

Abstract
Background and Purpose
Oxaliplatin is a platinum‐based chemotherapeutic drug used as a first‐line therapy for colorectal cancer. However, its use is associated with severe gastrointestinal side‐effects resulting in dose limitations and/or cessation of treatment. In this study, we tested whether oxidative stress, caused by chronic oxaliplatin treatment, induces enteric neuronal damage and colonic dysmotility.
Experimental Approach
Oxaliplatin (3 mg·kg−1 per day) was administered in vivo to Balb/c mice intraperitoneally three times a week. The distal colon was collected at day 14 of treatment. Immunohistochemistry was performed in wholemount preparations of submucosal and myenteric ganglia. Neuromuscular transmission was studied by intracellular electrophysiology. Circular muscle tone was studied by force transducers. Colon propulsive activity studied in organ bath experiments and faeces were collected to measure water content.
Key Results
Chronic in vivo oxaliplatin treatment resulted in increased formation of reactive oxygen species (O2ˉ), nitration of proteins, mitochondrial membrane depolarisation resulting in the release of cytochrome c, loss of neurons, increased inducible NOS expression and apoptosis in both the submucosal and myenteric plexuses of the colon. Oxaliplatin treatment enhanced NO‐mediated inhibitory junction potentials and altered the response of circular muscles to the NO donor, sodium nitroprusside. It also reduced the frequency of colonic migrating motor complexes and decreased circular muscle tone, effects reversed by the NO synthase inhibitor, Nω‐Nitro‐L‐arginine.
Conclusion and Implications
Our study is the first to provide evidence that oxidative stress is a key player in enteric neuropathy and colonic dysmotility leading to symptoms of chronic constipation observed in oxaliplatin‐treated mice.
Abbreviations
- CMMC
colonic migrating motor complex
- EJPs
excitatory junction potentials
- ENS
enteric nervous system
- fIJPs
fast inhibitory junction potentials
- iNOS
inducible NOS
- IR
immunoreactive
- L‐NNA
Nω‐nitro‐L‐arginine
- nNOS
neuronal NOS
- sIJPs
slow inhibitory junction potentials
- SNP
sodium nitroprusside
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| Muscarinic receptors |
| P2Y1 receptors |
| Enzymes b |
| nNOS, neuronal NOS |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Oxaliplatin, usually administered together with 5‐fluorouracil and leucovorin, is a third generation platinum‐based agent used as the first line of treatment for tumours resistant to the first and second generation platinum‐based agents, cisplatin and carboplatin (Raymond et al., 1998). Combinations containing oxaliplatin have shown great therapeutic potential, improving progression‐free and overall survival rates of patients with colorectal cancer (André et al., 2004; De Gramont et al., 2007).
Oxaliplatin exerts its cytotoxic effects via formation of platinum‐DNA adducts that trigger immobilization of the mitotic cell cycle and stimulate apoptosis of dividing cells (Graham et al., 2000; Goodisman et al., 2006). At clinically recommended doses, oxaliplatin is reported to be less toxic for the auditory, haematological and renal systems than cisplatin and carboplatin (Raymond et al., 1998). However, oxaliplatin‐induced neurotoxicity differs from that induced by other platinum compounds, triggering both acute and delayed peripheral neuropathies as well as gastrointestinal dysfunctions (Stojanovska et al., 2015).
Gastrointestinal side‐effects are a predominant cause of dose limitation, presenting a constant challenge for efficient and tolerable treatment of cancer (Verstappen et al., 2003; McQuade et al., 2014). About 40% of patients receiving standard dose chemotherapy and nearly all patients receiving high dose chemotherapy exhibit pain, ulceration, bloating, vomiting, diarrhoea and/or constipation throughout the course of treatment (McQuade et al., 2014). Platinum‐based agents specifically are linked to a heightened incidence of gastrointestinal side‐effects with up to 90% of patients experiencing vomiting, nausea and/or diarrhoea during the course of treatment (Sharma et al., 2005). Addition of platinum‐based chemotherapeutics to combination regimes (such as FOLFOX – a combination of 5‐fluorouracil, leucovorin and oxaliplatin) increases the incidence of chronic diarrhoea and treatment‐related death (Souglakos et al., 2006). Gastrointestinal side‐effects can persist up to 10 years after the treatment has ceased (Denlinger and Barsevick, 2009).
Gastrointestinal functions are controlled by the enteric nervous system (ENS) embedded in the wall of the gastrointestinal tract (Furness, 2012). Damage to the ENS underlies gastrointestinal dysfunction in many pathophysiological conditions (Furness, 2012). To date, few studies have investigated the effects of platinum‐based chemotherapeutics on enteric neurons in animal models (Vera et al., 2011; Wafai et al., 2013; Pini et al., 2016). These studies reported significant reductions in the number of myenteric neurons in the colon and stomach, alongside altered expression of neuronal NOS (nNOS) following in vivo treatment with cisplatin (Vera et al., 2011; Pini et al., 2016) and oxaliplatin (Wafai et al., 2013). These changes in the ENS are correlated with reductions in gastrointestinal transit and colonic propulsive activity in oxaliplatin‐treated animals, which may be related to symptoms of constipation or diarrhoea.
NO, synthesizedby the NOS enzymes, is a highly reactive and widely distributed transmitter found throughout both the central and peripheral nervous systems. In the gastrointestinal tract, NO is a well‐established mediator of vasodilation and gastrointestinal relaxation (Takahashi, 2003; Bornstein et al., 2004). Two types of NOS participate in normal physiological responses, nNOS localized to neurons and endothelial NOS (eNOS) localized to the endothelium. Increased expression of inducible NOS (iNOS) occurs during times of cellular stress. Neurons containing nNOS are the primary source of NO in the ENS and represent approximately 30% of the neuronal population in the myenteric plexus of the mouse small intestine and colon (Qu et al., 2008; Wafai et al., 2013). In enteric ganglia, nNOS is expressed by descending interneurons and by inhibitory motor neurons supplying the intestinal smooth muscle (Lecci et al., 2002). Moreover, a myogenic nNOS isoform is expressed by gastrointestinal smooth muscle cells (Daniel et al., 2000). NO released from both neurons and smooth muscle is essential for sphincter relaxation and generation of complex gastrointestinal motor patterns, which contribute to propulsion during digestion (Sarna et al., 1993; Roberts et al., 2007; Roberts et al., 2008). Altered expression of nNOS in the ENS has been linked to several pathological conditions, and increased levels of nNOS in the ENS can be indicative of oxidative stress (Rivera et al., 2011a). The effects of oxidative stress on post mitotic cells, including enteric neurons, can be cumulative, resulting in neuronal loss and deterioration of neuronal function impairing gastrointestinal motility patterns (Chandrasekharan et al., 2011). The role of oxidative stress in chemotherapy‐induced gastrointestinal dysfunction has not been explored. This study investigated the role of oxidative stress in enteric neuronal damage and colonic dysmotility caused by in vivo oxaliplatin treatment.
Methods
Animals
All animal care and experimental procedures were approved by the Victoria University Animal Experimentation Ethics Committee and performed in accordance with the guidelines of the National Health and Medical Research Council Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male Balb/c mice aged 6–8 weeks (18–25 g) supplied from the Animal Resources Centre (Perth, Australia) were used for all experiments. Mice had free access to food and water and were kept under a 12 h light/dark cycle in a well‐ventilated room at an approximate temperature of 22°C. Mice acclimatized for a minimum of 5 days and a maximum of 7 days prior to the commencement of in vivo intraperitoneal injections. A total of 70 mice were used for this study.
In vivo oxaliplatin injections
Mice were randomly assigned into two groups: oxaliplatin‐treated and sham‐treated. Mice received intraperitoneal injections of oxaliplatin (Tocris Bioscience, UK), 3 mg·kg−1 per dose, 3 times a week via a 26 gauge needle. Oxaliplatin was dissolved in sterile water in order to make 10−2 M·L−1 stock solutions and refrigerated at −20°C. The stock was then defrosted and further diluted with sterile water to make 10−3 M·L−1 solutions for injections. The oxaliplatin dose was calculated to be equivalent to standard human dose per body surface area (Renn et al., 2011). Sham‐treated mice received sterile water via intraperitoneal injection 3 times a week via a 26 gauge needle. The volume injected did not exceed 200 μL per injection. Mice were killed via cervical dislocation 14 days after the first injection. Colons were collected for in vitro experiments.
Assessment of mitochondrial superoxide production
MitoSOX™ Red M36008 (Invitrogen, Australia) was used to visualize mitochondrially‐derived superoxide in whole‐mount preparations of submucosal and myenteric ganglia of the distal colon. Freshly excised distal colon preparations were dissected to expose submucosal and myenteric ganglia. Preparations were incubated in oxygenated physiological saline with MitoSOX™ Red M36008 (5 μM) in a gently shaking incubator Unimax 1010 (Heidolph Instruments, Germany) at a constant temperature of 37°C for 40 min. Tissues were washed (2 × 30 min) with oxygenated physiological saline (composition in mM: NaCl 118, KCl 4.6, CaCl2 3.5, MgSO4 1.2, NaH2PO4 1, NaHCO3 25 and d‐Glucose 11; bubbled with 95%O2 and 5% CO2) and fixed in 4% paraformaldehyde overnight at 4°C. The following day tissues were washed (2 × 30 min) with physiological saline and mounted on glass slides with DAKO fluorescent mounting medium for imaging. All images were captured at identical acquisition exposure‐time conditions, calibrated to standardized minimum baseline fluorescence, converted to binary and changes in fluorescence from baseline were measured in arbitrary units (arb. units) using Image J software (NIH, MD, USA). The corrected total fluorescence was calculated as previously described (Burgess et al., 2010) in 32 5 × 5 μm2 boxes within myenteric ganglia from each preparation to exclude fluorescence outside the ganglia.
Mitochondrial membrane potential assay
Mitochondrial membrane potential changes in cells may be detected with the cationic, lipophilic JC‐10 dye. In normal cells, JC‐10 concentrates in the mitochondrial matrix where it forms red fluorescent aggregates (JC aggregates). In contrast, apoptotic cells stain in green fluorescent colour (JC monomeric form) due to the JC‐10‐labelled release of cytochrome c diffusing out of the mitochondria as a result of mitochondrial depolarisation and increased permeability. JC‐10 fluorescent mitochondrial membrane potential microplate assay kit (Abcam, MA, USA) was used to detect mitochondrial membrane potential changes, and the release of cytochrome c from damaged mitochondria in the myenteric ganglia of the distal colon. Freshly excised distal colon preparations from sham and oxaliplatin‐treated mice were bathed in oxygenated physiological saline and dissected to expose the myenteric ganglia. Immediately following dissection, preparations were incubated for 20 min with 500 μL of JC‐10 dye solution (buffer A) in a gently shaking incubator Unimax 1010 (Heidolph Instruments, Germany) at a constant temperature of 37°C. After 20 min, 500 μL of buffer B solution was added to tissue preparations and allowed to incubate for another 20 min in a gently shaking incubator at a constant temperature of 37°C. Immediately following final incubation, tissues were mounted on glass slides with DAKO fluorescent mounting medium for imaging under a Nikon Eclipse Ti laser scanning microscope (Nikon, Japan).
Immunohistochemistry
Collected tissues (distal and proximal colon) were placed in oxygenated PBS (pH 7.2) containing nicardipine (3 μM) (Sigma‐Aldrich, Australia) for 20 min to inhibit smooth muscle contraction. Samples were cut open along the mesenteric border, cleared of their contents, maximally stretched and dissected mucosa down to expose either the submucosal plexus (distal colon) or myenteric plexus (distal and proximal colon). Tissues were then fixed with Zamboni's fixative (2% formaldehyde, 0.2% picric acid) overnight at 4°C. Preparations were cleared of fixative by washing three times for 10 min with DMSO (Sigma‐Aldrich, Australia) followed by 3 × 10 min washes with PBS. Fixed tissues were stored at 4°C in PBS for a maximum of 5 days.
