

Abstract
Background and Purpose
3,4‐Methylenedioxypyrovalerone (MDPV) is a synthetic cathinone with stimulatory cardiovascular effects that can lead to serious medical complications. Here, we examined the pharmacological mechanisms underlying these cardiovascular actions of MDPV in conscious rats.
Experimental Approach
Male Sprague–Dawley rats had telemetry transmitters surgically implanted for the measurement of BP and heart rate (HR). On test days, rats were placed individually in standard isolation cubicles. Following drug treatment, cardiovascular parameters were monitored for 3 h sessions.
Key Results
Racemic MDPV (0.3–3.0 mg·kg−1) increased BP and HR in a dose‐dependent manner. The S(+) enantiomer (0.3–3.0 mg·kg−1) of MDPV produced similar effects, while the R(−) enantiomer (0.3–3.0 mg·kg−1) had no effects. Neither of the hydroxylated phase I metabolites of MDPV altered cardiovascular parameters significantly from baseline. Pretreatment with the ganglionic blocker chlorisondamine (1 and 3 mg·kg−1) antagonized the increases in BP and HR produced by 1 mg·kg−1 MDPV. The α1‐adrenoceptor antagonist prazosin (0.3 mg·kg−1) attenuated the increase in BP following MDPV, while the β‐adrenoceptor antagonists propranolol (1 mg·kg−1) and atenolol (1 and 3 mg·kg−1) attenuated the HR increases.
Conclusions and Implications
The S(+) enantiomer appeared to mediate the cardiovascular effects of MDPV, while the metabolites of MDPV did not alter BP or HR significantly; MDPV increased BP and HR through activation of central sympathetic outflow. Mixed‐action α/β‐adrenoceptor antagonists may be useful as treatments in counteracting the adverse cardiovascular effects of MDPV.
Abbreviations
- 3,4‐catechol‐PV
3,4‐dihydroxypyrovalerone
- 4‐OH‐3‐MeO‐PV
4‐hydroxy‐3‐methoxypyrovalerone
- DAT
dopamine transporter
- HR
heart rate
- MDPV
3,4‐methylenedioxypyrovalerone
- mephedrone
4‐methyl‐N‐methylcathinone
- NET
noradrenaline transporter
- NPS
new psychoactive substances
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Transporters b |
| α1 Adrenoceptor | DAT, dopamine transporter |
| β‐Adrenoceptor | NET, noradrenaline transporter |
| Nicotinic receptors |
| LIGANDS |
|---|
| Atenolol |
| Propranolol |
| Prazosin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Synthetic cathinones are among the most prevalent new psychoactive substances (NPS) worldwide. Despite legislation in the US, Europe and elsewhere banning their use, many synthetic cathinones continue to be available in the recreational drug marketplace (Seely et al., 2013; Drug Enforcement Administration, 2014). The misuse of synthetic cathinones can lead to life‐threatening medical consequences. In this regard, the synthetic cathinone 3,4‐methylenedioxypyrovalerone (MDPV) is one of the most frequently detected NPS in individuals presenting for emergency medical care. In one of the first studies of patients reported to US poison control centres for ‘bath salts’ overdose, the majority of subjects with blood and urine toxicology data were found to be positive for MDPV (Spiller et al., 2011). A more recent report, which interrogated a clinical toxicology database in the US, found that all patients with confirmed synthetic cathinone exposure tested positive for MDPV (Froberg et al., 2015). Collectively, the clinical data point to MDPV as a major cause of the serious medical consequences.
The most commonly reported adverse effects of MDPV intoxication include psychiatric complications such as agitation, aggression and violent behaviour (Spiller et al., 2011; Ross et al., 2012; Beck et al., 2015; Froberg et al., 2015). However, cardiovascular symptoms are also prominent and include marked hypertension and tachycardia (Spiller et al., 2011; Ross et al., 2012; Beck et al., 2015; Froberg et al., 2015). While the number of clinical cases involving MDPV continues to grow, little is known about the pharmacological mechanisms responsible for the cardiovascular effects of the drug. Baumann et al. (2013) showed that MDPV produces dose‐dependent increases in both BP and heart rate (HR) in rats, which mirrors the effects of the drug in human subjects. The abused formulation of MDPV is a racemic mixture consisting of S(+) and R(−) enantiomers, and recent evidence indicates that neurochemical and behavioural effects reside primarily with the S(+) enantiomer. Kolanos et al. (2015a,b) reported that S(+)‐MDPV is a potent blocker of [3H]neurotransmitter uptake at the dopamine transporter (DAT) and noradrenaline transporters (NET), similar to the racemic mixture, whereas the R(−) enantiomer is much less potent. In the same report, in vivo results showed the S(+) enantiomer produces facilitation of intracranial self‐stimulation in rats, but the R(−) enantiomer does not. In mice, S(+)‐MDPV fully substitutes for cocaine in drug discrimination studies and is more potent than R(−)‐MDPV in producing locomotor stimulation (Gannon et al., 2016). To date, no studies have examined the cardiovascular effects of MDPV stereoisomers.
It is now well established that MDPV is extensively metabolized by hepatic mechanisms in rodents and humans (Meyer et al., 2010; Anizan et al., 2014; Novellas et al., 2015). Anizan et al. (2016) examined MDPV pharmacokinetics and metabolism in rats, and found that plasma concentrations of MDPV were positively correlated with the extent of locomotor activation. By contrast, plasma concentrations of the hydroxylated metabolites, 3,4‐dihydroxypyrovalerone (3,4‐catechol‐PV) and 4‐hydroxy‐3‐methoxypyrovalerone (4‐OH‐3‐MeO‐PV) were negatively correlated with motor activity, suggesting an inhibitory action. Alternatively, lower 4‐OH‐3‐MeO‐PV levels may reflect reduced MDPV metabolism resulting in higher MDPV exposure and effects, as shown for the similar metabolism of 3,4‐methylenedioxymethamphetamine (MDMA) in humans (Schmid et al., 2016). A previous investigation reported that 3,4‐catechol‐PV is an inhibitor of DAT and NET with potency similar to MDPV (Meltzer et al., 2006), but the bioactivity of MDPV metabolites in vivo has not been explored. Few studies have addressed the pharmacological mechanisms underlying the cardiovascular effects produced by MDPV or any other synthetic cathinones. In a study examining the cardiovascular effects of cathinone itself, Alsufyani and Docherty (2015) showed that tachycardia produced by the drug involves indirect actions at β‐adrenoceptors.
