

Abstract
In moderate‐to‐severe asthma, adding an inhaled long‐acting β2‐adenoceptor agonist (LABA) to an inhaled corticosteroid (ICS) provides better disease control than simply increasing the dose of ICS. Acting on the glucocorticoid receptor (GR, gene NR3C1), ICSs promote anti‐inflammatory/anti‐asthma gene expression. In vitro, LABAs synergistically enhance the maximal expression of many glucocorticoid‐induced genes. Other genes, including dual‐specificity phosphatase 1(DUSP1) in human airways smooth muscle (ASM) and epithelial cells, are up‐regulated additively by both drug classes. Synergy may also occur for LABA‐induced genes, as illustrated by the bronchoprotective gene, regulator of G‐protein signalling 2 (RGS2) in ASM. Such effects cannot be produced by either drug alone and may explain the therapeutic efficacy of ICS/LABA combination therapies. While the molecular basis of synergy remains unclear, mechanistic interpretations must accommodate gene‐specific regulation. We explore the concept that each glucocorticoid‐induced gene is an independent signal transducer optimally activated by a specific, ligand‐directed, GR conformation. In addition to explaining partial agonism, this realization provides opportunities to identify novel GR ligands that exhibit gene expression bias. Translating this into improved therapeutic ratios requires consideration of GR density in target tissues and further understanding of gene function. Similarly, the ability of a LABA to interact with a glucocorticoid may be suboptimal due to low β2‐adrenoceptor density or biased β2‐adrenoceptor signalling. Strategies to overcome these limitations include adding‐on a phosphodiesterase inhibitor and using agonists of other Gs‐coupled receptors. In all cases, the rational design of ICS/LABA, and derivative, combination therapies requires functional knowledge of induced (and repressed) genes for therapeutic benefit to be maximized.
Abbreviations
- AHR
airways hyperreactivity
- AP‐1
activator protein 1
- ASM
airways smooth muscle
- C/EBP
CCAAT‐enhancer binding protein
- COPD
chronic obstructive pulmonary disease
- CRE
cAMP response element
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- HBE
human bronchial epithelial
- HRV
human rhinovirus
- ICS
inhaled corticosteroid
- LABA
long‐acting β2‐adrenoceptor agonist
- PEPCK
phosphoenolpyruvate carboxykinase
- PKA
protein kinase A
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| Adenosine A2B receptor | Phosphodiesterase 4 (PDE4) |
| β2‐adrenoreceptor | |
| Prostacyclin IP receptor | |
| Nuclear hormone receptors b | |
| Glucocorticoid receptor (GR) / NR3C1 | |
| LIGANDS | |
|---|---|
| Beclomethasone dipropionate | Forskolin |
| Budesonide | Mifepristone (RU486) |
| Ciclesonide | Roflumilast |
| Dexamethasone | Salmeterol |
| Fluticasone furoate | |
| Fluticasone propionate | |
| Formoterol | |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
In 1994, a clinical study was published that fundamentally transformed the treatment algorithm for the management of asthma (Greening et al., 1994). The salient finding was that asthmatic subjects who were symptomatic despite maintenance therapy with a standard dose of the inhaled corticosteroid (ICS), beclomethasone dipropionate, were controlled by the addition of the long‐acting β2‐adrenoceptor agonist (LABA), salmeterol, but not by increasing the dose of ICS (Greening et al., 1994). The clinical superiority of an ICS and LABA in combination relative to ICS alone, irrespective of dose, was subsequently and independently corroborated using several outcome measures including, symptom score, lung function, use of rescue medication and exacerbation frequency (Pauwels et al., 1997; Shrewsbury et al., 2000; O'Byrne et al., 2001; Frois et al., 2009; Ducharme et al., 2010a; Ducharme et al., 2010b; Sears, 2011). Today, ICS/LABA combination therapies are entrenched in all national and international asthma treatment guidelines and are a recommended option for patients whose symptoms remain uncontrolled by ICS monotherapy (www.ginasthma.org). Furthermore, patients with chronic obstructive pulmonary disease (COPD), who present with a high level of inflammation and suffer frequent exacerbations, may also respond more favourably to an ICS/LABA combination therapy relative to a LABA alone (Celli et al., 2008; Nannini et al., 2013; Kew et al., 2014).
Despite these clinical data, a mechanistic basis for the superiority of ICS/LABA combination therapies remains unclear. In 2008, we reviewed ways that LABAs and ICSs may interact to deliver superior clinical outcomes in asthma and COPD (Giembycz et al., 2008). In particular, we discussed evidence that LABAs, in addition to directly improving airway calibre, may enhance the anti‐inflammatory activity of ICSs (Giembycz et al., 2008). Furthermore, as glucocorticoids act via the glucocorticoid receptor (GR; gene NR3C1) (Newton, 2000; Clark and Belvisi, 2012), a transcription factor, we argued that LABAs enhance GR‐dependent gene expression above the maximum level achievable by ICS alone (Giembycz et al., 2008). However, like other cAMP‐elevating agents (Mayr and Montminy, 2001; Sands and Palmer, 2008), LABAs also have a major impact on gene expression, and this necessitates refinement of our original hypothesis. While the LABA‐induced transcriptome is not, per se, anti‐inflammatory, the ability of ICSs to enhance the expression of LABA‐inducible genes may be clinically relevant. Thus, the therapeutic activity of ICS/LABA combination therapies will reflect the ability of each component to promote gene expression alone, as well as to enhance, or modify, gene expression produced by its respective companion.
ICS/LABA interactions
Non‐interacting, independent effects of glucocorticoids and LABAs
Multiple mechanisms could account for the clinical efficacy of ICS/LABA combination therapies. Perhaps the most straightforward is that LABAs and ICSs each elicit a collection of independent, non‐interacting responses that combine additively to provide clinical benefit (Figure 1A). At a basic level, this is illustrated by the ability of LABAs and ICSs to promote bronchoprotection and suppress inflammation respectively (Johnson, 2004). However, as LABAs lack anti‐inflammatory activity in asthma, additivity is unlikely to account for their superiority when they are combined with an ICS.
Figure 1.
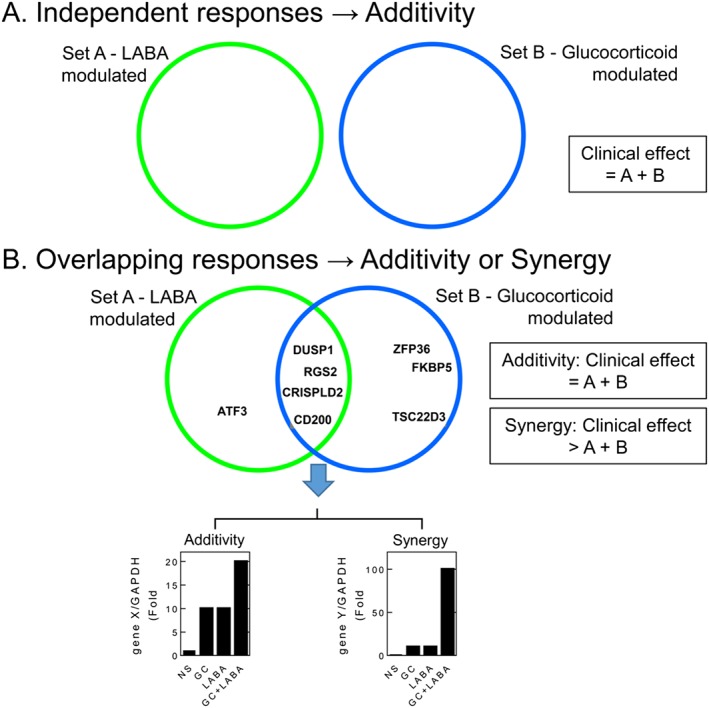
Additive and/or synergistic effector responses induced by LABAs and glucocorticoids (GCs). A. LABAs and GCs may each induce a set of responses (sets A and B respectively) that do not interact. The net effect of a LABA/GC combination treatment would be the sum of the responses produced by each drug alone (A + B). B. While LABAs and GCs each induce a set of responses (sets A and B respectively), a number of these responses may be modulated by both LABAs and GCs. In these situations, where A ∩ B, there may be no interaction between each response, and the net effect is one of simple additivity. This is shown for a hypothetical gene (gene X) that was induced by LABA or GC. In this situation, the overall effect is the sum of the responses produced by each drug (A + B). Alternatively, responses in the overlap region, A ∩ B, may show interaction between the two drugs. This is shown for a hypothetical gene, gene Y, which is modestly induced by maximally effective concentrations of a LABA and by a GC, but together, there is a large induction of gene expression. As this is greater than the sum of the components, this can be described as synergy. In this situation, the overall effect of each drug is greater than the simple sum of A + B.
Additive effects of glucocorticoids and LABAs
Independent, additive effects may occur for any response that is regulated by both a glucocorticoid and a LABA (Figure 1B). Induction of the dual‐specificity phosphatase, DUSP1, which inactivates MAPKs (Abraham and Clark, 2006), provides an example (Figure 1B). DUSP1 mRNA is rapidly and transiently induced by LABAs and other cAMP‐elevating agents (Burgun et al., 2000; Kaur et al., 2008; Korhonen et al., 2013). Similarly, GR binding sites in the DUSP1 promoter allow glucocorticoids to induce, or transactivate, DUSP1 transcription (Johansson‐Haque et al., 2008; Shipp et al., 2010; Tchen et al., 2010). This mediates inhibition of MAPK signalling and inflammatory gene expression (Kassel et al., 2001; Lasa et al., 2002; Abraham et al., 2006; Issa et al., 2007; Shah et al., 2014). However, LABAs and glucocorticoids independently promote DUSP1 mRNA expression, and their effects combine in an essentially additive manner (Kaur et al., 2008; Manetsch et al., 2012; Manetsch et al., 2013; BinMahfouz et al., 2015). Similar effects may also be observed in respect of the repression of inflammatory gene expression (see below). Thus, responses induced by LABAs and glucocorticoids can overlap (Figure 1B), but may result in simple additivity (Figure 1B and 2A).
Figure 2.
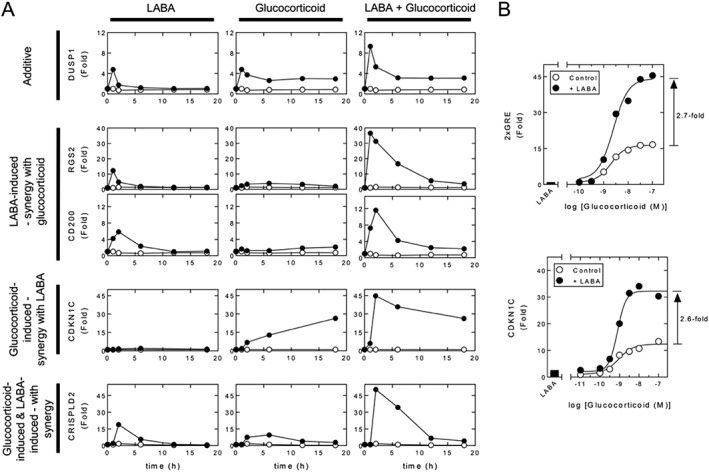
Patterns of mRNA expression induced by LABAs and GCs. A. Overview real‐time PCR data showing the effect of maximally effective concentrations of LABA (10 nM, formoterol) and GC (1 μM, dexamethasone) on the mRNA expression of various genes in human bronchial epithelial, BEAS‐2B, cells. Experimental data from BinMahfouz et al. (2015) represent expression of the indicated gene/GAPDH expressed as fold of untreated (at 1 h) plotted as a mean. B. A maximally effective concentration of LABA (indacaterol, 100 nM) enhanced the activation of a simple 2 × GRE reporter induced by GC (fluticasone furoate) (upper panel). Luciferase data expressed as fold of untreated are plotted as means. Data are taken from Joshi et al. (2015b). Alternatively (lower panel), BEAS‐2B cells were harvested for RNA and gene expression of CDKN1C and GAPDH analysed by real‐time PCR. Data for CDKN1C/GAPDH expressed as fold of untreated are plotted as means.
However, glucocorticoids and LABAs activate their cognate receptors to produce adverse effects in off‐ and on‐target tissues. These could be exaggerated by combination therapies and is, again, illustrated by DUSP1. Thus, osteoblast proliferation and the restoration, or maintenance, of bone mass involve ERK, and the up‐regulation of DUSP1, by inactivating ERK, may promote osteopaenia (Horsch et al., 2007). Such effects may be relevant in patients taking high dose combination therapy, where systemic exposure is likely.
Opposing effects of glucocorticoids and LABAs
A further example of interaction is where responses induced by one component of an ICS/LABA combination therapy are diminished by the other. In cell‐based assays, using airway smooth muscle (ASM) and either transformed or primary bronchial epithelial cells, LABAs induce, and/or potentiate, the expression of certain inflammatory genes, including IL6, CXCL5 and CXCL8, and these effects are suppressed by glucocorticoids (Ammit et al., 2000; Korn et al., 2001; Ammit et al., 2002; Faisy et al., 2002; Edwards et al., 2007; Holden et al., 2010). Likewise, expression of TRPV1 receptors and several GPCRs that mediate inflammatory responses or bronchoconstriction are up‐regulated by β2‐adrenoceptor agonists and down‐regulated by glucocorticoids (Katsunuma et al., 1999; Mak et al., 2000; Faisy et al., 2004; Liu et al., 2015). Thus, in airway diseases, many undesirable effects of LABAs are attenuated by glucocorticoids. Conversely, it is also possible that desirable effects elicited by one component of an ICS/LABA therapy could be attenuated with the combination.
Synergistic induction of gene expression by glucocorticoids and LABAs
Clinical data in asthma suggest that ICSs and LABAs can interact in a synergistic manner, such that outcome measures, including improved lung function or reduced exacerbation frequency produced by the two drugs in combination, are superior to the sum of their individual effects. Indeed, LABAs enhance ICS efficacy and lead to asthma control, whereas increasing the dose of ICS may not (Frois et al., 2009; Ducharme et al., 2010a; Ducharme et al., 2010b). While rationalization of these data requires that ICSs and LABAs interact in a way that cannot be reproduced by merely increasing the dose of ICS (Giembycz et al., 2008; Newton et al., 2010b), descriptions of such interactions have been inconsistent. Nevertheless, an increasing number of responses are expressed in a synergistic manner when a glucocorticoid is combined with a LABA. One example is induction of the regulator of G‐protein signalling, RGS2 (Holden et al., 2011; Holden et al., 2014) (Figure 2A). Thus, in ASM and bronchial epithelial cells, RGS2 mRNA and protein are elevated by LABAs, and other cAMP‐elevating agents, as well as by glucocorticoids (Pepperl et al., 1998; Tsingotjidou et al., 2002; Wang et al., 2004; Chivers et al., 2006; Holden et al., 2011; Holden et al., 2014). In combination, the response to maximally effective concentrations of LABA and glucocorticoid produces profound synergy at the level of both RGS2 mRNA and protein (Figures 2A and 3A–B) (Holden et al., 2011; Greer et al., 2013; Moodley et al., 2013; Holden et al., 2014; BinMahfouz et al., 2015; Joshi et al., 2015b). Thus, the level of RGS2 expression achieved by maximally effective concentrations of LABA plus glucocorticoid is considerably greater than the simple sum of the responses produced by each component alone (Figures 2A and 3A–B). Functionally, RGS2 is a GTPase‐activating protein that terminates signalling from GPCRs that signal via Gq (Heximer, 2004; Kimple et al., 2009) (Figure 3C). Such receptors include those mediating ASM contraction and pulmonary leukocyte recruitment. Thus, by reducing Gq‐dependent signalling, RGS2 expression should be beneficial in asthma and COPD. Indeed, up‐regulation of RGS2 in mice is bronchoprotective, whereas RGS2‐deficiency promotes airways hyperreactivity (AHR), mucin expression and airways remodelling (Holden et al., 2011; Xie et al., 2012; Liu et al., 2013; Jiang et al., 2014). Thus, the synergy that is achieved by LABAs and glucocorticoids at inducing RGS2 expression not only provides some explanation for the therapeutic activity of ICS/LABA combination therapies, but the relatively modest response to glucocorticoid alone clearly illustrates why this effect may not be achieved by simply increasing the dose of ICS (Figure 3).
Figure 3.
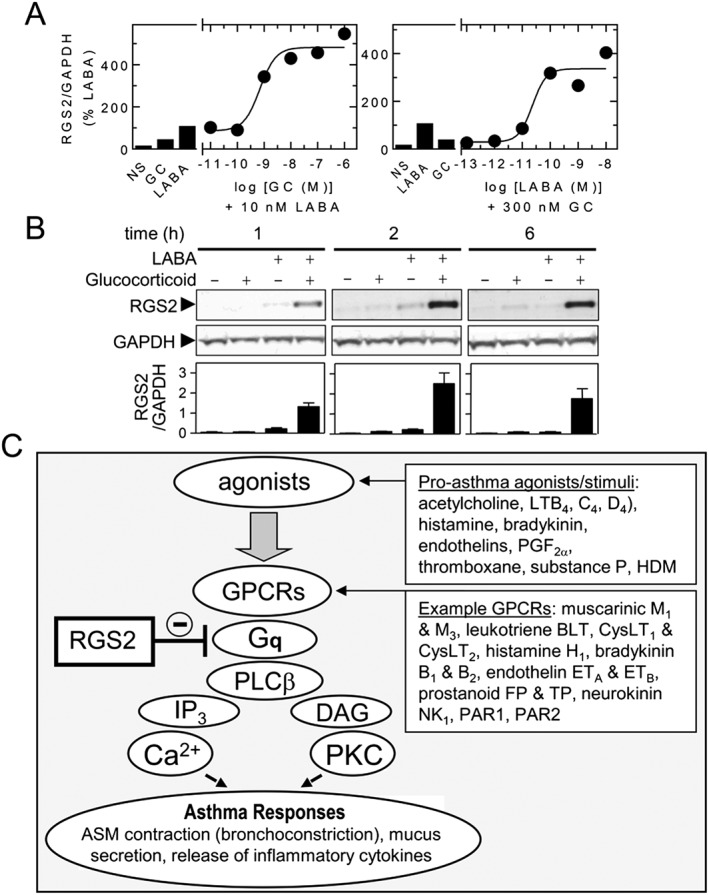
RGS2 expression is synergistically induced by LABA and GC. A. BEAS‐2B cells were treated with: in the left panel, GC alone (budeosnide, 1 μM), LABA alone (formoterol, 10 nM) or the indicated concentrations of GC (budesonide) in the presence of LABA (formoterol, 10 nM); in the right panel, LABA alone (formoterol, 10 nM), GC alone (budeosnide, 300 nM) or the indicated concentrations of LABA (formoterol) in the presence of GC (budesonide, 300 nM). After 2 h, the cells were harvested for RNA and real‐time PCR analysis of RGS2 and GAPDH mRNA. Data, normalized as RGS2/GAPDH, are expressed as a percentage of the LABA (formoterol, 10 nM) alone and are plotted as means. Data are derived from experiments in Holden et al. (2014). B Human bronchial epithelial, BEAS‐2B, cells were treated with LABA (salmeterol, 100 nM) and GC (dexamethasone, 1 μM) for the times indicated prior to western blot analysis of RGS2 and GAPDH. Data are expressed as RGS2/GAPDH and plotted as means ± SEM. Data are derived from experiments in Holden et al. (2014). C. Schematic showing possible roles for RGS2 in reducing ‘pro‐asthma’ responses. Agonists/activators of GPCRs that couple via Gq lead to phospholipase Cβ activation and the production of inositol(1,4,5)trisphosphate (IP3) and DAG. This increases [Ca2 +]c and activates protein kinase C (PKC) to promote ASM contraction, mucus secretion, release of inflammatory mediators, cytokines and other pro‐asthma responses. RGS2 is a GTPase activating protein that enhances the GTP hydrolysis of Gαq and thereby inactivates signalling by Gq. HDM = house dust mite.