Whole‐mount preparations were incubated with 10% normal donkey serum (Chemicon, USA) for 1 h at room temperature. Tissues were then washed (2 × 5 min) with PBS and incubated with primary antibodies against β‐tubulin class III (TuJ1) (chicken, 1:1000, Abcam, MA, USA), nitrotyrosine (rabbit, 1:1000, Millipore, CA, USA), nNOS (goat, 1:500, Abcam, MA, USA) and cleaved caspase‐3 (rabbit, 1:500, Cell Signalling Technologies, MA, USA) overnight at 4°C. Tissues were then washed in PBS (3 × 10 min) before incubation with species‐specific secondary antibodies labelled with different fluorophores: donkey anti‐chicken Alexa 594 (1:200, Jackson Immuno research Laboratories, PA, USA), donkey anti‐goat Alexa 488 (1:200, Jackson Immuno research Laboratories, PA, USA) and donkey anti‐rabbit Alexa 488 and 647 (1:200, Jackson Immuno research Laboratories, PA, USA) for 2 h at room temperature. Double immunohistochemical labelling was performed using mixtures of antibodies raised in different species and using species specific secondary antibodies labelled with different fluorophores. Tissues were given a further 3 × 10 min washes with PBS, followed by a 2 min incubation with the fluorescent nucleic acid stain, 4′‐6‐diamidino‐2‐phenylindole (DAPI; 14 nM) (Invitrogen, Australia). Whole‐mount preparations were given three final 10 min washes in PBS and then mounted on glass slides using fluorescent mounting medium (DAKO, Australia). Whole‐mount preparations were observed under a Nikon Eclipse Ti laser scanning microscope (Nikon, Japan); eight randomly chosen images from each preparation were captured with a 20× objective and processed using NIS Elements software (Nikon, Japan). The numbers of β‐tubulin‐immunoreactive (IR) neurons, nNOS‐IR neurons and neurons displaying translocation of nitrated proteins to the nuclei were quantified in both submucosal and myenteric ganglia within a 2 mm2 area of each distal colon preparation. Proximal colon preparations were use to quantify the numbers of β‐tubulin‐IR and nNOS‐IR neurons. Images were then calibrated to standardized minimum baseline fluorescence, converted to binary, and changes in caspase‐3 fluorescence were detected using Image J software (NIH, MD, USA). Quantitative analyses were conducted blindly.
Histology
The distal colon was harvested and placed in a 10% formalin solution overnight and then transferred into 70% ethanol the following day. Paraffin embedded colon sections were cut 5 μm thick and de‐waxed in a 60°C oven for 30 min. To examine the morphological changes to the colon, a standard haematoxylin and eosin staining protocol was followed (26, 27). Ten sections per preparation were analysed. All images were analysed blindly.
Imaging
Three dimensional (z‐series) images of wholemount preparations were taken using a Nikon Eclipse Ti laser scanning microscope (Nikon, Japan. Fluorophores were visualized using excitation filters for Alexa 594 Red (excitation wavelength 559 nm), Alexa 488 (excitation wavelength 473 nm) and Alexa 405 (excitation wavelength 405 nm). Z‐series images were taken at step size of 1.75 μm (1600 × 1200 pixels).
Western blotting
The whole colon was harvested, cut along the mesenteric border and pinned mucosa side up to a silicone‐based petri dish containing physiological saline. The colon was flushed of its contents, and the mucosa, submucosa and the circular muscle layers were dissected and discarded. The remaining longitudinal muscle‐myenteric plexus (LMMP) whole‐mount preparations were then snap frozen using liquid nitrogen.
Frozen tissues were homogenized with a Polytron homogeniser (Kinematica AG, Lucerne, Switzerland) for 20 s in ice‐cold WB buffer (40 mM Tris, pH 7.5; 1 mM EDTA; 5 mM EGTA; 0.5% Triton X‐100; 25 mM β‐glycerophosphate; 25 mM NaF; 1 mM Na3VO4; 10 μg·mL−1 leupeptin; and 1 mM PMSF), and the whole homogenate was used for Western blot analysis. Sample protein concentrations were determined with a DC protein assay kit (Bio‐Rad Laboratories, Hercules, CA, USA), and equivalent amounts of protein from each sample were dissolved in Laemmli buffer and subjected to electrophoretic separation on SDS‐PAGE acrylamide gels. Following electrophoretic separation, proteins were transferred to a PVDF membrane, blocked with 5% powdered milk in Tris‐buffered saline containing 0.1% Tween 20 (TBST) for 1 h followed by an overnight incubation at 4°C with rabbit anti‐iNOS primary antibody (D6B6S, Cell Signalling Technology, Danvers, MA, USA) dissolved in TBST containing 1% BSA. The following day membranes were washed for 30 min in TBST and then probed with a peroxidase‐conjugated anti‐rabbit secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. Following 30 min of washing in TBST, the blots were developed with a DARQ CCD camera mounted to a Fusion FX imaging system (Vilber Lourmat, Germany) using ECL Prime reagent (Amersham, Piscataway, NJ, USA). Densitometric measurements of the protein of interest were carried out using Fusion CAPT Advance software (Vilber Lourmat, Germany). Membranes were then stained with Coomassie Blue, scanned and total protein loaded quantified using Image J software. The signal intensity of the protein of interest was normalized to the signal for total protein loaded.
Intracellular recordings
Segments of the distal colon proximal to the pelvic brim were collected from day 14 sham and oxaliplatin‐treated mice and placed in physiological saline bubbled with 95% O2 and 5% CO2 at room temperature. The physiological saline contained the L‐type Ca2⁺ channel blocker nicardipine (3 μM) (Sigma‐Aldrich, Australia) to limit contractions. The distal colon was opened along the mesenteric border and pinned in a Sylgard‐lined (Dow Corning, USA) Petri dish. The mucosa and submucosa were removed revealing the circular muscle layer. A 20 mm long, full circumference segment was transferred into a Sylgard‐lined recording chamber and pinned with 50 μm gold‐plated tungsten pins. The recording chamber was placed on the stage of an inverted microscope fitted with fluorescent optics (Zeiss Axiovert 200), and was continuously superfused with physiological saline (3 mL·min−1) pre‐heated to yield a bath temperature of 35°C. Following 2 h recovery from dissection (Carbone et al., 2012), circular smooth muscle cells were impaled with conventional intracellular borosilicate glass capillary electrodes filled with 5% 5,6‐carboxyfluorescein in 20 mM Tris buffer (pH 7.0) in 1 M KCl. Electrode resistances ranged from 60–120 MΩ. Recordings were made using an Axoclamp 2B amplifier (Axon Instruments, USA), digitised at 1–10 kHz via a Digidata 1440A interface (Molecular Devices, USA) and stored using PClamp 10.0 (Molecular Devices) on a PC. Carboxyfluorescein‐labelled cells were identified as circular muscle cells in situ from their morphology (Carbone et al., 2012). Intracellular hyperpolarising current pulses (duration 500 ms, intensity 100–500 pA) were used to determine input resistance (R in). A tungsten electrode (10–50 mm tip diameter, placed 1 mm circumferential to the recording microelectrode) was connected to an ISO‐Flex stimulator controlled by a Master‐8 pulse generator (AMPI, Israel). Single pulse stimuli (20 V, 0.4 ms duration) and short trains of high frequency pulses (20 V, 3 pulses, 40 ms interval, 0.4 ms duration) were used to activate nerve fibres. Junction potential responses were recorded in the impaled smooth muscle cells. Responses in 3–4 cells were averaged per test condition, in each animal. Data were analysed using AxoGraph 10 software.
Contraction force
Freshly excised distal colon was cut into 3 mm rings, cleaned of connective tissue and fat and then placed in a custom built organ‐bath chamber containing physiological saline oxygenated with 95% O2 and 5% CO2 and maintained at a constant temperature of 37°C and pH of 7.4. Colonic rings were mounted between two small metal hooks attached to force displacement transducers (Zultek Engineering, Australia), and stretched to a resting tension of 0.2 g. After 30 min, rings were returned to resting tension and allowed to stabilize for 2 h with physiological saline changed every 20 min (Habiyakare et al., 2014). Following stabilization, 10 μM sodium nitroprusside (SNP) was added to organ bath. Baseline values were obtained by averaging 60 s of data 5 min prior to drug application; maximum relaxation was calculated as absolute change from the baseline values.
Colonic motility experiments
The entire colon was removed from day 14 sham and oxaliplatin‐treated mice and set up in organ‐bath chambers to record motor patterns in vitro (Wafai et al., 2013). Briefly, the colon was placed into warmed (35°C), oxygenated physiological saline until the faecal pellets were expelled. The empty colon was cannulated at both ends and arranged horizontally in organ‐bath chambers. The proximal end of the colon was connected to a reservoir containing oxygenated physiological saline to maintain intraluminal pressure. The distal end was attached to an outflow tube that provided a maximum of 2 cm H2O back‐pressure. Organ baths were continuously superfused with oxygenated physiological saline solution, and preparations were left to equilibrate for 30 min. Contractile activity of each segment was recorded with a Logitech Quickcam Pro camera positioned 7–8 cm above the preparation. Videos (2 × 20 min) of each test condition were captured and saved in avi format using VirtualDub software (version 1.9.11).
Colonic migrating motor complexes (CMMCs) were defined as propagating contractions directed from the proximal to the distal end of the colon, which travelled more than 50% of the colon length (Roberts et al., 2007; Roberts et al., 2008). Contractions that propagated less than 50% of the length of the colon were considered to be short contractions. Another form of incomplete contraction was identified as fragmented contractions occurring simultaneously at different parts of the colon rather than propagating over the length of the colon. Recordings were used to construct spatiotemporal maps using in‐house edge detection software (Gwynne et al., 2004). Spatiotemporal maps plot the diameter of the colon at all points during the recording allowing contractile motor patterns to be analysed with Matlab software (version 12).
Faecal water content and colonic faecal content
Wet weight of faecal pellets was measured immediately upon pellet expulsion. Pellets were then dehydrated for 72 h at room temperature prior to measurement of the dry weight. Water content was calculated as the difference between the wet weight and dry weight. Total number of faecal pellets along the entire length of the colon was counted in freshly excised intact colons from day 14 sham and oxaliplatin‐treated mice.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM. Sample size was calculated based on our previous studies on the enteric neuropathy and intestinal dysmotility associated with chemotherapy (Wafai et al., 2013; McQuade et al., 2016). To detect a 30% change at a power 0.8 and α = 0.05 with 10% SD, the effect size should be minimum n = 5 animals per group as calculated by the GPOWER program. Data were assessed using two‐way ANOVA, Welch's two‐tailed t‐test and Student's two‐tailed t‐test. Analyses were performed using Graph Pad Prism (Graph Pad Software Inc., CA, USA). Differences between group means were considered statistically significant at P < 0.05.
Materials
MRS2500 [(1R,2S,4S,5S)‐4‐[2‐iodo‐6‐(methylamino)purin‐9‐yl]‐2‐phosphonooxy‐1‐bicyclo [3.1.0]hexanyl]methyl dihydrogen phosphate ) was from Tocris, UK; atropine, carbachol, Nω‐nitro‐L‐arginine (L‐NNA), nicardipine and SNP were all from Sigma‐Aldrich, Australia. These compounds were prepared as stock solutions and diluted in physiological saline daily before addition to preparations.