Given the evidence discussed above, it is tempting to speculate that cardiovascular actions of MDPV and other synthetic cathinones may involve stimulation of central or peripheral noradrenergic systems, perhaps secondary to blockade of NET. In the present study, we sought to explore the underlying mechanisms responsible for the stimulatory effects of MDPV on HR and BP in conscious rats. Specifically, we examined the dose‐dependent effects of acute administration of racemic MDVP, its stereoisomers, and its metabolites, in rats fitted with indwelling telemetric transmitters responsive to HR and BP. We used ganglionic blockade to determine whether the effects of racemic MDPV on cardiovascular function are mediated via central or peripheral sites of action. Finally, we pretreated rats with α‐ and β‐adrenoceptor antagonists prior to MDPV administration to investigate the receptor mechanisms responsible for cardiovascular effects of the drug.
Methods
Animals and housing
All animal care and experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Care and Use Committee of the NIDA/IRP. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All animals were maintained in facilities fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Thirteen adult male Sprague–Dawley rats (Charles River, Frederick, MD, USA) were used in the following experiments. Rats were chosen for study as this species has been used extensively and successfully in the study of abused drugs, particularly cathinone and its derivatives, in our laboratory and in others (Meng et al., 2012; Baumann et al., 2013; Varner et al., 2013; Alsufyani and Docherty, 2015). Prior to arrival in the laboratory, the rats received surgically implanted telemetry transmitters at Data Sciences International (St. Paul, MN, USA). Briefly, rats were anaesthetised with isoflurane. Following a midline incision of the abdominal wall, the catheter of the telemetry transmitter was implanted into the descending aorta and fixed in place. The transmitter was then secured to the abdominal cavity and the abdomen closed. Six of the rats were implanted with PA‐C40 transmitters, and seven were implanted with a newer replacement version of the PA‐C40, the HD‐S10. The PA‐C40 transmitters were no longer available for refurbishment from Data Sciences following our initial dose–response studies, which necessitated the switch to the HD‐S10 transmitters. Rats implanted with PA‐C40 transmitters were given 1 mg·kg−1 meloxicam s.c. and 0.25 mg·kg−1 buprenorphine s.c. on the day of surgery for post‐operative analgesia. Rats implanted with the HD‐S10 transmitters were only given s.c. meloxicam. All rats were given meloxicam for 3 days following surgery.
Rats were then shipped to the NIDA/IRP in Baltimore where they underwent 7 days of quarantine. The rats were individually housed in ventilated racks (One Cage, Lab Products, Seaford, DE), in cages with hardwood bedding (Teklad sani‐chips, Envigo, Indianapolis, IN) in a room (both housing and experimental rooms were maintained at 22.2 ± 1.1oC and 45 ± 10% humidity) with a 12 h reverse light/dark cycle (lights off at 7 a.m.). Water was available at all times except during training sessions. Over the course of the experiments, groups of rats bearing PA‐C40 or HD‐S10 transmitters were tested with 1 mg·kg−1 MDPV on multiple occasions, and the results were similar. The transmitters were only turned on during testing sessions.
Telemetric measurements
From 2 ‐ 4 weeks following arrival, experimental procedures began in a room separate from the housing room. The initial adaptation procedures and basic testing procedures were identical to those detailed in Schindler et al. (2014). Dose‐effect testing of MDPV, its isomers or metabolites was performed in the six rats with the PA‐C40 transmitters. Pretreatment time was 5 min. Prior to testing with MDPV and related drugs, the rats had been tested with a variety of cannabinoids and cannabinoid antagonists. These rats were allowed a minimum of 3 weeks washout before being used for the present work. The testing for these rats occurred when the rats were approximately 9 to 11 months of age. Rats implanted with the HD‐S10 transmitters were tested with pretreatment of prazosin (5 min), propranolol (5 min), atenolol (5 min) or chlorisondamine (10 min) prior to MDPV. The testing for these rats occurred when the rats were approximately 6 to 8 months of age, with the exception of testing with atenolol, which occurred when the rats were approximately 1 year old. The atenolol experiments were carried out in response to reviewer comments to an earlier version of the manuscript, which resulted in testing the rats when they were older. Test drugs were given s.c. no more frequently than twice a week, usually on Tuesdays and Fridays, with control vehicle injections given typically once or twice a month on Thursdays. Food (Teklad global 18% protein rodent diet, Envigo, Indianapolis, IN) intake was slightly restricted to maintain constant, or slowly increasing, body weight over the course of the experimental testing period (approximately 400–500 g). Order of drug treatment and dose administered was non‐systematic. However, all animals in a group were tested with the same dose on any given test day. That is, drug test order was not randomized over subjects. Over the course of the experiment, the rats were repeatedly tested with MDPV 1 mg·kg−1. In general, the results of each of these tests were comparable and they were always significantly different from saline treatment for both BP and HR, indicating that age and duration of drug testing did not affect the results.
Data analysis and statistics
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). BP and HR were averaged over 10 min periods (from the 1 min samples) for the time course data. Time course data were then analysed by two‐factor (dose × time) ANOVA (GraphPad Prism ver 5.0f, GraphPad Software). At individual time points, effects of drug treatment were compared with saline control by Bonferroni post hoc tests. Because time course data showed that effects of MDPV generally persisted throughout most of the 3 h session, data for the dose‐effect statistical comparisons among MDPV, its isomers and metabolites were performed using mean values across the entire session. As BP clearly peaked in the first 60 min of the session (Figure 1), and to make sure the pretreatment drugs were at their peak of effectiveness, data from the first 60 min of the session were averaged and compared for statistical analysis of the pretreatment studies. Statistical comparisons for the mean dose‐effect and pretreatment data were made using ANOVA, followed by Dunnett's post hoc tests to compare treatments with appropriate controls. Where appropriate, Tukey tests were used to compare across treatments. Because some of the pretreatments had effects on their own, we also calculated change scores to make appropriate comparisons. For example, prazosin alone decreased BP and increased HR. So we took the effects of prazosin alone and subtracted them from the effects of prazosin plus MDPV. We then compared this change score to the change score determined by the difference between MDPV alone and saline control. Similar change scores were calculated for chlorisondamine and propranolol.
Figure 1.