Other genes with anti‐inflammatory potential, including the surface marker, CD200 and CRISPLD2, are also synergistically up‐regulated by glucocorticoid/LABA combinations (Moodley et al., 2013; BinMahfouz et al., 2015; Joshi et al., 2015b) (Figure 2A). CD200 is a transmembrane glycoprotein expressed on haematopoietic and non‐haematopoietic cells, which by binding its cognate receptor, CD200R, on alveolar macrophages, down‐regulates cytokine production (Snelgrove et al., 2008). This interaction also suppresses AHR in a murine model of allergic asthma (Vaine and Soberman, 2014; Lauzon‐Joset et al., 2015). Similarly, the secreted protein, CRISPLD2, attenuates gene toll‐like receptor‐4‐mediated, pro‐inflammatory signalling by binding and inactivating lipopolysaccharide/lipid A (Wang et al., 2009; Himes et al., 2014; Vasarhelyi et al., 2014).
The widespread changes in gene expression produced by cAMP and glucocorticoids will also promote expression of adverse‐effect genes (Gonzales et al., 2002; Wade et al., 2006). In cell‐based experiments, cAMP‐elevating stimuli, including LABAs, synergize with glucocorticoids to up‐regulate expression of pyruvate dehydrogenase kinase, isoform 4 (gene PDK4) and phosphoenolpyruvate carboxykinase (PEPCK, gene PCK2), which encode proteins involved in glucose and carbohydrate metabolism (Hanson and Reshef, 1997; Jitrapakdee, 2012; Oh et al., 2013; Joshi et al., 2015b). This illustrates a concern that combination therapies enhance known glucocorticoid‐induced adverse effects (Schacke et al., 2002). Furthermore, such data highlight the potential advantage of selecting each component of an ICS/LABA combination therapy to maximally induce the expression of clinically beneficial genes, in relevant target tissues, over those genes responsible for adverse effects.
Patterns of cooperatively regulated gene expression
The superiority of ICS/LABA combination therapy requires each component to contribute to overall clinical efficacy in a way that is more than simple additivity (Giembycz et al., 2008). In this respect, three, possibly four, general patterns of glucocorticoid/LABA‐dependent gene expression can be envisaged. (i) As shown by RGS2 and CD200, there are genes whose expression are primarily induced by the LABA, but are not, or are only modestly induced, by the glucocorticoid. However, in combination, the glucocorticoid enhances, and/or prolongs, expression induced by the LABA (Figure 2A) (Holden et al., 2011; Holden et al., 2014; BinMahfouz et al., 2015; Joshi et al., 2015b). (ii) There are genes induced by glucocorticoids, which are either not, or are only minimally, induced by the LABA, yet show LABA‐dependent enhancement of the glucocorticoid response (Figure 2A). Luciferase expression from a simple, albeit artificial, 2 × glucocorticoid response element (GRE)‐dependent reporter provides a good example (Figure 2B) (Kaur et al., 2008; Joshi et al., 2015b). Similarly, the cell cycle kinase inhibitor, CDKN1C (aka p57KIP2), and the metabolic gene, PDK4, show this pattern of regulation in BEAS‐2B cells (Figure 2B) (Kaur et al., 2008; Rider et al., 2011; Joshi et al., 2015b; Rider et al., 2015b). Evidence for such behaviour is also observed functionally. In T‐lymphocytes, LABAs augment GR‐mediated apoptosis in a protein kinase A (PKA)‐dependent manner (Ji et al., 2007; Ji et al., 2008), but are, by themselves, inactive (Pace et al., 2004). Because glucocorticoid‐induced apoptosis may involve GR‐mediated gene expression (Wu et al., 2013), such data are consistent with LABAs enhancing glucocorticoid‐induced transactivation. (iii) Genes, such as CRISPLD2, show mRNA expression that is independently induced by glucocorticoids and LABAs and reveal synergism when both drug classes are combined (Figure 2A) (BinMahfouz et al., 2015; Joshi et al., 2015b). Within this group is, arguably, a subset of genes (iv), where expression is induced by a glucocorticoid and LABA in combination, but not by either component individually (Giembycz and Newton, 2015). Currently, genes regulated in this way have not been described.
Such groupings represent extremes of what should be considered as a continuum of gene expression. Furthermore, the exact details of expression will depend on multiple external factors, including cell line or type and the treatment time. For example, in bronchial epithelial BEAS‐2B and ASM cells, the glucocorticoid‐dependent induction of RGS2 mRNA is modest (Holden et al., 2011; Holden et al., 2014), whereas in A549 type II adenocarcinoma cells and primary human bronchial epithelial (HBE) cells glucocorticoids promote robust RGS2 mRNA expression (Chivers et al., 2006; Holden et al., 2014). Likewise, the patterns of synergy may also differ. In BEAS‐2B and ASM cells, LABA‐induced expression of RGS2 is enhanced and prolonged by glucocorticoids (Holden et al., 2011; Holden et al., 2014), whereas in primary HBE cells, LABAs only modestly increased RGS2 expression, yet significantly enhanced the maximum response achieved by a glucocorticoid (Holden et al., 2014).
Repression of gene expression by glucocorticoids and LABAs
The effects of LABAs and glucocorticoids may also extend, in an apparently additive manner, to the repression of inflammatory genes. These include adhesion molecules, cytokines and chemokines induced by multiple inflammatory stimuli from structural cells of the airways (Korn et al., 2001; Pang and Knox, 2001; Silvestri et al., 2001; Spoelstra et al., 2002; Newton et al., 2010b). In contrast to ASM and epithelial cells (above), apparently additive repression can also occur for CXCL8 release from cigarette smoke‐treated macrophage and neutrophils (Sarir et al., 2007; Mortaz et al., 2008). Furthermore, additive effects of formoterol and budesonide were reported on the repression of ICAM‐1 expression as well as cytokine and chemokine expression measured following human rhinovirus (HRV) infection of epithelial cells (Yamaya et al., 2014). While the mechanistic basis for these effects remains to be established, it is equally clear that opposing effects, particularly of the LABA, for example on CXCL8 expression, are observed in a cell type‐ and potentially stimulus‐dependent manner.
Evidence for synergistic, repressive effects is also available. Glucocorticoids and LABAs combine to produce more than an additive repression of TNF‐induced CXCL8 release from human ASM cells (Pang and Knox, 2000), and of various mediators from airway epithelial cells in response to HRV infection (Edwards et al., 2006; Volonaki et al., 2006; Sarir et al., 2007; Skevaki et al., 2009). Although glucocorticoids and LABAs share the ability to recruit histone deacetylases to inflammatory gene promoters (Ito et al., 2001; Nie et al., 2005), the molecular mechanism(s) involved in any synergistic repression is unclear. Nevertheless, the glucocorticoid‐dependent repression of many inflammatory genes shows a requirement for ongoing gene expression, which implies a role for GR‐mediated transactivation (Newton and Holden, 2007; Clark and Belvisi, 2012). Furthermore, genes that appear to be repressed via mechanisms involving GR‐mediated transactivation show a greater level of repression and a greater sensitivity (i.e. lower EC50) to glucocorticoid compared with those inflammatory genes where direct GR‐mediated transrepression is implicated (King et al., 2013). In this context, it is salient that glucocorticoid‐mediated repression can show marked differences in potency and overall repression depending on the inflammatory stimulus and/or the response measured (Fernandes et al., 1999; Tran et al., 2005). Such data are consistent with the concept of multiple mechanisms of glucocorticoid action involving different GR effectors that could show differential dependence on GR transactivation and transrepression. Notwithstanding such complexity, many glucocorticoid‐induced genes, including DUSP1 and glucocorticoid‐induced leucine zipper (gene TSC22D3), which reduce NF‐κB and activator protein 1 (AP‐1)‐dependent transcription, are implicated in the repression of inflammatory gene expression (Newton and Holden, 2007; Ayroldi and Riccardi, 2009; Clark and Belvisi, 2012). As increased expression of glucocorticoid‐inducible genes, for example TSC22D3, occurs in the airways of normal individuals and in asthmatic subjects taking ICS monotherapy (Kelly et al., 2012; Leigh et al., 2016), clinically relevant roles for GR transactivation appear to be likely. Furthermore, as these effects may be enhanced by LABAs, this review focuses on transcriptional activation as a mechanism of synergy between glucocorticoids and LABAs that relates to asthma therapy.
LABA‐induced responses and modulation by glucocorticoids
Promotion of bronchoprotection by glucocorticoids
Chronic β2‐adrenoceptor agonist exposure can impair β2‐adrenoceptor signalling and functional responsiveness via mechanisms that are collectively referred to as desensitization (Lefkowitz, 2007; Vasudevan et al., 2011). However, it is well established that glucocorticoids offset such effects by increasing β2‐adrenoceptor number, Gs expression and/or coupling to adenylyl cyclase (Giembycz et al., 2008; Newton et al., 2010b).
An unanticipated effect of glucocorticoids and LABAs alone, and in combination, is their ability to protect against agonist‐induced bronchoconstriction via mechanisms that involve gene expression. For example, the pro‐contractile activity of MAPKs in ASM (Gerthoffer et al., 1997; Hedges et al., 2000; Dowell et al., 2010; Sakai et al., 2010) should be countered by a glucocorticoid via the induction of DUSP1 expression (Issa et al., 2007; Kang et al., 2008; Quante et al., 2008). Indeed, DUSP1 −/− mice exhibit enhanced airway contractile responses to ozone and a loss of glucocorticoid protection (Li et al., 2011). Despite there being no apparent protection by DUSP1 using chronic ozone exposure in a mouse model of glucocorticoid resistance (Pinart et al., 2014), it is possible that, by further enhancing DUSP1 expression with a LABA to increase glucocorticoid‐induced DUSP1 expression (Kaur et al., 2008; Manetsch et al., 2012), protection could have been re‐established. However, such effects remain to be tested.
Similarly, LABAs induce Rgs2 expression in murine airways to protect against agonist‐induced bronchoconstriction (Holden et al., 2011). Furthermore, analysis of cytosolic free calcium concentration ([Ca2 +]c) as a surrogate of contraction in human ASM cells shows RGS2 can account for the ability of a glucocorticoid to prolong LABA‐induced bronchoprotection (Holden et al., 2011). The additional finding that the ICS, budesonide, induces DUSP1 and RGS2 mRNA in vivo in human airways (Leigh et al., 2016) provides strong evidence that expression of these genes could attenuate bronchoconstriction in asthma and COPD. Clearly, this would be most pronounced with ICS/LABA combination therapies, where the LABA would not only promote direct smooth muscle relaxation, but also interact additively and synergistically with the glucocorticoid on DUSP1 and RGS2 expression, respectively (Kaur et al., 2008; Holden et al., 2011; Manetsch et al., 2012). Such data are consistent with enhanced gene expression contributing to the clinical superiority of ICS/LABA combination therapies in moderate‐to‐severe asthma.
Transcriptional activation by LABAs and modulation by glucocorticoids
Studies from the late 1980s document PKA‐dependent phosphorylation of cAMP‐activated transcription factors, such as cAMP response element (CRE) binding protein, CREB1 (Mayr and Montminy, 2001; Sands and Palmer, 2008). At the simplest level, CREB1 binds CRE sites in the promoters of target genes and, when phosphorylated at Ser133, recruits the co‐activator gene CREB‐binding protein to confer transcriptional competency (Chrivia et al., 1993; Mayr and Montminy, 2001). However, glucocorticoids often antagonize cAMP‐dependent transcription (Diaz‐Gallardo et al., 2010). Indeed, in bronchial epithelial cells, activation of CRE‐dependent transcription by LABAs was modestly repressed by glucocorticoids (Kaur et al., 2008). Thus, the enhancement of CRE‐dependent transcription, per se, is unlikely to explain how LABAs and glucocorticoids synergistically induce gene transcription. Indeed, CREB1, acting at conventional CRE sites, may account for the actions of LABAs on IL6 and CXCL8 expression and is therefore consistent with repressive effects of glucocorticoids (Ammit et al., 2002; Yin et al., 2006; Tan et al., 2007). Conversely, while the proximal promoter region for RGS2 harbours a simple CRE that binds CREB1 and responds to cAMP (Song et al., 2010; Xie et al., 2011), this may not explain synergy with a glucocorticoid. Rather, a putative GR binding site has been localized to a region within 10 kb of the mouse Rgs2 transcription start site (So et al., 2008). Thus, notwithstanding the involvement of other cAMP‐ and/or glucocorticoid‐activated transcription factors (Mayr and Montminy, 2001; Chinenov et al., 2014), a simple, but untested, hypothesis is that CREB1/GR interactions at the RGS2 promoter lead to transcriptional synergy.
CCAAT‐enhancer binding factors are cAMP and glucocorticoid regulated
Many members of the CCAAT‐enhancer binding protein (C/EBP) transcription factor family, including C/EBPα (gene CEBPA), C/EBPβ (gene CEBPB) and C/EBPδ (gene CEBPD), are activated and/or induced by cAMP‐elevating stimuli, glucocorticoids and by pro‐inflammatory stimuli (Cardinaux and Magistretti, 1996; Croniger et al., 1998; Poli, 1998; Vogel et al., 2004; Tsukada et al., 2011). Thus, CEBPB, a PKA target, is implicated in the transcriptional activation of genes, such as IL6 or CXCL8, by inflammatory stimuli, and this could account for the induction and/or enhancement of their expression by β2‐adrenoceptor agonists (Akira et al., 1990; Mukaida et al., 1990; Matsusaka et al., 1993; Kunsch et al., 1994; Trautwein et al., 1994). However, CEBPB expression is also induced by cAMP‐dependent activation of CREB1, and this is partly responsible for up‐regulating metabolic genes, such as PEPCK (Park et al., 1993; Niehof et al., 1997). C/EBPs are also glucocorticoid‐inducible proteins (Park et al., 1990; Baumann et al., 1991; Cao et al., 1991). For example, CEBPB and CEBPD expression is induced by glucocorticoids in bronchial epithelial cells, and up‐regulation of CEBPD occurs in the airways of human subjects following budesonide inhalation (Berg et al., 2005; Zhang et al., 2007a; Leigh et al., 2016). Thus, while C/EBPs are implicated in metabolic and inflammatory gene expression, and controlling proliferation (Nerlov, 2007; Tsukada et al., 2011), they are also involved in the maintenance of inflammation‐induced innate immune genes in the context of glucocorticoids (Zhang et al., 2007a; Didon et al., 2011). While such effects could be enhanced by LABAs, it is unclear whether this would be a desirable outcome in the context of chronic inflammatory diseases, such as asthma or COPD. Furthermore, CREB1 and CEBPB can synergistically promote gene transcription (Park et al., 1993; Niehof et al., 1997; Niehof et al., 2001), an effect that could explain the ability of glucocorticoids to enhance cAMP‐mediated responses from promoters driven by CREs that act in conjunction with C/EBP sites. Finally, as C/EBPs can act as pioneer factors to promote GR‐dependent transcription (Grontved et al., 2013), key roles are likely in respect of the regulation of glucocorticoid/LABA co‐regulated genes. However, the complex relationship between pro‐inflammatory, metabolic and potentially other more desirable effects of CEBP transcription factors requires careful examination.
Modulation of gene expression by glucocorticoids and the effect of LABAs
Transrepression and transactivation in the anti‐inflammatory effects of glucocorticoids
To understand how LABAs enhance the therapeutic activity of glucocorticoids, it is pertinent to consider mechanisms by which glucocorticoids act in diseases such as asthma. Glucocorticoids suppress various indices of inflammation, and their clinical efficacy in mild‐to‐moderate asthma relies on the ability to reduce inflammatory gene expression (Rhen and Cidlowski, 2005; Barnes, 2008). However, how glucocorticoids operate at a molecular level to exert repression of inflammatory gene expression remains controversial, and there are currently three main possibilities (Clark and Belvisi, 2012). One mechanism, often referred to as ‘transrepression’, is where activated GR undergoes nuclear translocation to interact with and inhibit those transcription factors (NF‐κB, AP‐1) responsible for driving inflammatory gene transcription (De Bosscher et al., 2003; Sundahl et al., 2015). In this scenario, GR may not directly bind DNA, but is tethered via the inflammatory transcription factor to recruit repressive factors, including histone deacetylases, which reduce gene transcription (Ito et al., 2000; Ito et al., 2006; Hua et al., 2016a). In a second mechanism of transrepression, GR binds DNA at negative GRE sites to inhibit gene transcription of inflammatory and other genes that are potentially associated with side effects (Surjit et al., 2011; Hua et al., 2016b). This effect appears to involve monomeric GR, which binds DNA sites with a lower affinity than GR dimers at conventional positive GREs (Hudson et al., 2013).
