

Abstract
Tumor cell survival in the hostile distant organ is a rate-limiting step in cancer metastasis. Bone marrow-derived myeloid cells can form a premetastatic niche and provide a tumor–promoting microenvironment. However, it is unclear whether these myeloid cells in the premetastatic site have any direct effect on tumor cell survival. Here we report that chemokine CCL9 was highly induced in Gr-1+CD11b+ immature myeloid cells and in premetastatic lung in tumor-bearing mice. Knockdown of CCL9 in myeloid cells decreased tumor cell survival and metastasis. Importantly, CCL9 overexpression in myeloid cells lacking TGF-β signaling rescued the tumor metastasis defect observed in mice with myeloid-specific Tgfbr2 deletion. The expression level of CCL23, the human orthologue for CCL9, in peripheral blood mononuclear cells correlated with progression and survival of cancer patients. Our study demonstrates that CCL9 could serve as a good candidate for anti-metastasis treatment by targeting the rate-limiting step of cancer cell survival. Additionally, targeting CCL9 may avoid the adverse effects of TGF-β-targeted therapy.
Keywords: metastasis, TGF-β, cytokines, myeloid cells, tumor microenvironment, breast cancer
Introduction
Tumor cell survival is a rate-limiting step in cancer metastasis (1). The majority of the cancer cells die in the peripheral blood and distant hostile organs during the multiple-step processes including invasion, intravasation, extravasation, and colonization (1). Recent evidence strongly suggests a critical role of host cells and tumor microenvironment in supporting tumor cell survival and metastasis. One mechanism is through formation of premetastatic niche (2, 3). Myeloid cells play a critical role in this process (2, 4). These include VEGFR1 myeloid progenitor cells (5), Mac-1+ myeloid cells (6), and Gr-1+CD11b+ myeloid derived suppressor cells (MDSCs) (7) as well as Ly6G+Ly6C+ granulocytes (8). Gr-1+CD11b+ immature myeloid cells constitute the majority of the immature myeloid cells under tumor conditions. They are heterogeneous immature myeloid cells that consist of Ly6G+CD11b+ granulocytes, Ly6C+CD11b+ monocytes and F4/80+CD11b+ macrophages (9). Gr-1+CD11b+ cells are over-produced in cancer patients, negatively regulate host anti-tumor immunity (10-12), and promote tumor angiogenesis (13). These immature myeloid cells are also found in premetastatic organ of tumor-bearing mice to modulate host inflammatory/immune microenvironment (7, 14) including NK (Natural Killer) cell activities (15, 16) and promoting mesenchymal to epithelial transition of tumor cells (17). However, it is not known whether these myeloid cells can directly support tumor cell survival in the premetastatic organs.
One important molecular mechanism underlying metastasis-promoting function of myeloid cells is myeloid-specific TGF-β signaling (18). TGF-β is a pleiotropic molecule and it possesses both tumor suppressor and tumor promoter functions (19, 20). The mechanisms underlying the dual role of TGF-β are very intricate and are poorly understood. Recent work reveals that TGF-β regulates production of chemokine/chemokine receptors in tumor cells that are important for inflammatory cell recruitment in the tumor microenvironment (21-23). This includes stromal derived factor 1 (SDF-1 or CXCL12) (24) and CCL9 (25). CXCL12 mediates its effects through CXCR4, a receptor that is highly expressed on putative stem and progenitor cells (26, 27). CCL9 signals through CCR1+ and recruits myeloid progenitor cells for tumor cell invasion (25). In addition, TGF-β suppresses CXCL1 and CXCL5, and deletion of TGF-β signaling in cancer cells significantly increases the expression of both chemokines (21). These chemokines are responsible for the recruitment of Gr-1+CD11b+ myeloid cells to the tumor microenvironment (21), where they produce large quantities of matrix metalloproteases (21). A chemokine signature generated due to a loss of TGF-β signaling correlates with poor prognosis of human breast cancer (22).
CCL9, a chemokine also known as macrophage inflammatory protein-1 gamma (MIP-1γ) (25, 28-30), is often produced in macrophages, osteoclasts, and functions as a cell survival factor (31, 32). CCL9/CCR1 signaling has been shown to be important for the recruitment of myeloid progenitors to intestinal tumors, leading to enhanced invasion (25, 30). However, it remains to be determined if CCL9 promotes cancer cell survival, and if TGF-β regulates CCL9 production. Here we report that CCL9 is highly induced in immature myeloid cells in the premetastatic lung of tumor-bearing mice or in co-culture with tumor cells, and it plays a critical role in tumor cell survival at the metastatic site. We further identify CCL9 as a critical mediator of the metastasis-promoting effects of myeloid-specific TGF-β signaling. Our study provides insight for understanding TGF-β cancer biology and suggests a new target for cancer treatment.
Materials and Methods
Cells and Mice
Murine mammary tumor cell lines 67NR, 168FARN, 4T07, 4T1, 67NR-GFP, 4T1-GFP, 4T1-luciferase and melanoma B16F1, B16BL6, B16F10, murine macrophage cell line RAW264.7 and neutrophil cell line 32DCl3, as well as human breast tumor cell line MDA-MB-231 and human melanoma cell lines A375S2, SK-MEL-28 were maintained per standard cell culture techniques. Cells labeled with GFP were obtained through pSico-GFP virus infection as published (42) and followed by GFP+ cell sorting. B16F1 and B16F10 were provided by Drs. Glenn Merlino and Shioko Kimura, NCI. All Gr-1+CD11b+ cells were sorted through FACS (fluorescence activated cell sorting) or MACS (magnetic activated cell sorting) from spleens of tumor-bearing wild type, Tgfbr2MyeKO and Tgfbr2Flox mice unless otherwise specified. Tgfbr2MyeKO and Tgfbr2Flox control mice were produced as previously described (18). P38 dominant negative mice are precious gifts from Dr. Albert J. Fornace Jr (Georgetown University). All animal studies are approved by the National Cancer Institute Animal Care and Use Committee.
Spontaneous and Experimental Metastasis
For orthotopic metastasis, mammary tumor 4T1 cells (5×105) were injected into the #2 mammary fat pad (MFP); B16F10 melanoma cells (1×106) were injected subcutaneously. For experimental metastasis, 67NR-control/67NR-CCR1 cells (5 or 10×105), 4T1 cells (5×105), B16F1-vec control/B16F1-CCR1 cells (10×105) were injected into the tail vein (TVI) of the recipient mice. For co-injection experiment, the tumor cells were injected with Gr-1+CD11b+ (1.5×106) or RAW264.7 cells (2×105). Tumor size was measured twice a week using calipers and the tumor volume was calculated as Volume= Length × (Width)2. The number of lung metastasis was evaluated using whole lung mounting procedure as described (73) or by H&E staining of butterfly sections of lungs. One butterfly section for each mouse was obtained, and a total of at least 5 mice were evaluated for each experimental group.
In Vivo Bioluminascence Imaging
Mice were received an intraperitoneal injection of D-luciferin (Caliper Life Sciences, 100ul of 10mg/ml) 15 minutes before image. Luminescent signals were detected using an IVIS-SPECTRUM in vivo photon-counting device (Caliper Life Sciences). Images were quantified as photon counts/second using the Living Image software (Caliper Life Sciences).
