectrum Green and the 5’ clones (CTD-2312O10, RP11-879C17, RP11-454I6 and RP11-135O16) were labeled with Spectrum Orange (Abbott Molecular, Des Plaines, IL). Abbott Molecular CEP 10 (D10Z1) labeled in Spectrum Aqua was added to the FGFR2 break-apart assay to help identify the chromosome 10s. Labeled clones were combined to create a dual-color, break-apart probe set. FISH was performed using standard FISH methodologies (Brockman et al. 2003). Chromosome 10 identification was performed using a whole chromosome paint probe from Abbott Molecular.
2.4 Drug studies
An initial study was performed to determine the optimal dose of ponatinib that the mice could tolerate. A previous study reported that ponatinib showed a wide therapeutic range (5–50 mg/kg) which was well tolerated in both SCID and nude mouse models [19]. Gozgit et al. showed that a dose of 30 mg/kg ponatinib resulted in more than 95% inhibition of FGFR2 and FRS2 phosphorylation in gastric cancer models with FGFR2 amplification [20]. In the same model, 10mg/kg ponatinib only cause partial inhibition and 3 mg/kg showed no inhibition of FGFR2 and FRS2 phosphorylation. We therefore first treated the mice with 30 mg/kg ponatinib, however the mice developed skin exfoliation at this dose, thus the dose of ponatinib was decreased to 25 or 20 mg/kg, which was well-tolerated without adverse skin reactions.
Chopped fresh tumor obtained from NSG mice was suspended in 0.1 mL Matrigel and injected subcutaneously into the right flank of athymic female nude mice aged 6-8 weeks (Harlan Laboratories, Indianapolis, IN). Tumor volume and body weight were recorded weekly. Tumor volume was calculated using the equation: Volume= LxS2/2 where L and S represent longest and shortest diameter of the tumor respectively. When the tumor volumes reached 140-200 mm3, animals were randomly assigned into 4 groups (n=10 per group, for the ponatinib group 9 animals were evaluable) and treated with vehicle (Citrate Buffer, pH 2.75), ponatinib (20 mg/kg, orally daily administered by gavage dissolved in Citrate Buffer) alone, gemcitabine (50 mg/kg, intraperitoneally weekly) and cisplatin (2.5 mg/kg, intraperitoneally weekly), or ponatinib in combination with gemcitabine and cisplatin at the aforementioned doses. The dose of 20 mg/kg ponatinib was selected based on prior experiments to ensure that the mice would tolerate the combination of the three drugs (data not shown). When the tumors reached a size of 2,000 mm3 or after 9 weeks of treatment, the mice were sacrificed and tumors were removed for further analysis.
For the study comparing the efficacy of the different FGFR inhibitors, ponatinib, BGJ398, and dovitinib on iCCA tumor growth, nude mice bearing LIV31 PDX tumors (n=10-13 per group) were treated via daily oral gavage with vehicle (1:1 ratio of PEG 400 and 50 mmol/L acetic acid/acetate buffer pH 4.68), 25 mg/kg ponatinib, in Citrate Buffer as described before, 15 mg/kg BGJ398 (from a 6 mg/mL solution in 1:1 PEG400 and acetic acid/acetate buffer, pH 4.68), or 30 mg/kg dovitinib (in distilled water). For this experiment, a dose of 25 mg/kg ponatinib was chosen because the dose of 20 mg/kg ponatinib was very well tolerated in the prior study. Pharmacokinetic studies indicated that 30 mg/kg of dovitinib administered daily by gavage in mice resulted in similar peak and overall exposures in humans [21]. Pharmacodynamic studies showed that 15 mg/kg dose BGJ398 completely suppressed FGFR2 tyrosine phosphorylation 3h after dosing in a mouse tumor model, consistent with its pharmacokinetic profile [22; 23]. Mice were sacrificed when the tumors reached 2,000 mm3 or after 9 weeks of treatment.
2.5 Immunohistochemistry (IHC) and TUNEL assay
Tumor tissue from 3-4 mice randomly selected from each group was sectioned and stained with H&E or by immunohistochemical staining for p-FGFR, p-FRS2, p-AKT, p-ERK, MMP2, MMP3 and MMP9. Tissues were also stained for CD31 to assess vascular density and for Ki-67 to assess tumor proliferation rates. The TUNEL assay and IHC for cleaved caspase-3 were performed to assess apoptosis.
Five μm sections were cut from formalin-fixed, paraffin-embedded tissues. Following deparaffinization, sections were rehydrated and blocked endogenous peroxidase activity for 10 min by 0.3% hydrogen peroxide, then were subjected to antigen retrieval by microwaving in 0.01 M sodium citrate (pH 6) for 5 minutes. After blocking for 10 min in 10% goat serum, sections were incubated at 4°C overnight with polyclonal antibodies against p-FGFR (1:100), p-FRS2 (1:200), p-AKT (1:200), p-ERK (1:400), MMP2 (1:500), MMP3 (1:500), MMP9 (1:500) cleaved caspase-3 (1:400), CD31 (1:600), and Ki-67 (1:400) as mentioned above. Secondary antibodies biotinylated goat anti-polyvalent were applied for 30 minutes. Immunostaining was performed using mouse/rabbit specific HRP/DAB Detection IHC Kit according to the manufacturer's instructions. Subsequently, sections were counterstained with hematoxylin and mounted in dimethyl benzene.