A third mechanism of action involves the repression of gene expression via GR‐dependent transactivation. Initial evidence for this comes from long‐standing observations that the repressive effects of glucocorticoids on the expression of inflammatory genes, including cyclooxygenase 2 (gene PTGS2), CXCL8, inducible gene NOS2 and GM‐CSF (gene CSF2) (Ristimaki et al., 1996; Newton et al., 1998; Chang et al., 2001; Korhonen et al., 2002; Chivers et al., 2006; Newton et al., 2010a), are largely prevented by inhibitors of transcription and translation (Newton, 2000; Stellato, 2004; Newton and Holden, 2007; Clark and Belvisi, 2012). In this model, ligand‐bound GR behaves as a positive transcriptional activator to induce expression of anti‐inflammatory genes. Indeed, direct GR‐mediated induction of the NF‐κB inhibitor, IκBα (gene NFKBIA), has been suggested to mediate glucocorticoid‐induced gene repression (Auphan et al., 1995; Scheinman et al., 1995). Similarly, DUSP1 is glucocorticoid‐induced and inhibits MAPK activity (Kassel et al., 2001; Lasa et al., 2002). This may affect multiple MAPK‐dependent processes, including activation of inflammatory transcription factors, such as AP‐1 and NF‐κB, mRNA stability and protein expression of inflammatory genes (Lasa et al., 2001; Abraham et al., 2006; Furst et al., 2007; Issa et al., 2007; Diefenbacher et al., 2008; Kang et al., 2008; King et al., 2009a; Shah et al., 2014). Likewise, TSC22D3 expression is increased by glucocorticoids in HBE cells, ASM, mast cells and macrophage, as well as in asthmatic patients taking ICS (Berrebi et al., 2003; Godot et al., 2006; Eddleston et al., 2007; Kelly et al., 2012). As TSC22D3 represses NF‐κB and AP‐1‐dependent transcription, roles in limiting inflammatory gene expression and inflammation are also implied (Mittelstadt and Ashwell, 2001; Di Marco et al., 2007; Eddleston et al., 2007; Ayroldi and Riccardi, 2009).
The relevance of GR‐mediated transactivation is increasingly suggested by the range of glucocorticoid‐induced effector genes [e.g. CD200, CRISPLD2, DUSP1, NFKBIA, RGS2 and TSC22D3; (Newton et al., 2010b)] that have now been identified. Importantly, many of these effector genes are up‐regulated by LABA and/or glucocorticoids in primary human ASM cells (Misior et al., 2009). Furthermore, the fact that the expression of many such genes is increased in the human airways following budesonide inhalation, as well as in COPD patients taking inhaled fluticasone propionate/salmeterol, supports the concept that such effects are therapeutically relevant and may be enhanced in the presence of a LABA (Kelly et al., 2012; Lee et al., 2016; Leigh et al., 2016).
Co‐operation between GR and inflammatory transcription factors to maintain repression
Many glucocorticoid‐induced genes are also involved in the normal regulatory processes that control inflammatory signalling and gene expression (Newton, 2014). In this regard, A20 (gene TNFAIP3) (Altonsy et al., 2014) and IRAK‐M (gene IRAK3) (Miyata et al., 2015) are informative. These genes are NF‐κB‐dependent. This allows their expression to be rapidly induced by inflammatory stimuli and, thereby, provides negative feedback control of NF‐κB‐dependent gene expression. However, rather than agonist‐bound GR causing repression of these genes, as is routinely established for other NF‐κB‐dependent inflammatory genes (De Bosscher et al., 2003), there is a robust, positive, transcriptional cooperativity between NF‐κB and GR that leads to enhanced TNFAIP3 and IRAK3 expression (Altonsy et al., 2014; Miyata et al., 2015). Such positive NF‐κB/GR interactions, which have been previously demonstrated (Hofmann and Schmitz, 2002), provide a mechanism by which glucocorticoids enhance gene expression in the context of an inflammatory stimulus and are remarkably common (Kadiyala et al., 2016). Thus, by increasing (or at least maintaining) feedback control by TNFAIP3 and IRAK3, positive interactions between GR and NF‐κB may play important anti‐inflammatory roles (Altonsy et al., 2014; Newton, 2014; Miyata et al., 2015). Similarly, expression of the NF‐κB inhibitor, NFKBIA, is glucocorticoid‐induced and is up‐regulated by inflammatory stimuli acting via NF‐κB to reduce NF‐κB signalling (Le Bail et al., 1993; Auphan et al., 1995; Scheinman et al., 1995). Likewise, while DUSP1 and the mRNA destabilizing protein, tristetraprolin (gene ZFP36), are rapidly and highly induced by pro‐inflammatory stimuli to provide negative regulation of MAPK pathways, or AUUUA‐containing mRNAs, such as IL6, CXCL8 or CSF2, they are also up‐regulated by glucocorticoids (Abraham et al., 2006; Smoak and Cidlowski, 2006; King et al., 2009b; Prabhala and Ammit, 2015). Such data are consistent with AP‐1 also co‐operating with GR at composite elements containing both AP‐1 and GR binding sites (So et al., 2007). Furthermore, transcription factors, including AP‐1, may modify the local chromatin structure, for example by co‐operating with GR to displace nucleosomes, and thereby play key roles in enhancing GR access and action at responsive promoters (Biddie et al., 2011; He et al., 2013). Thus, an emerging principle is that glucocorticoid‐bound GR co‐operates with core inflammatory transcription factors to promote expression of regulatory genes and maintain, or enhance, inhibition of inflammatory signalling and gene expression.
GR‐dependent transactivation is enhanced by LABAs
The exact contribution of direct, GR‐mediated transrepression in the anti‐inflammatory effects of glucocorticoids is currently unclear. While some studies fail to show the presence of GR at repressed genes and others report the widespread presence of negative GREs (So et al., 2008; Reddy et al., 2009; Surjit et al., 2011; Kadiyala et al., 2016), it is also plausible that both transrepression and transactivation play important roles in the glucocorticoid‐dependent repression of inflammatory gene expression (King et al., 2013). However, while we are unaware of data reporting enhancement of GR‐mediated transrepression, whether via tethering or nGRE mechanisms, by LABAs, there is unequivocal evidence that LABAs can augment glucocorticoid‐dependent transcription (Giembycz et al., 2008; Newton et al., 2010b) (Figure 2).
In HBE BEAS‐2B cells and primary human ASM cells, simple GRE‐dependent transcription is synergistically enhanced by the co‐administration of agonists that increase cAMP (Kaur et al., 2008; Wilson et al., 2009; Greer et al., 2013; Moodley et al., 2013; Joshi et al., 2015b). Thus, LABAs, such as salmeterol or formoterol, which alone have no obvious effect on a simple 2 × GRE‐driven luciferase reporter, increase by 2–3 fold the maximum effect (E max; Box 1) of a glucocorticoid (Figure 2B). Similarly, any given level of response to glucocorticoid is achieved at a lower concentration in the presence of a LABA, which is therefore glucocorticoid‐sparing (Figure 2B) (Giembycz et al., 2008). Significantly, this effect is reproduced with real genes, including CDKN1C (Figure 2B) (Joshi et al., 2015b), and occurs in primary cells (Kaur et al., 2008; Wilson et al., 2009; Moodley et al., 2013). Given roles for CDKN1C in cell cycle control (Samuelsson et al., 1999), and possibly the regulation of c‐Jun N‐terminal kinases (Chang et al., 2003), these synergistic and steroid‐sparing interactions may translate into enhanced therapeutic effects.
Box 1 – Glossary of Pharmacodynamic Terms
Affinity. (aka the equilibrium association constant): The tenacity to which a ligand binds to a receptor. Arithmetically, it is the molar concentration of ligand that at equilibrium binds to 50% of the receptor population and is equal to the reciprocal of the equilibrium dissociation constant, K A.
Biased Agonism. Receptors can couple to multiple signalling pathways. Biased agonism is a term that describes ligand‐dependent selectivity whereby a given ligand stabilizes a particular receptor conformation to preferentially activate certain downstream signalling pathways relative to others.
Intrinsic Efficacy. A term used to quantify the ability of an agonist to produce response. It is solely an agonist‐dependent parameter. However, response is also dependent upon receptor number and efficiency of receptor‐effector coupling, which are furnished by a tissue. Thus, the efficacy of an agonist to produce response in a given tissue is the product of intrinsic efficacy and receptor number.
Law of Mass Action. A law that states the rate of a simple reaction is proportional to the concentration (mass) of initial reactants. It was originally used to explain chemical reactions, but has been applied to pharmacology because in many cases an agonist interacts with a receptor in a simple, reversible manner. Thus, [A] + [R] ↔ [AR], where [A], [R] and [AR] are the concentrations of agonist, receptor and agonist‐receptor complexes respectively.
Maximum Response (E max ). The response produced by a maximally effective concentration of an agonist. For a full agonist E max = E m whereas E max < Em for a partial agonist.
Receptor Reserve. (aka spare receptors). A term used to describe a system where an agonist produces the maximum tissue response by activating only a fraction of the total receptor population. In such systems, all receptors will be bound and activated by the agonist, but the stimulus produced by only a fraction of the receptor population is sufficient to produce the maximum response. The remaining receptors will produce stimulus that is surplus to requirement and constitute a ‘reserve’ or are said to be ‘spare’. The K A/EC50 ratio is a measure of receptor reserve; the greater this ratio, the larger is the receptor reserve and the occupation of fewer receptors is required to elicit a particular level of response.
System Maximum (E m ). A term that refers to the maximum pharmacological response that can be produced in a given system. Typically, a response equivalent to the E m is produced by full agonists.
Mechanisms underlying enhanced GR‐mediated transactivation by LABAs
How LABAs enhance glucocorticoid‐dependent transcription is subject to considerable debate. Data from the 1990s revealed that the cAMP‐PKA pathway could enhance GR‐mediated transcription (Rangarajan et al., 1992; Gruol and Altschmied, 1993; Nordeen et al., 1993; Zhang et al., 1993). At that time, there was a lack of clarity as to the mechanism(s) of action. Some studies suggested enhanced GR DNA binding, increased GR expression and/or the ability of LABA to prevent the loss of GR that occurs following glucocorticoid exposure (Dong et al., 1989; Rangarajan et al., 1992; Korn et al., 1998). Conversely, other investigators showed no effect on GR expression, ligand binding, nuclear translocation or GR DNA binding (Gruol and Altschmied, 1993; Moyer et al., 1993; Zhang et al., 1993). More recently, such controversy has been perpetuated with claims that LABAs may enhance GR translocation to the nucleus (Eickelberg et al., 1999; Roth et al., 2002; Usmani et al., 2005; Mortaz et al., 2008), along with reports that this does not occur (Loven et al., 2007; Rider et al., 2015a). Intriguingly, the LABA, formoterol, was reported to increase GR DNA binding by low‐dose budesonide in sputum macrophages from mild asthmatics and COPD patients (Essilfie‐Quaye et al., 2011; Haque et al., 2013). Furthermore, the ability of formoterol to restore impaired GR activity in U937 cells treated with combined IL2/IL4 was explained by an effect on GR phosphorylation that was mediated by the protein phosphatase, PP2A (Kobayashi et al., 2012). However, this was neither blocked by the β2‐adrenoceptor antagonist, ICI 118551, nor mimicked by forskolin, which raises questions over mechanism. Conversely, enhancement of glucocorticoid action by a PDE4 inhibitor was shown not to involve altered GR translocation (Grundy et al., 2016).
In considering mechanisms underlying the ability of LABAs, or the cAMP pathway, to enhance GR activity, the possibility of cell‐ and system‐dependent effects cannot be ignored (Rider et al., 2015b). Nevertheless, it is clear that, within a given system, the expression of some glucocorticoid‐induced genes are not enhanced by LABAs, whereas others show either additivity or synergy (Kaur et al., 2008; BinMahfouz et al., 2015; Rider et al., 2015b). Such observations correspondingly require explanations that allow for gene‐specific enhancements, rather than generic mechanisms that should produce global changes in GR‐mediated gene expression (Giembycz et al., 2008; Newton et al., 2010b) (Table 1). Using a simple GRE reporter in BEAS‐2B cells, LABAs enhance transcription in the absence of any change in the potency, affinity or efficacy of the GR agonist (Joshi et al., 2015b) (Box 1). As this effect is mimicked by forskolin and other cAMP‐elevating agents, and is blocked by a selective inhibitor of PKA (Kaur et al., 2008; Wilson et al., 2009; Moodley et al., 2013; BinMahfouz et al., 2015), these data suggest that activation of the canonical cAMP signalling cascade augments the transcriptional competency of liganded GR and, therefore, the magnitude of GR‐mediated gene expression. This concept is consistent with findings that transcriptional co‐activators involved in GR‐mediated transcriptional responses are PKA substrates and provides a mechanism for gene‐dependent enhancements (Rowan et al., 2000; Constantinescu et al., 2004; Hoang et al., 2004; Fenne et al., 2008). As noted above, CEBP family members are glucocorticoid‐induced and up‐regulated by PKA‐dependent processes (Yeagley and Quinn, 2005). Thus, the presence of CEBP sites or, alternatively, co‐localization with CREs or other factors including co‐activators could all produce transcriptional synergies at target genes.
Table 1.
LABA‐dependent mechanisms of enhancing GR‐mediated gene transcription
| Possible mechanism of LABA action | Effect on GR‐mediated gene expression | Predicted nature of the expected effect on GR‐mediated gene expression | |
|---|---|---|---|
| General | Gene‐specific | ||
| Enhanced ligand affinity for GR | Yes | No | Leftward‐shift in the concentration–response curve for all responses. |
| Enhanced GR expression | Yes | No | Leftward shift in the concentration–response curves for all responses that were occurring at the system maximum (E m; Box 1). For responses below the system maximum, E max would increase until the system maximum was reached. |
| Enhanced GR translocation | Yes | No | Leftward shift in the concentration–response curves for all responses that were occurring at the system maximum. For responses below the system maximum, E max would increase until the system maximum was reached. |
| Enhanced DNA binding of GR | Yes | Yes | Enhanced DNA binding by GR could result in global effects on gene expression. Alternatively, LABA‐induced enhancements in GR binding could be gene specific. |
| Phosphorylation/other modification of GR | Yes | Yes | Changes in GR phosphorylation, or other modification, have the capacity to affect all, or just a specific group, of GR responsive genes. |
| Phosphorylation/modification of transcriptional apparatus | Yes | Yes | Changes in phosphorylation, or other modification, of any part of the transcriptional apparatus have the capacity to affect all, or just a specific group, of GR responses genes. |
| Interaction with cAMP‐activated TF | No | Yes | Genes with binding sites for other transcriptional activators will have the potential for interaction with GR, if GR is also recruited to this same promoter. |
| Activation/induction of a co‐activator or repression/inactivation/degradation of a co‐repressor | Yes | Yes | Activation or repression of coactivator, or co‐repressors may have effects on all or a population of GR regulated genes. Alternatively, gene‐dependent requirement for a co‐activator or repressor could allow gene‐specific regulation by a LABA |
Optimizing ICS/LABA combination therapy by exploiting pharmacodynamics
Potential for biased agonism
Early concepts of drug action assumed an agonist‐bound receptor existed in a single conformation and that the magnitude of response was due to the strength of signal. However, we now know that a GPCR does not exist in a single, static conformation, and, consequently, the intrinsic efficacy (Box 1) of a ligand can be response‐specific depending on the particular coupling, or transducer, involved (Figure 4). Indeed, GPCRs adopt multiple, interchangeable conformations (or activation states) that are differentially stabilized in an agonist‐dependent manner (Spengler et al., 1993; Pantaloni et al., 1996; Onaran and Costa, 1997; Liu et al., 2012). This leads to the concept of biased agonism (also known as stimulus trafficking, functional selectivity and ligand‐directed signalling) whereby a ligand stabilizes a particular receptor conformation to preferentially activate certain downstream signalling pathways relative to others (Kenakin, 1995; Kenakin, 2009; Evans et al., 2010; Seifert, 2013; Kenakin, 2015). For example, biased agonism was first shown for the pituitary adenylyl cyclase‐activating polypeptide 1 receptor 1 (gene ADCYAP1R1) at which the agonists PACAP1–27 and PACAP1–38 elevate cAMP (via Gs) and inositol phosphates (via Gq), respectively, with opposite orders of potency (Spengler et al., 1993) (Figure 4). In the sections below, we discuss biased agonism in the context of β2‐adrenoceptor agonists and consider whether ligands that bind GR could produce biased gene expression.
Figure 4.
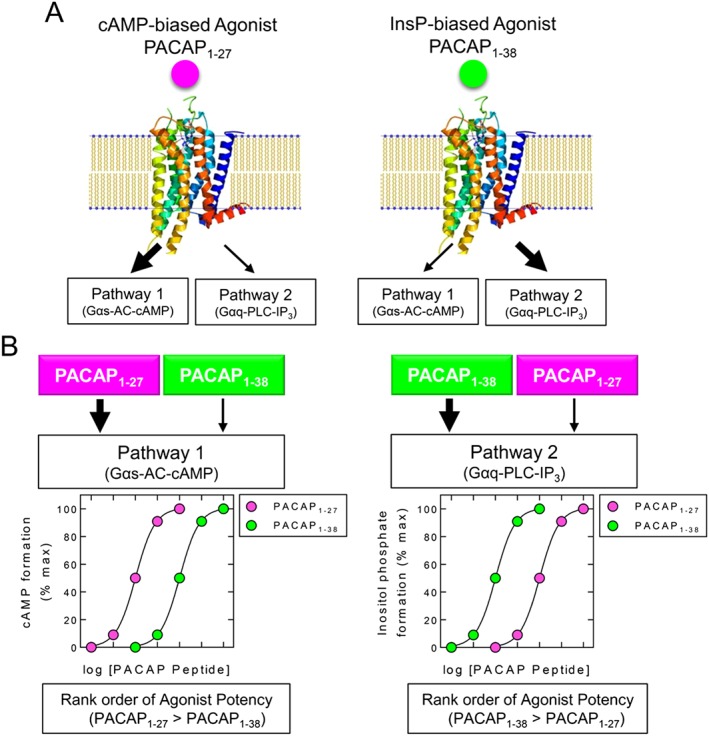
Biased agonism at the pituitary adenylate cyclase‐activating polypeptide type I (PAC1) receptor. Schematic representation of data taken from Spengler et al. (1993) where the PAC1 receptor was over‐expressed in LLC PK1 porcine kidney epithelial cells. A. PACAP1–27 preferentially promotes cAMP formation (Pathway 1) via a Gαs/adenylyl cyclase‐dependent mechanism. PACAP1–38 preferentially promotes inositol phosphate production (Pathway 2) through a Gαq/phospholipase Cβ activation pathway. Large and small arrows indicate the major and minor pathways activated by each PACAP peptide. B. Activation of each pathway is shown by PACAP1–27 and PACAP1–38, and the effect on the main downstream second messenger (cAMP or inositol phosphates) is depicted. Figure generated from data reported by Spengler et al. (1993) using the ribbon diagram found at https://commons.wikimedia.org/wiki/File:A2A_receptor_bilayer.png
Pharmacodynamics of GR‐mediated gene expression
Cooperative interactions of ligand‐bound GR with tissue‐dependent factors, including sequence‐specific transcription factors, co‐repressors and co‐activators (Szapary et al., 1999; Simons, Jr., 2006), combined with the linear arrangement of transcription factor binding sites and their actual DNA sequence will, along with the specific chromatin structure and any epigenetic modifications, result in a very specific three‐dimensional architecture for each individual gene promoter (Keenan et al., 2016). As a consequence, the ability of any given ligand‐induced conformation of GR to interact with each gene promoter will be unique and may, therefore, give rise to gene‐specific, patterns of transactivation, as is now reported (Tanigawa et al., 2002; Reddy et al., 2009; Simons, Jr., 2010; Uings et al., 2013; Joshi et al., 2015a; Rider et al., 2015b). For example, an analysis of dexamethasone‐induced genes in A549 cells revealed an average EC50 of ~3 nM, with the potency of the most and least sensitive genes varying by a factor of ~20‐fold (Reddy et al., 2009).