Single Cell Videomicroscopy (SCVM) for Evaluating Tumor Cell Survival
Mice were euthanized and lungs were removed 6 hours after tail vein injection of GFP labeled tumor cells (5×105) together with Gr-1+CD11b+ (1.5×106) or RAW264.7 cells (2×105). Images were normalized to the basal signal obtained from lungs 1 hour after tail vein injection. Lungs were observed under the fluorescent microscope, 10 random pictures under 10X magnification were taken and further analyzed as fluorescence signal per field by OpenLab software (Improvision) or ImageJ. For extravasation experiment, mice were injected with 100ul of 12.5ug/ul 70,000MW tetramethylrhodamine (Invitrogen). Lungs were imaged under Zeiss 510 NLO confocal microscope. Images were analyzed with Zeiss ZEN software.
Ex vivo Pulmonary Metastasis Assay (PuMA)
GFP-labeled tumor cells (5×105) were co-injected with sorted Gr-1+CD11b+ cells (1.5×106) or RAW264.7 cells (2×105) through the tail vein. Mice were euthanized 5 minutes after injection, and the lungs were infused with an agarose medium mixture as described (40). Lung sections (1-2mm thick) were placed on Gelfoam (Pfizer-Pharmacia & Upjohn Co.) for culture for 1-2 weeks. LEICA-DM IRB fluorescent inverted microscope (Leica) and Retiga-EXi Fast 1394 Mono Cooled CCD camera (QImaging) were used to capture GFP positive cells at 10 × or 2.5 × magnification. The GFP fluorescence pixels were obtained and analyzed using OpenLab software (Improvision) or ImageJ (40). The fluorescence intensity per field was quantified and normalized to day 0 signal and presented as metastasis survival index. Three to six lung sections for each mouse, and a total of 3-4 mice were evaluated for each experimental group.
Flow Cytometry and Cell Sorting
Single cell suspensions were made from spleens or peripheral blood of normal and 4T1 tumor-bearing mice (13), as well as lung tissues (74). Cells were labeled with fluorescence-conjugated antibodies: Gr-1, CD11b, Ly6G, Ly6C, F4/80, AnnexinV, 7AAD (BD Pharmingen), and CCR1 (R&D system). For flow cytometry analysis, cells were run on a FACS Calibur or Fortessa flow cytometer (BD, San Jose, CA) and analyzed on FlowJo. For sorting, Gr-1+CD11b+ cells, CD11b+Ly6G+ cells, CD11b+Ly6C+ cells, and CD11b+F4/80+ cells were sorted from spleens of 4T1 tumor-bearing mice by FACSAria flow cytometer (BD) or MACS (Magnetic-activated cell sorting) according to manufacturer protocol (Miltenyi Biotec). For sorting human CD33+ myeloid cells, normal human whole blood was obtained from NIH blood bank in clinical center. Myeloid cells were enriched by Ficoll-Paque™ (GE Healthcare), then labeled with CD33 antibody and sorted with MACS (Miltenyi Biotec).
Immunofluorescence (IF) Staining and TUNEL Assay
Paraffin-embedded lung sections or chamber slides with tumor cell culture were incubated with primary antibodies for GFP (Santa Cruz) or PAR (BD Pharmingen). Alexa flour 488 or 594 secondary antibodies were used for detection (Invitrogen). For TUNEL (Roche Applied Science) assay, lungs were taken out 6 hours after tail vein co-injection of GFP labeled tumor cells (5×105) with Gr-1+CD11b+ (1.5×106) or RAW264.7 cells (2×105). The lungs were fixed and Paraffin-embedded sections were obtained. TUNEL was performed according to manufactory protocol. The slides were then mounted with Prolong Gold antifade reagent with DAPI (Invitrogen) and examined using fluorescence microscopy.
Co-culture of Immature Myeloid Cells with Tumor Cells or in Tumor-conditioned Media, and Collection of Conditioned Media for Mice Injection, for myeloid-tumor co-culture, 5×105 tumor cells were co-cultured with 1×106 RAW264.7 or 32DCl3 cell lines, Gr-1+CD11b+ myeloid cells, Ly6G+CD11b+ neutrophiles, Ly6C+ CD11b+ monocytes, and F4/80+CD11b+ macrophages in 2 ml 5% FBS RPMI media in 6 well plate in 37C incubator for 24 hours. For myeloid cell culture in tumor-conditioned media, myeloid cells in 6-well plate were added 2 mls of tumor culture supernatant and cultured in 37C incubator for 24 hours. For p38 inhibition experiments, sorted Gr-1+CD11b+ cells from spleen of tumor-bearing mice were treated with p38 inhibitor SB203580 (Cell Signaling, 0, 5, 10 15 nM) in 10%FBS RPMI for 40 minutes. Tumor-conditioned media were then added to the culture for 6 hours to induce CCL9 expression. The cells were then collected and tested for CCL9 expression. For the effect of CCL9 neutralization on tumor cell or myeloid cell apoptosis, 10ug/ml CCL9 neutralizing antibody (R&D system) was added to myeloid-tumor co-culture supernatant (CoSN) and incubated in room temperature for 1 hour. Tumor cells were starved under 1% FBS for 24hs or myeloid cells that sorted from spleen were then cultured overnight in CoSN with or without CCL9 neutralization before apoptotic analysis. To collect 4T1 conditioned media for mice injection, the cells were cultured 24 hours in 0.1% oxygen, 5%CO2, and 94.9% Nitrogen conditions; the media was then intraperitoneally injected in mice.
Cytokine Antibody Array, ELISA of CCL9, CCL23, or TGF-β1
For Cytokine Antibody Array, culture supernatant was collected and processed per manufacturer protocol (Raybiotech, AAM-CYT-1000). The expression levels of cytokines/chemokines were measured by dot density using the ImageJ software, which were then normalized to control dot density, and presented as relative expression levels. For CCL9, CCL23, or TGF-β1 ELISA, cell culture supernatants, or protein extractions from cells or mouse organs were collected and processed per manufacturer protocol (R&D).
RT-qPCR
Total RNA was extracted using an RNeasy Mini Kit (Qiagen, CA) and cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystem). Relative gene expression was determined using SYBR Green PCR Master Mix (Applied Biosystem). The comparative threshold cycle method was used to calculate gene expression and normalized to GAPDH as a gene reference. RT-qPCR primers were as follow: CCR1 forward: GTGTTCATCATTGGAGTGGTGG, and reverse: GGTTGAACAGGTAGATGCTGGTC; GAPDH forward: AATGTGTCCGTCGTGGATCTGA, and reverse: GATGCCTGCTTCACCACCTTCT.
MTT Assay
4T1 cells (3×103) starved under 1% FBS for 24hs were plated in each well of 96-well plate for overnight. The cells were then cultured in mrCCL9 containing media for 2 hours followed by treatment of low serum (1%) or glucose free conditions, or doxorubicin (LC Laboratories, 1, 2, 10, 100, 250 nM). MTT assays were performed 24 hours later following manufacturer recommendations (Sigma).
Western Blotting
Protein extractions from tumor cells or Gr-1+CD11b+ cells were analyzed. For TGF-β signaling pathway in regulating CCL9 production, the sorted myeloid cells were directly subjected to protein extraction (basal state) or starved under 1% FBS for 5 hrs and treated with TGFβ-1 (5ng/ml, for one hour). Primary antibodies include TβRII, P-p38, p38, P-smad2, smad2, PARP, caspase 3, P-AKT, AKT, (Cell Signaling, 1:1000 dilution), BCL-2 (Santa Cruz, 1:500 dilution), and β-actin (Sigma). Anti-mouse/rabbit secondary antibodies were purchased from Bio-Rad (1:3000 dilution).