Results
Loss of enteric neurons and increase in subpopulations of nNOS‐immunoreactive neurons following oxaliplatin treatment
To investigate changes to the total number of myenteric neurons, whole‐mount preparations of the distal and proximal colon were labelled with β‐tubulin antibody to count neurons within a 2 mm2 area (Figure 1). Repeated in vivo administration of oxaliplatin caused myenteric neuronal loss in both the proximal and distal colon when compared to sham (Figure 2A). Significant neuronal loss was also observed in the submucosal plexus in the distal colon from oxaliplatin‐treated mice, compared to sham (Figure 2AI).
Figure 1.
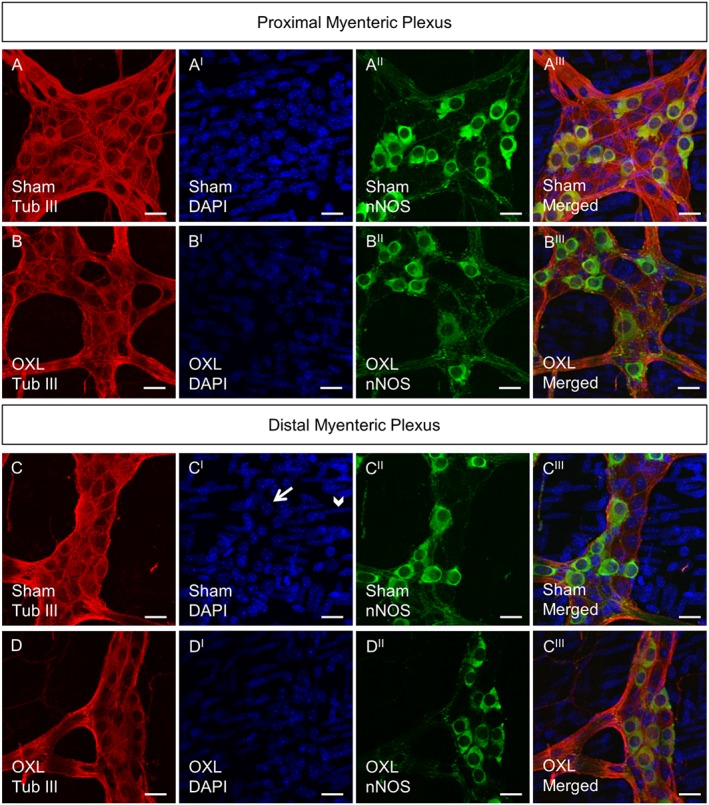
Whole‐mount preparations of myenteric neurons in the proximal and distal colon following 14 days of in vivo oxaliplatin treatment. Myenteric neurons labelled with anti‐β‐tubulin III antibody (Tub III, red) counterstained with DAPI (blue) that labels neuronal nuclei within the ganglion (arrow) and smooth muscle cell nuclei outside the ganglion (arrowhead) (CI). nNOS‐IR neurons (green). Scale bar = 20 μm.
Figure 2.
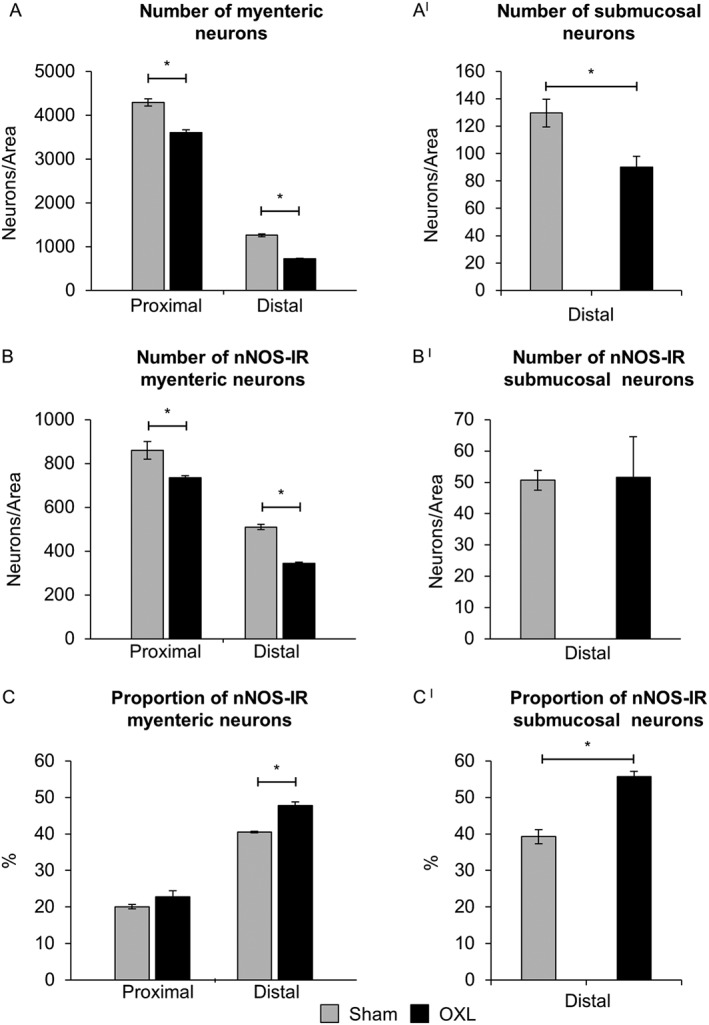
Effect of in vivo oxaliplatin treatment on total number of neurons and average number and proportion of nNOS‐IR enteric neurons. Average number of myenteric neurons in the proximal and distal colon and submucosal neurons was counted per 2 mm2 in the distal colon from day 14 sham and oxaliplatin‐treated mice (A,AI). Average number of nNOS‐IR neurons (B,BI) in myenteric and submucosal ganglia counted within 2 mm2 area. Proportion of nNOS‐IR neurons to the total number of myenteric and submucosal neurons (C,CI). Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 6 mice per group per time point.
To determine if oxaliplatin administration was associated with changes in subpopulations of myenteric neurons, inhibitory muscle motor and interneurons IR for nNOS were analysed. Fewer nNOS‐IR neurons were observed in the myenteric plexus of the both the proximal and distal colon following oxaliplatin administration when compared with sham (Figure 2B), but not in the submucosal plexus. The proportion of nNOS‐IR neurons was, however, significantly increased in the myenteric and submucosal plexus of the distal colon, but not the myenteric plexus of the proximal colon from oxaliplatin‐treated mice, when compared to sham‐treated mice (Figure 2C,CI).
Reactive oxygen species, iNOS expression and protein nitration in the submucosal and myenteric ganglia of the colon following in vivo oxaliplatin treatment
To evaluate production of ROS following long‐term oxaliplatin treatment, distal colon samples were probed with a fluorescent mitochondrial superoxide marker MitoSOX™ Red M36008. Increased MitoSOX fluorescence was found in both the submucosal (Figure 3A,AI and B,BI) and myenteric (Figure 3C,CI and D,DI) plexuses of the distal colon from oxaliplatin‐treated mice compared to sham‐treated animals (Figure 3E).
Figure 3.
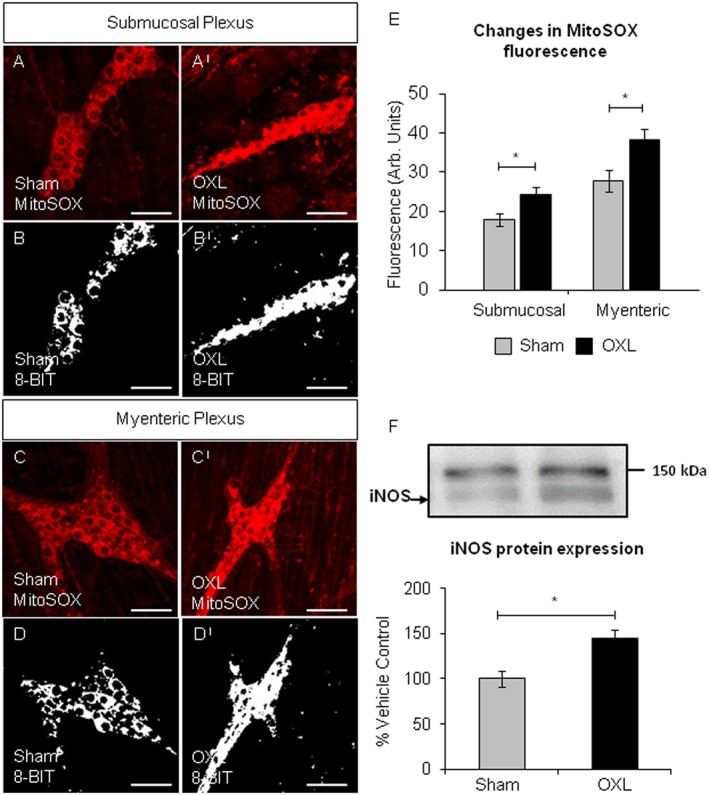
Mitochondrial superoxide in the colonic submucosal and myenteric ganglia and iNOS protein expression. Fluorescent and binary images of wholemount preparations of submucosal (A,AI and B,BI) and myenteric (C,CI and D,DI) ganglia labelled with MitoSOX™ Red in the colons from day 14 sham and oxaliplatin‐treated mice. Scale bar = 50 μm. (E) Quantification of the levels of mitochondrial superoxide production visualized by fluorescent probe in submucosal and myenteric ganglia in colonic preparations from day 14 sham and oxaliplatin‐treated animals. *P < 0.05, significantly different as indicated; n = 6 mice per group. (F) Representative images and quantification of the Western blot analysis for iNOS in LMMP tissue from day 14 sham and oxaliplatin‐treated mice. iNOS protein was normalized to total protein values obtained from the Coomassie Blue membrane staining (see Methods section). All values are expressed as a percentage of the values obtained from sham‐treated mice. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 5 mice per group.
Western blot analysis revealed a 40% increase in the expression of iNOS in LMMP preparations from the colon of oxaliplatin‐treated mice (n = 5 mice/group, Figure 3F).
An antibody against nitrotyrosine was used to label nitrated proteins within the submucosal (Figure 4A–AII and B–BII) and myenteric (Figure 4C–CII and D–DII) plexuses. The number of neurons per 2 mm2 displaying translocation of nitrated proteins to the nuclei was higher in both the submucosal and myenteric plexuses of the colon from oxaliplatin‐treated ,compared with sham‐treated (Figure 4E).
Figure 4.