Tunnel staining was performed using the standard protocol of In Situ Cell Death Detection Kit (Fluorescein). TUNEL-positive cells were counted in 6 random high-power fields per section. Quantification of microvessel density (MVD), Ki-67, and cleaved caspase-3 positive cell percent were peformed as previously reported [24]. The IHC score was calculated by multiplying the intensity score with the percentage of positive cells [25]. Protein staining was evaluated under a light microscope at 400× magnification. Staining intensity was scored manually by an independent pathologist blinded to the treatment applied as 0=no staining, 1=weak staining, 2=moderate staining, and 3=strong staining. The percentage of positively stained tumor cells in five fields was counted (0-100%).
2.6 Western immunoblotting
Western immunoblots for FGFR2, p-FGFR, AKT, p-AKT, ERK, p-ERK, FRS2, and p-FRS2were performed on lysates from mouse cholangiocarcinoma tissues. Twenty to sixty micrograms of protein was separated by denaturing gel electrophoresis. After transfer to PVDF membrane, membranes were blocked with 5% BSA and incubated overnight with primary antibodies against p-FGFR (1:1000), FGFR2 (1:200), p-FRS2 (1:1,000), FRS2 (1:1,000), p-ERK (1:1,000), ERK (1:1,000), p-AKT (1:1,000), AKT (1:1,000), and β-actin (1:5,000). Blots were then incubated with secondary antibodies conjugated with HRP, and signals were visualized using the HyGLO HRP detection kit. β-actin was measured as the loading control. Quantitation of the signal was performed using Image J software.
2.7 RNA extraction and RT–PCR for verification of FGFR2-CCDC6 fusion
RT-PCR was performed to confirm the presence of the FGFR-CCDC6 fusion in cholangiocarcinoma tissues from 3 mice in each group. Total RNA from the LIV31 PDX tissues was extracted using the RNAeasy mini Kit (Qiagen, Valencia, CA). cDNA synthesis was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Grand Island, NY) to transcribe 2 μg of total RNA. PCR conditions using Platinum Taq High Fidelity Polymerase (Invitrogen – ThermoFisher Scientific, Waltham, MA) were 95°C for 5 min; followed by 34 cycles of 95°C for 20 sec, 53.2°C for 20 sec, and 68°C for 3 min 45 sec for detection of the FGFR2-CCDC6 fusion transcript. The PCR products were directly sequenced by Sanger sequencing using the BigDye terminator kit (Life Technologies). The amplified products were further extended by additional incubation at 68°C for 10 min. PCR products were then loaded on a 1% agarose gel containing ethidium bromide. Amplification of GAPDH was used as a control to verify cDNA integrity.
FGFR2-CCDC6 fusion primers were as follows:
Forward: AGGACCGGGGATTGGTACCGTAAC,
Reverse: TAATGAATTCTTCTTCCTGCTC
We used the recombinant FGFR2-CCDC6 plasmid: pENTR 5’ FGFR2-CCDC6 clone mp# 1.7 as a positive control.
2.8 Short tandem repeat genotyping of LIV31 cells by the American Type Culture Collection (ATCC)
Seventeen short tandem repeat (STR) loci plus the gender determining locus, Amelogenin, were amplified using the commercially available PowerPlex® 18D Kit from Promega. The cell line sample was processed using the ABI Prism® 3500xl Genetic Analyzer. Data were analyzed using GeneMapper® ID-X v1.2 software (Applied Biosystems - ThermoFisher Scientific, Waltham, MA). Appropriate positive and negative controls were run and confirmed for each sample submitted. (ATCC Sales Order Number: SOJ32770).
2.9 Statistics
Data are expressed as the mean ±SEM from at least three independent experiments. All statistical tests were performed using GraphPad Prism 5.0. Differences between groups were compared using an unpaired two-tailed Student t test. The tumor growth curves were compared using a non-linear comparison of curves. Survival curves were compared using the log rank test. P<0.05 was considered statistically significant.
3. Results
3.1 Successful establishment and characterization of a novel patient derived xenograft from a lung metastases of intrahepatic cholangiocarcinoma
A novel PDX mouse model designated LIV31 was successfully established from fresh tumor tissue from a metastatic lung nodule resected from a patient with stage IV iCCA (Fig. 1A-1B). The patient initially presented with a large mass involving segments V and VI of the right lobe in a non-cirrhotic liver. Surgical resection was performed with curative intent. Pathologic examination of the resected tissue showed grade 3 of 4 adenocarcinoma consistent with cholangiocarcinoma forming an 8 × 7.5 × 6.5 cm mass. There were two adjacent satellite nodules that were 2.5 cm and 1 cm in greatest diameter. There was venous invasion noted. The surgical margins were negative for tumor, with a minimum tumor free margin of 2 cm. Five resected lymph nodes were negative for tumor. The tumor was staged as AJCCpT2bN0. Two months after resection, adjuvant chemotherapy was started with gemcitabine and cisplatin but discontinued four months later due to repeated episodes of afebrile neutropenia. Nine months after resection of the liver mass, a lung metastasis was found on follow up imaging. The patient was then started on capecitabine and oxaliplatin. Four months later the patient developed evidence of progression of the lung metastasis and was switched to sorafenib. After six months, progression of two lung metastases was again noted. The metastases were resected and a portion of the tissue implanted in the flanks of NSG mice. Histopathology of the liver tumor and metastatic lung nodule showed typical glandular proliferation with abundant stromal reaction characteristic of cholangiocarcinoma (Fig.1C-1D).
Figure 1.