Transfection and Electroporation
FUGW plasmid (Gift from Dr. Zheng-Gen Jin, University of Rochester) was used for generating CCL9/CCR1 over-expression vectors; PLKO.1 plasmid (Gift from Dr. Jing Huang, NCI) was used for CCL9 and CCR1 knockdown vectors. Tumor cells (67NR, B16F1) and RAW264.7 cells were transfected with Lipofectamin2000 per manufacturer protocol (Invitrogen). Gr-1+CD11b+ cells sorted from spleens of tumor-bearing wild type, Tgfbr2MyeKO, and Tgfbr2Flox mice, were transfected using an Amaxa mouse macrophage nuclefector kit per manufacturer instructions (Lonza).
Statistical Analysis
Graphpad Prism v5.04 was used for graphs and for statistics. Unless otherwise indicated, data was expressed as mean ± SE. All data were analyzed using the Student’s t-test for comparison of two groups or One-way ANOVA for three groups or more. Differences were considered statistically significant when the p-value was < 0.05.
Results
CCL9 was induced in Gr-1+CD11b+ immature myeloid cells and the premetastatic lung
Host derived myeloid cells form a premetastatic niche and support metastatic colony formation (5-7, 33). We suspected that tumor-educated myeloid cells might secrete factors that could directly promote metastasis of the tumor cells at the metastatic site. To address this question, we used 4T1 mammary tumor model that shares many characteristics with human breast cancer, particularly its ability to spontaneously metastasize to the lungs. Additionally, the 4T1 cell line has related cell lines derived from the same spontaneous tumor that represent different degree of metastasis capability, from localized tumor (67NR) to fully metastatic tumors (4T1). We sorted Gr-1+CD11b+ cells from spleens of 4T1 tumor-bearing mice as they showed very close cytokine profiling from that of Gr-1+CD11b+ cells derived from the lungs (Fig. S1A). We then co-cultured sorted Gr-1+CD11b+ cells with 4T1 mammary tumor cells, and performed a cytokine protein array analysis on culture supernatants. Interestingly, CCL9 was highly induced in the myeloid-tumor co-culture supernatant but not in the supernatant of tumor cell or myeloid cells cultured alone (Fig. 1A, red box, and red bar on the right panel). In contrast, other CC family members were poorly expressed or not changed. CCL2, another CCL9 family member was mostly produced in tumor cells (Fig. 1A, yellow rectangle), in agreement with a previous finding (34). Pro-MMP9, a key regulator for tumor vascular remodeling (35), was also expressed in these myeloid cells (Fig. 1A, blue rectangle), consistent with our earlier studies (13).
Fig. 1.
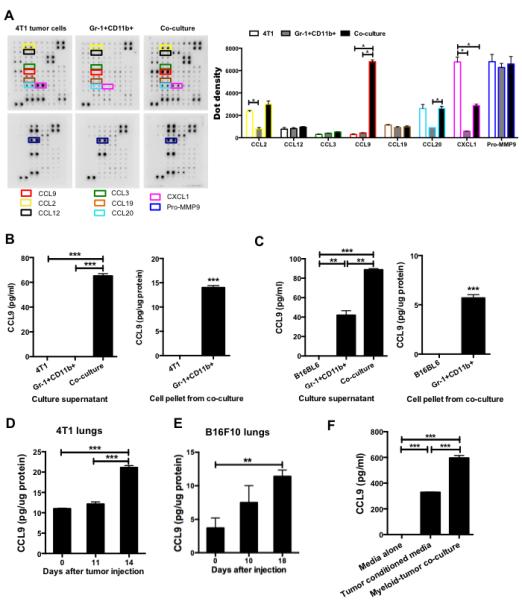
CCL9 is up-regulated in the myeloid-tumor co-culture supernatant and premetastatic lung. (A) Cytokine protein array, CCL9 and family members are highlighted with colored boxes. One of two experiments performed is shown here. Right upper panel: semi-quantitative data from dot density. (B-C) CCL9 ELISA of culture supernatant from myeloid-tumor co-culture, myeloid or tumor cell single culture, as well as protein extraction from pellet of 4T1 (B) or B16BL6 (C) tumor cells. For both (B) and (C), Gr-1+CD11b+ cells were pooled from 3 mice, with technical triplicates per treatment group. (D-E) CCL9 ELISA of premetastatic lung protein extraction from 4T1 tumor-bearing mice (D) and B16F10 tumor-bearing mice (E). n=3 mice for each group. Equal amount of proteins extracted from lungs were loaded to the plate and the results were presented as pg CCL9/ug protein. (F) CCL9 expression in myeloid cells cultured in 4T1 tumor-conditioned media or in co-culture with 4T1 tumor cells, ELISA assay with triplicates of samples. The experiment was repeated 2 times with different biological samples. (G) CCL9 ELISA of sorted Gr-1+CD11b+ cells and lungs from mice received intraperitoneally injection of hypoxic 4T1 conditioned medium for 14 days (n=5 mice). *P<0.05, **P<0.01, ***P<0.001.
We therefore focused our effort on CCL9. Using ELISA, we found that CCL9 was only present in myeloid-tumor co-culture supernatants but not in supernatants from tumor cells or myeloid cells cultured alone, in both systems of 4T1 mammary tumor cells and B16BL6 melanoma cells (Fig. 1B and 1C, left panels). Adherent tumor cells were separated from non-adherent myeloid cells before protein extraction and CCL9 ELISA. Only myeloid cells but not tumor cells showed detectable CCL9 expression (Fig. 1B and 1C, right panels), confirming that tumor cells did not produce CCL9 in our system. The basal level of CCL9 in B16BL6 model is likely due to different subset composition of myeloid cells, which will be addressed later. As Gr-1+CD11b+ cells or other immature myeloid cells are present in the lung prior to tumor cell arrival (5-7), we further tested whether CCL9 was up-regulated in premetastatic lungs. As expected, we found that CCL9 expression was significantly higher in premetastatic lungs compared with normal lungs in both 4T1 and B16F10 models (Fig. 1D and E). The 4T1 and B16F10 models have been reported for the existence of a premetastatic niche establishment prior to tumor cell arrival, day 14 for 4T1 model and day 18 for B16 model after tumor cell injection (5, 7, 8). This elevated CCL9 production seen in the premetastatic lungs was not observed in other organs at the premetastatic phase of mice bearing 4T1 (Fig. S1B) and B16F10 tumors (data not shown), suggesting an organ specificity for premetastatic niche formation.
To ask if direct contact between tumor cells and Gr-1+CD11b+ cells was necessary for CCL9 expression, we cultured Gr-1+CD11b+ cells in tumor cell-conditioned media, or in direct contact with tumor cells, and measured CCL9 concentrations in the supernatant. Interestingly, Gr-1+CD11b+ cell cultured in tumor-conditioned media also produced high levels of CCL9 expression (Fig. 1F). However, CCL9 was produced at a higher level in Gr-1+CD11b+ cells co-cultured with tumor cells (Fig. 1F), suggesting that although cell-cell contact was not necessary, proximity enhanced CCL9 expression. Lastly, to investigate CCL9 production in Gr-1+CD11b+ cells and lungs under premetastatic conditions, we performed intraperitoneal injection of hypoxic tumor culture supernatant. An induction of CCL9 in Gr-1+CD11b+ cells and premetastatic lungs was observed by CCL9 ELISA (Fig. 1G). Together, our data suggest that CCL9 is produced by Gr-1+CD11b+ cells and in the premetastatic lung. We therefore hypothesized that CCL9 might play a critical role in metastatic colony formation.