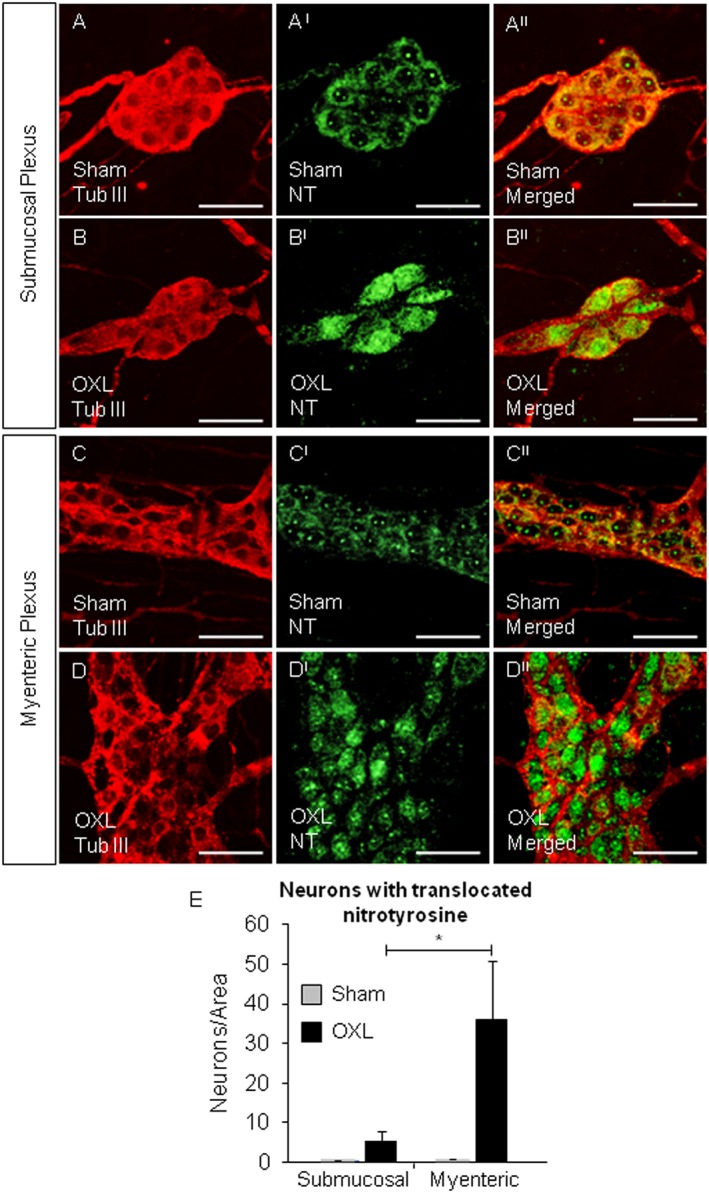
Translocation of nitrotyrosine to the nuclei of submucosal and myenteric neurons. Whole‐mount preparations of colonic submucosal ganglia from day 14 sham (A–AII) and oxaliplatin‐treated (B–BII) mice and myenteric ganglia from sham (C–CII) and oxaliplatin‐treated (D–DII) mice. Scale bar = 50 μm. Neurons and ganglia were labelled with anti‐β‐tubulin III antibody (Tub III, red). Nitrotyrosine within the ganglia was labelled with anti‐nitrotyrosine antibody (NT, green). *P < 0.05, significantly different as indicated; n = 6 mice per group.
Apoptosis of submucosal and myenteric neurons in the colon
Diffusion of cytochrome c out of the mitochondria as a result of mitochondrial membrane depolarisation and increased permeability was measured via fluorescence (green) of monomeric JC‐10. Increased monomeric JC‐10 fluorescence was found in both submucosal (Figure 5A,AI and B,BI) and myenteric (Figure 5C,CI and D,DI) plexuses of the distal colon from oxaliplatin‐treated mice compared with sham‐treated animals (Figure 5E). Both tissues from both sham and oxaliplatin‐treated mice experienced the same dissection and therefore the same level of cellular stress associated with dissection. Cellular stress associated with dissection can induce short‐term mitochondrial depolarisation, resulting in modest increases in JC‐10 fluorescence in tissues from both sham and oxaliplatin‐treated mice. However, the level of JC‐10 fluoresce in tissue from oxaliplatin‐treated mice was significantly higher (Figure 5) due to long‐term mitochondrial depolarisation associated with neurotoxicity.
Figure 5.
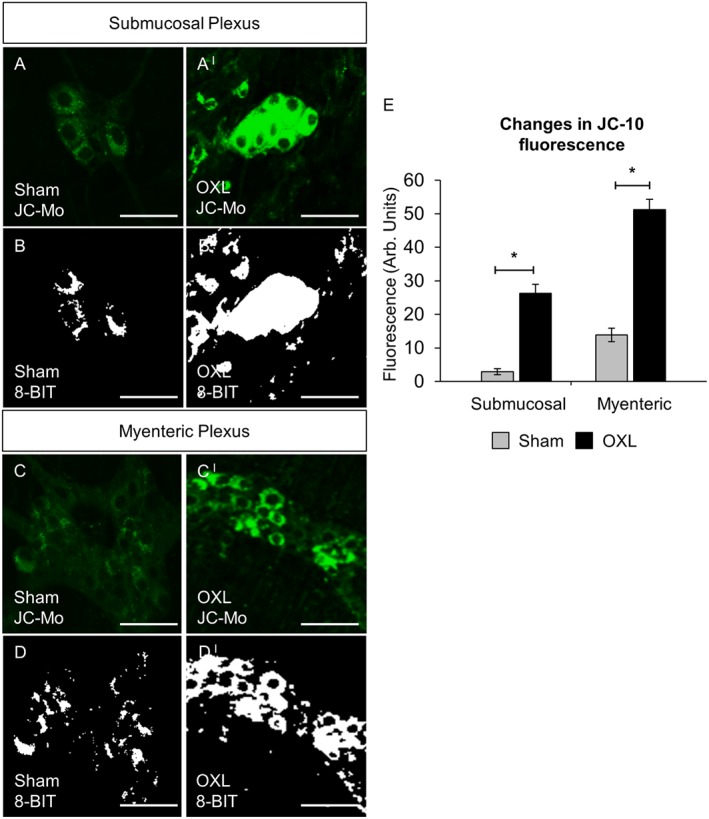
Changes in neuronal mitochondrial membrane potential indicative of cytochrome c release in submucosal and myenteric plexuses. Fluorescent and binary wholemount preparations of submucosal (A,AI and B,BI) and myenteric (C,CI and D,DI) ganglia labelled with JC‐10 dye in the colons from day 14 sham and oxaliplatin‐treated mice. Green fluorescent colour (JC monomeric form) is due to the JC‐10‐labelled release of cytochrome c diffusing out of the mitochondria as a result of mitochondrial depolarisation and increased permeability. Scale bar = 50 μm. (E) Quantification of the levels of monomeric JC‐10 production visualized by fluorescent probe in submucosal and myenteric ganglia in colonic preparations from sham and oxaliplatin‐treated animals. *P < 0.05, significantly different as indicated; n = 6 mice per group.
Caspase‐3 immunoreactivity was absent in both the submucosal and myenteric ganglia in the colon preparations from sham‐treated mice, but was observed in both the submucosal and myenteric ganglia in the colon preparations from oxaliplatin‐treated mice indicating neuronal apoptosis (not shown, n = 6 mice/group). Caspase‐3 labelling appeared both within the neuronal cell body as well as smaller, irregular patches of apoptotic debris that did not colocalise with β‐tubulin III immunoreactivity.
Effects of oxaliplatin treatment on neuromuscular transmission in the colon
Neuromuscular transmission in the distal colon was investigated using intracellular electrophysiology. Electrical stimulation of nerve fibres innervating the circular muscle in distal colon segments evoked junction potentials in single smooth muscle cells that were defined as fast inhibitory junction potentials (fIJPs), slow inhibitory junction potentials (sIJPs) and excitatory junction potentials (EJPs) using pharmacological blockers.
In control physiological saline, the resting membrane potential (RMP) of smooth muscle cells from sham‐treated mice averaged −38 ± 1.0 mV (n = 6 mice) and did not differ from that of smooth muscle cells from oxaliplatin‐treated mice (−39 ± 1.0 mV, n = 6 mice). RMPs in both groups were unaffected by the selective P2Y1 receptor antagonist MRS2500 (1 μM) (sham: −39 ± 1.4 mV, oxaliplatin: −37 ± 1.3 mV), L‐NNA (1 mM) (sham: −39 ± 1.3 mV, oxaliplatin: −38 ± 0.9 mV) or the muscarinic antagonist, atropine (1 μM) (sham: −38 ± 1.4 mV, oxaliplatin: −41 ± 3.3 mV). The input resistance of smooth muscle cells from sham‐treated mice averaged 8.3 ± 0.9 MΩ (n = 6 mice) in control physiological saline; this did not differ significantly from the input resistance of smooth muscle cells from oxaliplatin‐treated mice (8.7 ± 1.0 MΩ, n = 6 mice).
Single pulse and compound stimuli (trains of 20 V, 40 ms interval, 0.4 ms pulse duration) evoked fIJPs in colonic smooth muscle cells (Figure 6A, AI). The amplitudes of fIJPs recorded in smooth muscle cells from the distal colon of sham‐treated mice following a 20 V compound stimulus did not differ significantly from those recorded from the distal colon of oxaliplatin‐treated mice (Figure 6B). The duration of fIJPs (width at half amplitude) was not different between (sham: 397 ± 17 ms) and oxaliplatin‐treated (422 ± 27 ms) groups. The amplitudes of fIJPs increased with increasing stimulus strength, but no significant differences were found in responses between sham and oxaliplatin‐treated mice (Figure 6C). Fast IJPs were inhibited by MRS2500 (1 μM).
Figure 6.
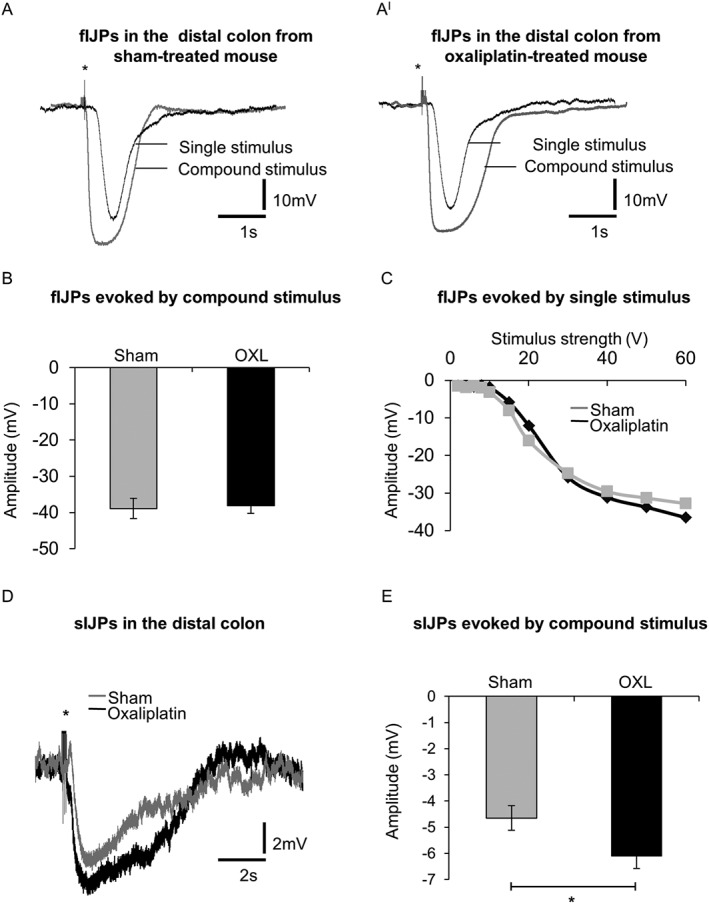
Intracellular recordings of fast and sIJPs from colonic smooth muscle cells. Single pulse electrical stimulus (40 V, 0.4 ms duration) and high frequency compound stimulus (20 V, 0.04 ms interval, 0.4 ms duration) evoked fIJPs in smooth muscle cells from day 14 sham (A) and oxaliplatin‐treated (AI) mice (* stimulus artefact). (B) The mean amplitude of fIJPs in response to compound stimuli from smooth muscle cells of sham and oxaliplatin‐treated mice (n = 4 mice/group). (C) Amplitudes of fIJPs to increasing strength of the electrical stimuli (2–60 V) in smooth muscle cells of sham and oxaliplatin‐treated mice. (D) Blocking fIJPs with a selective antagonist of P2Y1 receptors, MRS2500 (1 μM) revealed sIJPs in smooth muscle cells from both sham and oxaliplatin‐treated mice. (E) Comparison of the amplitude of sIJPs in smooth muscle cells from sham and oxaliplatin‐treated mice. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 6 mice per group.