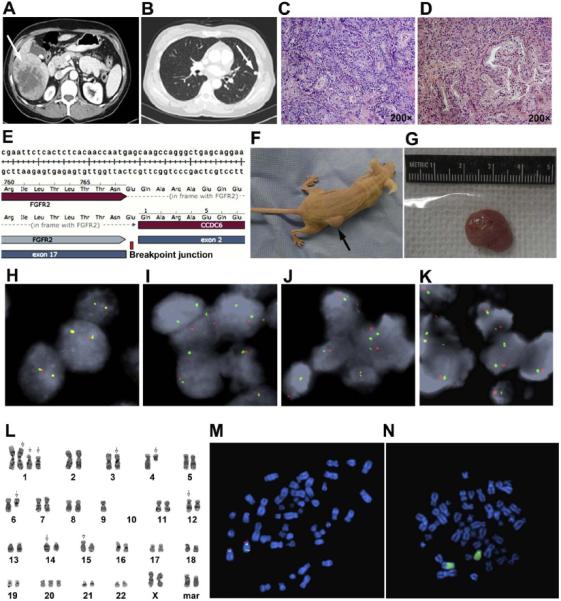
Establishment of a novel cholangiocarcinoma patient derived xenograft (LIV31) endogenously harboring an FGFR2-CCDC6 fusion protein. (A) Contrast-enhanced abdominal CT image of intrahepatic CCA bearing an FGFR2-CCDC6 fusion, demonstrates a right liver lobe mass. (B) Contrast-enhanced chest CT image of the same patient shows a metastasis in the left lung which developed 9 months after resection of the liver mass. (C) H&E staining of the primary CCA shows a grade 3 of 4 moderately-differentiated adenocarcinoma. (D) H&E staining of lung metastasis shows a similar moderately differentiated adenocarcinoma (magnification, x200). (E) Sanger sequencing of the RT-PCR product validates in-frame fusion transcript. (F) LIV31 grows subcutaneously in the flanks of nude mice. (G) Representative excised tumor from a LIV31 nude mouse xenograft. (H-K) FGFR2 break-apart FISH results from control tissue (H), primary liver tumor (I), lung metastasis (J), and patient derived xenograft (K) confirming that the primary tumor harbors a gene rearrangement involving the FGFR2 gene that is faithfully conserved in the LIV31 patient derived xenograft. (L) G-Banded karyogram of the LIV31 cell line. (M) Metaphase spread of LIV31 cell line with the FGFR2 BAC and D10Z1 probes applied. The image shows two derivative chromosome 10s, one with loss of the 3’ FGFR2 and one with a rearrangement of the FGFR2 gene. (N) Metaphase spread of the LIV31 cell line with the whole chromosome paint 10 probe applied. The image shows that one of the derivative chromosome 10s is made up of entirely chromosome 10 material and the other chromosome 10 is involved in an unbalanced translocation with an unidentified chromosome.
The LIV31 iCCA PDX tumor was subsequently successfully passaged in the flanks of nude mice (Fig. 1F-1G). LIV31 cells implanted into nude mice reached a tumor size of 1,000 mm3 in approximately 7-8 weeks.
RNA sequencing of the LIV31 tumor revealed a fusion between exon 17 of the FGFR2 gene and exon 2 of the CCDC6 gene, resulting in a chimeric mRNA, which was subsequently confirmed by RT-PCR and Sanger sequencing (Fig. 1E) and validated with a break apart FISH assay in cholangiocarcinoma tissue from the primary tumor in the liver the metastatic lung nodule, and the PDX tissue established as a subcutaneous tumor in mice (Fig. 1H-1K). The FGFR2 fusion in LIV31 was also validated using a break apart assay and G-banding (Fig. 1L-1N).
3.2 Ponatinib significantly inhibits growth of LIV31 xenograft tumors
First, we determined whether ponatinib had therapeutic efficacy compared to vehicle in vivo using the LIV31 PDX model. Ponatinib alone administered by oral gavage at 20 mg/kg/day significantly reduced tumor growth as compared to control mice treated with vehicle (P<0.0001) (Fig. 2A and Supplementary Fig. 1A). At day 63 of treatment, tumors in ponatinib treated mice had significantly less tumor volume than control mice treated with vehicle, with mean tumor volumes of 619.3 ± 119.5 mm3 vs 1417 ± 206.3 mm3, respectively. Survival curves showed significantly improved survival (assessed by number of mice with tumor volume <400 mm3) with treatment using ponatinib alone. Four of 9 (44%) mice treated with ponatinib had tumor volumes of <400 mm3 whereas none of the 10 mice treated with vehicle had a tumor volume of <400 mm3 at the end of treatment (P<0.05) (Fig. 2B). H&E staining of xenografts from each group showed similar moderately-differentiated adenocarcinoma (Fig. 2C). The gross tumor size was substantially decreased after ponatinib treatment (Fig. 2D). Ki-67 staining showed decreased cell proliferation in tumors in the ponatinib group compared to controls (Fig. 2C and 2E). CD31 staining showed no significant difference in microvascular density in LIV31 tumors treated with ponatinib compared to controls (Fig. 2C and 2E). Cleaved caspase-3 staining and TUNEL staining assays showed significantly increased apoptosis in the ponatinib-treated tumors as compared to the controls (Fig. 2C and 2E). Western blot and IHC indicate that treatment with ponatinib decreased phosphorylation of FGFR and its downstream signaling markers FRS2, AKT, and ERK (Fig. 3A-3D). RT-PCR shows the expression of the FGFR-CCDC6 fusion persists after treatment with ponatinib (Supplemental Fig. 2). This experiment confirms the efficacy of ponatinib against growth of LIV31 iCCA tumors.
Figure 2.