Deletion of CCL9 in RAW264.7 macrophages or primary Gr-1+CD11b+ cells decreased cancer cell metastasis
Tumor associated myeloid cells have been shown to promote cancer metastasis (5-7, 33, 34). Consistent with this, 4T1 mammary tumor cells when co-injected with Gr-1+CD11b+ cells showed an increased metastasis (Fig. 2A). Moreover, 67NR, a relatively non-metastatic cell line derived from the same mammary tumor as 4T1, showed a three-fold increase in metastasis when co-injected with Gr-1+CD11b+ cells by tail vein (Fig. 2B). To determine whether CCL9 is a critical mediator in the myeloid-mediated promotion of cancer metastasis, we first explored whether RAW264.7, a widely used macrophage cell line, could be used for CCL9 knockdown. When cultured in tumor-conditioned media, RAW264.7 macrophages showed a similar cytokine profile with primary Gr-1+CD11b+ cells especially in their high level expression of CCL9, which was not seen in the 32Dcl3 neutrophil cell line (Fig. 2C). In addition, RAW264.7 cells showed a metastasis-promoting effect similar to Gr-1+CD11b+ cells when co-injected with tumor cells through tail vein (Fig. S1C). Importantly, 67NR tumor cells co-injected with CCL9-deficient RAW264.7 cells showed a decrease in lung metastasis compared with those co-injected with the control RAW264.7 cells (Fig. 2D-E, Fig. S1D). This result was further validated with CCL9 knockdown in primary Gr-1+CD11b+ cells (Fig. 2F-G). CCL9 knockdown did not show any effect on the primary tumor growth (Fig. S1E). Together, our data suggest a unique function of CCL9 in myeloid cells to promote cancer metastasis.
Fig. 2.

Deletion of CCL9 in myeloid cells decreased cancer metastasis. (A) Luciferase Imaging of lung metastasis with tumor cells co-injected with Gr-1+CD11b+ cells. IVIS live image 7 days after tail vein co-injection of 5×105 4T1 tumor cells expressing luciferase (4T1-luc) with 1×106 Gr-1+CD11b+ cells. Mice n=5 for each group. Quantitative data is on the right. (B) Number of lung metastasis of 67NR tumor cells co-injected with Gr-1+CD11b+ cells. 5×105 67NR cells were co-injected with 1×106 Gr-1+CD11b+ cells through tail vein, lungs were harvested after 21 days. n=8 for each group. (C) Cytokine protein array of primary Gr-1+CD11b+ cells (sorted from spleens of tumor-bearing mice), RAW264.7 macrophage cell line, and 32Dcl3 neutrophil cell line, cultured in tumor-conditioned media for 24 hrs. The culture supernatant was used for array. Red boxes on blots indicate CCL9 expression. Heat map was generated based on semi-quantitative data from dot density then normalized to control dots. Blue rectangle indicates CCL9 expression. (D) CCL9 knockdown in RAW264.7 cells, by ELISA. (E) Lung metastasis of 67NR tumor cells co-injected with RAW264.7 cells deficient in CCL9 expression. (F) CCL9 knockdown in primary Gr-1+CD11b+ cells. (G) Lung metastasis of 67NR tumor cells co-injected with Gr-1+CD11b+ cells deficient in CCL9 expression. For both (D) and (F), CCL9 expression was detected by ELISA. One experiment from two with triplicates was shown. For (E) and (G), mice received tail vein co-injection of 5×105 67NR and 2×105 RAW264.7 cells or 1.5 ×106 Gr-1+CD11b+ cells, and were sacrificed 4 weeks after cell injection. Mice n=8-9 for each group. *P<0.05, **P<0.01, ***P<0.001.
CCR1 mediates CCL9 signaling and promotes tumor metastasis
CCR1 is the sole receptor mediating CCL9 signaling (25, 28-30). We next examined the correlation between CCR1 expression in tumor cells with cancer metastasis. We used the 4T1 mammary tumor and B16 melanoma models comprised of cell lines with different degree of metastatic capability. Flow cytometry analysis revealed approximately 8% of highly metastatic 4T1 cells express CCR1 protein, which was higher than that of 67NR, 168FARN and 4T07 cells (Fig. 3A). Similarly, less metastatic B16F1 cells had lower level of CCR1 mRNA expression compared to more metastatic B16F10 cells (Fig. S2A). Interestingly, tumor cells from metastatic lung nodules showed higher CCR1 expression than those from the primary tumor tissues and cells in culture (Fig. 3B). To examine whether CCR1 is important for lung metastasis, we over-expressed CCR1 in low metastatic 67NR and B16F1 cells (Fig. S2B and S2C). Mice bearing 67NR-CCR1 or B16F1-CCR1 tumor cells showed significantly increased lung metastasis (Fig. 3C-D). To further investigate whether CCL9 signals through CCR1 to promote cancer metastasis, we co-injected CCL9-deficient RAW264.7 cells with 67NR-CCR1 cells. Consistent with our earlier observations, CCL9 knockdown in RAW264.7 cells decreased tumor metastasis in 67NR cells, as well as 67NR-CCR1 cells (Fig. 3E). Interestingly, CCR1 overexpression in 67NR cells did not rescue the metastasis deficiency in the context of myeloid-specific CCL9 knockdown (Fig. 3E), suggesting that CCL9, but not the other family members, is the major cytokine signaling through the CCR1 receptor to promote metastasis. This finding is consistent with our earlier observation that CCL9 is the most up-regulated cytokine in the CCL9 family members (Fig. 1A).
Fig. 3.
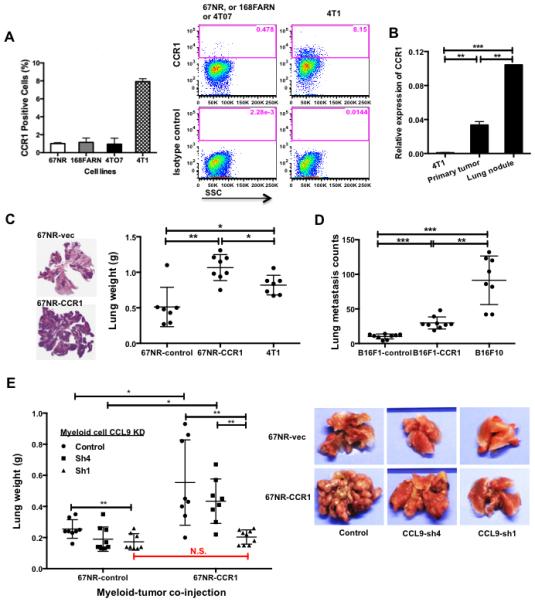
CCL9/CCR1 signaling promotes tumor metastasis. (A) Flow cytometry showing CCR1+ cells in cell line derivatives of 4T1 tumor. One experiment is shown from two performed. Quantitative data is on the left. (B) RT-qPCR of CCR1 expression in cultured 4T1 cells, primary tumors, and metastatic lung tumor nodules. Three biological samples were pooled, with triplicates for each group used in experiments. (C) Number of lung metastatic nodules upon CCR1 over-expression in 67NR cells (67NR-CCR1). Mice received tail vein injection of 106 67NR-CCR1, 67NR-control, and 4T1 cells; the lungs were harvested 21 days later. Left panel: representative H&E sections of mouse lungs; Right panel: number of lung metastatic nodules, n=7-8 mice for each group. (D) Number of lung metastasis in mice received tail vein injection of B16F1-CCR1, B16F1-vec, and B16F10 cells. Lungs were collected 21 days after tail vein injection and subjected to whole lung mounting procedure. n=8-9 mice for each group. (E) Number of lung metastatic nodules of tumor-bearing mice received co-injection of 67NR-CCR1 and its control with CCL9 knockdown RAW264.7 cells. Mice were euthanized 4 weeks after injection. n=9 mice for each group. Representative pictures on the right. *P<0.05, **P<0.01, ***P<0.001.