Extending this concept to different ligands, which are known to stabilize GR in distinct conformations (Allan et al., 1992; Wagner et al., 1996; Biggadike et al., 2009a; Biggadike et al., 2009b; Edman et al., 2014), raises the possibility of agonist‐directed gene expression (Figure 5). Such agonists, by stabilizing different GR conformations, may differentially affect GR's ability to interact productively at different gene promoters and may thereby lead to agonist‐specific programmes of gene expression (Keenan et al., 2016). Accordingly, gene expression changes within the glucocorticoid‐induced transcriptome will be described by a family of concentration–response curves that are unique to the agonist of interest. Central to this concept is that the intrinsic efficacy for an agonist is governed by the relationship between the specific agonist‐induced conformation of GR and differences in the promoter context for each gene (Mercier et al., 1983; Newton et al., 2010b; Joshi et al., 2015a). Thus, in A549 cells, dexamethasone and the dissociated steroidal ligand, RU24858 (Vayssiere et al., 1997), showed differential abilities to induce the expression of glucocorticoid‐inducible genes (Chivers et al., 2006). Moreover, the abundance and stoichiometry of GR relative to other factors necessary for gene expression are not invariant across cell types. Therefore, tissue‐dependent heterogeneity in agonist‐induced, gene expression signatures is predicted (Simons, Jr., 2010). Although evidence for such differences in gene expression is sparse, Tanigawa and colleagues reported that dexamethasone was a robust activator of a mouse mammary tumour virus (MMTV)‐based reporter transfected into HeLa cells under conditions where RU24858 was a very weak agonist (Tanigawa et al., 2002). In contrast, these ligands were almost equi‐effective in driving the same reporter transfected into CV‐1 cells (Tanigawa et al., 2002). Thus, a cell‐dependent factor, or factors, is responsible for this discrepant behaviour. Consistent with this idea, the rank order of potency of three glucocorticoids for activating a different (3 × GRE) reporter transfected into both HeLa cells and CV‐1 cells was distinct (dexamethasone > prednisolone > RU24858 versus dexamethasone > RU24858 > prednisolone respectively). Tanigawa and colleagues also reported that substituting the MMTV‐based construct in HeLa cells with the 3 × GRE reporter transformed RU24858 from a very partial agonist into one that was as effective as dexamethasone (Tanigawa et al., 2002). Thus, the conformation adopted by the RU24858:GR complex was more favourable for driving the 3 × GRE reporter than for the MMTV‐based construct, and this establishes the principle that differential signalling from GR can occur in different cells (Figure 5). Further evidence for this proposal is that the potency of dexamethasone differed by ~10‐fold in driving transcription of either the MMTV‐ and 3 × GRE reporters in both HeLa and CV‐1 cells, and the genes encoding glutamine synthetase (glutamate‐ammonia ligase; gene: Glul) and tyrosine aminotransferase (gene Tat) in FU5‐5 rat hepatoma cells (Mercier et al., 1983; Tanigawa et al., 2002).
Figure 5.
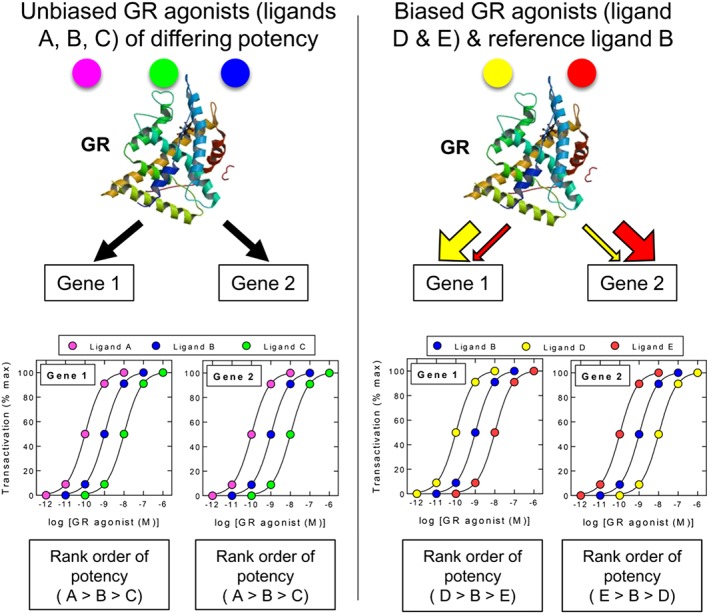
Biased agonism at GR. The possibility that nuclear hormone receptors such as GR can adopt different active conformations in a ligand‐dependent manner could provide a means to promote gene expression bias in a given tissue. According to this hypothetical model, unbiased ligands (A, B and C) generate a GR conformation(s) that promotes the expression of the GC transcriptome (shown here as Gene 1 and Gene 2) with the same rank order of potency where, in the figure, ligand A is more potent than ligand B, which is more potent that ligand C on all genes. In contrast, biased agonists (D and E) lead to the stabilization of different GR conformers that more favourably promote the transcription of one gene (or gene population) over another relative to the reference ligand B. Gene expression bias could result from either a change in the way GR interacts with essential co‐factors and/or its ability to bind DNA and effect transcription. In each case, this will be dictated by the promoter context of each target gene. The rank order of agonist potency can therefore vary in a gene‐dependent manner as depicted by the concentration–response curves describing ligands D and E relative to the reference ligand B. Figure generated using the GR ribbon diagram found at http://www.rcsb.org/pdb/explore/explore.do?pdbId=1P93
Recent studies using BEAS‐2B bronchial epithelial cells also show striking differences in the ability of a panel of GR ligands to induce expression of CDKN1C, CRISPLD2, PDK4 and TSC22D3 (Joshi et al., 2015a; Joshi et al., 2015b). In those studies, the GR agonists tested were equi‐effective at inducing TSC22D3 and CRISPLD2, whereas marked differences were apparent for CDKN1C and PDK4. Notably, GW870086X and desisobutyryl‐ciclesonide behaved as weak and very weak partial agonists on CDKN1C and PDK4, respectively, relative to a reference agonist, dexamethasone. Similar data were also reported for GW870086X in A549 cells, where intrinsic activity values (Box 1) varied from 0.1 to 0.9 across eight genes with EC50 values that differed by 20‐fold (Uings et al., 2013). Collectively, these data are consistent with agonist intrinsic efficacy varying in a gene‐dependent manner that is dictated by sequence and structural differences between promoters in a given cell (Figure 5). This finding has significant implications for drug development as it may be possible to design GR agonists that selectively enhance the expression of some genes, while leaving others relatively unaffected.
GR number as a determinant of efficacy
The Law of Mass Action (Box 1) predicts that increases or decreases in the density of a given receptor will increase or decrease, respectively, agonist potency in that cell type. This is an important concept because GR expression varies considerably across structural and immune cells in the lungs, which are the intended sites of action for ICS, to off‐target tissues responsible for side‐effects (Bourgeois and Newby, 1979; Vanderbilt et al., 1987; Lowy, 1989; Spencer et al., 1991; Miller et al., 1998). This effect is shown in over‐expression studies where the maximal response is enhanced and/or the agonist concentration–response curves are displaced to higher potency (Robertson et al., 2013). For example, mifepristone (also known as RU486) appears to function as a full agonist in promoting the translocation of GR to the nucleus (Schaaf and Cidlowski, 2003; Chivers et al., 2004; Lewis‐Tuffin et al., 2007). However, mifepristone is generally inactive, or poorly active, in glucocorticoid‐sensitive systems that rely on endogenous levels of GR (i.e. no over‐expression), where it behaves as a GR antagonist. However, with GR over‐expression, mifepristone displays partial agonist activity in reporter assays of transcriptional activation and repression (Zhao et al., 2003; Zhang et al., 2007b). Thus, increasing GR expression allows mifepristone to show some agonist activity. This finding suggests that the conformation of GR stabilized by mifepristone, while capable of binding DNA, does so in a manner that is only poorly favourable for activating gene transcription. In other words, mifepristone is a very weak partial agonist. Likewise, RU24858‐bound GR adopts a conformation that is suboptimal for the recruitment of steroid receptor co‐activator 1 (gene NCOA1) (Dezitter et al., 2014). As this is necessary for GR‐mediated transactivation, the failure to recruit sufficient NCOA1 results in transactivation becoming highly dependent on GR number (Dezitter et al., 2014). Collectively, such data show that GR density, the specific‐conformation of ligand‐bound GR and the promoter context of a gene, determines ‘transcriptional competency’. In pharmacological terms, this defines a given ligand as a full agonist, partial agonist or antagonist at any particular gene promoter.
As individual genes interpret equivalent degrees of GR occupancy differently, the concept described above suggests that the relationship between the ligand occupancy of GR and transcription is not uniform. Pharmacologically, the relationship between receptor occupancy and measured response is described by the receptor reserve, of which the affinity (K A)/EC50 ratio (Box 1) is a typical measure. The greater this ratio (i.e. where K A > EC50), the larger is the receptor reserve, and the occupation of fewer receptors is required to elicit a particular level of response Conversely, as the EC50 approaches the K A, the reserve declines toward zero where all receptors must be occupied to achieve the measured response. The concept of receptor reserve is traditionally associated with agonists that act at GPCRs. However, recently, this was applied to GR‐mediated gene expression. In BEAS‐2B cells, the potency of the glucocorticoid fluticasone furoate was found to vary in a gene‐dependent manner (Joshi et al., 2015a). Receptor occupancies necessary for half‐maximal gene induction were calculated as 21, 24, 29 and 39% for the glucocorticoid‐induced genes, TSC22D3, CRISPLD2, CDKN1C and PDK4 respectively (Joshi et al., 2015a) (Figure 6A). Thus, engagement of an equivalent fraction of GR by fluticasone furoate produced different degrees of gene activation with TCS22D32 and PDK4 being the most and least sensitive genes respectively. These results were reproduced in cells subjected to fractional GR inactivation, confirming that agonist intrinsic efficacy varies in a gene‐dependent manner (Joshi et al., 2015a). Likewise, GW870086X, which has lower intrinsic efficacy relative to fluticasone furoate, revealed increasing partial agonist behaviour that correlated inversely with the receptor reserve for each gene (Figure 6B).
Figure 6.
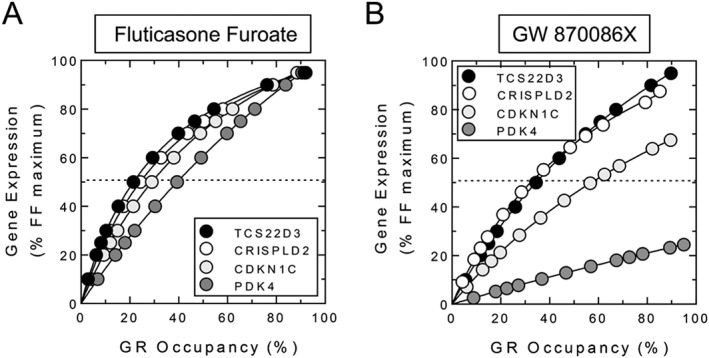
Relationship between GR occupancy and gene expression in human airway epithelial, BEAS‐2B, cells. The data show that gene induction expressed as a function of GR occupancy is not uniform across genes. This indicates that a given cell type interprets the same degree of GR occupancy differently for each gene. In panel A, half maximal TSC22D3, CRISPLD2, CDKN1C and PDK4 induction was achieved with a concentration of fluticasone furoate that produced 21, 24, 29 and 39% GR occupancy respectively. In panel B, a more extreme profile is shown with a different GC, GW 870086X. Relative to fluticasone furoate, half maximal expression of GILZ and CRISPLD2 required ~23% GR occupancy. However, GW 870086X was a partial agonist on both CDKN1C and PDK4 such that 100% GR occupancy induced these genes to a level that was only 70 and 30% of the same responses produced by fluticasone furoate. Data re‐drawn from Joshi et al. (2015a).
Differences in receptor reserve have particular significance in tissues in which GR number is limiting as two agonists, one with low and the other with a relatively high intrinsic efficacy, will not generate the same gene expression signature. In tissues with low GR expression, agonists with low intrinsic efficacy, such as RU24858 or GW870086X, fail to appreciably transactivate certain populations of genes, and this may contribute to their improved therapeutic ratio in some models (Vayssiere et al., 1997; Uings et al., 2013). However, whether tissue‐dependent variation in GR number affects the anti‐inflammatory activity of glucocorticoids used in clinical practice is unknown. Furthermore, there is no published precedent to suggest that GR number in target tissues is considered when selecting glucocorticoids for development. Nevertheless, it is noteworthy that the active metabolite of the GR agonist, ciclesonide, behaves in BEAS‐2B cells similarly to GW870086X (Joshi et al., 2015a). Collectively, these data suggest that the exploitation of partial GR agonists for enhanced therapeutic benefit requires an assessment of their ability to induce therapeutically desirable and undesirable response genes in target and off‐target tissues.
Biased agonism and GR‐mediated gene expression
While differential binding of agonists to nuclear hormone receptors is established (Allan et al., 1992; Wagner et al., 1996; Biggadike et al., 2009a; Biggadike et al., 2009b; Edman et al., 2014), it is unclear whether this can lead to biased gene expression (Figure 5) or merely different degrees of partial agonism. Certainly, GR effector functions and sub‐cellular localization of GR can occur in a ligand‐specific manner (Croxtall et al., 2002; Schaaf et al., 2005). Furthermore, unique patterns of ligand‐directed GR stabilization, differences in cofactor recruitment and agonist‐dependent heterogeneity of GR‐mediated gene expression all support the possibility of selectively inducing one gene (or gene population) over another (Bledsoe et al., 2002; Coghlan et al., 2003; Kauppi et al., 2003; Miner et al., 2007; Ronacher et al., 2009; Edman et al., 2014). This idea is supported by data derived with non‐steroidal GR ligands, such AL‐438 and LGD5552, which retain anti‐inflammatory activity with reduced induction of, at least some, side‐effect genes (Coghlan et al., 2003; Miner et al., 2007). Similarly, mifepristone preferentially recruits the co‐repressor, NCoR1 (Schulz et al., 2002; Ronacher et al., 2009), thereby establishing the principle that GR ligands can differentially modify gene expression by affecting co‐activator/co‐repressor recruitment. However, one attempt to identify biased agonism showed that fluticasone furoate and GW870086X induced TSC22D3, CRISPLD2, CDKN1C and PDK4 in BEAS‐2B cells, but there was no evidence of agonist potency reversal; fluticasone furoate was always 5–7‐times more potent than GW870086X for each gene (Joshi et al., 2015a). Nevertheless, that study was limited by the number of ligands and genes examined, as well as the structural similarity of the agonists. Clearly, comprehensive testing will be necessary to determine if different conformations of GR are associated with selective transcriptional signatures and whether these result from partial or, possibly, biased agonism. Furthermore, the relationship between any such biased agonism and the enhancement by LABA will need to be carefully quantified and assessed functionally.
Pharmacodynamics of β2‐adrenoceptor signalling
Tissue‐dependent variation in β2‐adrenoceptor number, coupling efficiency to adenylyl cyclase and differences in agonist intrinsic efficacy also have the potential to influence the magnitude of cAMP‐induced responses. This applies to the enhancement of GR‐mediated gene transcription and the direct induction of cAMP‐inducible genes by a LABA. In the latter case, the promoter context of the gene of interest will dictate whether PKA‐phosphorylated transcription factors, such as CREB1 or ATF1, bind to DNA consensus sequences and effect transcription. In humans, pulmonary β2‐adrenoceptor number varies by approximately 50‐fold. For example, ASM cells express a high number of β2‐adrenoceptors (30 000–40 000/cell), whereas T‐lymphocytes express relatively few (~750/cell) (Johnson, 2002). Therefore, the ability of LABAs to promote cAMP‐dependent transcription could be compromised in cell types where β2‐adrenoceptor number is limiting and/or when coupling to adenylyl cyclase is weak. Likewise, the LABA enhancement of GR‐mediated gene expression and resultant anti‐inflammatory activity could also be impaired. This concept is illustrated by studies with eosinophils, where salmeterol failed to suppress granule protein secretion and activation of the NADPH oxidase under conditions where the higher intrinsic activity agonist, formoterol, was effective (Rabe et al., 1993; Munoz et al., 1995).
A number of mutually inclusive approaches could improve the therapeutic activity of a LABA in cells that express low β2‐adrenoceptor number: (i) combine a GR agonist with a LABA that has very high intrinsic efficacy, (ii) add‐on a PDE4 inhibitor to an ICS/LABA combination therapy, and (iii) add‐on, or replace with, a ligand for another Gs‐coupled receptor that is either more highly expressed or couples to adenylyl cyclase with higher efficiency than the β2‐adrenoceptor. With the exception of salmeterol, most LABAs described to date already have high intrinsic efficacy, and it is unclear to what extent this property can be improved. In contrast, attenuating cAMP hydrolysis with a PDE4 inhibitor might transform a pro‐inflammatory or immune cell that is normally insensitive to a LABA into one that now generates a cAMP signal of sufficient magnitude to promote cAMP‐dependent transcriptional responses, and/or enhance GR‐mediated gene transcription. Likewise, agonists of GPCRs, other than the β2‐adrenoceptor, which also increase cAMP, will synergize with glucocorticoids. For example, activation of the prostacyclin receptor (gene PTGIR) or the adenosine A2B receptor (gene ADORA2B) enhanced simple GRE‐dependent transcription and synergized with glucocorticoids to induce gene expression (Wilson et al., 2009; Greer et al., 2013).