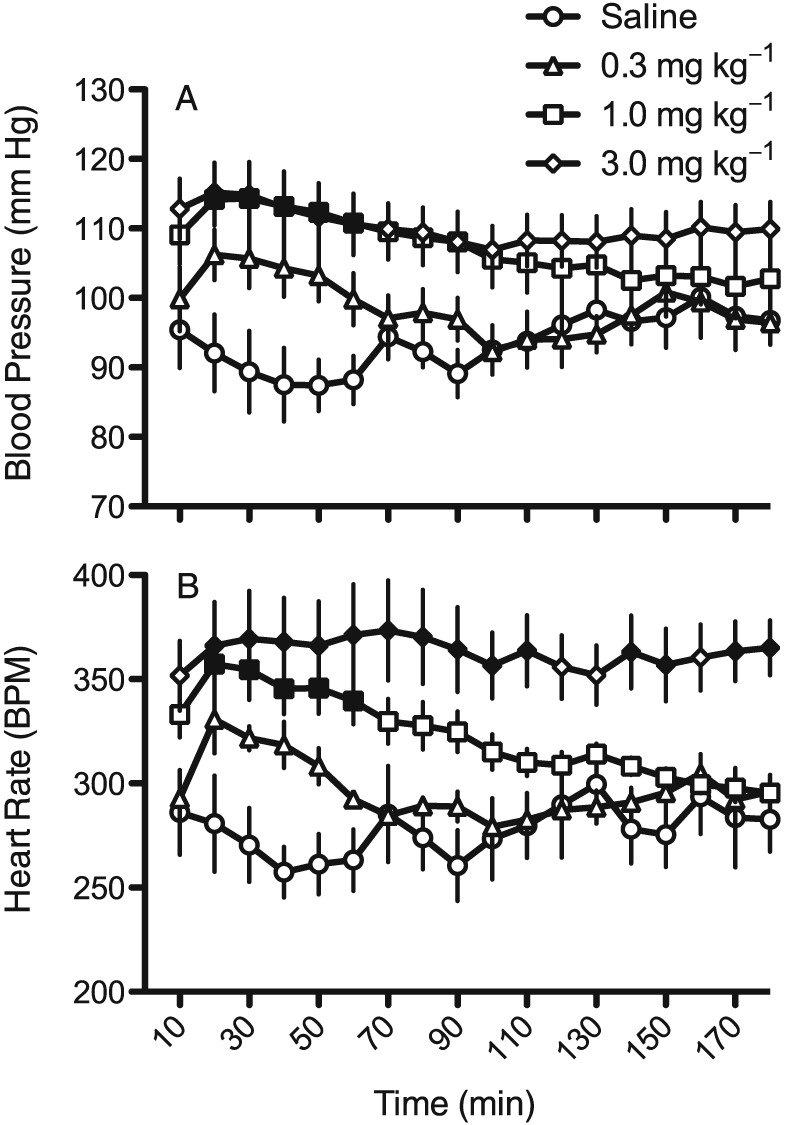
Time course for the effects of saline and three doses of racemic MDPV on BP (A) and in (B) HR (as beats min‐1; BPM). Each point represents a 10 min mean ± SEM (n = 6), and solid symbols are significantly different (P<0.05) compared with the same time point for saline administration.
Materials
(±)‐3,4‐Methylenedioxypyrovalerone HCl (from here on referred to as MDPV), S(+)‐3,4‐methylenedioxypyrovalerone HCl [S(+)‐MDPV], R(−)‐3,4‐methylenedioxypyrovalerone HCl [R(−)‐MDPV], 3,4‐catechol‐PV and 4‐OH‐3‐MeO‐PV were synthesized and analysed for purity in the Design and Synthesis Section of the National Institutes on Drug Abuse. Propranolol HCl, prazosin HCl and atenolol were purchased from Sigma Chemical (St. Louis, MO, USA). Chlorisondamine diiodide was purchased from Tocris (Ellisville, MO, USA). All drugs were dissolved in saline except prazosin, which was dissolved in sterile water. All drugs were given s.c. in a volume of 1 mL·kg−1 of body weight. All doses are expressed as the salt.
Results
Rats injected with saline showed gradual decreases in BP and HR over the first hour after being placed in the experimental chambers (Figure 1). These initial drops in BP and HR were followed by small, gradual increases in both parameters throughout the last 2 h of the session. Racemic MDPV produced clear dose‐dependent increases in both BP and HR. The effects on BP peaked in the first 60 min of the session and returned towards the saline baseline by the end of the 3 h session (F51,340 = 5.1, P < 0.05). With lower doses of MDPV a similar pattern of transient HR increases was seen (F51,340 = 3.2, P < 0.05), however, at the 3.0 mg·kg−1 dose, tachycardia was sustained throughout the 3 h session.
Figure 2 shows the dose‐effect function for MDPV for BP and HR averaged across the 3 h session. The dose‐dependent increase in BP is clearly evident (F3,23 = 4.3, P < 0.05), with the two highest doses being significantly different from saline. For HR, a dose‐dependent increase is also seen (F3,23 = 8.6, P < 0.05), but only the increase at 3.0 mg·kg−1 MDPV was significant. The S(+) isomer of MDPV also increased BP (F3,23 = 6.1, P < 0.05) and HR (F3,23 = 9.5, P < 0.05). The effect of the S(+) isomer on BP was slightly greater than MDPV, while the highest dose of S(+)‐MDPV showed a slight downturn in the HR increase from that seen at 1.0 mg·kg−1. The R(−) isomer of MDPV did not significantly affect either parameter. Neither of the hydroxylated phase I metabolites of MDPV (3,4‐catechol‐PV or 4‐OH‐3‐MeO‐PV) produced increases in BP or HR. The effect of 4‐OH‐3‐MeO‐PV on HR rate approached significance (F3,23 = 2.9, P = 0.06), with the 3.0 mg·kg−1 dose slightly reducing HR when compared with saline.
Figure 2.