CCL9/CCR1 correlates with metastasis and decreased survival in cancer patients
To establish the clinical significance of our findings, we next looked into the correlation between CCL9 expression levels and metastasis using publicly available patient databases. CCL23 has been identified as the human orthologue for mouse CCL9 (25, 30). They shared 92-99% sequence identity (Fig. S3) and cluster together by Treefam analysis (Fig. S3A). They are predicted orthologs by MetaPhOrs online tool (Fig. S3B) and show similar chemoattractant functionality (25, 29, 36). In addition, human CCL23 and mouse CCL9 are two of the four NC6 subfamily members in β chemokines (37). Analysis of publically available database (Oncomine, GSE20685) showed a negative correlation between CCL23 or CCR1 expression levels and metastatic status three years after diagnosis in breast cancer patients (Fig. 4A, heat map), or with a decreased metastasis-free survival (Fig. 4A). Interestingly, meta-analysis of 1577 breast cancer patients in GOBO breast cancer database (38) revealed that patients with low expression of CCL23/CCR1 have a better survival among 166 PAM50 HER2+ patients (Fig. 4B), which was not observed in the HER2− patients (data not shown). PAM50 (Prediction Analysis of Microarray using 50-gene classifier) is a 50-gene signature that adds further prognostic and predictive information to standard parameters for breast cancer patients (39). These data suggest a negative correlation of CCL23/CCR1 with metastasis-free survival of cancer patients. To examine CCL23 induction in human myeloid cells under cancer conditions, we sorted human CD33+ myeloid cells from peripheral blood mononuclear cells (PBMC) of healthy blood donors and co-cultured them with human breast cancer cell line MDA-MB-231 or human melanoma cell lines A375S2 and SK-MEL-28. Culture supernatant from CD33+ myeloid cells alone, tumor cells alone, or co-culture was examined by CCL23 ELISA. Consistent with our mouse findings, CCL23 was highly induced in myeloid-tumor co-culture supernatants but not in supernatants from tumor cells or myeloid cells cultured alone (Fig. 4C). In addition, CCL23 was mostly produced by myeloid cells but not tumor cells, consistent with our mouse data. This observation was made with separation of adherent tumor cells from non-adherent myeloid cells for protein extraction and CCL23 ELISA (Fig. 4D). Lastly, MDA-MB-231 tumor-conditioned media induced CCL23 production, however in co-culture, CCL23 was produced at a higher level in CD33+ myeloid cells (Fig. 4E), suggesting cell-cell proximity enhanced CCL9 expression, similar to our findings in mouse. Together, our data support that CCL23 was induced in myeloid cells and correlates with human cancer metastasis.
Fig. 4.
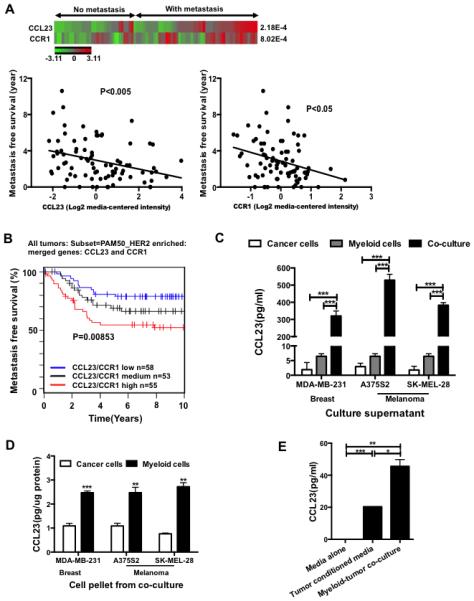
CCL23/CCR1 correlates with metastatic status or survival of cancer patients. (A) Correlation of CCL23 or CCR1 expression levels with breast cancer metastatic status 3 years after diagnosis (heat map), or with a decreased overall metastasis free survival. Oncomine database (GSE20685) was used for the analysis. (B) Kaplan–Meier survival curve of CCL23/CCR1 expression in 166 PAM50 HER2+ breast cancer patients in GOBO database (38). (C) CCL23 ELISA in sorted human CD33+ PBMC co-cultured with human cancer cell lines of breast and melanoma. CD33+ myeloid cells were sorted from normal donors, were pooled, with triplicates per treatment group. Culture supernatant from CD33+ PBMC cells alone, tumor cells alone, or in co-culture was tested. (D) CCL23 ELISA of protein extraction from adherent tumor cells or non-adherent CD33+ PBMC cells after separation from co-culture. Tumor cells or CD33+ PBMC were pooled, with triplicates per treatment group. (E) CCL23 ELISA of sorted CD33+ PBMC cultured in MDA-MB-231 tumor-conditioned media or in co-culture with MDA-MB-231 cells, ELISA assay with triplicates of samples. *P<0.05, **P<0.01, ***P<0.001.
CCL9/CCR1 promotes tumor cell survival
Tumor cell survival in the hostile distant organ is a rate-limiting step in the metastatic process (1). Indeed, when 4T1 cells were introduced into the venous circulation of mice, the majority of 4T1 tumor cells die within 24 hours after arrest at the secondary site (Fig. S4A). To investigate the effect of myeloid-derived CCL9 on tumor cell survival in the premetastatic lung, we utilized fluorescence imaging of single cell video microscopy (SCVM) and a pulmonary metastasis assay (PUMA) (40). SCVM detects single live tumor cells hours after tail vein injection, which allows us to examine the effect of myeloid CCL9 on tumor cell survival in vivo. PUMA evaluates tumor cell colonization ex vivo in cultured lung slice 7-14 days after tail vein co-injection of the tumor cells and myeloid cells. We used 67NR-GFP or B16F1-GFP cells in these assays, as they are more vulnerable to CCL9 deletion in myeloid cells. Using SCVM and TUNEL assays, we found that CCL9 knockdown in RAW264.7 cells decreased the survival of 67NR-CCR1 and increased apoptosis (Fig. 5A left panel) and B16F1 tumor cells (Fig. 5A right panel). CCL9 knockdown in RAW264.7 cells also decreased metastasis colony formation of 67NR-CCR1 (Fig. 5B, left and middle panels) or B16F1 cells (Fig. 5B, right panel) as demonstrated by PUMA. These results were further validated through CCL9 knockdown in primary Gr-1+CD11b+ myeloid cells that increased tumor cell apoptosis (Fig. S4B, upper panel) and decreased metastatic colony formation (Fig. S4B, lower panel). Deletion of CCL9 in Gr-1+CD11b+ cells did not affect tumor cell extravasation (Fig. S4C). This was evaluated through counting green colored 67NR-GFP tumor cells against a red vascular map (Dextran labeled) under confocal microscope. The B16F1 cells seemed to be more sensitive to changes in CCL9 concentration than 67NR (Fig. S4D), likely due to higher CCR1 expression in B16F1 cells (Fig. S4E). To determine the effect of CCL9/CCR1 on tumor cell survival in vitro, we continued to use the above low metastatic 67NR mammary tumor cells and B16F1 melanoma cells that were more sensitive for apoptosis induction upon serum starvation and glucose free culture condition. Recombinant mouse CCL9 (rmCCL9) decreased and neutralization of CCL9 increased the number of 7AAD+/AnnexinV+ cells (Fig. S4D). rmCCL9 also increased the survival of 67NR cells cultured in glucose-free conditions (Fig. 5C, left panel). Furthermore, 4T1 cells treated with doxorubicin (67NR were not used as they were mostly killed under the same conditions) showed an increased survival upon rmCCL9 treatment (Fig. 5C, right panel, and Fig. S4F).