Growth inhibitory effects of ponatinib, gemcitabine and cisplatin, or ponatinib in combination with gemcitabine and cisplatin on LIV31 xenografts established subcutaneously in nude mice were studied in vivo. (A) Mice bearing LIV31 xenografts were randomized into four groups: vehicle, ponatinib, combination gemcitabine (Gem) and cisplatin (Cis), or ponatinib plus Gem and Cis. Tumor growth curves were compared. Data are mean±SEM (n=9-10 mice per group). (B) Survival curves showing percent of mice with tumor volume less than 400 mm3. (C) H&E staining and immunohistochemistry of xenografts from mice treated as indicated and evaluated for proliferation (Ki-67), microvascular density (CD31), and apoptosis (cleaved caspase-3 (CC3) and TUNEL). H&E and CD31 (magnification, x200); Ki-67 and cleaved caspase-3 (magnification, x400), TUNEL stain (magnification, x630). (D) Representative tumors harvested from each group. (E) Statistical quantification of the immunohistochemical staining (Ki-67, CD31, cleaved caspase-3, and TUNEL) between four groups. The values are calculated as the mean percentage (Ki67, cleaved caspase-3) or number (CD31, TUNEL) of positive staining cells in six HPFs per slide from 3 animals for each condition. **, P< 0.01 compared to the control group.
Figure 3.

Effects of ponatinib, gemcitabine and cisplatin, or ponatinib in combination with gemcitabine and cisplatin on FGFR signaling pathway activation in LIV31 xenografts. (A) Total proteins were extracted from the xenograft tumors and downstream markers of FGFR signaling were analyzed by Western blotting. (B) Quantitations of Western blots are shown. Data are mean±SEM. n=4 per group (C) Representative immunohistochemical staining of xenografts from mice treated as indicated and evaluated for the downstream markers of FGFR signaling p-FGFR, p-FSR2, p-AKT, and p-ERK (magnification, x400). (D) Statistical quantitation of immunohistochemical staining for p-FGFR, p-FSR2, p-AKT, and p-ERK. Data are mean±SEM. **, P< 0.01 compared to the control group.
3.3 Ponatinib does not show a synergistic effect with combination gemcitabine and cisplatin
Standard of care treatment with cisplatin (2.5 mg/kg/week i.p) and gemcitabine (50 mg/kg/week i.p) in combination significantly inhibited tumor growth (P<0.0001) and was significantly more potent than ponatinib alone (P<0.0001). (Fig. 2A-2B). However, the combination of ponatinib with gemcitabine and cisplatin was not more potent than the combination of gemcitabine and cisplatin (P=0.7). These results indicate that ponatinib exerts an anticancer effect on cholangiocarcinoma in vivo, but has no additive or synergistic effect when given in combination with gemcitabine and cisplatin. The body weights of the mice treated with ponatinib vs. cisplatin and gemcitabine vs. ponatinib, cisplatin and gemcitabine were not different from those of the mice treated with vehicle (Supplemental Fig. 1C). Ki-67 staining showed decreased cell proliferation in the tumor after gemcitabine and cisplatin treatment (Fig. 2C and 2E). Microvascular density in cholangiocarcinoma tumor did not decrease when treated with gemcitabine and cisplatin. Cleaved caspase-3 staining and TUNEL assays showed significant apoptotic activity in the combined gemcitabine and cisplatin-treated tumors as compared to the controls (Fig. 2C and 2E). Treatment with gemcitabine and cisplatin did not decrease phosphorylation of FGFR or the downstream signaling molecules FRS2, AKT, and ERK (Fig. 3A-3D). The expression of the FGFR-CCDC6 fusion persists after treatment with gemcitabine and cisplatin, as assessed by non-quantitative RT-PCR (Supplemental Fig. 2).
3.4 BGJ398 is more potent than ponatinib and dovitinib in inhibiting growth of LIV31 xenograph tumors
Lastly, we compared the efficacy of different FGFR inhibitors in inhibiting cholangiocarcinoma growth. After 63 days of treatment, mean (SEM) tumor volumes were 412.8 ± 53.82 mm3, 269.7 ± 24.98 and 204.2 ± 30.13 mm3 (N=10) for mice treated with ponatinib, dovitinib, and BGJ398, respectively, significantly less than tumor volumes in control mice treated with vehicle 1210 ± 127.9 mm3 (Fig. 4A). The tumor growth curves suggest that at the doses administered, BGJ398 was the most potent of the tested FGFR inhibitors for inhibiting growth of the LIV31 xenografts (Fig. 4A). Survival curves showed significantly improved survival (assessed by number of mice with tumor volume <400 mm3) with treatment using ponatinib, dovitinib or BGJ398, compared to control mice (Fig. 4B, P<0.001). Furthermore, all three inhibitors were well tolerated and did not result in weight loss at the administered dose (Supplemental Fig. 1D).
Figure 4.
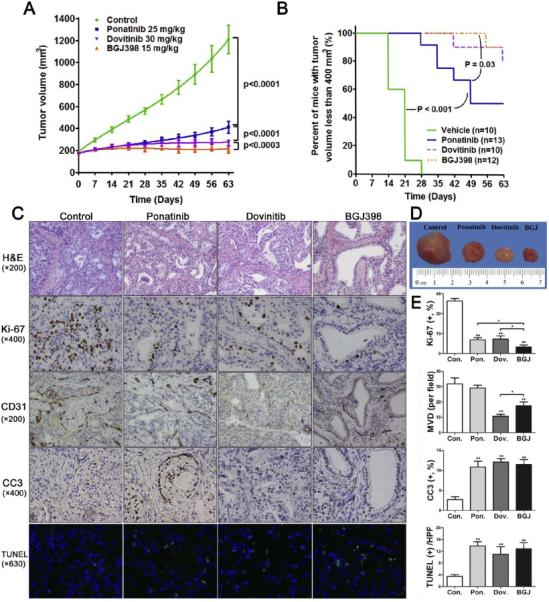
Growth inhibitory effects of ponatinib, dovitinib, and BGJ398 on LIV31 xenografts in nude mice in vivo. (A) Tumor growth curves were compared. Data are mean±SEM (n=10-13 mice per group). (B) Percent of mice with tumor volume less than 400 mm3. (C) H&E staining and immunohistochemistry of xenografts from mice treated as indicated and evaluated for proliferation (Ki-67,) microvascular density (CD31), and apoptosis (cleaved caspase-3 (CC3) and TUNEL). (D) Representative tumors harvested from each group. (E) Statistical quantitation of immunohistochemical staining (Ki-67, CD31, cleaved caspase-3, and TUNEL) between four groups. *, P< 0.05; **, P< 0.01 compared to the control group or as indicated in the figure.