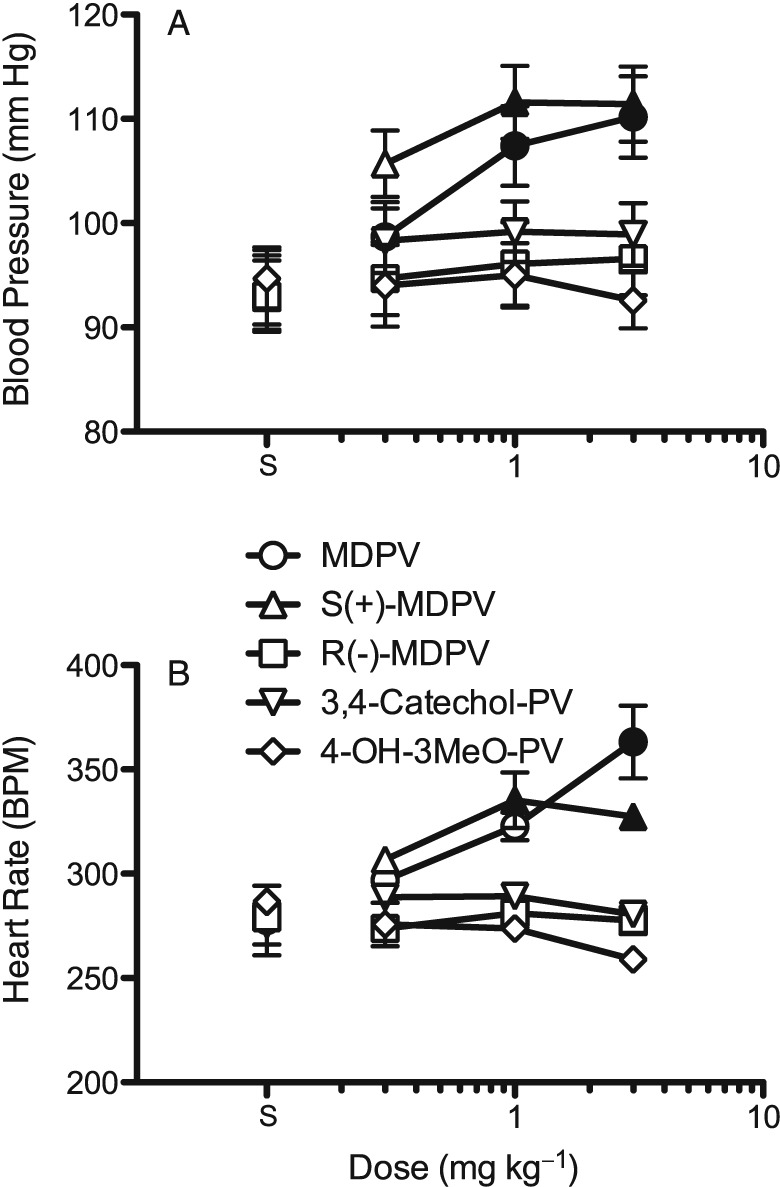
Dose‐effect functions for racemic MDPV, the MDPV enantiomers and the MDPV metabolites on BP (A) and in (B) HR (as beats min‐1; BPM). Each point represents the mean ± SEM (n = 6) for the entire 3 h session. Solid symbols indicate significant difference (P<0.05) from the respective saline controls (points above ‘S’). Only racemic MDPV and the S(+) enantiomer increased BP and HR.
Because the effect of racemic MDPV (1 mg·kg−1) on BP was most robust during the first 60 min of the session (Figure 1), for the drug interaction studies, only data from the first 60 min of the session were analysed. We also only investigated racemic MDPV, as its effects were similar to S(+)‐MDPV, whereas the R(−) isomer and the metabolites did not increase BP or HR at any dose tested. The ganglionic blocker chlorisondamine decreased BP on its own (Figure 3A), and combinations of chlorisondamine and MDPV also showed lower BP than saline (F5,41 = 100.8, P < 0.05). MDPV alone produced a significant increase in HR (Figure 3C, F5,41 = 7.1, P < 0.05), and pretreatment with 1 mg·kg−1 chlorisondamine did not reverse this effect. However, rats pretreated with 3 mg·kg−1 chlorisondamine prior to 1 mg·kg−1 MDPV did not show a significant increase in HR when compared with saline.
Figure 3.
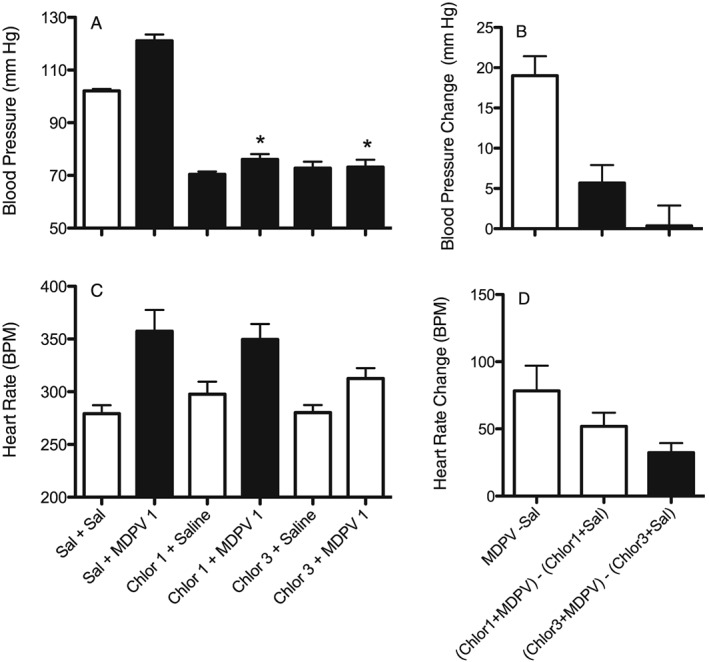
Effects of MDPV (1 mg·kg−1) on BP (A and B) and in (C and D) HR (as beats min‐1; BPM), alone and after pretreatment with the ganglionic blocker chlorisondamine (1 and 3 mg·kg−1). A and C show actual results for each individual treatment. Dark bars are treatments that are significantly different (P < 0.05) from the saline + saline control (Sal + Sal). *P<0.05, significant difference between chlorisondamine + MDPV compared with MDPV alone. B and D show change scores whereby effects of saline or chlorisondamine + saline are subtracted from the effects of the corresponding MDPV treated group. Dark bars are significantly different (P<0.05) from MDPV 1 – Saline. Each bar is mean ± SEM (n = 7) for the first hour of the session.
To account for the drop in pressure produced by chlorisondamine, we calculated change scores for each rat between the MDPV treatment and the appropriate control baseline (Figure 3B). MDPV produced an increase in BP of about 20 mm Hg when compared with saline controls (MDPV – Saline). When pretreated with chlorisondamine 1 mg·kg−1, MDPV produced an increase in BP of approximately 5 mm Hg [(chlorisondamine 1 mg·kg–1 + MDPV 1 mg·kg−1) – (chlorisondamine 1 mg·kg−1 + saline)], which was significantly reduced compared with MDPV alone (F2,20 = 16.1, P < 0.05). When pretreated with a higher dose of chlorisondamine (3 mg·kg−1), there was no change in BP following MDPV when compared with chlorisondamine alone [(chlorisondamine 3 mg‐kg−1 + MDPV 1) – (chlorisondamine 3 mg·kg−1 + saline)]. For HR change scores (Figure 3D), MDPV alone produced an increase in HR in the first 60 min of the session of approximately 75 beats/min (F2,20 = 5.2, P = 0.07). Pretreatment with 1 mg·kg−1 chlorisondamine reduced the increase in HR to around 50 beats/min, but that was not significantly different from MDPV alone. Pretreatment with the higher dose of chlorisondamine (3 mg·kg−1) reduced the HR increase following MDPV even more, and this increase was significantly different from MDPV alone. While there was a tendency for MDPV to increase mean pressure more than diastolic pressure and to increase systolic pressure more than mean pressure, chlorisondamine was able to antagonize the effects on diastolic and systolic pressure as well as on mean pressure (data not shown).