Adverse effects of β2‐adrenoceptor agonists, non‐canonical signalling and response to glucocorticoids
In asthma, β2‐adrenoceptor agonists alleviate bronchoconstriction, but fail to combat the underlying inflammation. For this reason, chronic use of β2‐adrenoceptor agonists, in the 1970s, was identified as a risk factor contributing to asthma deaths (Tattersfield, 2006). Consequently, asthma treatment guidelines now recommend that β2‐adrenoceptor agonists, as a class, should only be used on an as‐needed basis or, in the case of LABAs, in combination with an ICS (Cates and Cates, 2008).
Mechanistically, β2‐adrenoceptor agonists may be harmful in asthma by promoting or enhancing pro‐inflammatory gene expression through direct, cAMP‐dependent signalling mechanisms. The β2‐adrenoceptor may also mediate G‐protein‐independent signalling that involves the recruitment of β‐arrestin‐2 and the activation of ERK (Shenoy et al., 2006; Billington et al., 2013; Walker and DeFea, 2014; Pera and Penn, 2016). In murine models of asthma, this pathway is linked to pro‐inflammatory effects of certain β2‐adrenoceptor agonists in the airways (Nguyen et al., 2009; Walker et al., 2011; Thanawala et al., 2013; Penn et al., 2014; Zhou et al., 2014). For example, in allergen‐sensitized β‐arrestin‐2 knockout mice, the typical asthma‐like phenotype (AHR, pulmonary leukocyte burden, histological features of inflammation and mucus hyper‐secretion) resulting from allergen challenge was decreased relative to effects in wild‐type animals (Walker et al., 2003; Hollingsworth et al., 2010; Nichols et al., 2012; Chen et al., 2015). Similar results were obtained in sensitized and challenged mice deficient in either the β2‐adrenoceptor (Nguyen et al., 2009) or phenylethanolamine N‐methyltransferase (Gene PNMT; an enzyme central to adrenaline biosynthesis) (Thanawala et al., 2013). Collectively, these data suggest that, in mice, the cardinal features of asthma are dependent on the release of adrenaline and its ability to activate β2‐adrenoceptors. Significantly, chronic administration of β2‐adrenoceptor agonist to PNMT −/− and wild‐type mice reconstitutes or exacerbates, respectively, these features of asthma (Lin et al., 2012; Thanawala et al., 2013). Thus, β2‐adrenoceptor agonists may exert pro‐inflammatory effects in the mouse lung, via a mechanism that is independent of cAMP generation.
If clinical evidence for such β2‐adrenoceptor‐mediated biased agonism is established, it would raise important questions about existing LABAs and how the next generation should be designed. For example, to what extent are currently marketed LABAs biased toward β‐arrestin recruitment and signalling? Can the effect of LABA bias towards β‐arrestin‐dependent effectors be reduced or mitigated? Currently, there is some evidence that formoterol and salmeterol display a degree of bias for β‐arrestin (Rajagopal et al., 2011; van der Westhuizen et al., 2014). If confirmed, this highlights the need to understand the structural elements in these ligands that preferentially stabilize the β2‐adrenoceptor in this unwanted conformation. One means to compensate for adverse‐effects mediated by β‐arrestin‐dependent ERK activation is to offset bias by enhancing LABA‐induced, cAMP signalling with a PDE4 inhibitor (Forkuo et al., 2016; Pera and Penn, 2016). Indeed, in sensitized and challenged PNMT‐deficient mice, roflumilast and rolipram prevented formoterol and salmeterol from restoring the asthma‐like phenotype when compared with wild‐type animals. Similarly, the ability of glucocorticoids, and LABAs, to induce DUSP1 expression, for example in primary human ASM or bronchial epithelial cells (Quante et al., 2008; Manetsch et al., 2012; Rider et al., 2015b; Shah et al., 2016), would independently inactivate ERK. This effect may not only protect against MAPK‐dependent desensitization (Nino et al., 2010) but should also reduce unwanted signalling due to biased agonism from the β2‐adrenoceptor. Thus, reducing the effect of β2‐adrenoceptor‐mediated, β‐arrestin signalling with a PDE4 inhibitor and/or a glucocorticoid could tip the balance away from ‘pro‐asthma’ effects towards more favourable ‘therapeutic’ outcomes (Forkuo et al., 2016; Pera and Penn, 2016).
These concepts of biased agonism are relevant to a recent reappraisal of the PDE4 inhibitor, roflumilast, in mild‐to‐moderate asthma (Bardin et al., 2015; Bateman et al., 2015; Meltzer et al., 2015). Clinical efficacy (measured by improvement in forced expiratory volume in 1 s, time to first exacerbation, rescue medication consumption and symptoms) was found, perhaps surprisingly, not to be inferior to an ICS (Bateman et al., 2015). If these findings are confirmed, a strong case for a β2‐adrenoceptor agonist/PDE4 inhibitor combination therapy can be made based on their ability to increase airway calibre, suppress inflammation and offset unwanted effects that may be associated with signal bias. Moreover, this approach would necessarily require combination with an ICS (Meltzer et al., 2015), which should further mitigate adverse effects of LABAs that occur via canonical and non‐canonical β2‐adrenoceptor‐mediated signalling. The possibility that LABAs and PDE4 inhibitors interact synergistically on target cells and tissues with an attendant improvement in clinical outcomes is also predicted. Logic dictates that this would be most effective in cells and tissues where β2‐adrenoceptor density is limiting and/or coupling efficiency to adenylyl cyclase is weak (Giembycz and Newton, 2014). It is noteworthy, that this approach has been adopted for the treatment of COPD with the development of the novel, bifunctional ligand, GS‐5759, which is formulated for inhaled delivery (Salmon et al., 2014; Tannheimer et al., 2014). GS‐5759 is a single chemical entity composed of a PDE4 inhibitor linked via a spacer to a β2‐adrenoceptor agonist. This provides greater lung retention, low oral bioavailability, reduced systemic exposure and an improved therapeutic ratio that is typical of inhaled, high molecular weight molecules (Phillips and Salmon, 2012; Robinson et al., 2013). Furthermore, the identical deposition characteristics of the two pharmacophores maximize the opportunity for synergistic interaction at the same target tissues.
Outlook and opportunity for drug discovery
The clinical superiority of ICS/LABA combination therapy is well established and, for some time, has been the standard‐of‐care where ICS monotherapy fails to provide adequate disease control. While this enhanced effect seems likely to have arisen as a consequence of the natural interactions between endogenous stress response mechanisms, for example between glucocorticoids and catecholamines, the molecular basis remains unclear. Indeed, although several explanations have been proposed to explain how LABAs enhance ICS action, their ability to account for gene‐specific effects is questionable (Table 1). It is clear that glucocorticoids and LABAs independently and in combination induce gene transcription. This is important given the increasing appreciation that GR‐mediated transactivation contributes to the mechanism of action of ICS. The ability of LABAs to enhance glucocorticoid‐dependent transcription and for LABAs and glucocorticoids to synergistically enhance the expression of genes, such as RGS2, provides a compelling explanation for the improved asthma control seen in clinical practice. Therefore, understanding the consequences of LABA/glucocorticoid‐induced gene expression is central to the development of improved combination therapies. Knowing which genes are desirable, and which are not, is necessary to comprehend integrated biological function in complex disease networks. Equally, knowing that the enhancement of GR‐mediated gene expression is cAMP‐dependent provides additional opportunities to improve anti‐inflammatory glucocorticoid therapies. For example, adding‐on a PDE4 inhibitor may prove useful by augmenting the effects of a LABA in target cells where β2‐adrenoceptor density is low. This approach has the additional advantage of reducing the potential undesirable consequences of biased agonism from the β2‐adrenoceptor. Alternatively, the identification of non‐biased, or perhaps Gs‐biased, β2‐adrenoceptor agonists or ligands for other GPCRs that are more highly expressed and/or better coupled to adenylyl cyclase on target cells may be highly effective in combination with an ICS. In each case, such effects may be further optimized by combining multiple drugs into single molecules that display unique polypharmacological characteristics.
A further, and currently underexplored, avenue for improving therapeutic efficacy lies with the realization that every GR responsive gene, each with its own unique promoter environment, may be maximally activated only by specific GR conformations (Keenan et al., 2016). Thus, each gene is effectively an independent signal transducer that responds to ligand‐activated GR. Furthermore, as each different GR ligand has the potential to preferentially activate gene expression in manner that is dictated by the ligand‐induced conformation that is adopted by GR, there is the potential for biased GR‐dependent gene expression. However, approaches to improve glucocorticoid effectiveness have generally rested on the concept that GR transactivation is undesirable, whereas transrepression, for example of NF‐κB, is beneficial. This may have unwittingly resulted in the identification of partial agonists of GR. Thus, the repression of NF‐κB‐dependent transcription or genes, such as IL6 and CXCL8, that are NF‐κB‐dependent versus the ability to activate simple GR‐responsive reporters have been used to screen for candidate ligands. However, the repression of NF‐κB‐dependent reporters and inflammatory gene expression is routinely observed at lower glucocorticoid concentrations compared with transactivation of simple GRE reporters (Jonat et al., 1990; Chivers et al., 2004; Chivers et al., 2006; King et al., 2013). Thus, partial agonists of GR will inevitably produce separation of repression from transactivation. Indeed, ligands such as RU24858, GW870086X and ciclesonide can behave as partial GR agonists (Joshi et al., 2015a). While this may produce some improvement in therapeutic benefit, the details and functions of genes showing differential expression to agonists with high and low intrinsic efficacy remain to be fully explored. Nevertheless, we submit that the possibility of biased GR agonism offers greater therapeutic potential compared with partial GR agonists.
In conclusion, GR‐mediated transactivation of the numerous response genes that contribute to the therapeutic activity of ICS in asthma offers new opportunity for the identification and selection of ligands that may transactivate more desirable groups of effector genes. At present, this area of GR biology is relatively underdeveloped, but could contribute substantially to the discovery of novel therapeutics. Exploiting gene expression bias to design improved anti‐inflammatory GR ligands will require changes in gene expression to be correlated with an extensive understanding of their function in on‐ and off‐target tissues. While this is necessarily an empirical approach, modern high‐throughput technologies now make the initial part of this objective an achievable goal. Probably more challenging will be to accurately assign gene function as beneficial or undesirable in asthma management. Finally, the identification of biased GR agonists will need to be considered in the context of combination therapies. While this substantially increases the level of complexity, there are correspondingly more opportunities for drug optimization and discovery.
Conflict of interest
The authors declare no conflicts of interest.
Newton, R. , and Giembycz, M. A. (2016) Understanding how long‐acting β2‐adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids in asthma – an update. British Journal of Pharmacology, 173: 3405–3430. doi: 10.1111/bph.13628.
References
- Abraham SM, Clark AR (2006). Dual‐specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans 34: 1018–1023. [DOI] [PubMed] [Google Scholar]
- Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J et al. (2006). Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 203: 1883–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y et al. (1990). A nuclear factor for IL‐6 expression (NF‐IL6) is a member of a C/EBP family. EMBO J 9: 1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan GF, Leng X, Tsai SY, Weigel NL, Edwards DP, Tsai MJ et al. (1992). Hormone and antihormone induce distinct conformational changes which are central to steroid receptor activation. J Biol Chem 267: 19513–19520. [PubMed] [Google Scholar]
- Altonsy MO, Sasse SK, Phang TL, Gerber AN (2014). Context‐dependent cooperation between nuclear factor kappaB (NF‐kappaB) and the glucocorticoid receptor at a TNFAIP3 intronic enhancer: a mechanism to maintain negative feedback control of inflammation. J Biol Chem 289: 8231–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammit AJ, Hoffman RK, Amrani Y, Lazaar AL, Hay DW, Torphy TJ et al. (2000). Tumor necrosis factor‐alpha‐induced secretion of RANTES and interleukin‐6 from human airway smooth‐muscle cells. Modulation by cyclic adenosine monophosphate. Am J Respir Cell Mol Biol 23: 794–802. [DOI] [PubMed] [Google Scholar]
- Ammit AJ, Lazaar AL, Irani C, O'Neill GM, Gordon ND, Amrani Y et al. (2002). Tumor necrosis factor‐alpha‐induced secretion of RANTES and interleukin‐6 from human airway smooth muscle cells: modulation by glucocorticoids and beta‐agonists. Am J Respir Cell Mol Biol 26: 465–474. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M (1995). Immunosuppression by glucocorticoids: inhibition of NF‐kappa B activity through induction of I kappa B synthesis. Science 270: 286–290. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Riccardi C (2009). Glucocorticoid‐induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J 23: 3649–3658. [DOI] [PubMed] [Google Scholar]
- Bardin P, Kanniess F, Gauvreau G, Bredenbroker D, Rabe KF (2015). Roflumilast for asthma: Efficacy findings in mechanism of action studies. Pulm Pharmacol Ther 35 (Suppl): S4–S10. [DOI] [PubMed] [Google Scholar]
- Barnes PJ (2008). Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183–192. [DOI] [PubMed] [Google Scholar]
- Bateman ED, Bousquet J, Aubier M, Bredenbroker D, O'Byrne PM (2015). Roflumilast for asthma: Efficacy findings in non‐placebo‐controlled comparator and dosing studies. Pulm Pharmacol Ther 35 (Suppl): S11–S19. [DOI] [PubMed] [Google Scholar]
- Baumann H, Jahreis GP, Morella KK, Won KA, Pruitt SC, Jones VE et al. (1991). Transcriptional regulation through cytokine and glucocorticoid response elements of rat acute phase plasma protein genes by C/EBP and JunB. J Biol Chem 266: 20390–20399. [PubMed] [Google Scholar]
- Berg T, Didon L, Barton J, Andersson O, Nord M (2005). Glucocorticoids increase C/EBPbeta activity in the lung epithelium via phosphorylation. Biochem Biophys Res Commun 334: 638–645. [DOI] [PubMed] [Google Scholar]
- Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet‐Delbos L et al. (2003). Synthesis of glucocorticoid‐induced leucine zipper (GILZ) by macrophages: an anti‐inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL‐10. Blood 101: 729–738. [DOI] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL et al. (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K, Bledsoe RK, Coe DM, Cooper TW, House D , Iannone MA et al. (2009a). Design and x‐ray crystal structures of high‐potency nonsteroidal glucocorticoid agonists exploiting a novel binding site on the receptor. Proc Natl Acad Sci U S A 106: 18114–18119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K, Caivano M, Clackers M, Coe DM, Hardy GW, Humphreys D et al. (2009b). Highly tractable, sub‐nanomolar non‐steroidal glucocorticoid receptor agonists. Bioorg Med Chem Lett 19: 4846–4850. [DOI] [PubMed] [Google Scholar]
- Billington CK, Ojo OO, Penn RB, Ito S (2013). cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther 26: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BinMahfouz H, Borthakur B, Yan D, George T, Giembycz MA, Newton R (2015). Superiority of combined phosphodiesterase PDE3/PDE4 inhibition over PDE4 inhibition alone on glucocorticoid‐ and long‐acting beta2‐adrenoceptor agonist‐induced gene expression in human airway epithelial cells. Mol Pharmacol 87: 64–76. [DOI] [PubMed] [Google Scholar]
- Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD et al. (2002). Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110: 93–105. [DOI] [PubMed] [Google Scholar]
- Bourgeois S, Newby RF (1979). Correlation between glucocorticoid receptor and cytolytic response of murine lymphoid cell lines. Cancer Res 39: 4749–4751. [PubMed] [Google Scholar]
- Burgun C, Esteve L, Humblot N, Aunis D, Zwiller J (2000). Cyclic AMP‐elevating agents induce the expression of MAP kinase phosphatase‐1 in PC12 cells. FEBS Lett 484: 189–193. [DOI] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL (1991). Regulated expression of three C/EBP isoforms during adipose conversion of 3 T3‐L1 cells. Genes Dev 5: 1538–1552. [DOI] [PubMed] [Google Scholar]
- Cardinaux JR, Magistretti PJ (1996). Vasoactive intestinal peptide, pituitary adenylate cyclase‐activating peptide, and noradrenaline induce the transcription factors CCAAT/enhancer binding protein (C/EBP)‐beta and C/EBP delta in mouse cortical astrocytes: involvement in cAMP‐regulated glycogen metabolism. J Neurosci 16: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cates CJ, Cates MJ (2008). Regular treatment with salmeterol for chronic asthma: serious adverse events. Cochrane Database Syst Rev: CD006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli BR, Thomas NE, Anderson JA, Ferguson GT, Jenkins CR, Jones PW et al. (2008). Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med 178: 332–338. [DOI] [PubMed] [Google Scholar]
- Chang MM, Juarez M, Hyde DM, Wu R (2001). Mechanism of dexamethasone‐mediated interleukin‐8 gene suppression in cultured airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 280: L107–L115. [DOI] [PubMed] [Google Scholar]
- Chang TS, Kim MJ, Ryoo K, Park J, Eom SJ, Shim J et al. (2003). p57KIP2 modulates stress‐activated signaling by inhibiting c‐Jun NH2‐terminal kinase/stress‐activated protein Kinase. J Biol Chem 278: 48092–48098. [DOI] [PubMed] [Google Scholar]
- Chen M, Hegde A, Choi YH, Theriot BS, Premont RT, Chen W et al. (2015). Genetic Deletion of beta‐Arrestin‐2 and the Mitigation of Established Airway Hyperresponsiveness in a Murine Asthma Model. Am J Respir Cell Mol Biol 53: 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y, Coppo M, Gupte R, Sacta MA, Rogatsky I (2014). Glucocorticoid receptor coordinates transcription factor‐dominated regulatory network in macrophages. BMC Genomics 15: 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivers JE, Cambridge LM, Catley MC, Mak JC, Donnelly LE, Barnes PJ et al. (2004). Differential effects of RU486 reveal distinct mechanisms for glucocorticoid repression of prostaglandin E release. Eur J Biochem 271: 4042–4052. [DOI] [PubMed] [Google Scholar]