Short compound stimuli (trains of 20 V, 3 pulses, 40 ms interval, 0.4 ms pulse duration) applied in the presence of MRS2500 evoked sIJPs (Figure 6D), whose amplitudes were greater in smooth muscle cells from the distal colon of mice treated with oxaliplatin than from sham‐treated mice (Figure 6E). The sIJPs were abolished by addition of L‐NNA (1 mM).
Short compound stimuli (trains of 20 V, 3 pulses, 40 ms interval, 0.4 ms pulse duration) of nerve fibres in the presence of both MRS2500 and L‐NNA, inhibiting fast and slow IJPs respectively, evoked EJPs (data not shown). EJPs were inhibited by atropine (1 μM). The EJPs recorded in smooth muscle cells from the distal colon of sham‐treated mice (6 ± 0.4 mV) did not differ from those of oxaliplatin‐treated mice (6 ± 0.7 mV). Atropine reduced the amplitude of responses in sham‐treated mice from 6 ± 0.4 to 0.8 ± 0.3 mV (P < 0.05) and from 6 ± 0.7 to 0.8 ± 0.2 mV in oxaliplatin‐treated mice (P < 0.05, n = 6 animals per group).
Effects of oxaliplatin treatment on colonic smooth muscles
Smooth muscle tone of the distal colon was studied in organ bath experiments using force transducers. We measured the force produced by 3 mm wide circular muscle rings. Application of SNP (10 μM) to organ baths containing distal colon segments resulted in a decrease in circular muscle tone, that is relaxation, in tissues from both sham (Figure 7A) and oxaliplatin‐treated (Figure 7AI) mice. The reduction in tension produced by SNP was greater in the colon segments from sham‐treated mice than in segments from oxaliplatin‐treated mice (Figure 7B). Thus, the relaxation force in response to SNP was reduced in colonic circular muscles from oxaliplatin‐treated mice.
Figure 7.
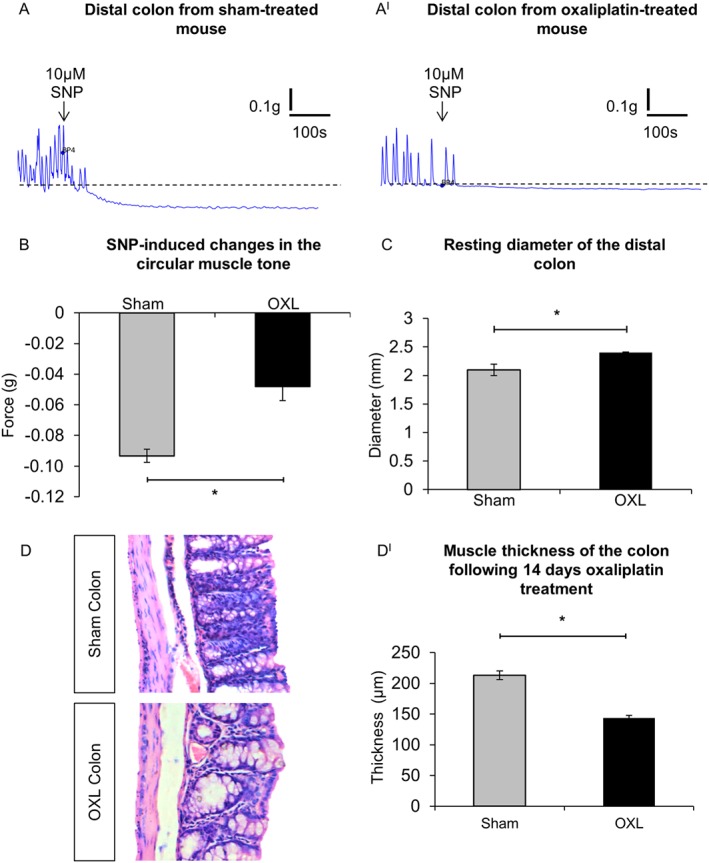
Effects of oxaliplatin treatment on colonic smooth muscles. (A,AI) Smooth muscle relaxation following application of the NO donor, SNP (10 μM) to the colon from day 14 sham (A) and oxaliplatin‐treated (AI) mice. (B) Comparison of the maximum relaxation produced by circular muscles in response to SNP in colonic preparations from sham and oxaliplatin‐treated mice quantified as an absolute change in the force transduction from the basal values. *P < 0.05, significantly different as indicated; n = 6 mice per group. (C) Resting diameter of the distal colon from sham and oxaliplatin‐treated mice. *P = 0.05, significantly different as indicated; n = 7 mice per group. (D) Gross morphological changes in the colon following repeated in vivo oxaliplatin administration. Colonic crypt length was shorter in oxaliplatin‐treated mice and muscle thickness was reduced in comparison to the sham‐treated animals. (DI) Statistical analysis of the muscle layer thickness in the colon preparations from sham and oxaliplatin‐treated mice. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 6 mice per group, 10 sections per preparation from each animal.
The resting colon diameter measured in organ‐bath experiments between contractions was larger in preparations from mice treated with oxaliplatin (2.4 ± 0.1 mm) compared with sham‐treated mice (2.1 ± 0.1 mm) (n = 7 animals per group) (Figure 7C). Histological analysis of colon sections revealed thinning of the colonic muscle layer in oxaliplatin‐treated mice when compared with sham (Figure 7D). Quantitative analysis confirmed that this reduction was statistically significant (Figure 7DI).
Colonic motility
Colonic motor patterns
Analysis of the total number of contractions included all types of motor patterns in the colon (CMMCs, short and fragmented contractions) (Figure 8A). The frequency, speed of propagation and length of contractions were measured for each specific type of motor activity.The total number and rate of rise of contractions were reduced in the colons from oxaliplatin‐treated animals compared to sham‐treated mice (P < 0.05 for both, Table 1). The addition of L‐NNA did not alter the overall number of contractions in either group (Figure 8B, Table 1). Further analysis revealed differences in occurrence of various types of colonic motor patterns in the oxaliplatin‐treated group with a significant decrease in the proportion of CMMCs (P < 0.05, Figure 8C) and increase in the proportion of fragmented contractions (P < 0.05, Figure 8E). Application of L‐NNA restored the proportion of CMMCs in oxaliplatin‐treated colons to the levels comparable with sham‐treated colons (Figure 8C), but had no significant effect on the proportion of either short or fragmented contractions (Figure 8D,E).
Figure 8.
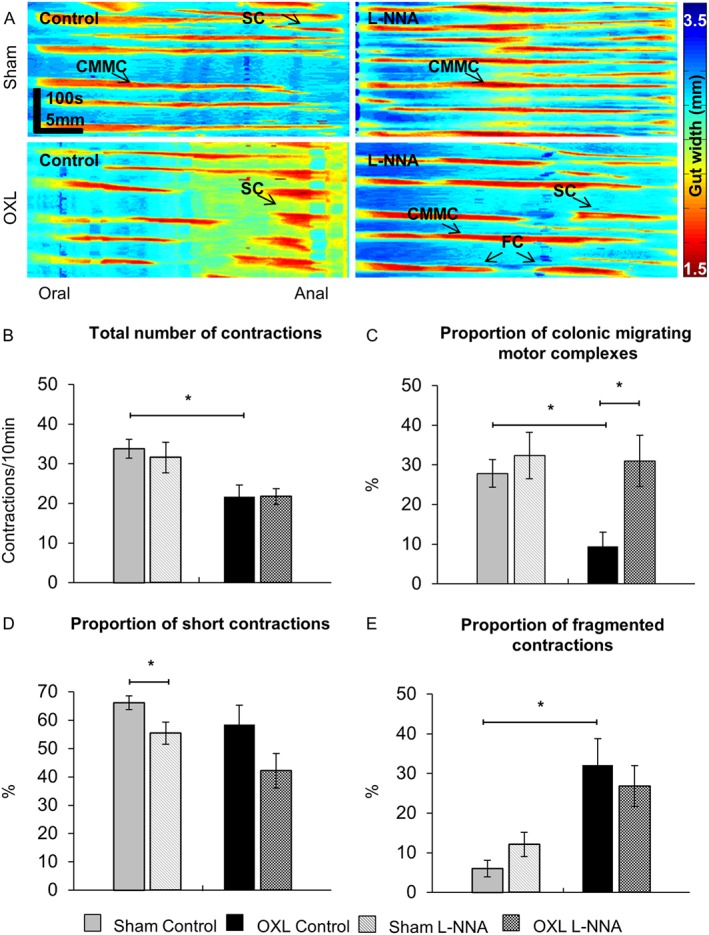
Total number of contractions and proportion of different types of contractile activity in the colon before and after application of L‐NNA. (A) Examples of spatiotemporal maps generated from digital video recordings of colonic motility from day 14 sham and oxaliplatin‐treated mice before (control) and after addition of L‐NNA. Each contraction can be seen as a reduction in the gut width (red), while relaxation as an increase in the gut width (blue). CMMCs propagate >50% of the colon length, short contractions (SCs) propagate <50% of the colon length and fragmented contractions (FCs) are interrupted by period(s) of relaxation during contraction. (B) The total number of contractions including all types of contractile activity in the colons from sham and oxaliplatin‐treated mice. The proportion of CMMCs (C), short contractions (D) and fragmented contractions (E) to the total number of contractions was calculated in maps from sham‐treated and oxaliplatin‐treated mice before and after addition of L‐NNA. Data are presented as an average of contractions per 10 min from a total of 40 min of video recording at baseline intraluminal pressure prior to and after the addition of L‐NNA. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 10 mice per group.
Table 1.
Parameters of different types of colonic contractions
| Sham‐treated (n = 10 mice) | Oxaliplatin‐treated (n = 10 mice) | ||
|---|---|---|---|
| All contractions | |||
| Frequency (contractions/10 min) | Control | 33.8 ± 2.4 | 21.7 ± 2.9a |
| L‐NNA | 31.6 ± 3.9 | 21.8 ± 3.9 | |
| Speed (mm/s) | Control | 1.8 ± 0.1 | 1.3 ± 0.2 |
| L‐NNA | 3.4 ± 0.5 | 2.1 ± 0.4 | |
| CMMCs | |||
| Proportion (%) | Control | 27.8 ± 3.5 | 9.4 ± 3.7a |
| L‐NNA | 32.4 ± 5.9 | 31.0 ± 6.4b | |
| Frequency (contractions/10 min) | Control | 9.0 ± 0.9 | 2.5 ± 1.0a |
| L‐NNA | 9.8 ± 2.2 | 6.0 ± 1.2b | |
| Speed (mm/s) | Control | 2.2 ± 0.3 | 2.0 ± 0.5 |
| L‐NNA | 3.5 ± 0.8a | 1.9 ± 0.5 | |
| Short contractions | |||
| Proportion (%) | Control | 66.2 ± 2.4 | 58.5 ± 6.9 |
| L‐NNA | 55.5 ± 3.9a | 42.2 ± 6.2 | |
| Frequency (contractions/10 min) | Control | 22.7 ± 2.4 | 13.0 ± 2.0a |
| L‐NNA | 17.6 ± 2.3 | 9.8 ± 1.9 | |
| Speed (mm·s−1) | Control | 1.6 ± 0.1 | 0.8 ± 0.1a |
| L‐NNA | 2.8 ± 0.6a | 1.2 ± 0.2 | |
| Distal anterograde short contraction frequency (per 10 min) | Control | 14.5 ± 1.5 | 6.4 ± 1.5a |
| L‐NNA | 10.3 ± 1.5 | 4.8 ± 1.4 | |
| Distal retrograde short contraction frequency (per 10 min) | Control | 2.0 ± 0.6 | 0.1 ± 0.1a |
| L‐NNA | 1.0 ± 0.3 | 0.3 ± 0.1 | |
| Fragmented contractions | |||
| Proportion (%) | Control | 6.0 ± 2.1 | 32.1 ± 6.1a |
| L‐NNA | 12.1 ± 3.1 | 26.8 ± 5.2 | |
| Frequency (contractions/10 min) | Control | 2.1 ± 0.7 | 6.2 ± 1.2a |
| L‐NNA | 4.2 ± 1.2 | 6.0 ± 1.1 | |
P < 0.05 compared with sham‐control.