When compared with saline, neither 0.3 mg·kg−1 prazosin nor 1 mg·kg−1 propranolol altered BP. However, prazosin pretreatment did block the BP increase following 1 mg·kg−1 MDPV (Figure 4A, F5,41 = 27.9, P < 0.05). Prazosin also blocked the increase seen in diastolic and systolic pressure (data not shown). Rats pretreated with propranolol continued to show an increase in BP following MDPV. For the change scores (Figure 4B), MDPV alone produced an increase in pressure of just under 20 mm Hg when compared with saline (F2,20 = 13.7, P < 0.05), while in rats pretreated with prazosin the increase in pressure following MDPV was less than 5 mm Hg. Rats pretreated with propranolol continued to show an increase in BP similar to MDPV alone. As with chlorisondamine, the effects of prazosin and propranolol on mean pressure were mimicked on systolic and diastolic pressure.
Figure 4.
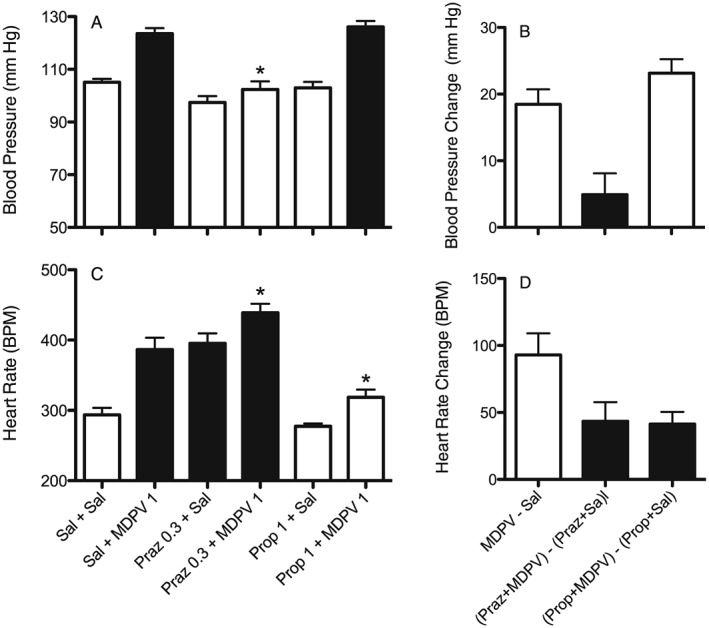
Effects of MDPV (1 mg·kg−1, s.c.) on BP (A and B) and HR (C and D) alone and after pretreatment with the α‐adrenoceptor antagonist prazosin (Praz 0.3 mg·kg−1, s.c.) or the β‐adrenoceptor antagonist propranolol (Prop 1.0 mg·kg−1, s.c.). A and C show actual results for each individual treatment. Dark bars are treatments that are significantly different (P<0.05) from the saline + saline control (Sal + Sal). * P<0.05, significant differences between pretreatment + MDPV compared with MDPV alone. B and D show change scores whereby effects of saline, prazosin + saline or propanolol + saline are subtracted from the effects of the corresponding MDPV treated group. Dark bars are significantly different (P<0.05) from MDPV 1 – Saline. Each bar is mean ± SEM (n = 7) for the first hour of the session.
In contrast to the BP results, prazosin alone produced a significant increase in HR (Figure 4C, F5,41 = 28.2, P < 0.05), and the tachycardia following MDPV was enhanced even further in those rats pretreated with prazosin. Propranolol produced a small, non‐significant decrease in HR and pretreatment with propranolol blocked the increase in HR following MDPV. For the change scores (Figure 4D), MDPV alone produced an increase in HR of about 100 beats min‐1 (F2,20 = 18.8, P < 0.05). When compared with prazosin alone, the effect of MDPV on HR was reduced following prazosin. However, since prazosin alone increased HR, the effect of MDPV following prazosin was still significantly above saline levels. Pretreatment with propranolol also significantly decreased the effect of MDPV on HR, with the overall effect being no different from saline.
As propranolol is a non‐selective β‐adrenoceptor antagonist, we also tested the β1‐selective adrenoceptor antagonist atenolol. As observed with propranolol, pretreatment with atenolol at doses of 1 or 3 mg·kg−1 did not affect the BP increase (F5,40 = 30.2, P < 0.05) seen with 1 mg·kg−1 MDPV (Figure 5A). However, both doses of atenolol did reduce the HR increase seen with MDPV (Figure 5B, F5,40 = 17.3, P < 0.05).
Figure 5.
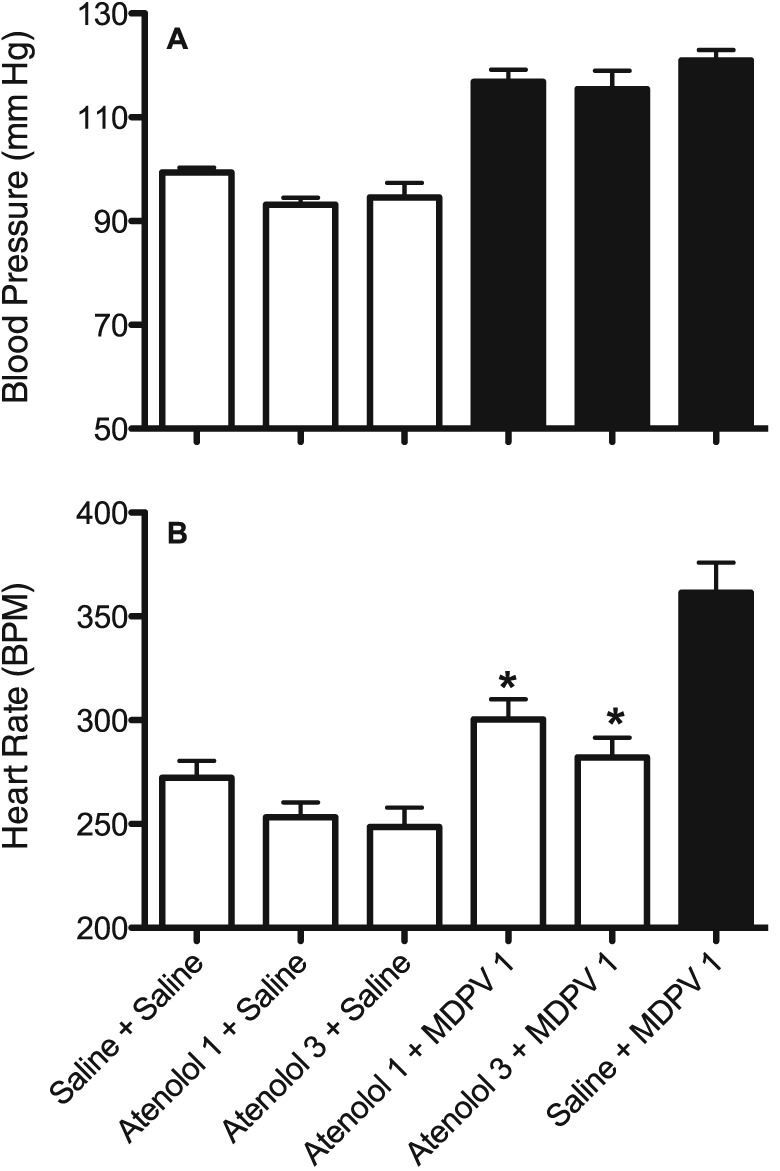
Effects of MDPV (1 mg·kg−1, s.c.) on BP (A) and HR (B) alone and after pretreatment with the β1‐adrenoceptor antagonist atenolol (1.0 and 3.0 mg/kg, s.c.). Dark bars are treatments that are significantly different (P<0.05) from the saline + saline control (Sal + Sal). * P<0.05, significant differences between pretreatment + MDPV compared with MDPV alone;. Each bar is mean ± SEM (n = 7, except for the atenolol 3 + MDPV group that was n = 6 due to the loss of data from one rat) for the first hour of the session.