- Chivers JE, Gong W, King EM, Seybold J, Mak JC, Donnelly LE et al. (2006). Analysis of the dissociated steroid, RU24858, does not exclude a role for inducible genes in the anti‐inflammatory actions of glucocorticoids. Mol Pharmacol 70: 2084–2095. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH (1993). Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365: 855–859. [DOI] [PubMed] [Google Scholar]
- Clark AR, Belvisi MG (2012). Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther 134: 54–67. [DOI] [PubMed] [Google Scholar]
- Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW et al. (2003). A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol 17: 860–869. [DOI] [PubMed] [Google Scholar]
- Constantinescu A, Wu M, Asher O, Diamond I (2004). cAMP‐dependent protein kinase type I regulates ethanol‐induced cAMP response element‐mediated gene expression via activation of CREB‐binding protein and inhibition of MAPK. J Biol Chem 279: 43321–43329. [DOI] [PubMed] [Google Scholar]
- Croniger C, Leahy P, Reshef L, Hanson RW (1998). C/EBP and the control of phosphoenolpyruvate carboxykinase gene transcription in the liver. J Biol Chem 273: 31629–31632. [DOI] [PubMed] [Google Scholar]
- Croxtall JD, Van Hal PT, Choudhury Q, Gilroy DW, Flower RJ (2002). Different glucocorticoids vary in their genomic and non‐genomic mechanism of action in A549 cells. Br J Pharmacol 135: 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G (2003). The interplay between the glucocorticoid receptor and nuclear factor‐kappaB or activator protein‐1: molecular mechanisms for gene repression. Endocr Rev 24: 488–522. [DOI] [PubMed] [Google Scholar]
- Dezitter X, Fagart J, Taront S, Fay M, Masselot B, Hetuin D et al. (2014). A structural explanation of the effects of dissociated glucocorticoids on glucocorticoid receptor transactivation. Mol Pharmacol 85: 226–236. [DOI] [PubMed] [Google Scholar]
- Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E et al. (2007). Glucocorticoid‐induced leucine zipper (GILZ)/NF‐kappaB interaction: role of GILZ homo‐dimerization and C‐terminal domain. Nucleic Acids Res 35: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Gallardo MY, Cote‐Velez A, Charli JL, Joseph‐Bravo P (2010). A rapid interference between glucocorticoids and cAMP‐activated signalling in hypothalamic neurones prevents binding of phosphorylated cAMP response element binding protein and glucocorticoid receptor at the CRE‐Like and composite GRE sites of thyrotrophin‐releasing hormone gene promoter. J Neuroendocrinol 22: 282–293. [DOI] [PubMed] [Google Scholar]
- Didon L, Barton JL, Roos AB, Gaschler GJ, Bauer CM, Berg T et al. (2011). Lung epithelial CCAAT/enhancer‐binding protein‐beta is necessary for the integrity of inflammatory responses to cigarette smoke. Am J Respir Crit Care Med 184: 233–242. [DOI] [PubMed] [Google Scholar]
- Diefenbacher M, Sekula S, Heilbock C, Maier JV, Litfin M, van Dam H et al. (2008). Restriction to Fos family members of Trip6‐dependent coactivation and glucocorticoid receptor‐dependent trans‐repression of activator protein‐1. Mol Endocrinol 22: 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Aronsson M, Gustafsson JA, Okret S (1989). The mechanism of cAMP‐induced glucocorticoid receptor expression. Correlation to cellular glucocorticoid response. J Biol Chem 264: 13679–13683. [PubMed] [Google Scholar]
- Dowell ML, Lavoie TL, Lakser OJ, Dulin NO, Fredberg JJ, Gerthoffer WT et al. (2010). MEK modulates force‐fluctuation‐induced relengthening of canine tracheal smooth muscle. Eur Respir J 36: 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme FM, Ni CM, Greenstone I, Lasserson TJ (2010a). Addition of long‐acting beta2‐agonists to inhaled corticosteroids versus same dose inhaled corticosteroids for chronic asthma in adults and children. Cochrane Database Syst Rev: CD005535. [DOI] [PMC free article] [PubMed]
- Ducharme FM, Ni CM, Greenstone I, Lasserson TJ (2010b). Addition of long‐acting beta2‐agonists to inhaled steroids versus higher dose inhaled steroids in adults and children with persistent asthma. Cochrane Database Syst Rev: CD005533. [DOI] [PMC free article] [PubMed]
- Eddleston J, Herschbach J, Wagelie‐Steffen AL, Christiansen SC, Zuraw BL (2007). The anti‐inflammatory effect of glucocorticoids is mediated by glucocorticoid‐induced leucine zipper in epithelial cells. J Allergy Clin Immunol 119: 115–122. [DOI] [PubMed] [Google Scholar]
- Edman K, Ahlgren R, Bengtsson M, Bladh H, Backstrom S, Dahmen J et al. (2014). The discovery of potent and selective non‐steroidal glucocorticoid receptor modulators, suitable for inhalation. Bioorg Med Chem Lett 24: 2571–2577. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Johnson MW, Johnston SL (2006). Combination therapy: Synergistic suppression of virus‐induced chemokines in airway epithelial cells. Am J Respir Cell Mol Biol 34: 616–624. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Haas J, Panettieri RA Jr, Johnson M, Johnston SL (2007). Corticosteroids and beta2 agonists differentially regulate rhinovirus‐induced interleukin‐6 via distinct Cis‐acting elements. J Biol Chem 282: 15366–15375. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M et al. (1999). Ligand‐independent activation of the glucocorticoid receptor by beta2‐adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 274: 1005–1010. [DOI] [PubMed] [Google Scholar]
- Essilfie‐Quaye S, Ito K, Ito M, Kharitonov SA, Barnes PJ (2011). Comparison of Symbicort(R) versus Pulmicort(R) on steroid pharmacodynamic markers in asthma patients. Respir Med 105: 1784–1789. [DOI] [PubMed] [Google Scholar]
- Evans BA, Sato M, Sarwar M, Hutchinson DS, Summers RJ (2010). Ligand‐directed signalling at beta‐adrenoceptors. Br J Pharmacol 159: 1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisy C, Naline E, Diehl JL, Emonds‐Alt X, Chinet T, Advenier C (2002). In vitro sensitization of human bronchus by beta2‐adrenergic agonists. Am J Physiol Lung Cell Mol Physiol 283: L1033–L1042. [DOI] [PubMed] [Google Scholar]
- Faisy C, Naline E, Rouget C, Risse PA, Guerot E, Fagon JY et al. (2004). Nociceptin inhibits vanilloid TRPV‐1‐mediated neurosensitization induced by fenoterol in human isolated bronchi. Naunyn Schmiedebergs Arch Pharmacol 370: 167–175. [DOI] [PubMed] [Google Scholar]
- Fenne IS, Hoang T, Hauglid M, Sagen JV, Lien EA, Mellgren G (2008). Recruitment of coactivator glucocorticoid receptor interacting protein 1 to an estrogen receptor transcription complex is regulated by the 3′,5′‐cyclic adenosine 5′‐monophosphate‐dependent protein kinase. Endocrinology 149: 4336–4345. [DOI] [PubMed] [Google Scholar]
- Fernandes D, Guida E, Koutsoubos V, Harris T, Vadiveloo P, Wilson JW et al. (1999). Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular‐regulated kinases in human cultured airway smooth muscle. Am J Respir Cell Mol Biol 21: 77–88. [DOI] [PubMed] [Google Scholar]
- Forkuo GS, Kim H, Thanawala VJ, Al‐Sawalha N, Valdez D, Joshi R et al. (2016). PDE4 Inhibitors Attenuate the Asthma Phenotype Produced by beta‐adrenoceptor Agonists in PNMT‐KO Mice. Am J Respir Cell Mol Biol 55: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frois C, Wu EQ, Ray S, Colice GL (2009). Inhaled corticosteroids or long‐acting beta‐agonists alone or in fixed‐dose combinations in asthma treatment: a systematic review of fluticasone/budesonide and formoterol/salmeterol. Clin Ther 31: 2779–2803. [DOI] [PubMed] [Google Scholar]
- Furst R, Schroeder T, Eilken HM, Bubik MF, Kiemer AK, Zahler S et al. (2007). MAPK phosphatase‐1 represents a novel anti‐inflammatory target of glucocorticoids in the human endothelium. FASEB J 21: 74–80. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT, Yamboliev IA, Pohl J, Haynes R, Dang S, McHugh J (1997). Activation of MAP kinases in airway smooth muscle. Am J Physiol 272: L244–L252. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Newton R (2014). How Phosphodiesterase 4 Inhibitors Work in Patients with Chronic Obstructive Pulmonary Disease of the Severe, Bronchitic, Frequent Exacerbator Phenotype. Clin Chest Med 35: 203–217. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Newton R (2015). Potential mechanisms to explain how LABAs and PDE4 inhibitors enhance the clinical efficacy of glucocorticoids in inflammatory lung diseases. F1000Prime Rep 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz MA, Kaur M, Leigh R, Newton R (2008). A Holy Grail of asthma management: toward understanding how long‐acting beta(2)‐adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids. Br J Pharmacol 153: 1090–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godot V, Garcia G, Capel F, Arock M, Durand‐Gasselin I, Asselin‐Labat ML et al. (2006). Dexamethasone and IL‐10 stimulate glucocorticoid‐induced leucine zipper synthesis by human mast cells. Allergy 61: 886–890. [DOI] [PubMed] [Google Scholar]
- Gonzales LW, Guttentag SH, Wade KC, Postle AD, Ballard PL (2002). Differentiation of human pulmonary type II cells in vitro by glucocorticoid plus cAMP. Am J Physiol Lung Cell Mol Physiol 283: L940–L951. [DOI] [PubMed] [Google Scholar]
- Greening AP, Ind PW, Northfield M, Shaw G (1994). Added salmeterol versus higher‐dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Allen and Hanburys Limited UK Study Group. Lancet 344: 219–224. [DOI] [PubMed] [Google Scholar]
- Greer S, Page CW, Joshi T, Yan D, Newton R, Giembycz MA (2013). Concurrent Agonism of Adenosine A2B and Glucocorticoid Receptors in Human Airway Epithelial Cells Cooperatively Induces Genes with Anti‐Inflammatory Potential: A Novel Approach to Treat Chronic Obstructive Pulmonary Disease. J Pharmacol Exp Ther 346: 473–485. [DOI] [PubMed] [Google Scholar]
- Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C et al. (2013). C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32: 1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy S, Plumb J, Kaur M, Ray D, Singh D (2016). Additive anti‐inflammatory effects of corticosteroids and phosphodiesterase‐4 inhibitors in COPD CD8 cells. Respir Res 17: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DJ, Altschmied J (1993). Synergistic induction of apoptosis with glucocorticoids and 3′,5′‐cyclic adenosine monophosphate reveals agonist activity by RU 486. Mol Endocrinol 7: 104–113. [DOI] [PubMed] [Google Scholar]
- Hanson RW, Reshef L (1997). Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem 66: 581–611. [DOI] [PubMed] [Google Scholar]
- Haque R, Hakim A, Moodley T, Torrego A, Essilfie‐Quaye S, Jazrawi E et al. (2013). Inhaled long‐acting beta2 agonists enhance glucocorticoid receptor nuclear translocation and efficacy in sputum macrophages in COPD. J Allergy Clin Immunol 132: 1166–1173. [DOI] [PubMed] [Google Scholar]
- He X, Chatterjee R, John S, Bravo H, Sathyanarayana BK, Biddie SC et al. (2013). Contribution of nucleosome binding preferences and co‐occurring DNA sequences to transcription factor binding. BMC Genomics 14: 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT (2000). Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am J Physiol Cell Physiol 278: C718–C726. [DOI] [PubMed] [Google Scholar]
- Heximer SP (2004). RGS2‐mediated regulation of Gqalpha. Methods Enzymol 390: 65–82. [DOI] [PubMed] [Google Scholar]
- Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B et al. (2014). RNA‐Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One 9: e99625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T, Fenne IS, Cook C, Borud B, Bakke M, Lien EA et al. (2004). cAMP‐dependent protein kinase regulates ubiquitin‐proteasome‐mediated degradation and subcellular localization of the nuclear receptor coactivator GRIP1. J Biol Chem 279: 49120–49130. [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Schmitz ML (2002). The promoter context determines mutual repression or synergism between NF‐kappaB and the glucocorticoid receptor. Biol Chem 383: 1947–1951. [DOI] [PubMed] [Google Scholar]
- Holden NS, Rider CF, Bell MJ, Velayudhan J, King EM, Kaur M et al. (2010). Enhancement of inflammatory mediator release by beta(2)‐adrenoceptor agonists in airway epithelial cells is reversed by glucocorticoid action. Br J Pharmacol 160: 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden NS, Bell MJ, Rider CF, King EM, Gaunt DD, Leigh R et al. (2011). beta2‐Adrenoceptor agonist‐induced RGS2 expression is a genomic mechanism of bronchoprotection that is enhanced by glucocorticoids. Proc Natl Acad Sci U S A 108: 19713–19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden NS, George T, Rider CF, Chandrasekhar A, Shah S, Kaur M et al. (2014). Induction of Regulator of G‐Protein Signaling 2 Expression by Long‐Acting beta2‐Adrenoceptor Agonists and Glucocorticoids in Human Airway Epithelial Cells. J Pharmacol Exp Ther 348: 12–24. [DOI] [PubMed] [Google Scholar]
- Hollingsworth JW, Theriot BS, Li Z, Lawson BL, Sunday M, Schwartz DA et al. (2010). Both hematopoietic‐derived and non‐hematopoietic‐derived {beta}‐arrestin‐2 regulates murine allergic airway disease. Am J Respir Cell Mol Biol 43: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsch K, de Wet H, Schuurmans MM, Allie‐Reid F, Cato AC, Cunningham J et al. (2007). Mitogen‐activated protein kinase phosphatase 1/dual specificity phosphatase 1 mediates glucocorticoid inhibition of osteoblast proliferation. Mol Endocrinol 21: 2929–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Ganti KP, Chambon P (2016a). Glucocorticoid‐induced tethered transrepression requires SUMOylation of GR and formation of a SUMO‐SMRT/NCoR1‐HDAC3 repressing complex. Proc Natl Acad Sci U S A 113: E635–E643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Paulen L, Chambon P (2016b). GR SUMOylation and formation of an SUMO‐SMRT/NCoR1‐HDAC3 repressing complex is mandatory for GC‐induced IR nGRE‐mediated transrepression. Proc Natl Acad Sci U S A 113: E626–E634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Youn C, Ortlund EA (2013). The structural basis of direct glucocorticoid‐mediated transrepression. Nat Struct Mol Biol 20: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa R, Xie S, Khorasani N, Sukkar M, Adcock IM, Lee KY et al. (2007). Corticosteroid Inhibition of Growth‐Related Oncogene Protein‐{alpha} via Mitogen‐Activated Kinase Phosphatase‐1 in Airway Smooth Muscle Cells. J Immunol 178: 7366–7375. [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM (2000). Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin‐1beta‐induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 20: 6891–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Jazrawi E, Cosio B, Barnes PJ, Adcock IM (2001). p65‐activated histone acetyltransferase activity is repressed by glucocorticoids: mifepristone fails to recruit HDAC2 to the p65‐HAT complex. J Biol Chem 276: 30208–30215. [DOI] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie‐Quaye S, Cosio B, Ito M, Barnes PJ et al. (2006). Histone deacetylase 2‐mediated deacetylation of the glucocorticoid receptor enables NF‐kappaB suppression. J Exp Med 203: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Mei FC, Johnson BH, Thompson EB, Cheng X (2007). Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J Biol Chem 282: 37370–37377. [DOI] [PubMed] [Google Scholar]
- Ji Z, Mei FC, Miller AL, Thompson EB, Cheng X (2008). Protein kinase A (PKA) isoform RIIbeta mediates the synergistic killing effect of cAMP and glucocorticoid in acute lymphoblastic leukemia cells. J Biol Chem 283: 21920–21925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xie Y, Abel PW, Wolff DW, Toews ML, Panettieri RA Jr et al. (2014). RGS2 Repression Exacerbates Airway Hyperresponsiveness and Remodeling in Asthma. Am J Respir Cell Mol Biol 53: 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitrapakdee S (2012). Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int J Biochem Cell Biol 44: 33–45. [DOI] [PubMed] [Google Scholar]
- Johansson‐Haque K, Palanichamy E, Okret S (2008). Stimulation of MAPK‐phosphatase 1 gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J Mol Endocrinol 41: 239–249. [DOI] [PubMed] [Google Scholar]
- Johnson M (2002). Effects of beta2‐agonists on resident and infiltrating inflammatory cells. J Allergy Clin Immunol 110: S282–S290. [DOI] [PubMed] [Google Scholar]
- Johnson M (2004). Interactions between corticosteroids and beta2‐agonists in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 1: 200–206. [DOI] [PubMed] [Google Scholar]
- Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H et al. (1990). Antitumor promotion and antiinflammation: down‐modulation of AP‐1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62: 1189–1204. [DOI] [PubMed] [Google Scholar]
- Joshi T, Johnson M, Newton R, Giembycz M (2015a). An analysis of glucocorticoid receptor‐mediated gene expression in BEAS‐2B human airway epithelial cells identifies distinct, ligand‐directed, transcription profiles with implications for asthma therapeutics. Br J Pharmacol 172: 1360–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T, Johnson M, Newton R, Giembycz MA (2015b). The long‐acting beta2 ‐adrenoceptor agonist, indacaterol, enhances glucocorticoid receptor‐mediated transcription in human airway epithelial cells in a gene‐ and agonist‐dependent manner. Br J Pharmacol 172: 2634–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiyala V, Sasse SK, Altonsy MO, Berman R, Chu HW, Phang TL et al. (2016). Cistrome‐based Cooperation Between Airway Epithelial Glucocorticoid Receptor and NF‐kappaB Orchestrates Anti‐inflammatory Effects. J Biol Chem 291: 12673–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BN, Jude JA, Panettieri RA Jr, Walseth TF, Kannan MS (2008). Glucocorticoid regulation of CD38 expression in human airway smooth muscle cells: role of dual specificity phosphatase 1. Am J Physiol Lung Cell Mol Physiol 295: L186–L193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC (2001). Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP‐1. EMBO J 20: 7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsunuma T, Roffel AF, Elzinga CR, Zaagsma J, Barnes PJ, Mak JC (1999). beta(2)‐adrenoceptor agonist‐induced upregulation of tachykinin NK(2) receptor expression and function in airway smooth muscle. Am J Respir Cell Mol Biol 21: 409–417. [DOI] [PubMed] [Google Scholar]
- Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M et al. (2003). The three‐dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand‐binding domain: RU‐486 induces a transconformation that leads to active antagonism. J Biol Chem 278: 22748–22754. [DOI] [PubMed] [Google Scholar]