Ki-67 staining showed that BGJ398, dovitinib, and ponatinib all decreased cell proliferation in the xenografts (Fig. 4C and 4E). BGJ398 was more potent at suppressing cell proliferation than the other two drugs. Microvascular density of tumors treated with dovitinib or BGJ398 was less than for the control and ponatinib groups, and dovitinib caused the most profound decrease in the MVD (Fig. 4C and 4E). Cleaved caspase-3 staining and TUNEL assays showed increased apoptosis in the ponatinib, dovitinib, and BGJ398-treated tumors (Fig. 4C and 4E). In studies of the downstream signaling pathways, all three inhibitors decreased phosphorylation of FGFR, with BGJ398 being the most potent. All three inhibitors also signficiantly decreased phosphorylation of the downstream signaling molecules FRS2, AKT, and ERK (Fig. 5A-5D). The expression of the FGFR-CCDC6 fusion also persisted after treatment with all three inhibitors (Supplemental Fig. 2). These experiments suggest that of the three tested FGFR inhibitors tested, BGJ398 is the most potent in inhibiting tumor growth and dovitinib is most effective in inhibiting vascular proliferation, perhaps due to the efficacy of dovitinib as an inhibitor of VEGF receptor signaling [26].
Figure 5.
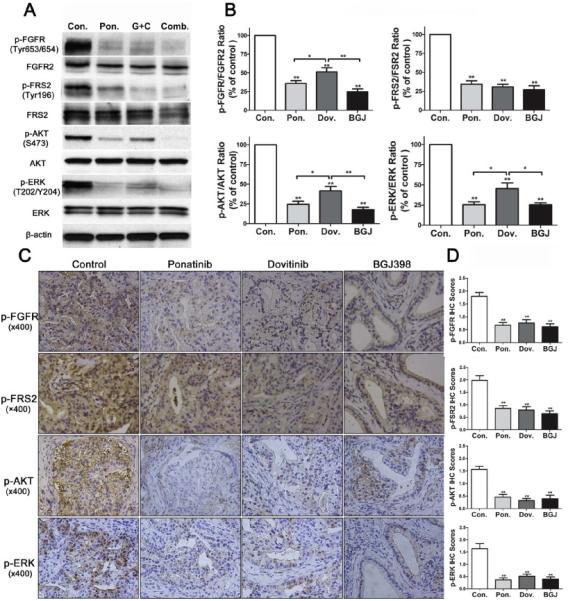
Effects of ponatinib, dovitinib, and BGJ398 on FGFR signaling pathway activation. (A) Total protein lysates were prepared from the xenografts and downstream markers of FGFR signaling were analyzed by Western blotting. (B) Quantitations of Western blots are shown. Data are mean±SEM. n=4 per group. (C) Representative immunohistochemical staining of xenografts from mice treated as indicated and evaluated for the downstream markers of FGFR signaling p-FGFR, p-FRS2, p-AKT, and p-ERK. (D) Statistical quantitation of immunohistochemical staining (p-FGFR, p-FRS2, p-AKT, and p-ERK). Data are mean±SEM. *, P< 0.05; **, P< 0.01 compared to the control group or as indicated in the figure.
3.5 Evaluation of MMP2, MMP3, and MMP9 in tumor tissues
MMPs, whose hyperactivity promotes tumor cell proliferation and metastasis, have been implicated in signaling by some combinations of FGFs and FGFRs [27; 28; 29; 30]. We therefore evaluated the expression of MMP2, MMP3, and MMP9 by IHC in the control, ponatinib, dovitinib, and BGJ398-treated tumors. There were no significant changes in expression of MMP2, MMP3, or MMP9 after treatment with the FGFR2 inhibitors, suggesting that these matrix metalloproteases are not direct mediators of the inhibitory effect of the FGFR2 inhibitors on tumorigenesis (Supplementary Figure 3).
3.6 STR profile of LIV31
The submitted sample STRA3186 (LIV31) profile is human and does not match any cell lines within the ATCC database or Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ) database (Supplemental Table 1).
4. Discussion
FGFR fusions have been identified as a novel and druggable oncogenic target in a number of cancers including bladder cancer (FGFR3) [31], glioblastoma (FGFR1 and FGFR3) [32], breast cancer (FGFR2) [12], and most recently CCA (FGFR2). Gene fusions of FGFR2 with multiple partners have been uncovered in cholangiocarcinoma as well as other cancers [10; 11]. These partners provide dimerization domains that facilitate oligomerization, leading to constitutive activation of the FGFR kinase and a variety of downstream signaling pathways including the RAS/MAPK and PI3K/AKT pathways [10; 11]. Therefore, small molecule inhibitors of FGFR2 may serve as rational therapies for this patient population.