Discussion
MDPV is a widely abused synthetic cathinone analogue, and here, we aimed to determine the mechanisms underlying the cardiovascular stimulation produced by the drug. In agreement with previous results (Baumann et al., 2013), we found that MDPV produces substantial dose‐dependent increases in both BP and HR in conscious male rats. In this regard, MDPV is similar to other psychomotor stimulant drugs of abuse such as cocaine (Tella et al., 1992, 1993), methamphetamine (Varner et al., 2013) and MDMA ecstasy (Schindler et al., 2014). The stimulatory actions of MDPV on BP and HR are also similar to structurally related compounds such as cathinone (Yanagita, 1979; Alsufyani and Docherty, 2015) and 4‐methyl‐N‐methylcathinone (mephedrone) (Meng et al., 2012; Varner et al., 2013).
Here, we report stereo‐selective cardiovascular effects of MDPV for the first time. Analogous to the behavioural and neurochemical effects of racemic MDPV (Kolanos et al., 2015a,b; Gannon et al., 2016), the BP and HR increases produced by the drug reside primarily with the S(+) enantiomer. While not significantly different from the effects of racemic MDPV, the pressor effects of the S(+) enantiomer were greater in magnitude than MDPV at lower doses. Interestingly, at the highest dose, S(+)‐MDPV produced a smaller increase in HR than the racemate. Such a paradoxical effect is consistent with more potent effects of the S(+) enantiomer at higher doses, which triggers baroreceptor reflex responses to lower HR (i.e. reflex bradycardia). Stereo‐selective pharmacological effects have been reported for other cathinone analogues. Kalix (1986) reported that the naturally occurring S(−)‐enantiomer of cathinone is more potent than the R(+)‐enantiomer in stimulating central dopamine release, although both enantiomers were equipotent in releasing peripheral noradrenaline. Glennon et al. (1995) reported that S(−)‐methcathinone is more potent that the R(+)‐methcathinone for increasing locomotor activity and generalizing to cocaine and amphetamine in drug discrimination studies. By contrast, Gregg et al. (2015) recently demonstrated that R(+)‐mephedrone (i.e. the 4‐position ring‐substituted analogue of methcathinone) is more potent in producing locomotor activity and locomotor sensitization than S(−)‐mephedrone, and only R(+)‐mephedrone produces conditioned place preference. Hutsell et al. (2016) reported that R(+)‐4‐methylcathinone is DAT‐selective and also produces intracranial self‐stimulation (ICSS) facilitation, an effect common to drugs of abuse, while the S(−) enantiomer produces ICSS depression. Thus, it appears that ring‐substituted cathinones have inverted stereo‐selectivity, where R(+) isomers are more potent stimulants than S(−)‐isomers, when compared with the effects of cathinone and methcathinone.
Metabolites of psychomotor stimulant drugs can contribute significantly to their overall cardiovascular effects, particularly when drugs are used in binge patterns where additional doses of drug may be administered during a time when metabolite concentrations from the initial drug administration are increasing. For example, a metabolite of cocaine produced when cocaine is used in combination with alcohol, cocaethylene, has potent cardiovascular effects in rats (Erzouki et al., 1993; Morishima et al., 1999). The dihydroxy metabolite of MDMA has stimulatory effects on HR that exceed the effects of the parent compound (Schindler et al., 2014). In this regard, the hepatic metabolism of MDPV is analogous to that of MDMA, where O‐demethylenation of MDPV forms the primary phase I metabolite, 3,4‐catechol‐PV. Once formed, 3,4‐catechol‐PV is rapidly O‐methylated to form 4‐OH‐3‐MeO‐PV (Anizan et al., 2016). Because 3,4‐catechol‐PV is a potent blocker of DAT and NET in vitro (Meltzer et al., 2006), it seemed possible that peripheral actions of this metabolite at NET may affect cardiovascular function. However, here we provide convincing evidence that neither of the hydroxylated metabolites of MDPV significantly altered BP or HR responses at doses equivalent to those of MDPV. Our findings support the idea that metabolites of MDPV do not contribute to its cardiovascular effects.
One fundamental question regarding the cardiovascular effects of MDPV and other stimulants relates to the role of peripheral versus central noradrenergic mechanisms. Chlorisondamine is a ganglionic blocker that serves as a useful tool to disrupt central sympathetic outflow (see Kiritsy‐Roy et al., 1990). In our experiments, chlorisondamine eliminated the BP responses to MDPV and greatly reduced the accompanying increase in HR. Furthermore, the α1‐adrenoceptorantagonist prazosin blocked the BP increase, while the non‐selective β‐adrenoceptor antagonist propranolol and the β1‐selective adrenoceptor antagonist atenolol blocked the HR increase following MDPV. Collectively, these results suggest that the cardiovascular effects of MDPV are mediated via central sympathetic outflow. The finding that prazosin also attenuated the HR response following MDPV could be related to the effect of prazosin alone on baseline HR. Prazosin alone had a profound tachycardic effect, and increased HR by about 100 beats min‐1. Following pretreatment with prazosin, MDPV produced a further increase of approximately 40 beats min‐1, but this may have been reduced from MDPV alone due to a ceiling effect on HR. Similar to the present findings with MDPV, blockade of α‐adrenoceptors lowered BP, but enhanced HR increases in response to MDMA (Hysek et al., 2013). Blockade of β‐adrenoceptors lowered tachycardia, but enhanced the pressure response to cocaine (Ramoska and Sacchetti, 1985; Schindler et al., 1992) or MDMA (Hysek et al., 2010). Blockade of both α‐ and β‐adrenoceptors reduces the increases in BP and HR responses produced by stimulant drugs (Schindler et al., 1992; Boehrer et al., 1993; Hysek et al., 2012).