- Kaur M, Chivers JE, Giembycz MA, Newton R (2008). Long‐acting beta2‐adrenoceptor agonists synergistically enhance glucocorticoid‐dependent transcription in human airway epithelial and smooth muscle cells. Mol Pharmacol 73: 203–214. [DOI] [PubMed] [Google Scholar]
- Keenan CR, Lew MJ, Stewart AG (2016). Biased signalling from the glucocorticoid receptor: Renewed opportunity for tailoring glucocorticoid activity. Biochem Pharmacol 112: 6–12. [DOI] [PubMed] [Google Scholar]
- Kelly MM, King EM, Rider CF, Gwozd C, Holden NS, Eddleston J et al. (2012). Corticosteroid‐induced gene expression in allergen‐challenged asthmatic subjects taking inhaled budesonide. Br J Pharmacol 165: 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (1995). Agonist‐receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16: 232–238. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2009). Biased agonism. F1000 Biol Rep 1: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2015). Gaddum Memorial Lecture 2014: receptors as an evolving concept: from switches to biased microprocessors. Br J Pharmacol 172: 4238–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew KM, Dias S, Cates CJ (2014). Long‐acting inhaled therapy (beta‐agonists, anticholinergics and steroids) for COPD: a network meta‐analysis. Cochrane Database Syst Rev 3: CD010844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple AJ, Soundararajan M, Hutsell SQ, Roos AK, Urban DJ, Setola V et al. (2009). Structural determinants of G‐protein alpha subunit selectivity by regulator of G‐protein signaling 2 (RGS2). J Biol Chem 284: 19402–19411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EM, Holden NS, Gong W, Rider CF, Newton R (2009a). Inhibition of NF‐kappaB‐dependent transcription by MKP‐1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J Biol Chem 284: 26803–26815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EM, Kaur M, Gong W, Rider CF, Holden NS, Newton R (2009b). Regulation of tristetraprolin expression by interleukin‐1beta and dexamethasone in human pulmonary epithelial cells: roles for nuclear factor‐kappaB and p38 mitogen‐activated protein kinase. J Pharmacol Exp Ther 330: 575–585. [DOI] [PubMed] [Google Scholar]
- King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R (2013). Glucocorticoid Repression of Inflammatory Gene Expression Shows Differential Responsiveness by Transactivation‐ and Transrepression‐Dependent Mechanisms. PLoS One 8: e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Mercado N, Miller‐Larsson A, Barnes PJ, Ito K (2012). Increased corticosteroid sensitivity by a long acting beta2 agonist formoterol via beta2 adrenoceptor independent protein phosphatase 2 A activation. Pulm Pharmacol Ther 25: 201–207. [DOI] [PubMed] [Google Scholar]
- Korhonen R, Lahti A, Hamalainen M, Kankaanranta H, Moilanen E (2002). Dexamethasone inhibits inducible nitric‐oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide‐treated macrophages. Mol Pharmacol 62: 698–704. [DOI] [PubMed] [Google Scholar]
- Korhonen R, Hommo T, Keranen T, Laavola M, Hamalainen M, Vuolteenaho K et al. (2013). Attenuation of TNF production and experimentally induced inflammation by PDE4 inhibitor rolipram is mediated by MAPK phosphatase‐1. Br J Pharmacol 169: 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SH, Wouters EF, Wesseling G, Arends JW, Thunnissen FB (1998). Interaction between glucocorticoids and beta2‐agonists: alpha and beta glucocorticoid‐receptor mRNA expression in human bronchial epithelial cells. Biochem Pharmacol 56: 1561–1569. [DOI] [PubMed] [Google Scholar]
- Korn SH, Jerre A, Brattsand R (2001). Effects of formoterol and budesonide on GM‐CSF and IL‐8 secretion by triggered human bronchial epithelial cells. Eur Respir J 17: 1070–1077. [DOI] [PubMed] [Google Scholar]
- Kunsch C, Lang RK, Rosen CA, Shannon MF (1994). Synergistic transcriptional activation of the IL‐8 gene by NF‐kappa B p65 (RelA) and NF‐IL‐6. J Immunol 153: 153–164. [PubMed] [Google Scholar]
- Lasa M, Brook M, Saklatvala J, Clark AR (2001). Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen‐activated protein kinase p38. Mol Cell Biol 21: 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR (2002). Dexamethasone causes sustained expression of mitogen‐activated protein kinase (MAPK) phosphatase 1 and phosphatase‐mediated inhibition of MAPK p38. Mol Cell Biol 22: 7802–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzon‐Joset JF, Langlois A, Lai LJ, Santerre K, Lee‐Gosselin A, Bosse Y et al. (2015). Lung CD200 Receptor Activation Abrogates Airway Hyperresponsiveness in Experimental Asthma. Am J Respir Cell Mol Biol 53: 276–284. [DOI] [PubMed] [Google Scholar]
- Le Bail O, Schmidt‐Ullrich R, Israel A (1993). Promoter analysis of the gene encoding the I kappa B‐alpha/MAD3 inhibitor of NF‐kappa B: positive regulation by members of the rel/NF‐kappa B family. EMBO J 12: 5043–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Machin M, Russell KE, Pavlidis S, Zhu J, Barnes PJ et al. (2016). Corticosteroid modulation of immunoglobulin expression and B‐cell function in COPD. FASEB J 30: 2014–2026. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ (2007). Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 190: 9–19. [DOI] [PubMed] [Google Scholar]
- Leigh R, Mostafa MM, King EM, Rider CF, Shah S, Dumonceaux C et al. (2016). An inhaled dose of budesonide induces genes involved in transcription and signaling in the human airways: enhancement of anti‐ and proinflammatory effector genes. Pharma Res Per 4: e00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis‐Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA (2007). Human glucocorticoid receptor beta binds RU‐486 and is transcriptionally active. Mol Cell Biol 27: 2266–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zhang M, Hussain F, Triantaphyllopoulos K, Clark AR, Bhavsar PK et al. (2011). Inhibition of p38 MAPK‐dependent bronchial contraction after ozone by corticosteroids. Eur Respir J 37: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Degan S, Theriot BS, Fischer BM, Strachan RT, Liang J et al. (2012). Chronic treatment in vivo with beta‐adrenoceptor agonists induces dysfunction of airway beta(2) ‐adrenoceptors and exacerbates lung inflammation in mice. Br J Pharmacol 165: 2365–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K (2012). Biased signaling pathways in beta2‐adrenergic receptor characterized by 19F‐NMR. Science 335: 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Q, Zhou X, Kolosov VP, Perelman JM (2013). Regulator of G‐protein signaling 2 inhibits acid‐induced mucin5AC hypersecretion in human airway epithelial cells. Respir Physiol Neurobiol 185: 265–271. [DOI] [PubMed] [Google Scholar]
- Liu YH, Wu SZ, Wang G, Huang NW, Liu CT (2015). A long‐acting beta2‐adrenergic agonist increases the expression of muscarine cholinergic subtype3 receptors by activating the beta2‐adrenoceptor cyclic adenosine monophosphate signaling pathway in airway smooth muscle cells. Mol Med Rep 11: 4121–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Svitacheva N, Jerre A, Miller‐Larsson A, Korn SH (2007). Anti‐inflammatory activity of beta2‐agonists in primary lung epithelial cells is independent of glucocorticoid receptor. Eur Respir J 30: 848–856. [DOI] [PubMed] [Google Scholar]
- Lowy MT (1989). Quantification of type I and II adrenal steroid receptors in neuronal, lymphoid and pituitary tissues. Brain Res 503: 191–197. [DOI] [PubMed] [Google Scholar]
- Mak JC, Roffel AF, Katsunuma T, Elzinga CR, Zaagsma J, Barnes PJ (2000). Up‐regulation of airway smooth muscle histamine H(1) receptor mRNA, protein, and function by beta(2)‐adrenoceptor activation. Mol Pharmacol 57: 857–864. [PubMed] [Google Scholar]
- Manetsch M, Ramsay EE, King EM, Seidel P, Che W, Ge Q et al. (2012). Corticosteroids and beta(2)‐agonists upregulate mitogen‐activated protein kinase phosphatase 1: in vitro mechanisms. Br J Pharmacol 166: 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetsch M, Rahman MM, Patel BS, Ramsay EE, Rumzhum NN, Alkhouri H et al. (2013). Long‐acting beta2‐agonists increase fluticasone propionate‐induced mitogen‐activated protein kinase phosphatase 1 (MKP‐1) in airway smooth muscle cells. PLoS One 8: e59635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T et al. (1993). Transcription factors NF‐IL6 and NF‐kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci U S A 90: 10193–10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001). Transcriptional regulation by the phosphorylation‐dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609. [DOI] [PubMed] [Google Scholar]
- Meltzer EO, Chervinsky P, Busse W, Ohta K, Bardin P, Bredenbroker D et al. (2015). Roflumilast for asthma: Efficacy findings in placebo‐controlled studies. Pulm Pharmacol Ther 35 (Suppl): S20–S27. [DOI] [PubMed] [Google Scholar]
- Mercier L, Thompson EB, Simons SS Jr (1983). Dissociation of steroid binding to receptors and steroid induction of biological activity in a glucocorticoid‐responsive cell. Endocrinology 112: 601–609. [DOI] [PubMed] [Google Scholar]
- Miller AH, Spencer RL, Pearce BD, Pisell TL, Azrieli Y, Tanapat P et al. (1998). Glucocorticoid receptors are differentially expressed in the cells and tissues of the immune system. Cell Immunol 186: 45–54. [DOI] [PubMed] [Google Scholar]
- Miner JN, Ardecky B, Benbatoul K, Griffiths K, Larson CJ, Mais DE et al. (2007). Antiinflammatory glucocorticoid receptor ligand with reduced side effects exhibits an altered protein–protein interaction profile. Proc Natl Acad Sci U S A 104: 19244–19249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misior AM, Deshpande DA, Loza MJ, Pascual RM, Hipp JD, Penn RB (2009). Glucocorticoid‐ and protein kinase A‐dependent transcriptome regulation in airway smooth muscle. Am J Respir Cell Mol Biol 41: 24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstadt PR, Ashwell JD (2001). Inhibition of AP‐1 by the glucocorticoid‐inducible protein GILZ. J Biol Chem 276: 29603–29610. [DOI] [PubMed] [Google Scholar]
- Miyata M, Lee JY, Susuki‐Miyata S, Wang WY, Xu H, Kai H et al. (2015). Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK‐M. Nat Commun 6: 6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley T, Wilson SM, Joshi T, Rider CF, Sharma P, Yan D et al. (2013). Phosphodiesterase 4 inhibitors augment the ability of formoterol to enhance glucocorticoid‐dependent gene transcription in human airway epithelial cells: a novel mechanism for the clinical efficacy of roflumilast in severe chronic obstructive pulmonary disease. Mol Pharmacol 83: 894–906. [DOI] [PubMed] [Google Scholar]
- Mortaz E, Rad MV, Johnson M, Raats D, Nijkamp FP, Folkerts G (2008). Salmeterol with fluticasone enhances the suppression of IL‐8 release and increases the translocation of glucocorticoid receptor by human neutrophils stimulated with cigarette smoke. J Mol Med 86: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer ML, Borror KC, Bona BJ, DeFranco DB, Nordeen SK (1993). Modulation of cell signaling pathways can enhance or impair glucocorticoid‐induced gene expression without altering the state of receptor phosphorylation. J Biol Chem 268: 22933–22940. [PubMed] [Google Scholar]
- Mukaida N, Mahe Y, Matsushima K (1990). Cooperative interaction of nuclear factor‐kappa B‐ and cis‐regulatory enhancer binding protein‐like factor binding elements in activating the interleukin‐8 gene by pro‐inflammatory cytokines. J Biol Chem 265: 21128–21133. [PubMed] [Google Scholar]
- Munoz NM, Rabe KF, Vita AJ, McAllister K, Mayer D, Weiss M et al. (1995). Paradoxical blockade of beta adrenergically mediated inhibition of stimulated eosinophil secretion by salmeterol. J Pharmacol Exp Ther 273: 850–854. [PubMed] [Google Scholar]
- Nannini LJ, Poole P, Milan SJ, Kesterton A (2013). Combined corticosteroid and long‐acting beta(2)‐agonist in one inhaler versus inhaled corticosteroids alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 8: CD006826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C (2007). The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol 17: 318–324. [DOI] [PubMed] [Google Scholar]
- Newton R (2000). Molecular mechanisms of glucocorticoid action: what is important? Thorax 55: 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R (2014). Anti‐inflammatory glucocorticoids: changing concepts. Eur J Pharmacol 724: 231–236. [DOI] [PubMed] [Google Scholar]
- Newton R, Holden NS (2007). Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol 72: 799–809. [DOI] [PubMed] [Google Scholar]
- Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ (1998). Repression of cyclooxygenase‐2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post‐transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem 273: 32312–32321. [DOI] [PubMed] [Google Scholar]
- Newton R, King EM, Gong W, Rider CF, Staples KJ, Holden NS et al. (2010a). Glucocorticoids inhibit IL‐1beta‐induced GM‐CSF expression at multiple levels: roles for the ERK pathway and repression by MKP‐1. Biochem J 427: 113–124. [DOI] [PubMed] [Google Scholar]
- Newton R, Leigh R, Giembycz MA (2010b). Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol Ther 125: 286–327. [DOI] [PubMed] [Google Scholar]
- Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ et al. (2009). Beta2‐adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci U S A 106: 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El‐Mays T et al. (2012). beta‐Arrestin‐2 mediates the proinflammatory effects of proteinase‐activated receptor‐2 in the airway. Proc Natl Acad Sci U S A 109: 16660–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Knox AJ, Pang L (2005). beta2‐Adrenoceptor agonists, like glucocorticoids, repress eotaxin gene transcription by selective inhibition of histone H4 acetylation. J Immunol 175: 478–486. [DOI] [PubMed] [Google Scholar]
- Niehof M, Manns MP, Trautwein C (1997). CREB controls LAP/C/EBP beta transcription. Mol Cell Biol 17: 3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehof M, Kubicka S, Zender L, Manns MP, Trautwein C (2001). Autoregulation enables different pathways to control CCAAT/enhancer binding protein beta (C/EBP beta) transcription. J Mol Biol 309: 855–868. [DOI] [PubMed] [Google Scholar]
- Nino G, Hu A, Grunstein JS, Grunstein MM (2010). Mechanism of glucocorticoid protection of airway smooth muscle from proasthmatic effects of long‐acting beta2‐adrenoceptor agonist exposure. J Allergy Clin Immunol 125: 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordeen SK, Bona BJ, Moyer ML (1993). Latent agonist activity of the steroid antagonist, RU486, is unmasked in cells treated with activators of protein kinase A. Mol Endocrinol 7: 731–742. [DOI] [PubMed] [Google Scholar]
- O'Byrne PM, Barnes PJ, Rodriguez‐Roisin R, Runnerstrom E, Sandstrom T, Svensson K et al. (2001). Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 164: 1392–1397. [DOI] [PubMed] [Google Scholar]
- Oh KJ, Han HS, Kim MJ, Koo SH (2013). CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep 46: 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onaran HO, Costa T (1997). Agonist efficacy and allosteric models of receptor action. Ann N Y Acad Sci 812: 98–115. [DOI] [PubMed] [Google Scholar]
- Pace E, Gagliardo R, Melis M, La GS, Ferraro M, Siena L et al. (2004). Synergistic effects of fluticasone propionate and salmeterol on in vitro T‐cell activation and apoptosis in asthma. J Allergy Clin Immunol 114: 1216–1223. [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ (2000). Synergistic inhibition by beta(2)‐agonists and corticosteroids on tumor necrosis factor‐alpha‐induced interleukin‐8 release from cultured human airway smooth‐muscle cells. Am J Respir Cell Mol Biol 23: 79–85. [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ (2001). Regulation of TNF‐alpha‐induced eotaxin release from cultured human airway smooth muscle cells by beta2‐agonists and corticosteroids. FASEB J 15: 261–269. [DOI] [PubMed] [Google Scholar]
- Pantaloni C, Brabet P, Bilanges B, Dumuis A, Houssami S, Spengler D et al. (1996). Alternative splicing in the N‐terminal extracellular domain of the pituitary adenylate cyclase‐activating polypeptide (PACAP) receptor modulates receptor selectivity and relative potencies of PACAP‐27 and PACAP‐38 in phospholipase C activation. J Biol Chem 271: 22146–22151. [DOI] [PubMed] [Google Scholar]
- Park EA, Roesler WJ, Liu J, Klemm DJ, Gurney AL, Thatcher JD et al. (1990). The role of the CCAAT/enhancer‐binding protein in the transcriptional regulation of the gene for phosphoenolpyruvate carboxykinase (GTP). Mol Cell Biol 10: 6264–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EA, Gurney AL, Nizielski SE, Hakimi P, Cao Z, Moorman A et al. (1993). Relative roles of CCAAT/enhancer‐binding protein beta and cAMP regulatory element‐binding protein in controlling transcription of the gene for phosphoenolpyruvate carboxykinase (GTP). J Biol Chem 268: 613–619. [PubMed] [Google Scholar]
- Pauwels RA, Lofdahl CG, Postma DS, Tattersfield AE, O'Byrne P, Barnes PJ et al. (1997). Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (FACET) International Study Group. N Engl J Med 337: 1405–1411. [DOI] [PubMed] [Google Scholar]
- Penn RB, Bond RA, Walker JK (2014). GPCRs and arrestins in airways: implications for asthma. Handb Exp Pharmacol 219: 387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperl DJ, Shah‐Basu S, VanLeeuwen D, Granneman JG, MacKenzie RG (1998). Regulation of RGS mRNAs by cAMP in PC12 cells. Biochem Biophys Res Commun 243: 52–55. [DOI] [PubMed] [Google Scholar]
- Pera T, Penn RB (2016). Bronchoprotection and bronchorelaxation in asthma: New targets, and new ways to target the old ones. Pharmacol Ther 164: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, Salmon M (2012). Bifunctional compounds for the treatment of COPD. Ann Rev Med Chem 472: 209–221. [Google Scholar]
- Pinart M, Hussain F, Shirali S, Li F, Zhu J, Clark AR et al. (2014). Role of mitogen‐activated protein kinase phosphatase‐1 in corticosteroid insensitivity of chronic oxidant lung injury. Eur J Pharmacol 744: 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli V (1998). The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem 273: 29279–29282. [DOI] [PubMed] [Google Scholar]
- Prabhala P, Ammit AJ (2015). Tristetraprolin and its role in regulation of airway inflammation. Mol Pharmacol 87: 629–638. [DOI] [PubMed] [Google Scholar]
- Quante T, Ng YC, Ramsay EE, Henness S, Allen JC, Parmentier J et al. (2008). Corticosteroids reduce IL‐6 in ASM cells via up‐regulation of MKP‐1. Am J Respir Cell Mol Biol 39: 208–217. [DOI] [PubMed] [Google Scholar]