In the current study, we developed a novel iCCA PDX model, designated LIV31, that endogenously expresses an FGFR2-CCDC6 fusion protein. Fusion of FGFR2 with CCDC6 has recently been identified in a breast cancer patient [12]. Overexpression of FGFR fusions proteins including FGFR-CCDC6 activates ERK signaling and induces cell proliferation in vitro [12]. The FGFR inhibitor ponatinib has shown potent antitumor activity in multiple human tumors with dysregulated FGFR activity [20; 33; 34; 35]. Of note, cells engineered to overexpress FGFR1-4 showed enhanced sensitivity to ponatinib compared to their parent cells [33; 34]. Ponatinib induced tumor shrinkage in patients with advanced iCCA carrying FGFR2 fusion proteins, including a patient who had previously responded to and then progressed on pazopanib, which preferentially targets the VEGFR and PDGFR receptors but also has modest efficacy against FGFR [14]. We therefore examined the antitumor effect of ponatinib in this FGFR2-CCDC6 driven CCA mouse model. At 20 mg/kg, ponatinib significantly inhibited growth of the LIV31 cholangiocarcinoma model. Further, IHC and Western blotting showed that ponatinib inhibited the activation of FGFR signaling, resulting in suppression of cell proliferation and induction of apoptosis in the LIV31 cholangiocarcinoma tumors. The potency of ponatinib in this model is similar to that previously observed in models of FGFR-amplified or -mutated tumors and BCR-ABL-driven tumors [19; 20]. Gemcitabine and cisplatin, the standard chemotherapy combination for advanced cholangiocarcinoma, only achieves a response rate of 26.1% and extends median overall survival from 8.1 to 11.7 months [8]. Thus, it is important to investigate whether FGFR inhibitors can enhance the anticancer effect of gemcitabine and cisplatin in FGFR2-fusion bearing cholangiocarcinomas. We found that at the doses administered, ponatinib did not enhance the effect of this combination therapy in vivo. The absence of synergy of ponatinib with gemcitabine and cisplatin may be due to the relatively high dose of gemcitabine and cisplatin we administered; or alternatively, could be due to the recently demonstrated ability of many tyrosine kinase inhibitors to block accumulation of co-administered gemcitabine in cancer cells, rendering the combination less effective [36]. As the combination of gemcitabine (50 mg/kg) and cisplatin (2.5 mg/kg) almost completely inhibited tumor growth, it was difficult to achieve an additional effect by combination with ponatinib. Therefore, further studies may be needed to explore the effects of combination therapy with FGFR inhibitors and lower dose of gemcitabine and cisplatin in vivo, including studies using sequential administration with gemcitabine being given before the FGFR inhibitors.
Therapeutic options are limited following progression on first-line combination chemotherapy as there is no standard second-line therapy. Targeted therapies against EGFR, VEGF, and MEK alone or in combination with chemotherapy have been tested in cholangiocarcinoma [37]. However, existing data from clinical trials do not support their use in cholangiocarcinoma.
Pazopanib, a multitargeted TKI against VEGFR, PDGFR, and FGFR, has been reported to induce tumor shrinkage in an advanced cholangiocarcinoma patient carrying an FGFR2-TACC3 fusion that progressed on chemotherapy with gemcitabine and cisplatin [14]. Therefore, FGFR inhibitors may be used as second-line therapies against cholangiocarcinoma with genetic alterations in FGFR, particularly FGFR2 fusion events.
We also investigated the differential antitumor effects of the FGFR inhibitors ponatinib, dovitinib, and BGJ 398 in the LIV31 PDX model. BGJ398 is a potent, highly selective pan-FGFR inhibitor with predominant activity against FGFR1-3 [22]. Dovitinib is a nonselective FGFR inhibitor, which has also been shown to inhibit VEGFR and PDGFR [26; 38]. Ponatinib is a non-selective pan-FGFR inhibitor that is also effective against BCR-ABL fusion protein [19]. All three inhibitors are orally bioavailable and in clinical trials. We showed that all three tested FGFR inhibitors, at dose levels that showed efficacy in other models, significantly inhibited the growth of the FGFR2-CCDC6 fusion mouse xenograft PDX tumors when compared with vehicle. 15 mg/kg BGJ398 and 30 mg/kg dovitinib were more potent than 25 mg/kg ponatinib and BGJ398 was superior to dovitinib. In fact, BGJ398 given over 63 days at doses of 15 mg/kg daily almost completely inhibited the growth of FGFR2-CCDC6 fusion cholangiocarcinoma tumors. This result was similar to a previous study where daily oral administration with BGJ398 led to substantial tumor growth inhibition resulting in tumor stasis and regression at doses of 15 mg/kg or more in human gastric tumors harboring FGFR2 amplification [23]. The IHC and TUNEL assay results showed that these inhibitors can also inhibit proliferation and induce apoptosis of cholangiocarcinoma xenografts. All three inhibitors strongly inhibited activation of well-established downstream markers of FGFR signaling, including p-FGFR, p-FRS2, p-AKT, and p-ERK. Taken together, cholangiocarcinoma xenografts with FGFR fusions were sensitive to the FGFR inhibitors. Thus, the FGFR fusion proteins may represent suitable predictive biomarkers for FGFR inhibitors. The clinical application of FGFR inhibitors in cholangiocarcinoma will to some extent depend on the frequency of FGFR2 fusions in cholangiocarcinoma. To date, only a limited number of cholangiocarcinoma patients have undergone evaluation for the presence of FGFR2 fusion events and additional studies are needed to better estimate the true prevalence.