Few studies have investigated the pharmacological mechanisms involved with cardiovascular actions of cathinones. Alsufyani and Docherty (2015) reported that cathinone itself increases both BP and HR, similar to the effects observed here for MDPV. Both effects of cathinone are blocked in chemically‐sympathectomized rats, and propranolol pretreatment blocked HR increases, suggesting that cardiovascular effects of cathinone are mediated via the sympathetic nervous system. Meng et al. (2012) showed that the synthetic cathinone mephedrone also increased BP and HR, but they found little evidence for a direct effect of mephedrone on the heart. Clearly, more experiments are warranted to address the central and peripheral mechanisms responsible for cardiovascular effects of mephedrone and other cathinone analogues.
The results of the present study with MDPV are in general agreement with the findings for other psychostimulants such as cocaine, where adrenoceptor antagonists and ganglionic blockers can reverse increases in both BP and HR (Tella et al., 1992; Tella et al., 1993). This should not be surprising as the molecular mechanism of action for MDPV closely mimics that of cocaine. Specifically, MDPV is a potent blocker of DAT and NET, which inhibits the reuptake of dopamine and noradrenaline at central synapses (Baumann et al., 2013; Simmler et al., 2013). In contrast to cocaine, however, MDPV has weak activity as a blocker of SERT. Unlike the effects of amphetamines, MDPV is not a transporter substrate, so the drug does not induce transporter‐mediated release of dopamine or noradrenaline. Other synthetic cathinones, like mephedrone and methylone, function more like amphetamine and are capable of inducing transporter‐mediated release of dopamine, noradrenaline and 5‐HT (Baumann et al., 2012).
To summarize, we found that the synthetic cathinone MDPV produced significant increases in both BP and HR. Such cardiovascular stimulation would be expected to increase the workload on the heart, and could potentially lead to adverse consequences in certain vulnerable individuals. The cardiovascular effects of racemic MDPV are mediated by the S(+) enantiomer, while the metabolites of MDPV do not appear to contribute to its cardiovascular effects. The mechanism responsible for stimulation of BP and HR after MDPV most likely involves an increase in central sympathetic outflow. Therefore, treatment with adrenoceptor antagonists, particularly mixed‐action α/β‐adrenoceptor antagonists, may be useful in counteracting the adverse cardiovascular effects of MDPV in emergency situations. Treatment with α‐adrenoceptor antagonists alone would not be recommended as these compounds may worsen the HR response to MDPV.
Author contributions
C.W.S. and M.H.B. were responsible for the conception and the design of the study. C.W.S. wrote the first draft of the paper. C.W.S. and E.B.T. analysed the data, and C.W.S. and M.H.B. interpreted the data. M.S. and K.C.R. synthesized the enantiomers and metabolites of MDPV. E.B.T. handled the animals. C.W.S., E.B.T., M.S., K.C.R. and M.H.B. gave final approval of the article.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This research was supported by the Intramural Research Programs of the National Institute on Drug Abuse (NIDA) and National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institutes of Health.
Schindler, C. W. , Thorndike, E. B. , Suzuki, M. , Rice, K. C. , and Baumann, M. H. (2016) Pharmacological mechanisms underlying the cardiovascular effects of the “bath salt” constituent 3,4‐methylenedioxypyrovalerone (MDPV). British Journal of Pharmacology, 173: 3492–3501. doi: 10.1111/bph.13640.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsufyani HA, Docherty JR (2015). Direct and indirect cardiovascular actions of cathinone and MDMA in the anaesthetized rat. Eur J Pharmacol 758: 142–146. [DOI] [PubMed] [Google Scholar]
- Anizan S, Ellefsen K, Concheiro M, Suzuki M, Rice KC, Baumann MH et al. (2014). 3,4‐Methylenedioxypyrovalerone (MDPV) and metabolites quantification in human and rat plasma by liquid chromatography‐high resolution mass spectrometry. Anal Chim Acta 827: 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anizan S, Concheiro M, Lehner KR, Bukhari MO, Suzuki M, Rice KC et al. (2016). Linear pharmacokinetics of 3,4‐methylenedioxypyrovalerone (MDPV) and its metabolites in the rat: relationship to pharmacodynamic effects. Addict Biol 21: 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF et al. (2012). The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37: 1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M et al. (2013). Powerful cocaine‐like actions of 3,4‐methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology 38: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck O, Franzen L, Backberg M, Signell P, Helander A (2015). Intoxications involving MDPV in Sweden during 2010‐2014: results from the STRIDA project. Clin Toxicol (Phila) 53: 865–873. [DOI] [PubMed] [Google Scholar]
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA (1993). Influence of labetalol on cocaine‐induced coronary vasoconstriction in humans. Am J Med 94: 608–610. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drug Enforcement Administration (2014). National Forensic Laboratory Information System Special Report: Synthetic Cannabinoids and Synthetic Cathinones Reported in NFLIS, 2010–2013. U.S. Drug Enforcement Administration: Springfield, VA. [Google Scholar]
- Erzouki HK, Baum I, Goldberg SR, Schindler CW (1993). Comparison of the effects of cocaine and its metabolites on cardiovascular function in anesthetized rats. J Cardiovasc Pharmacol 22: 557–563. [DOI] [PubMed] [Google Scholar]
- Froberg BA, Levine M, Beuhler MC, Judge BS, Moore PW, Engebretsen KM et al. (2015). Acute methylenedioxypyrovalerone toxicity. J Med Toxicol 11: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon BM, Williamson A, Suzuki M, Rice KC, Fantegrossi WE (2016). Stereoselective effects of abused “bath salt” constituent 3,4‐methylenedioxypyrovalerone in mice: drug discrimination, locomotor activity, and thermoregulation. J Pharmacol Exp Ther 356: 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennon RA, Young R, Martin BR, Dal Cason TA (1995). Methcathione (“cat”): an enantiomeric potency comparison. Pharmacol Biochem Behav 50: 601–606. [DOI] [PubMed] [Google Scholar]