P < 0.05 compared with oxaliplatin‐control.
Colonic migrating motor complexes
In vivo treatment with oxaliplatin was associated with a lower CMMC frequency than in sham‐treated preparations (P < 0.05). Application of L‐NNA significantly increased the frequency of CMMCs in colons from oxaliplatin‐treated (P < 0.05), but not sham‐treated mice (Figure 9A, Table 1). The rate of rise of CMMCs was comparable between sham‐treated and oxaliplatin‐treated mice, but increased CMMC rate of rise following addition of L‐NNA was noted in the colons from sham (P < 0.05), but not oxaliplatin‐treated animals (Figure 9B, Table 1).
Figure 9.
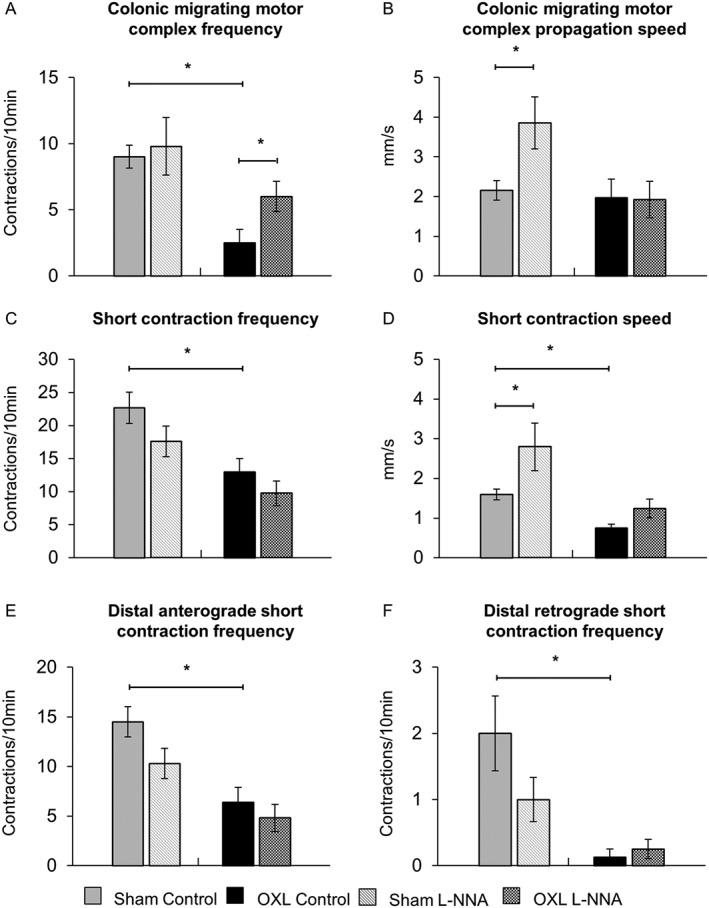
Effects of L‐NNA on CMMCs and short contractions. (A) The frequencies of CMMCs quantified in spatiotemporal maps from sham‐treated and oxaliplatin‐treated mice before and after L‐NNA application. (B) Speed of CMMCs in the colons from sham and oxaliplatin‐treated mice in both test conditions. *P < 0.05, significantly different as indicated; n = 10 mice per group. (C) The frequencies of short contractions were quantified in spatiotemporal maps from sham and oxaliplatin‐treated mice before and after L‐NNA application. (D) Speed of all short contractions analysed before and after L‐NNA application. Changes in the frequency of distal anterograde (E) and retrograde (F) short contractions in the colons from sham and oxaliplatin‐treated mice in both test conditions. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 10 mice per group).
Short contractions
Treatment with oxaliplatin resulted in a reduced frequency of short contractions in the colon (Figure 9C, Table 1). L‐NNA had no effect on the frequency of short contractions in either group. The rate of rise of short contractions was twofold slower in colons from oxaliplatin‐treated mice (Table 1). L‐NNA significantly enhanced the propagation speed of short contractions in the sham (P < 0.05), but not the oxaliplatin‐treated group. The frequency of short anterograde contractions in the distal region of the colon was significantly reduced in oxaliplatin‐treated animals compared with sham‐treated mice (Figure 9E, Table 1). Short contractions initiated in the distal colon were also found to travel in a retrograde direction. These retrograde short contractions occurred at a significantly reduced frequency in colons from oxaliplatin‐treated mice compared to sham‐treated mice (Figure 9F, Table 1). L‐NNA had no effects on anterograde or retrograde short distal contractions in either group.
Fragmented contractions
These were defined as incomplete contractions occurring simultaneously rather than propagating over the length of the colon (Figure 10A). The frequency of fragmented contractions was significantly higher in oxaliplatin‐treated than in sham‐treated mice (Figure 10B, Table 1). Application of L‐NNA did not affect the frequency of fragmented contractions in either oxaliplatin‐treated or sham‐treated mice.
Figure 10.
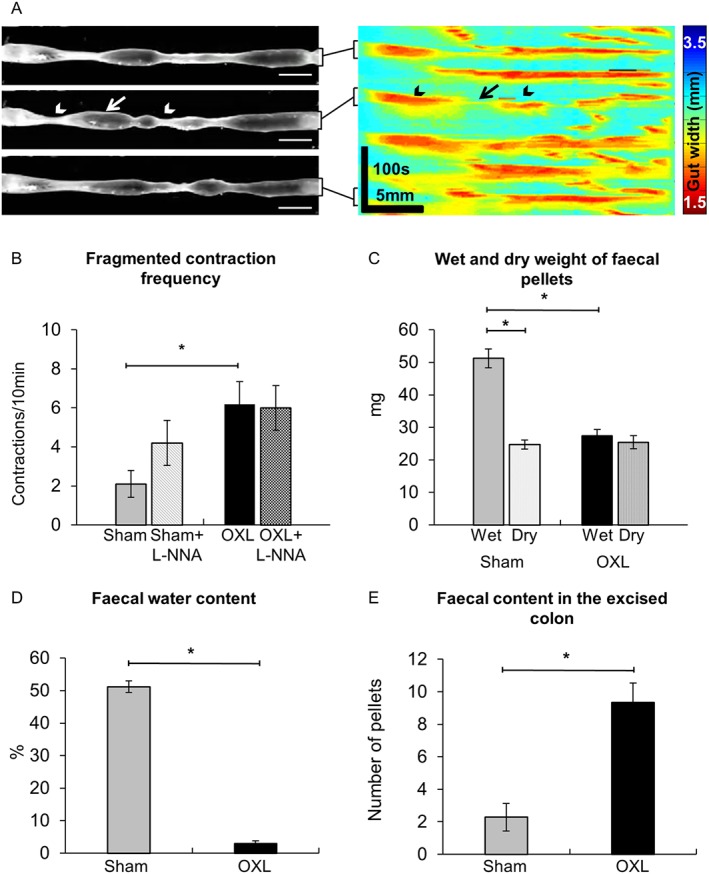
Effects of L‐NNA on fragmented contractions and colonic faecal content. (A) Fragmented contractions were defined as interrupted contractions consisting of period(s) of relaxation (arrow) and simultaneously occurring contractions (arrowheads). The frequency (B) of fragmented contractions in the colon from sham and oxaliplatin‐treated mice before and after L‐NNA application. (C) Wet weight of faecal pellets measured immediately upon pellet expulsion; dry weight of faecal pellets measured after 72 h of dehydration at room temperature. (D) Faecal water content calculated as the difference between the wet weight and dry weight. (E) Total number of faecal pellets along the entire length of the colon counted in freshly excised intact colons. Data presented as mean ± SEM. *P < 0.05, significantly different as indicated; n = 10 mice per group.
Colonic faecal content
To define the clinical symptoms resulting from the altered patterns of colonic motor activity, fresh faecal pellets were collected from both sham and oxaliplatin‐treated mice and faecal water content was calculated as the difference between wet and dry pellet weight (Figure 10C,D). The fresh pellets (wet weight) from oxaliplatin‐treated mice weighed significantly less than those of sham‐treated mice (Figure 10C). After dehydration, the dry weight of pellets from sham‐treated mice and from oxaliplatin‐treated mice were not different (Figure 10C). Thus, the water content was significantly lower in pellets collected from oxaliplatin‐treated mice than in those collected from sham‐treated mice (Figure 10D). Total number of pellets within the entire length of excised colons from oxaliplatin‐treated mice was significantly higher than the number in sham‐treated mice (Figure 10E).
Discussion
This study is the first to examine the role of oxidative stress in vivo on enteric neurons of the colon that may be involved in mechanisms underlying colonic dysmotility associated with oxaliplatin treatment. The results show that mitochondrial superoxide and nitrated protein levels are significantly increased in the myenteric and submucosal ganglia after oxaliplatin treatment. Mitochondrial depolarisation and increased mitochondrial permeability leading to release of cytochrome c from mitochondria were increased in myenteric ganglia in the colon from oxaliplatin‐treated mice. This presumably led to increased expression of an ‘executioner’ caspase‐3 in both the submucosal and myenteric ganglia in the colon preparations from oxaliplatin‐treated mice. Oxidative stress and neuronal apoptosis resulted in functional changes in the colon (increased nitrergic neuromuscular transmission, decreased smooth muscle tone, changes in the patterns and parameters of colonic motility) leading to symptoms of constipation in mice after long‐term treatment with oxaliplatin.
Oxaliplatin‐induced oxidative stress and neuronal apoptosis
Oxidative stress in enteric neurons following oxaliplatin treatment was shown by increased mitochondrial superoxide and translocation of nitrated proteins in submucosal and myenteric plexuses as well as the increase in expression of iNOS observed in our study. Mitochondria are responsible for most of the ROS burden under both normal and pathological conditions. The increased level of mitochondrial superoxide observed in enteric neurons is consistent with findings showing increased mitochondrial ROS in neuronal cultures from dorsal root ganglia of oxaliplatin‐treated rats (Kelley et al., 2014). Inhibition of mitochondrial electron transport chain complexes I and III involved in mitochondrial ROS formation improves sensory hyperalgesia in a rat model of peripheral neuropathy induced by another platinum‐based chemotherapeutic drug, cisplatin (Joseph and Levine, 2009). Mitotoxicity affecting mitochondrial respiration and ATP production were found in sciatic nerve from oxaliplatin‐treated rats and linked to oxaliplatin‐induced peripheral neuropathy (Zheng et al., 2011), which could be prevented by peroxynitrite decomposition catalyst (Janes et al., 2013). Thus, reduction of oxidative stress‐related biomarkers in peripheral neurons may be a promising avenue for alleviation of chemotherapy‐induced neuropathies.