Fig. 5.
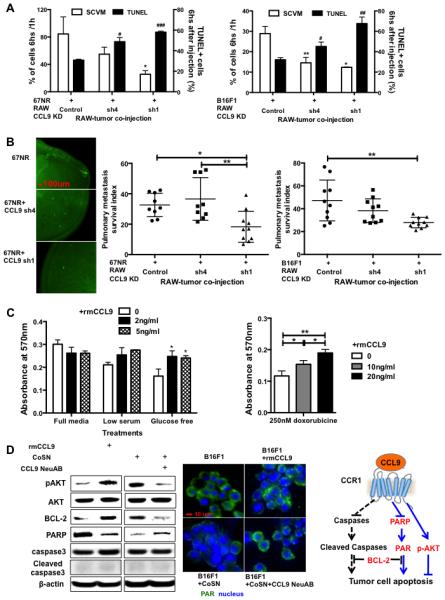
CCL9 promotes cancer cell survival and colony formation. (A) Tumor cell survival and apoptosis after co-injection with CCL9-deficient RAW264.7 cells. Fluorescence signal from SCVM indicating tumor cell survival was plotted on the left Y-axis; and percentage of TUNEL positive cells indicating apoptosis was on the right Y-axis. Left panel: 67NR-CCR1 mammary tumor model; Right panel: B16F1 melanoma model. n=3-4 mice per group. * or # P<0.05, ** or ## P<0.01, ###P<0.001 compared to the controls. (B) PUMA for tumor cell survival and metastasis. GFP labeled 67NR-CCR1 or B16F1 cells (5×105) were co-injected with RAW264.7 cells with or without CCL9 knockdown. Fluorescence imaging was obtained 7 days after lung section culture. The GFP florescence pixels were obtained on day 7 and were normalized with those from day 0 (baseline), and presented as pulmonary metastasis index. Representative images are on the left panel; quantitative data for 67NR are in the middle and B16F1 on the right. Three mice each group, 3-4 lung pieces each mouse. (C) MTT assay for tumor cell survival with addition of rmCCL9. The cells were cultured under glucose free or low serum conditions (left panel), and with treatment of anti-cancer drug doxorubicine (right panel). (D) Left panels: Western blot of P-AKT, AKT, BCL2, PARP, caspase 3 and cleaved caspase 3, as well as β-actin; Middle panels: Fluorescence staining of PAR (in green, with nuclei in blue) in starved B16F1 cells treated rmCCL9 or in B16F1 cells cultured in myeloid-tumor co-culture supernatant and treated with CCL9 neutralizing antibody. Right panel: schematic pathways for tumor cell survival and apoptosis regulated by CCL9. * P<0.05, ** P<0.01. Abbreviation: mrCCL9: mouse recombinant CCL9; CoSN: myeloid-tumor co-culture supernatant; CCL9 NeuAB: CCL9 neutralizing antibody.
We next addressed the molecular mechanisms underlying CCL9 promotion of tumor cell survival. We found that rmCCL9 increased phospho-AKT (P-AKT) and BLC-2, two well-described cell survival pathways and markers, whereas CCL9 neutralizing antibody decreased P-AKT and BLC-2 expression (Fig. 5D, left panel). There was no change in cleaved caspase 3 in cells treated with rmCCL9 or CCL9 neutralizing antibody. Rather, there was a change in the expression of poly (ADP-ribose) polymerase (PARP), an important non-canonical apoptosis mediator (41). rmCCL9 decreased PARP expression in starved 67NR cells whereas CCL9 neutralizing antibody elevated its expression in 67NR cells cultured in myeloid-tumor co-culture supernatant (Fig. 5D, left panel). Immunofluorescence staining of PAR, the substrate of PARP, showed rmCCL9 decreased PAR in starved B16F1 tumor cells, whereas CCL9 neutralizing antibody elevated PAR in B16F1 cells cultured in myeloid-tumor co-culture supernatant, supporting a role of PARP in CCL9 promotion of tumor cells survival (Fig. 5D, middle panel). Together, these data provide evidence for a role of CCL9/CCR1 signaling in tumor cell survival and metastasis as a result of interaction between myeloid cells and tumor cells.
CCL9 is critical in myeloid-specific TGF-β regulation of tumor metastasis
We recently reported that deletion of Tgfbr2 in myeloid cells (Tgfbr2MyeKO) decreased tumor metastasis through modulation of cytokine production (18). TβRII-deficient Gr-1+CD11b+ cells displayed significantly decreased CCL9 expression in a cytokine protein array (Fig. 6A). This result was further confirmed with ELISA using multiple TβRII-deficient or control tumor-bearing mice (Fig. 6B). Unlike wild type Gr-1+CD11b+ cells, TβRII-deficient Gr-1+CD11b+ cells failed to support tumor cell survival in vivo by SCVM (Fig. 6C). Instead these cells induced tumor cell apoptosis to a level similar to the tumor cell alone group (Fig. 6D). To investigate the importance of CCL9 in TGF-β regulation of myeloid cell function, we overexpressed CCL9 in TβRII-deficient Gr-1+CD11b+ cells (Fig. S5A). Tumor cells coinjected with CCL9-expressing TβRII-deficient myeloid cells showed an increased survival and metastasis when compared to the controls by PUMA (Fig. 6E). We next determined the downstream mediators of the TGF-β pathway that might regulate CCL9 expression. TGF-β signals through SMAD-dependent canonical pathways and SMAD-independent non-canonical pathways (20). Upon TGF-β treatment, a decreased level of P-p38 or P-smad2 (less pronounced) was observed in TβRII-deficient myeloid cells compared to that of wild type. This difference between TβRII-deficient and wild type myeloid cells was not observed in untreated conditions (Fig. 6F). To investigate the role of p-38 in TGF-β regulation of CCL9 production, we cultured control and TβRII-deficient Gr-1+CD11b+ cells in tumor-conditioned media that contained high level of TGF-β. These cells were treated with the p38 inhibitor SB203580 that inhibits p38 kinase activities, shown by inhibited phosphorylation of ATF2 (Fig. S5B). SB203580 reduced CCL9 production specifically in the control but not in TβRII-deficient Gr-1+CD11b+ cells (Fig. 6G). Importantly, P38 dominant negative Gr-1+CD11b+ cells showed a decreased CCL9 production (Fig. 6H). Lastly, TGF-β was produced in tumor-conditioned medium and the premetastatic lungs in mice received injection of conditioned medium (Fig. S5B). Together, our data suggest that CCL9 is regulated by p38 and is critical in TGF-β signaling in myeloid cells that promotes tumor cell survival in metastatic lungs.
Fig. 6.