- Rabe KF, Giembycz MA, Dent G, Perkins RS, Evans P, Barnes PJ (1993). Salmeterol is a competitive antagonist at beta‐adrenoceptors mediating inhibition of respiratory burst in guinea‐pig eosinophils. Eur J Pharmacol 231: 305–308. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen‐MacDonald W, Lam CM, Dewire SM et al. (2011). Quantifying ligand bias at seven‐transmembrane receptors. Mol Pharmacol 80: 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan PN, Umesono K, Evans RM (1992). Modulation of glucocorticoid receptor function by protein kinase A. Mol Endocrinol 6: 1451–1457. [DOI] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ et al. (2009). Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA (2005). Antiinflammatory action of glucocorticoids‐‐new mechanisms for old drugs. N Engl J Med 353: 1711–1723. [DOI] [PubMed] [Google Scholar]
- Rider CF, King EM, Holden NS, Giembycz MA, Newton R (2011). Inflammatory stimuli inhibit glucocorticoid‐dependent transactivation in human pulmonary epithelial cells: Rescue by long‐acting beta2‐adrenoceptor agonists. J Pharmacol Exp Ther 338: 860–869. [DOI] [PubMed] [Google Scholar]
- Rider CF, Miller‐Larsson A, Giembycz MA, Newton R (2015a). Long‐acting beta2‐adrenoceptor agonists enhance glucocorticoid receptor‐inducible gene expression via a rapidly acting pathway without affecting glucocorticoid receptor expression or translocation. Am J Respir Crit Care Med 191: A5026. [Google Scholar]
- Rider CF, Shah S, Miller‐Larsson A, Giembycz MA, Newton R (2015b). Cytokine‐induced loss of glucocorticoid function: effect of kinase inhibitors, long‐acting beta2‐adrenoceptor agonist and glucocorticoid receptor ligands. PLoS One 10: e0116773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Hla T (1996). Down‐regulation of cytokine‐induced cyclo‐oxygenase‐2 transcript isoforms by dexamethasone: evidence for post‐transcriptional regulation. Biochem J 318: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson S, Rohwer JM, Hapgood JP, Louw A (2013). Impact of glucocorticoid receptor density on ligand‐independent dimerization, cooperative ligand‐binding and basal priming of transactivation: a cell culture model. PLoS One 8: e64831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C, Zhang J, Newton GK, Perrior TR (2013). Nonhuman targets in allergic lung conditions. Future Med Chem 5: 147–161. [DOI] [PubMed] [Google Scholar]
- Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS Jr, Louw A et al. (2009). Ligand‐selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol 299: 219–231. [DOI] [PubMed] [Google Scholar]
- Roth M, Johnson PR, Rudiger JJ, King GG, Ge Q, Burgess JK et al. (2002). Interaction between glucocorticoids and beta2 agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet 360: 1293–1299. [DOI] [PubMed] [Google Scholar]
- Rowan BG, Garrison N, Weigel NL, O'Malley BW (2000). 8‐Bromo‐cyclic AMP induces phosphorylation of two sites in SRC‐1 that facilitate ligand‐independent activation of the chicken progesterone receptor and are critical for functional cooperation between SRC‐1 and CREB binding protein. Mol Cell Biol 20: 8720–8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Nishizawa Y, Nishimura A, Chiba Y, Goto K, Hanazaki M et al. (2010). Angiotensin II induces hyperresponsiveness of bronchial smooth muscle via an activation of p42/44 ERK in rats. Pflugers Arch 460: 645–655. [DOI] [PubMed] [Google Scholar]
- Salmon M, Tannheimer SL, Gentzler TT, Cui ZH, Sorensen EA, Hartsough KC et al. (2014). The in vivo efficacy and side effect pharmacology of GS‐5759, a novel bifunctional phosphodiesterase 4 inhibitor and long‐acting beta 2‐adrenoceptor agonist in preclinical animal species. Pharmacol Res Perspect 2: e00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson MK, Pazirandeh A, Davani B, Okret S (1999). p57Kip2, a glucocorticoid‐induced inhibitor of cell cycle progression in HeLa cells. Mol Endocrinol 13: 1811–1822. [DOI] [PubMed] [Google Scholar]
- Sands WA, Palmer TM (2008). Regulating gene transcription in response to cyclic AMP elevation. Cell Signal 20: 460–466. [DOI] [PubMed] [Google Scholar]
- Sarir H, Mortaz E, Karimi K, Johnson M, Nijkamp FP, Folkerts G (2007). Combination of fluticasone propionate and salmeterol potentiates the suppression of cigarette smoke‐induced IL‐8 production by macrophages. Eur J Pharmacol 571: 55–61. [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, Cidlowski JA (2003). Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol Cell Biol 23: 1922–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf MJ, Lewis‐Tuffin LJ, Cidlowski JA (2005). Ligand‐selective targeting of the glucocorticoid receptor to nuclear subdomains is associated with decreased receptor mobility. Mol Endocrinol 19: 1501–1515. [DOI] [PubMed] [Google Scholar]
- Schacke H, Docke WD, Asadullah K (2002). Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96: 23–43. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS Jr (1995). Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 270: 283–286. [DOI] [PubMed] [Google Scholar]
- Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R (2002). RU486‐induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem 277: 26238–26243. [DOI] [PubMed] [Google Scholar]
- Sears MR (2011). The addition of long‐acting beta‐agonists to inhaled corticosteroids in asthma. Curr Opin Pulm Med 17: 23–28. [DOI] [PubMed] [Google Scholar]
- Seifert R (2013). Functional selectivity of G‐protein‐coupled receptors: from recombinant systems to native human cells. Biochem Pharmacol 86: 853–861. [DOI] [PubMed] [Google Scholar]
- Shah S, King EM, Chandrasekhar A, Newton R (2014). Roles for the mitogen‐activated protein kinase (MAPK) phosphatase, DUSP1, in feedback control of inflammatory gene expression and repression by dexamethasone. J Biol Chem 289: 13667–13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Mostafa MM, McWhae A, Traves SL, Newton R (2016). Negative feed‐forward control of tumor necrosis factor (TNF) by tristetraprolin (ZFP36) is limited by the mitogen‐activated protein kinase phosphatase, dual‐specificity phosphatase 1 (DUSP1): implications for regulation by glucocorticoids. J Biol Chem 291: 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S et al. (2006). beta‐arrestin‐dependent, G protein‐independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281: 1261–1273. [DOI] [PubMed] [Google Scholar]
- Shipp LE, Lee JV, Yu CY, Pufall M, Zhang P, Scott DK et al. (2010). Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS One 5: e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrewsbury S, Pyke S, Britton M (2000). Meta‐analysis of increased dose of inhaled steroid or addition of salmeterol in symptomatic asthma (MIASMA). BMJ 320: 1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri M, Fregonese L, Sabatini F, Dasic G, Rossi GA (2001). Fluticasone and salmeterol downregulate in vitro, fibroblast proliferation and ICAM‐1 or H‐CAM expression. Eur Respir J 18: 139–145. [DOI] [PubMed] [Google Scholar]
- Simons SS Jr (2006). How much is enough? Modulation of dose–response curve for steroid receptor‐regulated gene expression by changing concentrations of transcription factor. Curr Top Med Chem 6: 271–285. [DOI] [PubMed] [Google Scholar]
- Simons SS Jr (2010). Glucocorticoid receptor cofactors as therapeutic targets. Curr Opin Pharmacol 10: 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skevaki CL, Christodoulou I, Spyridaki IS, Tiniakou I, Georgiou V, Xepapadaki P et al. (2009). Budesonide and formoterol inhibit inflammatory mediator production by bronchial epithelial cells infected with rhinovirus. Clin Exp Allergy 39: 1700–1710. [DOI] [PubMed] [Google Scholar]
- Smoak K, Cidlowski JA (2006). Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol 26: 9126–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L et al. (2008). A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol 9: 1074–1083. [DOI] [PubMed] [Google Scholar]
- So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR (2007). Determinants of Cell‐ and Gene‐Specific Transcriptional Regulation by the Glucocorticoid Receptor. PLoS Genet 3: e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Cooper SB, Feldman BJ, Manuchehri M, Yamamoto KR (2008). Conservation analysis predicts in vivo occupancy of glucocorticoid receptor‐binding sequences at glucocorticoid‐induced genes. Proc Natl Acad Sci U S A 105: 5745–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Altarejos J, Goodarzi MO, Inoue H, Guo X, Berdeaux R et al. (2010). CRTC3 links catecholamine signalling to energy balance. Nature 468: 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer RL, Miller AH, Stein M, McEwen BS (1991). Corticosterone regulation of type I and type II adrenal steroid receptors in brain, pituitary, and immune tissue. Brain Res 549: 236–246. [DOI] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH et al. (1993). Differential signal transduction by five splice variants of the PACAP receptor. Nature 365: 170–175. [DOI] [PubMed] [Google Scholar]
- Spoelstra FM, Postma DS, Hovenga H, Noordhoek JA, Kauffman HF (2002). Additive anti‐inflammatory effect of formoterol and budesonide on human lung fibroblasts. Thorax 57: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellato C (2004). Post‐transcriptional and nongenomic effects of glucocorticoids. Proc Am Thorac Soc 1: 255–263. [DOI] [PubMed] [Google Scholar]
- Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM (2015). Selective glucocorticoid receptor modulation: New directions with non‐steroidal scaffolds. Pharmacol Ther 152: 28–41. [DOI] [PubMed] [Google Scholar]
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D et al. (2011). Widespread negative response elements mediate direct repression by agonist‐liganded glucocorticoid receptor. Cell 145: 224–241. [DOI] [PubMed] [Google Scholar]
- Szapary D, Huang Y, Simons SS Jr (1999). Opposing effects of corepressor and coactivators in determining the dose–response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor‐regulated gene expression. Mol Endocrinol 13: 2108–2121. [DOI] [PubMed] [Google Scholar]
- Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM (2007). Beta2 adrenergic receptor activation stimulates pro‐inflammatory cytokine production in macrophages via PKA‐ and NF‐kappaB‐independent mechanisms. Cell Signal 19: 251–260. [DOI] [PubMed] [Google Scholar]
- Tanigawa K, Tanaka K, Nagase H, Miyake H, Kiniwa M, Ikizawa K (2002). Cell type‐dependent divergence of transactivation by glucocorticoid receptor ligand. Biol Pharm Bull 25: 1619–1622. [DOI] [PubMed] [Google Scholar]
- Tannheimer SL, Sorensen EA, Cui ZH, Kim M, Patel L, Baker WR et al. (2014). The in vitro pharmacology of GS‐5759, a novel bifunctional phosphodiesterase 4 inhibitor and long acting beta2‐adrenoceptor agonist. J Pharmacol Exp Ther 349: 85–93. [DOI] [PubMed] [Google Scholar]
- Tattersfield AE (2006). Current issues with beta2‐adrenoceptor agonists: historical background. Clin Rev Allergy Immunol 31: 107–118. [DOI] [PubMed] [Google Scholar]
- Tchen CR, Martins JR, Paktiawal N, Perelli R, Saklatvala J, Clark AR (2010). Glucocorticoid regulation of mouse and human dual specificity phosphatase 1 (DUSP1) genes: unusual cis‐acting elements and unexpected evolutionary divergence. J Biol Chem 285: 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala VJ, Forkuo GS, Al‐Sawalha N, Azzegagh Z, Nguyen LP, Eriksen JL et al. (2013). beta2‐Adrenoceptor agonists are required for development of the asthma phenotype in a murine model. Am J Respir Cell Mol Biol 48: 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T, Fernandes DJ, Schuliga M, Harris T, Landells L, Stewart AG (2005). Stimulus‐dependent glucocorticoid‐resistance of GM‐CSF production in human cultured airway smooth muscle. Br J Pharmacol 145: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein C, van der Geer P, Karin M, Hunter T, Chojkier M (1994). Protein kinase A and C site‐specific phosphorylations of LAP (NF‐IL6) modulate its binding affinity to DNA recognition elements. J Clin Invest 93: 2554–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsingotjidou A, Nervina JM, Pham L, Bezouglaia O, Tetradis S (2002). Parathyroid hormone induces RGS‐2 expression by a cyclic adenosine 3′,5′‐monophosphate‐mediated pathway in primary neonatal murine osteoblasts. Bone 30: 677–684. [DOI] [PubMed] [Google Scholar]
- Tsukada J, Yoshida Y, Kominato Y, Auron PE (2011). The CCAAT/enhancer (C/EBP) family of basic‐leucine zipper (bZIP) transcription factors is a multifaceted highly regulated system for gene regulation. Cytokine 54: 6–19. [DOI] [PubMed] [Google Scholar]
- Uings IJ, Needham D, Matthews J, Haase M, Austin R, Angell D et al. (2013). Discovery of GW870086: a potent anti‐inflammatory steroid with a unique pharmacological profile. Br J Pharmacol 169: 1389–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani OS, Ito K, Maneechotesuwan K, Ito M, Johnson M, Barnes PJ et al. (2005). Glucocorticoid receptor nuclear translocation in airway cells after inhaled combination therapy. Am J Respir Crit Care Med 172: 704–712. [DOI] [PubMed] [Google Scholar]
- Vaine CA, Soberman RJ (2014). The CD200‐CD200R1 inhibitory signaling pathway: immune regulation and host‐pathogen interactions. Adv Immunol 121: 191–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M (2014). Quantification of ligand bias for clinically relevant beta2‐adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85: 492–509. [DOI] [PubMed] [Google Scholar]
- Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR (1987). Intracellular receptor concentration limits glucocorticoid‐dependent enhancer activity. Mol Endocrinol 1: 68–74. [DOI] [PubMed] [Google Scholar]
- Vasarhelyi V, Trexler M, Patthy L (2014). Both LCCL‐domains of human CRISPLD2 have high affinity for lipid A. Biochimie 97: 66–71. [DOI] [PubMed] [Google Scholar]
- Vasudevan NT, Mohan ML, Goswami SK, Naga Prasad SV (2011). Regulation of beta‐adrenergic receptor function: an emphasis on receptor resensitization. Cell Cycle 10: 3684–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vayssiere BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C et al. (1997). Synthetic glucocorticoids that dissociate transactivation and AP‐1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol 11: 1245–1255. [DOI] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F (2004). Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem 279: 8886–8894. [DOI] [PubMed] [Google Scholar]
- Volonaki E, Psarras S, Xepapadaki P, Psomali D, Gourgiotis D, Papadopoulos NG (2006). Synergistic effects of fluticasone propionate and salmeterol on inhibiting rhinovirus‐induced epithelial production of remodelling‐associated growth factors. Clin Exp Allergy 36: 1268–1273. [DOI] [PubMed] [Google Scholar]
- Wade KC, Guttentag SH, Gonzales LW, Maschhoff KL, Gonzales J, Kolla V et al. (2006). Gene induction during differentiation of human pulmonary type II cells in vitro. Am J Respir Cell Mol Biol 34: 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BL, Pollio G, Leonhardt S, Wani MC, Lee DY, Imhof MO et al. (1996). 16 alpha‐substituted analogs of the antiprogestin RU486 induce a unique conformation in the human progesterone receptor resulting in mixed agonist activity. Proc Natl Acad Sci U S A 93: 8739–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JK, DeFea KA (2014). Role for beta‐arrestin in mediating paradoxical beta2AR and PAR2 signaling in asthma. Curr Opin Pharmacol 16: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA et al. (2003). Beta‐arrestin‐2 regulates the development of allergic asthma. J Clin Invest 112: 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JK, Penn RB, Hanania NA, Dickey BF, Bond RA (2011). New perspectives regarding beta(2) ‐adrenoceptor ligands in the treatment of asthma. Br J Pharmacol 163: 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR (2004). Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A 101: 15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Xing WM, Fan HH, Wang KS, Zhang HK, Wang QW et al. (2009). The novel lipopolysaccharide‐binding protein CRISPLD2 is a critical serum protein to regulate endotoxin function. J Immunol 183: 6646–6656. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Shen P, Rider CF, Traves SL, Proud D, Newton R et al. (2009). Selective prostacyclin receptor agonism augments glucocorticoid‐induced gene expression in human bronchial epithelial cells. J Immunol 183: 6788–6799. [DOI] [PubMed] [Google Scholar]
- Wu I, Shin SC, Cao Y, Bender IK, Jafari N, Feng G et al. (2013). Selective glucocorticoid receptor translational isoforms reveal glucocorticoid‐induced apoptotic transcriptomes. Cell Death Dis 4: e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Liu D, Liu S, Calderon L, Zhao G, Turk J et al. (2011). Identification of a cAMP‐response element in the regulator of G‐protein signaling‐2 (RGS2) promoter as a key cis‐regulatory element for RGS2 transcriptional regulation by angiotensin II in cultured vascular smooth muscles. J Biol Chem 286: 44646–44658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Jiang H, Nguyen H, Jia S, Berro A, Panettieri RA Jr et al. (2012). Regulator of G protein signaling 2 is a key modulator of airway hyperresponsiveness. J Allergy Clin Immunol 130: 968–976. [DOI] [PubMed] [Google Scholar]
- Yamaya M, Nishimura H, Nadine L, Kubo H, Nagatomi R (2014). Formoterol and budesonide inhibit rhinovirus infection and cytokine production in primary cultures of human tracheal epithelial cells. Respir Investig 52: 251–260. [DOI] [PubMed] [Google Scholar]
- Yeagley D, Quinn PG (2005). 3′,5′‐cyclic adenosine monophosphate response element‐binding protein and CCAAT enhancer‐binding protein are dispensable for insulin inhibition of phosphoenolpyruvate carboxykinase transcription and for its synergistic induction by protein kinase A and glucocorticoids. Mol Endocrinol 19: 913–924. [DOI] [PubMed] [Google Scholar]
- Yin F, Wang YY, Du JH, Li C, Lu ZZ, Han C et al. (2006). Noncanonical cAMP pathway and p38 MAPK mediate beta2‐adrenergic receptor‐induced IL‐6 production in neonatal mouse cardiac fibroblasts. J Mol Cell Cardiol 40: 384–393. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li YC, Young AP (1993). Protein kinase A activation of glucocorticoid‐mediated signaling in the developing retina. Proc Natl Acad Sci U S A 90: 3880–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Truong‐Tran QA, Tancowny B, Harris KE, Schleimer RP (2007a). Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol 179: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Jonklaas J, Danielsen M (2007b). The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids 72: 600–608. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Pang J, Favata MF, Trzaskos JM (2003). Receptor density dictates the behavior of a subset of steroid ligands in glucocorticoid receptor‐mediated transrepression. Int Immunopharmacol 3: 1803–1817. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang Y, Guo Y, Zhang Y, Xu M, He B (2014). beta2‐Adrenoceptor involved in smoking‐induced airway mucus hypersecretion through beta‐arrestin‐dependent signaling. PLoS One 9: e97788. [DOI] [PMC free article] [PubMed] [Google Scholar]