Oxaliplatin‐induced mitochondrial damage has also been proposed as a potential mechanism leading to activation of NOS (Areti et al., 2014). The direct toxicity of NO can be greatly enhanced by reacting with superoxide to form peroxynitrite, which modifies tyrosines in proteins to create nitrotyrosines. Nitration of structural proteins can have major pathological consequences, but can also be an indicator of oxidative stress (Beckman and Koppenol, 1996). We identified translocation of nitrated proteins in both submucosal and myenteric plexuses of the colon from oxaliplatin‐treated mice. Translocation of nitrotyrosine has been previously found in myenteric neurons following ischaemia and reperfusion in mice (Rivera et al., 2011b) and induction of colitis in rats (Zingarelli et al., 2003); both of which were correlated with increases in NO and ROS. The increased level of iNOS observed in the LMMP preparations in our study probably leads to increased production of NO contributing to the accumulation of nitrotyrosine in enteric neurons. Although nitration can be used as an indirect measure of peroxynitrite, accumulation of nitrotyrosine itself is correlated with increased susceptibility to NO‐induced apoptosis (Savidge, 2011). Accumulation of nitrotyrosine has been linked to protein misfolding, mitochondrial dysfunction and neuronal degeneration (Nakamura and Lipton, 2008). Increased ROS and concurrent oxidative stress are linked to damage in various cellular components, and neuronal death (Wei et al., 2000).
One of many by‐products of oxaliplatin metabolism is oxalate and increases in oxalate levels trigger intracellular Ca2 + influx (Webster et al., 2005). Increased cytoplasmic Ca2 + activates nNOS, potentially leading to excessive NO production as well as activating the release of cytochrome c (Yagihashi et al., 2000). Release of JC‐10‐labelled cytochrome c diffusing out of the mitochondria as a result of mitochondrial depolarisation and increased permeability was observed in both submucosal and myenteric neurons in our study. The increase in superoxide and NO production may be a direct consequence of the loss of mitochondrial cytochrome c (Düssmann et al., 2003). The rate of superoxide production is linked to the magnitude of mitochondrial membrane potential and mitochondrial permeability (Brand et al., 2004). The magnitude of mitochondrial depolarisation dictates cytochrome c release and hence the activation of apoptosis. Modest mitochondrial depolarisation is transient and reversible, and has been found to be protective against moderate tissue injury (Schweizer and Richter, 1994; Gottlieb et al., 2003). Apoptotic cells exhibit distinctive biochemical features, including the presence of specific cleaved proteins (Hengartner, 2000). Caspase‐3 is a key effector or ‘executioner’ caspase, cleaving various substrates to cause the morphological and biochemical changes seen in apoptotic cells (Slee et al., 2001; Elmore, 2007; McIlwain et al., 2013). Our results are consistent with previous studies using caspase‐3 antibody, which found both intracellular immunoreactivity and detected extracellular aggregates of apoptotic debris outside neuronal cells (Pasinelli et al., 2000). Due to the fact that the half‐life of activated caspase‐3 is 8 h, it is difficult to detect significant amounts of activated caspase 3 within enteric ganglia. It is possible that the activation of caspase‐3 might have occurred at the earlier stages of oxaliplatin treatment resulting into neuronal apoptosis; this would explain both neuronal loss and presence of apoptotic debris observed in our study at day 14. Increased expression of caspase‐3 observed in our study is indicative of oxaliplatin‐induced apoptosis which resulted in 43% loss of myenteric neurons in the distal colon, 16% of myenteric neurons in the proximal colon and 30% of submucosal neurons in the distal colon. Similarly, oxaliplatin treatment induced 12% loss of myenteric and 21% submucosal neurons in the mouse ileum at day 14 (Robinson et al., 2016). Although oxaliplatin causes neuronal death in all studied intestinal regions, it seems that neurons in the distal colon are more susceptible to oxaliplatin‐induced damage than neurons in the proximal colon and ileum. There may be many reasons for these regional differences, but it should be noted that the proportion of the nNOS‐IR neurons was significantly increased in the distal, but not proximal colon, consistent with a role for NO in oxaliplatin‐induced enteric neuronal loss. Moreover, repeated in vivo oxaliplatin administration induces decreases in glial fibrillary acidic protein‐IR enteric glia, contrasting with increases in s100β‐IR enteric glial cells in both the myenteric and submucosal ganglia at day 14 after commencement of treatment (Robinson et al., 2016).
Changes in neuromuscular transmission and smooth muscle tone
NO released from nNOS expressing inhibitory motor neurons supplying the intestinal smooth muscles is an integral inhibitory neurotransmitter in the gastrointestinal tract (Lecci et al., 2002; Takahashi, 2003). nNOS produces basal levels of NO to maintain the hyperpolarised state of the circular muscle cells leading to physiological tonic inhibition of the intestine ( Daniel et al., 1994). Excessive NO production and consequent nitrosylation has been linked to inhibition of dynamin‐related protein 1, resulting in synaptic impairment and disruption to synaptic transmission (Savidge, 2011). In our study, intracellular recordings from colonic circular muscles revealed changes in neuromuscular transmission with an increase in amplitude of NO‐mediated sIJPs potentials in the oxaliplatin‐treated group. No changes in ATP‐mediated fIJPs or ACh‐mediated EJPS were observed. These results differ from findings in inflamed guinea pig colon where oxidative stress leads to reduced purinergic neuromuscular transmission (Roberts et al., 2013). This may be due to differences in mechanisms of oxaliplatin‐induced neuronal damage from inflammation‐induced enteric neuropathy. Oxaliplatin exerts its cytotoxic effects via direct binding to nuclear and mitochondrial DNA and formation of platinum‐DNA adducts (Graham et al., 2000; Goodisman et al., 2006). Direct mitochondrial damage leads to excessive NO production and activation of apoptotic cascades as discussed above.
Direct oxaliplatin toxicity and increased NO signalling alter the functions of colonic circular muscles. The relaxation evoked by the NO donor, SNP, was reduced by about 50% in the distal colon from oxaliplatin‐treated mice. The baseline diameter of the distal colon was greater in oxaliplatin‐treated animals than in sham‐treated mice suggesting a reduction in the colonic muscle tone after oxaliplatin treatment. This was reversed by application of L‐NNA inhibiting all NOS isoforms including iNOS, which was increased in longitudinal muscle‐myenteric neuron preparations after oxaliplatin treatment. The reduced smooth muscle tone as well the reduced response to the NO donor could be a result of the relatively higher inhibitory activity compared with the excitatory due to the increased proportion of nNOS neurons observed in oxaliplatin‐treated mice. The reduction in muscle thickness that we observed after oxaliplatin treatment may also contribute to a reduction in muscle tone. Decreased basal tone and, thus, a larger resting diameter of the distal colon compromised the ability of smooth muscles to further relax. Similar findings have been observed following intestinal inflammation where smooth muscle relaxation in response to a NO donor was decreased (Van Bergeijk et al., 1998; Rajagopal et al., 2015). This compromised ability to relax was in part attributed to iNOS‐mediated protein nitrosylation (Rajagopal et al., 2015).
Oxaliplatin‐induced changes in colonic motility
To investigate the role of NO in colonic dysmotility resulting from oxaliplatin treatment, we performed experiments with and without application of the NOS inhibitor, L‐NNA, and analysed phases of contraction in various parts of the colon, overall colonic motor patterns and parameters of specific types of motor activity.
Alterations in the basal tone of the colon and augmented nitrergic transmission may be attributed to increased NO release providing enhanced inhibitory input to the smooth muscle and altering colonic contractile patterns. Analysis of spatiotemporal maps revealed reductions in the proportion and frequency of CMMCs as well as in frequency and speed of short contractions in the distal colon from oxaliplatin‐treated mice. Anterograde and retrograde short contractions were observed in the distal colon of both sham and oxaliplatin‐treated mice. Both types of short contractions were significantly reduced in the distal colon after oxaliplatin treatment. In contrast, the proportion and frequency of fragmented contractions were greater in colons from oxaliplatin‐treated mice. These findings are consistent with previous studies demonstrating inhibition of CMMCs in the colon of oxaliplatin‐treated mice (Wafai et al., 2013) and inhibition of intestinal transit in cisplatin‐treated rats (Vera et al., 2011).
Previous studies using NOS inhibitors have established a role of NO in the pathophysiology of gastrointestinal functional disorders (Rivera et al., 2011a). NO is a primary regulator of complex intestinal motor patterns and increased NO release can disrupt CMMCs (Sarna et al., 1993; Rodriguez‐Membrilla et al., 1995; Castro et al., 2012). In our study, application of the NOS inhibitor, L‐NNA, restored the proportion and frequency of CMMCs in oxaliplatin‐treated mice. NOS inhibition increased the rate of rise of all types of contractions (CMMCs, short and fragmented) in the colons from sham‐treated, but not oxaliplatin‐treated mice. This is consistent with previous studies reporting increased speed of contraction induced by L‐NNA in control mice (Rodriguez‐Membrilla et al., 1995). Inhibition of NOS by L‐NNA did not affect short and fragmented contractions in colons from oxaliplatin‐treated mice. This suggests that the augmented proportion and frequency of fragmented contractions and reduced frequency of short contractions observed in oxaliplatin‐treated mice are not due to increased NO release, but can be attributed to either disruption of enteric neural pathways or a direct effect on colonic smooth muscle function. Colonic dysmotility was associated with reduction in faecal water content and increase in number of pellets in the colon providing evidence that oxaliplatin‐treated mice had constipation.
In conclusion, oxaliplatin treatment induces severe gastrointestinal side‐effects such as nausea, vomiting, constipation and diarrhoea, which account for dose limitations and/or cessation of treatment. These symptoms can persist up to 10 years after the treatment has been ceased (Denlinger and Barsevick, 2009). Oxidative stress has been suggested as a potential mechanism underlying platinum neurotoxicity (Di Cesare Mannelli et al., 2012). Our study is the first providing evidence that oxidative stress is a key player in enteric neuronal death, changes in intestinal smooth muscle tone and neuromuscular transmission underlying colonic dysmotility and leading to chronic constipation associated with oxaliplatin treatment. Further studies elucidating mechanisms of oxaliplatin‐induced oxidative stress are warranted in order to develop strategies to alleviate gastrointestinal side‐effects of chemotherapy and achieve more effective anti‐cancer treatment.
Author contributions
R.M.M. conception and design, collection, analysis and interpretation of data, manuscript writing; S.E.C. collection and interpretation of data, manuscript revision; V.S. collection and interpretation of data, manuscript revision; A.R. analysis and interpretation of data, manuscript revision; R.M.G. collection and analysis of data; A.M.R. collection and analysis of data; C.A.G. collection and analysis of data, manuscript revision; J.C.B. interpretation of data, manuscript revision; K.N. conception and design, interpretation of data, manuscript revision. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Dr Paul Senior and Dr Anthony Zulli for assisting with training and troubleshooting of experimental protocols.
This work was supported by a research grant from Victoria University.