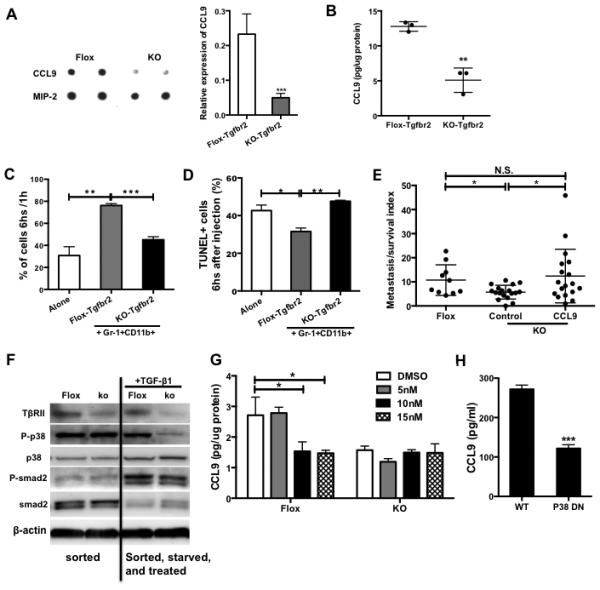
CCL9 is critical in myeloid-specific TGF-β regulation of tumor metastasis. (A) Cytokine protein array indicating CCL9 expression in floxed or TβRII-deficient Gr-1+CD11b+ cells sorted from peripheral blood of 4T1 tumor-bearing Tgfbr2flox or Tgfbr2MyeKO mice. The samples were combined from 3 mice, with duplicates for each sample. Semi-quantitative data of dot density is on the right. (B) CCL9 ELISA in myeloid cells sorted from peripheral blood of TβRII-deficient or control tumor-bearing mice (n=3). (C) SCVM for tumor cell survival. GFP labeled 67NR cells (5×105) were co-injected with floxed or TβRII-deficient Gr-1+CD11b+ cells (1×106). The lungs were taken out for images 1 and 6 hours after injection. Ten fields for each mouse lung were examined. Fluorescent signals at 6 hours were normalized with 1h signal. n=3 mice per group. (D) TUNEL assay for tumor cell apoptosis from lung sections from (C). TUNEL positive cells were counted and calculated and presented as percentage of GFP+ cells as shown at Y-axis. (E) PUMA for tumor cell survival and metastasis. 67NR-GFP cells were co-injected with TβRII-deficient Gr-1+CD11b+ cells with or without CCL9 over-expression. Fluorescence imaging was obtained 14 days after lung section culture. Fluorescence signal per field was quantified then normalized to day 0 signal and presented as metastasis survival index. Three mice each group, 3-4 lung pieces each mouse. (F) Western blot for P-p38 level in TβRII-deficient Gr-1+CD11b+ cells, compared with control cells, with or without TGF-β1 treatment. One representative experiment is shown here from two performed. (G) CCL9 ELISA of floxed or TβRII-deficient Gr-1+CD11b+ cells treated with p38 inhibitor for 6 hours. (H) CCL9 ELISA of Gr-1+CD11b+ cells sorted from spleen of P38 dominant negative or wild type mice. The cells were cultured in 4T1 tumor conditioned media for 24 hours. Spleens from 3 mice were pooled for sorting. Validation of P-p38 by Western is in the lower panel. *P<0.05, **P<0.01, ***P<0.001.
Autocrine effect of CCL9/CCR1 and myeloid subsets expressing CCL9
CCL9 was reported to decrease apoptosis in osteoclasts, one of the myeloid cell types (32). We therefore asked whether CCL9 has an effect on Gr-1+CD11b+ cell survival. CCL9 neutralization increased apoptosis of Gr-1+CD11b+ cells cultured in tumor-conditioned media (Fig. 7A). Knockdown of CCR1 in RAW264.7 cells decreased their CCL9 production (Fig. 7B), suggesting the existence of a CCL9-CCR1 autocrine effect.
Fig. 7.
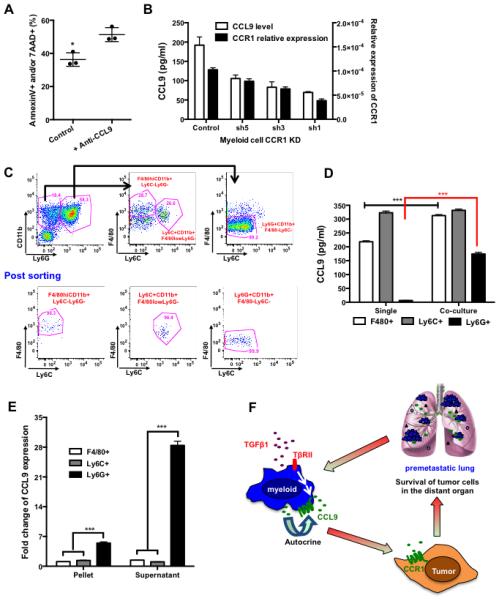
Autocrine effect of CCL9/CCR1 on myeloid cells and Ly6G+CD11b+ subset. (A) Flow cytometry analysis of AnnexinV and 7AAD of Gr-1+CD11b+ cells cultured in myeloid-tumor co-culture supernatant and treated with CCL9 neutralizing antibody. Three representative mice were used for each group. (B) Relative expression of CCL9 (ELISA, left Y-axis) and CCR1 (Q-PCR, right Y-axis) upon CCR1 knockdown in RAW264.7 cells. (C) Sorting strategy of Ly6G+, Ly6C+, and F4/80+ myeloid cell subsets. Lower panel: post-sorting flow cytometry analysis. (D) CCL9 ELISA of sorted F4/80+CD11b+, Ly6C+CD11b+, and Ly6G+CD11b+ cells upon co-culture with tumor cells. Three spleens from tumor-bearing mice were used for sorting. (E) Fold change of CCL9 production by myeloid cells with consideration of the fold increase in numbers of myeloid cell subsets in the premetastatic lung (Fig. S6). (F) Schematic hypothesis: TGF-β signals through TβRII on myeloid cells and stimulates CCL9 production; CCL9 then signals through CCR1 on tumor cells resulting an increased tumor cell survival and metastatic colony formation. CCL9 also signals through CCR1 on myeloid cells and mediates an autocrine effect that increases CCL9 production and myeloid cell survival. *P<0.05, ***P<0.001
Gr-1+CD11b+ cells consist of heterogeneous immature myeloid cells. To determine the cell subset(s) responsible for CCL9 production, FACS sorted myeloid subsets (Fig. 7C) were co-cultured with tumor cells, and CCL9 production was measured by ELISA. Although sorted Ly6G+CD11b+ cells produced lower levels of CCL9 than either Ly6C+CD11b+ or F4/80+CD11b+ subsets, upon co-culture with tumor cells, CCL9 production was elevated 20 fold in Ly6G+CD11b+ cells while remaining mostly unchanged in the Ly6C+CD11b+ and F4/80+CD11b+ subsets (Fig. 7D). In addition, only Ly6G+CD11b+ cells were significantly increased in the premetastatic lung (day 14 after 4T1 tumor injection), while the number of the other cell types was unchanged (Fig. S6). Taking this into consideration, the increase in CCL9 was far greater in the Ly6G+CD11b+ cells (Fig. 7E). Our studies suggest that CCL9 was produced in Ly6G+CD11b+ cells, and had an autocrine effect on myeloid cell survival and CCL9 production.
Discussion
We report here for the first time that CCL9 is a critical downstream mediator of myeloid TGF-β signaling, and that the myeloid CCL9/tumor CCR1 axis plays an important role in tumor cell survival and metastatic colony formation (Fig. 7F). CCL9 is highly induced in immature myeloid cells and in the premetastatic lung of tumor-bearing mice. It functions as a survival factor for tumor cells in the distant premetastatic site. Mechanistically, CCL9 is a critical downstream mediator of the metastasis-promoting effects of myeloid-specific TGF-β signaling. Our study suggests a novel cellular and molecular mechanism underlying the “seed and soil” hypothesis for cancer metastasis. Importantly, CCL23, the human orthologue for CCL9, and its receptor CCR1, correlate with progression and metastasis of breast cancers, indicating human relevance. Our work identifies a unique target that contributes to cancer cell survival and is situated downstream of the metastasis-promoting effects of TGF-β signaling in myeloid cells.