In summary, we report a novel cholangiocarcinoma PDX model expressing an FGFR2-CCDC6 fusion protein and demonstrate preclinical antitumor activity of the nonspecific multikinase inhibitors ponatinib and dovitinib and the FGFR-specific inhibitor BGJ389 against this tumor. Ponatinib did not appear to have an additive or synergistic effect with standard chemotherapy gemcitabine and cisplatin on this PDX model. Additionally, we were able to show a potential therapeutic advantage of BGJ398 over dovitinib and ponatinib which may be mediated by its more specific inhibitory effect on FGFR2.
Supplementary Material
Highlights.
We established a novel PDX mouse model bearing an FGFR2-CCDC6 fusion protein from a metastatic lung nodule of an iCCA patient.
FGFR inhibitors, ponatinib, dovitinib and BGJ398, inhibit FGFR signaling, and tumor growth in iCCAs harboring FGFR2 fusions.
Ponatinib did not appear to have an additive or synergistic effect with standard chemotherapy gemcitabine and cisplatin on this PDX model.
There was a potential therapeutic advantage of BGJ398 over dovitinib and ponatinib in this model.
5. Acknowledgments
This work was supported by, the National Institutes of Health (CA165076 to L.R.R.), the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), the Mayo Clinic Cancer Center (CA15083), ARIAD Pharmaceuticals, and The Cholangiocarcinoma Foundation. The authors thank Dr. Joseph M. Gozgit (ARIAD Pharmaceuticals Inc., Cambridge, MA) for helpful discussions and critical review of the manuscript. We also thank ARIAD Pharmaceuticals Inc. for providing ponatinib for this study. We thank the Mayo Cytogenetics Core and Darlene Knutson and Sara Kloft-Nelson for assistance with the FISH and G-banding analyses. The authors acknowledge the generous donation of the tissue sample from the patient without which this project would not be possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR DECLARATION
The authors have declared that no conflict of interest exists.
References
- 1.von Hahn T, Ciesek S, Wegener G, Plentz RR, Weismuller TJ, Wedemeyer H, Manns MP, Greten TF, Malek NP. Epidemiological trends in incidence and mortality of hepatobiliary cancers in Germany. Scandinavian journal of gastroenterology. 2011;46:1092–1098. doi: 10.3109/00365521.2011.589472. [DOI] [PubMed] [Google Scholar]
- 2.Yang JD, Kim B, Sanderson SO, Sauver JS, Yawn BP, Larson JJ, Therneau TM, Roberts LR, Gores GJ, Kim WR. Biliary tract cancers in Olmsted County, Minnesota, 1976-2008. The American journal of gastroenterology. 2012;107:1256–1262. doi: 10.1038/ajg.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertuccio P, Bosetti C, Levi F, Decarli A, Negri E, La Vecchia C. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2013;24:1667–1674. doi: 10.1093/annonc/mds652. [DOI] [PubMed] [Google Scholar]
- 4.Njei B. Changing pattern of epidemiology in intrahepatic cholangiocarcinoma. Hepatology. 2014;60:1107–1108. doi: 10.1002/hep.26958. [DOI] [PubMed] [Google Scholar]
- 5.Pinter M, Hucke F, Zielonke N, Waldhor T, Trauner M, Peck-Radosavljevic M, Sieghart W. Incidence and mortality trends for biliary tract cancers in Austria. Liver international : official journal of the International Association for the Study of the Liver. 2014;34:1102–1108. doi: 10.1111/liv.12325. [DOI] [PubMed] [Google Scholar]
- 6.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215–1229. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J, Investigators ABCT. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. The New England journal of medicine. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 9.Sandhu DS, Baichoo E, Roberts LR. Fibroblast growth factor signaling in liver carcinogenesis. Hepatology. 2014;59:1166–1173. doi: 10.1002/hep.26679. [DOI] [PubMed] [Google Scholar]
- 10.Parker BC, Engels M, Annala M, Zhang W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol. 2014;232:4–15. doi: 10.1002/path.4297. [DOI] [PubMed] [Google Scholar]
- 11.Borad MJ, Gores GJ, Roberts LR. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr Opin Gastroenterol. 2015;31:264–268. doi: 10.1097/MOG.0000000000000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, Cheng AJ, Kunju LP, Siddiqui J, Tomlins SA, Wyngaard P, Sadis S, Roychowdhury S, Hussain MH, Feng FY, Zalupski MM, Talpaz M, Pienta KJ, Rhodes DR, Robinson DR, Chinnaiyan AM. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, Ojima H, Furuta K, Shimada K, Okusaka T, Kosuge T, Shibata T. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59:1427–1434. doi: 10.1002/hep.26890. [DOI] [PubMed] [Google Scholar]
- 14.Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, McCullough AE, Barrett MT, Hunt K, Patel MD, Young SW, Collins JM, Silva AC, Condjella RM, Block M, McWilliams RR, Lazaridis KN, Klee EW, Bible KC, Harris P, Oliver GR, Bhavsar JD, Nair AA, Middha S, Asmann Y, Kocher JP, Schahl K, Kipp BR, Barr Fritcher EG, Baker A, Aldrich J, Kurdoglu A, Izatt T, Christoforides A, Cherni I, Nasser S, Reiman R, Phillips L, McDonald J, Adkins J, Mastrian SD, Placek P, Watanabe AT, Lobello J, Han H, Von Hoff D, Craig DW, Stewart AK, Carpten JD. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10:e1004135. doi: 10.1371/journal.pgen.1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, Murphy SJ, McWilliams RR, Hart SN, Halling KC, Roberts LR, Gores GJ, Couch FJ, Zhang L, Borad MJ, Kipp BR. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45:1630–1638. doi: 10.1016/j.humpath.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, Lee HJ, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Morosini D, Hawryluk M, Catenacci DV, Miller VA, Churi C, Ali S, Stephens PJ. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–242. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, Bonal D, Miltiadous O, Zhang Z, Hoshida Y, Cornella H, Castillo-Martin M, Pinyol R, Kasai Y, Roayaie S, Thung SN, Fuster J, Schwartz ME, Waxman S, Cordon-Cardo C, Schadt E, Mazzaferro V, Llovet JM. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nature communications. 2015;6:6087. doi: 10.1038/ncomms7087. [DOI] [PubMed] [Google Scholar]