McQuade, R. M. , Carbone, S. E. , Stojanovska, V. , Rahman, A. , Gwynne, R. M. , Robinson, A. M. , Goodman, C. A. , Bornstein, J. C. , and Nurgali, K. (2016) Role of oxidative stress in oxaliplatin‐induced enteric neuropathy and colonic dysmotility in mice. British Journal of Pharmacology, 173: 3502–3521. doi: 10.1111/bph.13646.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André T, Boni C, Mounedji‐Boudiaf L, Navarro M, Tabernero J, Hickish T et al. (2004). Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 350: 2343–2351. [DOI] [PubMed] [Google Scholar]
- Areti A, Yerra VG, Naidu V, Kumar A (2014). Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol 2: 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271: C1424–C1437. [DOI] [PubMed] [Google Scholar]
- Bornstein J, Costa M, Grider J (2004). Enteric motor and interneuronal circuits controlling motility. Neurogastroenterol Motil 16: 34–38. [DOI] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S et al. (2004). Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755–767. [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé J‐C, Lorca T, Castro A (2010). Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B‐Cdc2/PP2A balance. Proc Natl Acad Sci 107: 12564–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone SE, Wattchow DA, Spencer NJ, Brookes SJ (2012). Loss of responsiveness of circular smooth muscle cells from the guinea pig ileum is associated with changes in gap junction coupling. Am J Physiol Gastrointest Liver Physiol 302: G1434–G1444. [DOI] [PubMed] [Google Scholar]
- Castro M, Muñoz J, Arruebo M, Murillo M, Arnal C, Bonafonte J et al. (2012). Involvement of neuronal nitric oxide synthase (nNOS) in the regulation of migrating motor complex (MMC) in sheep. Vet J 192: 352–358. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan B, Anitha M, Blatt R, Shahnavaz N, Kooby D, Staley C et al. (2011). Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol Motil 23: 131–e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel E, Haugh C, Woskowska Z, Cipris S, Jury J, Fox‐Threlkeld J (1994). Role of nitric oxide‐related inhibition in intestinal function: relation to vasoactive intestinal polypeptide. Am J Physiol Gastrointest Liver Physiol 266: G31–G39. [DOI] [PubMed] [Google Scholar]
- Daniel EE, Wang YF, Salapatek AM, Mao YK, Mori M (2000). Arginosuccinate synthetise, arginosuccinate lyase and NOS in canine gastrointestinal tract: immunocytochemical studies. Neurogastroenterol Motil 12: 317–334. [DOI] [PubMed] [Google Scholar]
- De Gramont A, Boni C, Navarro M, Tabernero J, Hickish T, Topham C et al. (2007). Oxaliplatin/5FU/LV in adjuvant colon cancer: updated efficacy results of the MOSAIC trial, including survival, with a median follow‐up of six years. J Clin Oncol 25: 4007. [Google Scholar]
- Denlinger CS, Barsevick AM (2009). The challenges of colorectal cancer survivorship. J Natl Compr Canc Netw 7: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, Zanardelli M, Failli P, Ghelardini C (2012). Oxaliplatin‐induced neuropathy: oxidative stress as pathological mechanism. Protective effect of silibinin. J Pain 13: 276–284. [DOI] [PubMed] [Google Scholar]
- Düssmann H, Kögel D, Rehm M, Prehn JHM (2003). Mitochondrial membrane permeabilization and superoxide production during apoptosis. A single‐cell analysis. Biol Chem 278: 12645–12649. [DOI] [PubMed] [Google Scholar]
- Elmore S (2007). Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness JB (2012). The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol 9: 286–294. [DOI] [PubMed] [Google Scholar]
- Goodisman J, Hagrman D, Tacka KA, Souid A‐K (2006). Analysis of cytotoxicities of platinum compounds. Cancer Chemother Pharmacol 57: 257–267. [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Armour S, Harris M, Thompson C (2003). Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 10: 709–717. [DOI] [PubMed] [Google Scholar]
- Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E (2000). Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res 6: 1205–1218. [PubMed] [Google Scholar]
- Gwynne RM, Thomas E, Goh S, Sjövall H, Bornstein J (2004). Segmentation induced by intraluminal fatty acid in isolated guinea‐pig duodenum and jejunum. J Physiol 556: 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habiyakare B, Alsaadon H, Mathai ML, Hayes A, Zulli A (2014). Reduction of angiotensin A and alamandine vasoactivity in the rabbit model of atherogenesis: differential effects of alamandine and Ang (1‐7). Int J Exp Pathol 95: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO (2000). The biochemistry of apoptosis. Nature 407: 770–776. [DOI] [PubMed] [Google Scholar]
- Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J et al. (2013). Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy‐induced neuropathic pain resulting from peroxynitrite‐mediated post‐translational nitration of mitochondrial superoxide dismutase. Pain 154: 2432–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Levine JD (2009). Comparison of oxaliplatin‐and cisplatin‐induced painful peripheral neuropathy in the rat. J Pain 10: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR (2014). Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One 9: e106485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecci A, Santicioli P, Maggi CA (2002). Pharmacology of transmission to gastrointestinal muscle. Curr Opin Pharmacol 2: 630–641. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW (2013). Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5: a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade RM, Bornstein JC, Nurgali K (2014). Anti‐colorectal cancer chemotherapy‐induced diarrhoea: current treatments and side‐effects. Int J Clin Med 5: 393–406. [Google Scholar]
- McQuade RM, Stojanovska V, Donald E, Abalo R, Bornstein JC, Nurgali K (2016). Gastrointestinal dysfunction and enteric neurotoxicity following treatment with anticancer chemotherapeutic agent 5‐fluorouracil. Neurogastroenterol Motil. doi:10.1111/nmo.12890. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA (2008). Emerging roles of S‐nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid Redox Signal 10: 87–102. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown RH Jr, Cleveland DW (2000). Caspase‐1 and ‐3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase‐mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 97: 13901–13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pini A, Garella R, Idrizaj E, Calosi L, Baccari MC, Vannucchi MG (2016). Glucagon‐like peptide 2 counteracts the mucosal damage and the neuropathy induced by chronic treatment with cisplatin in the mouse gastric fundus. Neurogastroenterol Motil 28: 206–216. [DOI] [PubMed] [Google Scholar]
- Qu ZD, Thacker M, Castelucci P, Bagyánszki M, Epstein ML, Furness JB (2008). Immunohistochemical analysis of neuron types in the mouse small intestine. Cell Tissue Res 334: 147–161. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Nalli A, Kumar DP, Bhattacharya S, Hu W, Mahavadi S et al. (2015). Cytokine‐induced s‐nitrosylation of soluble guanylyl cyclase and expression of phosphodiesterase 1 A contribute to dysfunction of longitudinal smooth muscle relaxation. J Pharmacol Exp Ther 352: 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond E, Faivre S, Woynarowski JM, Chaney SG (1998). Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol 25: 4–12. [PubMed] [Google Scholar]
- Renn CL, Carozzi VA, Rhee P, Gallop D, Dorsey SG, Cavaletti G (2011). Multimodal assessment of painful peripheral neuropathy induced by chronic oxaliplatin‐based chemotherapy in mice. Mol Pain 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera L, Poole D, Thacker M, Furness J (2011a). The involvement of nitric oxide synthase neurons in enteric neuropathies. Neurogastroenterol Motil 23: 980–988. [DOI] [PubMed] [Google Scholar]
- Rivera LR, Thacker M, Pontell L, Cho H‐J, Furness JB (2011b). Deleterious effects of intestinal ischemia/reperfusion injury in the mouse enteric nervous system are associated with protein nitrosylation. Cell Tissue Res 344: 111–123. [DOI] [PubMed] [Google Scholar]
- Roberts JA, Durnin L, Sharkey KA, Mutafova‐Yambolieva VN, Mawe GM (2013). Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J Physiol 591: 3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RR, Bornstein JC, Bergner AJ, Young HM (2008). Disturbances of colonic motility in mouse models of Hirschsprung's disease. Am J Physiol Gastrointest Liver Physiol 294: G996–G1008. [DOI] [PubMed] [Google Scholar]
- Roberts RR, Murphy JF, Young HM, Bornstein JC (2007). Development of colonic motility in the neonatal mouse‐studies using spatiotemporal maps. Am J Physiol Gastrointest Liver Physiol 292: G930–G938. [DOI] [PubMed] [Google Scholar]
- Robinson AM, Stojanovska V, Rahman AA, McQuade RM, Senior PV, Nurgali K (2016). Effects of oxaliplatin treatment on the enteric glial cells and neurons in the mouse ileum. J Histochem Cytochem 64: 530–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Membrilla A, Martinez V, Jimenez M, Gonalons E, Vergara P (1995). Is nitric oxide the final mediator regulating the migrating myoelectric complex cycle? Am J Physiol Gastrointest Liver Physiol 268: G207–G214. [DOI] [PubMed] [Google Scholar]
- Sarna S, Otterson M, Ryan R, Cowles V (1993). Nitric oxide regulates migrating motor complex cycling and its postprandial disruption. Am J Physiol Gastrointest Liver Physiol 265: G749–G766. [DOI] [PubMed] [Google Scholar]
- Savidge TC (2011). S‐nitrosothiol signals in the enteric nervous system: lessons learnt from big brother. Front Neurosci 5: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer M, Richter C (1994). Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem Biophys Res Commun 204: 169–175. [DOI] [PubMed] [Google Scholar]
- Sharma R, Tobin P, Clarke SJ (2005). Management of chemotherapy‐induced nausea, vomiting, oral mucositis, and diarrhoea. Lancet Oncol 6: 93–102. [DOI] [PubMed] [Google Scholar]
- Slee EA, Adrain C, Martin SJ (2001). Executioner caspase‐3,‐6, and‐7 perform distinct, non‐redundant roles during the demolition phase of apoptosis. J Biol Chem 276: 7320–7326. [DOI] [PubMed] [Google Scholar]
- Souglakos J, Androulakis N, Syrigos K, Polyzos A, Ziras N, Athanasiadis A et al. (2006). FOLFOXIRI (folinic acid, 5‐fluorouracil, oxaliplatin and irinotecan) vs FOLFIRI (folinic acid, 5‐fluorouracil and irinotecan) as first‐line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). Br J Cancer 94: 798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovska V, Sakkal S, Nurgali K (2015). Platinum‐based chemotherapy: gastrointestinal immunomodulation and enteric nervous system toxicity. Am J Physiol Gastrointest Liver Physiol 308: G223–G232. [DOI] [PubMed] [Google Scholar]
- Takahashi T (2003). Pathophysiological significance of neuronal nitric oxide synthase in the gastrointestinal tract. J Gastroenterol 38: 421–430. [DOI] [PubMed] [Google Scholar]
- Van Bergeijk J, Van Westreenen H, Adhien P, Zijlstra F (1998). Diminished nitroprusside‐induced relaxation of inflamed colonic smooth muscle in mice. Mediators Inflamm 7: 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera G, Castillo M, Cabezos P, Chiarlone A, Martín M, Gori A et al. (2011). Enteric neuropathy evoked by repeated cisplatin in the rat. Neurogastroenterol Motil 23: 370–e163. [DOI] [PubMed] [Google Scholar]
- Verstappen CC, Heimans JJ, Hoekman K, Postma TJ (2003). Neurotoxic complications of chemotherapy in patients with cancer. Drugs 63: 1549–1563. [DOI] [PubMed] [Google Scholar]
- Wafai L, Taher M, Jovanovska V, Bornstein JC, Dass CR, Nurgali K (2013). Effects of oxaliplatin on mouse myenteric neurons and colonic motility. Front Neurosci 7: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Brain KL, Wilson RH, Grem JL, Vincent A (2005). Oxaliplatin induces hyperexcitability at motor and autonomic neuromuscular junctions through effects on voltage‐gated sodium channels. Br J Pharmacol 146: 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Chen C, Hou J, Xin W, Mori A (2000). Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim Biophys Acta 1498: 72–79. [DOI] [PubMed] [Google Scholar]
- Yagihashi N, Kasajima H, Sugai S, Matsumoto K, Ebina Y, Morita T et al. (2000). Increased in situ expression of nitric oxide synthase in human colorectal cancer. Virchows Arch 436: 109–114. [DOI] [PubMed] [Google Scholar]
- Zheng H, Xiao WH, Bennett GJ (2011). Functional deficits in peripheral nerve mitochondria in rats with paclitaxel‐and oxaliplatin‐evoked painful peripheral neuropathy. Exp Neurol 232: 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, O'Connor M, Hake PW (2003). Inhibitors of poly (ADP‐ribose) polymerase modulate signal transduction pathways in colitis. Eur J Pharmacol 469: 183–194. [DOI] [PubMed] [Google Scholar]