- Gregg RA, Baumann MH, Partilla JS, Bonano JS, Vouga A, Tallarida CS et al. (2015). Stereochemistry of mephedrone neuropharmacology: enantiomer‐specific behavioural and neurochemical effects in rats. Br J Pharmacol 172: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsell BA, Baumann MH, Partilla JS, Banks ML, Vekariya R, Glennon RA et al. (2016). Abuse‐related neurochemical and behavioral effects of cathinone and 4‐methylcathinone stereoisomers in rats. Eur Neuropsychopharmacol 26: 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hysek CM, Vollenweider FX, Liechti ME (2010). Effects of a beta‐blocker on the cardiovascular response to MDMA (Ecstasy). Emerg Med J 27: 586–589. [DOI] [PubMed] [Google Scholar]
- Hysek C, Schmid Y, Rickli A, Simmler LD, Donzelli M, Grouzmann E et al. (2012). Carvedilol inhibits the cardiostimulant and thermogenic effects of MDMA in humans. Br J Pharmacol 166: 2277–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hysek CM, Fink AE, Simmler LD, Donzelli M, Grouzmann E, Liechti ME (2013). alpha(1)‐Adrenergic receptors contribute to the acute effects of 3,4‐methylenedioxymethamphetamine in humans. J Clin Psychopharmacol 33: 658–666. [DOI] [PubMed] [Google Scholar]
- Kalix P (1986). The releasing effect of the isomers of the alkaloid cathinone at central and peripheral catecholamine storage sites. Neuropharmacology 25: 499–501. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsy‐Roy JA, Halter JB, Gordon SM, Smith MJ, Terry LC (1990). Role of the central nervous system in hemodynamic and sympathoadrenal responses to cocaine in rats. J Pharmacol Exp Ther 255: 154–160. [PubMed] [Google Scholar]
- Kolanos R, Partilla JS, Baumann MH, Hutsell BA, Banks ML, Negus SS et al. (2015a). Stereoselective actions of methylenedioxypyrovalerone (MDPV) to inhibit dopamine and norepinephrine transporters and facilitate intracranial self‐stimulation in rats. ACS Chem Nerosci 6: 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolanos R, Sakloth F, Jain AD, Partilla JS, Baumann MH, Glennon RA (2015b). Structural modification of the designer stimulant alpha‐pyrrolidinovalerophenone (alpha‐PVP) influences potency at dopamine transporters. ACS Chem Nerosci 6: 1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer PC, Butler D, Deschamps JR, Madras BK (2006). 1‐(4‐Methylphenyl)‐2‐pyrrolidin‐1‐yl‐pentan‐1‐one (Pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem 49: 1420–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Cao J, Kang J, Ying X, Ji J, Reynolds W et al. (2012). Mephedrone, a new designer drug of abuse, produces acute hemodynamic effects in the rat. Toxicol Lett 208: 62–68. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Du P, Schuster F, Maurer HH (2010). Studies on the metabolism of the alpha‐pyrrolidinophenone designer drug methylenedioxy‐pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC–MS and LC‐high‐resolution MS and its detectability in urine by GC–MS. J Mass Spectrom 45: 1426–1442. [DOI] [PubMed] [Google Scholar]
- Morishima HO, Whittington RA, Iso A, Cooper TB (1999). The comparative toxicity of cocaine and its metabolites in conscious rats. Anesthesiology 90: 1684–1690. [DOI] [PubMed] [Google Scholar]
- Novellas J, Lopez‐Arnau R, Carbo ML, Pubill D, Camarasa J, Escubedo E (2015). Concentrations of MDPV in rat striatum correlate with the psychostimulant effect. J Psychopharmacol 29: 1209–1218. [DOI] [PubMed] [Google Scholar]
- Ramoska E, Sacchetti AD (1985). Propranolol‐induced hypertension in treatment of cocaine intoxication. Ann Emerg Med 14: 1112–1113. [DOI] [PubMed] [Google Scholar]
- Ross EA, Reisfield GM, Watson MC, Chronister CW, Goldberger BA (2012). Psychoactive “bath salts” intoxication with methylenedioxypyrovalerone. Am J Med 125: 854–858. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Tella SR, Goldberg SR (1992). Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci 51: 653–660. [DOI] [PubMed] [Google Scholar]
- Schindler CW, Thorndike EB, Blough BE, Tella SR, Goldberg SR, Baumann MH (2014). Effects of 3,4‐methylenedioxymethamphetamine (MDMA) and its main metabolites on cardiovascular function in conscious rats. Br J Pharmacol 171: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid Y, Vizeli P, Hysek CM, Prestin K, Meyer Zu Schwabedissen HE, Liechti ME (2016). CYP2D6 function moderates the pharmacokinetics and pharmacodynamics of 3,4‐methylene‐dioxymethamphetamine in a controlled study in healthy individuals. Pharmacogenet Genomics 26: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seely KA, Patton AL, Moran CL, Womack ML, Prather PL, Fantegrossi WE et al. (2013). Forensic investigation of K2, Spice, and “bath salt” commercial preparations: a three‐year study of new designer drug products containing synthetic cannabinoid, stimulant, and hallucinogenic compounds. Forensic Sci Int 233: 416–422. [DOI] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J et al. (2013). Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller HA, Ryan ML, Weston RG, Jansen J (2011). Clinical experience with and analytical confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin Toxicol (Phila) 49: 499–505. [DOI] [PubMed] [Google Scholar]
- Tella SR, Schindler CW, Goldberg SR (1992). Cardiovascular effects of cocaine in conscious rats: relative significance of central sympathetic stimulation and peripheral neuronal monoamine uptake and release mechanisms. J Pharmacol Exp Ther 262: 602–610. [PubMed] [Google Scholar]
- Tella SR, Schindler CW, Goldberg SR (1993). Cocaine: cardiovascular effects in relation to inhibition of peripheral neuronal monoamine uptake and central stimulation of the sympathoadrenal system. J Pharmacol Exp Ther 267: 153–162. [PubMed] [Google Scholar]
- Varner KJ, Daigle K, Weed PF, Lewis PB, Mahne SE, Sankaranarayanan A et al. (2013). Comparison of the behavioral and cardiovascular effects of mephedrone with other drugs of abuse in rats. Psychopharmacology (Berl) 225: 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagita T (1979). Studies on cathinones: cardiovascular and behavioral effects in rats and self‐administration experiment in rhesus monkeys. NIDA Res Monogr 27: 326–327. [PubMed] [Google Scholar]