- 18.Ang C. Role of the fibroblast growth factor receptor axis in cholangiocarcinoma. J Gastroenterol Hepatol. 2015;30:1116–1122. doi: 10.1111/jgh.12916. [DOI] [PubMed] [Google Scholar]
- 19.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Molecular cancer therapeutics. 2012;11:690–699. doi: 10.1158/1535-7163.MCT-11-0450. [DOI] [PubMed] [Google Scholar]
- 21.Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T, McBride DJ, Gillen J, Wolff NC, Morlock L, Lotan Y, Raj GV, Sagalowsky A, Margulis V, Cadeddu JA, Ross MT, Bentley DR, Kabbani W, Xie XJ, Kapur P, Williams NS, Brugarolas J. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Science translational medicine. 2012;4:137ra175. doi: 10.1126/scitranslmed.3003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, Brueggen J, Jensen MR, Schnell C, Schmid H, Wartmann M, Berghausen J, Drueckes P, Zimmerlin A, Bussiere D, Murray J, Graus Porta D. Discovery of 3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. Journal of medicinal chemistry. 2011;54:7066–7083. doi: 10.1021/jm2006222. [DOI] [PubMed] [Google Scholar]
- 23.Guagnano V, Kauffmann A, Wohrle S, Stamm C, Ito M, Barys L, Pornon A, Yao Y, Li F, Zhang Y, Chen Z, Wilson CJ, Bordas V, Le Douget M, Gaither LA, Borawski J, Monahan JE, Venkatesan K, Brummendorf T, Thomas DM, Garcia-Echeverria C, Hofmann F, Sellers WR, Graus-Porta D. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer discovery. 2012;2:1118–1133. doi: 10.1158/2159-8290.CD-12-0210. [DOI] [PubMed] [Google Scholar]
- 24.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, Zhang ZF, VandenBeldt K, Duesbery NS, Resau JH, Teh BT. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer research. 2008;68:81–88. doi: 10.1158/0008-5472.CAN-07-5311. [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Liang L, Zhang XF, Jia HL, Qin Y, Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, Wei JW, Zhou HJ, Ren N, Ye QH, Dong QZ, Qin LX. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58:158–170. doi: 10.1002/hep.26305. [DOI] [PubMed] [Google Scholar]
- 26.Angevin E, Lopez-Martin JA, Lin CC, Gschwend JE, Harzstark A, Castellano D, Soria JC, Sen P, Chang J, Shi M, Kay A, Escudier B. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1257–1268. doi: 10.1158/1078-0432.CCR-12-2885. [DOI] [PubMed] [Google Scholar]
- 27.Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008;27:57–66. doi: 10.1007/s10555-007-9105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elewa MA, Al-Gayyar MM, Schaalan MF, Abd El Galil KH, Ebrahim MA, El-Shishtawy MM. Hepatoprotective and anti-tumor effects of targeting MMP-9 in hepatocellular carcinoma and its relation to vascular invasion markers. Clin Exp Metastasis. 2015;32:479–493. doi: 10.1007/s10585-015-9721-6. [DOI] [PubMed] [Google Scholar]
- 29.Ardi VC, Van den Steen PE, Opdenakker G, Schweighofer B, Deryugina EI, Quigley JP. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J Biol Chem. 2009;284:25854–25866. doi: 10.1074/jbc.M109.033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pintucci G, Yu PJ, Sharony R, Baumann FG, Saponara F, Frasca A, Galloway AC, Moscatelli D, Mignatti P. Induction of stromelysin-1 (MMP-3) by fibroblast growth factor-2 (FGF-2) in FGF-2−/− microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK-1/2). J Cell Biochem. 2003;90:1015–1025. doi: 10.1002/jcb.10721. [DOI] [PubMed] [Google Scholar]
- 31.Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Human molecular genetics. 2013;22:795–803. doi: 10.1093/hmg/dds486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chase A, Bryant C, Score J, Cross NC. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica. 2013;98:103–106. doi: 10.3324/haematol.2012.066407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren M, Qin H, Ren R, Cowell JK. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia. 2013;27:32–40. doi: 10.1038/leu.2012.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wynes MW, Hinz TK, Gao D, Martini M, Marek LA, Ware KE, Edwards MG, Bohm D, Perner S, Helfrich BA, Dziadziuszko R, Jassem J, Wojtylak S, Sejda A, Gozgit JM, Bunn PA, Jr., Camidge DR, Tan AC, Hirsch FR, Heasley LE. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:3299–3309. doi: 10.1158/1078-0432.CCR-13-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Damaraju VL, Kuzma M, Mowles D, Cass CE, Sawyer MB. Interactions of multitargeted kinase inhibitors and nucleoside drugs: Achilles heel of combination therapy? Molecular cancer therapeutics. 2015;14:236–245. doi: 10.1158/1535-7163.MCT-14-0337. [DOI] [PubMed] [Google Scholar]
- 37.Onesti CE, Romiti A, Roberto M, Falcone R, Marchetti P. Recent advances for the treatment of pancreatic and biliary tract cancer after first-line treatment failure. Expert review of anticancer therapy. 2015;15:1183–1198. doi: 10.1586/14737140.2015.1081816. [DOI] [PubMed] [Google Scholar]
- 38.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.