Tumor cell survival in the hostile distant organ is a critical rate-limiting step in cancer metastasis. Highly metastatic tumor cells have a significantly higher survival in the distant organ (42). Blocking tumor cell apoptosis increases metastasis (43, 44). In addition to the molecular mediators within tumor cells, factors produced by host cells, including chemokines and cytokines, also modulate tumor cell survival (2, 45-48). For example, the inflammatory chemoattractants S100A8 and S100A9 are produced in the premetastatic lung or brain and contribute to metastasis colony formation (6, 49, 50). CCL9 was clearly produced in the premetastatic lung and by the CD11b+LyG+ myeloid cells promoting tumor cell survival and metastasis. We showed that CCL9 was produced in myeloid cells and little CCL9 was found in tumor cells in both mouse models and human setting. As CCL9 expression levels were examined in myeloid cells and tumor cells at the same time so we could make a direct comparison (Fig. 1 and 4). Our data is in agreement with reports that CCL9 is secreted in macrophages and myeloid cell lines (31, 32). These data are different from previous reports in which the CCL9/CCR1 axis was implicated in the recruitment of CCR1+Gr-1-myeloid cells in response to CCL9 production by tumor cells (30, 51, 52). In contrast, our study reveals a new function of the CCL9/CCR1 axis in supporting tumor cell survival in the distant premetastatic organ. The observed CCL9 induction in intestinal tumor cells is likely due to the blockade of TGF-β signaling that has been shown to elevate the expression of a number of chemokines (21, 22).
Tumor-stroma interactions are indispensable participants in the metastatic process (53-57). Of interest, cancer cells often do not need additional genomic alterations in order to establish metastatic colonies when compared to the cancer cells at the primary tumor site (58). In fact, gene expression orchestrated by the stromal microenvironment cues could play a fundamental role in metastatic processes (57). Myeloid cells have been implicated in supporting cancer metastasis (5, 7, 34, 51, 59). The functionality of these cells involve suppression of host anti-tumor immunity (12, 18), promotion of tumor angiogenesis (13) or tumor cell extravasation (34), enhancement of tumor cell migration and invasion (51) as well as formation of the premetastatic niche (5, 7, 60, 61). We found that in the premetastatic lung, Ly6G+CD11b+ cells produce CCL9, which supports incoming metastatic tumor cell survival. CCL9 also has an autocrine effect on myeloid cells, which further amplifies CCL9 signaling and expression, likely enhancing myeloid cell survival in the premetastatic lung. These findings provide new molecular insights for the tumor-host interaction in the premetastatic organ site. Our results are consistent with the metastasis-promoting effect of Gr-1 positive inflammatory monocytes (34) as well as immature myeloid cells (6) or VEGFR1 myeloid progenitor cells (5), but different from a recent publication, which showed that Ly6G+CD11b+ or Gr-1+ cells have an anti-tumor function (62, 63). These differences may reflect the context-dependent functionality of myeloid cells such as time and spatial factors in mouse models.
Our data also provide molecular insight for CCL9 as a downstream mediator of the metastasis-promoting function of TGF-β. TGF-β is produced in high level by cancer cells and myeloid cells (18). It promotes cancer metastasis including breast cancer (20, 64, 65). One underlying mechanism is its role in premetastatic niche formation (6, 61, 66). However, TGF-β possesses both tumor suppressor and tumor promoter functions and has been rather puzzling. Multiple lines of evidence suggest that TGF-β signaling in epithelial cells, fibroblasts and T cells are tumor suppressing, as its deletion in these cell types promotes tumor development and progression (67, 68). Very interestingly, distinct from all cell types discussed above, myeloid-specific TGF-β has been shown to be metastasis-promoting, in that deletion of TβRII in these cells significantly decrease metastasis (18). Here we provide a mechanistic explanation for this observation, as we detected significantly lower CCL9 expression in TβRII-deficient Gr-1+CD11b+ cells. Overexpression of CCL9 in TβRII-deficient myeloid cells promoted tumor cell survival and metastasis capability (Fig. 6). Our data suggest that the chemokine CCL9 is a critical downstream mediator of the metastasis-promoting effects of myeloid-specific TGF-β signaling. This effect was regulated through P-p38 in the non-canonical TGF-β pathway (Fig. 6) in agreement with previous publications showing a role for p38 in CCL9 induction by other factors (69-71). Our study is consistent with previous reports that TGF-β signaling through BMPRII in mammary epithelial cells (52) and TGF-β-SMAD4 in colon epithelial cells regulate CCL9 expression (25). However, myeloid TGF-β promotes (Fig. 6), whereas epithelial TGF-β inhibits CCL9 expression (25, 52). The molecular mechanisms underlying this opposite mode of regulation might be due to the difference in downstream TGF-β signaling network between myeloid cells and tumor epithelial cells.
Chemokines have been implicated in TGF-β regulation of tumor microenvironment and tumor progression. Loss of TGF-β signaling in carcinoma cells enhances chemokine production and correlates with poor prognosis of human breast cancer patients (22). In mouse models, deletion of TGF-β signaling in cancer epithelial cells increases metastasis due to chemokine expression, which promotes the interaction of carcinoma cells and bone marrow derived cells, tumor cell migration and survival, and decreases tumor cell apoptosis (6, 21, 25, 49, 72). Our data demonstrate that CCL9 is the key regulator of the metastasis-promoting effects of myeloid TGF-β signaling and provide a new understanding of tumor-host interactions in TGF-β regulation of metastasis. Targeting CCL9 derived from myeloid cells through bone marrow manipulation (e.g. CCL9 shRNA driven by CD11b promoter) or CCL9 neutralizing antibody could be utilized for reprogramming premetastatic organ microenvironment, thus inhibiting metastasis. Additionally, targeting CCL9 as a downstream effector of the pro-metastatic effect of TGF-β might bypass some of the negative consequences of TGF-β neutralization.
Supplementary Material
Acknowledgements
We thank Drs. Howard Young, Lalage Wakefield, Glenn Merlino, Michael Lichton, and Zhenggang Liu for critical reading of the manuscript. We thank Barbara Taylor, Karen Wolcott, Subhadra Banerjee for technical assistance on FACS. We thank Dr. Arnulfo Mendoza for the technique support in PUMA. We are grateful for help from Drs. Anand Merchant, Aleksandra Michalowski, and Binwu Tang in bioinformatics. We appreciate the assistance of technicians from animal facility.
This work was supported by NCI intramural funding to Dr. Li Yang
Footnotes
The authors declare no competing financial interest.
Authors' Contributions
Conception and design: H.H. Yan, L. Yang
Development of methodology: H.H. Yan, M. Lizardo, C. Khanna
Acquisition of data: H.H. Yan, J. Jiang, Y. Pang, BR Achyut
Analysis and interpretation of data: H.H. Yan, J. Jiang, Y. Pang, BR Achyut, C. Hollander, K. Hunter, L. Yang
Writing, review and/or revision of the manuscript: H.H. Yan, L. Yang
Administrative, technical, or material support: H.H. Yan, M. Lizardo, X. Liang, C. Khanna
Study supervision: L. Yang
References
- 1.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6(6):449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 2.Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell. 2013;23(5):573–581. doi: 10.1016/j.ccr.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32(3-4):449–464. doi: 10.1007/s10555-013-9420-1. [DOI] [PubMed] [Google Scholar]
- 4.Keskinov AA, Shurin MR. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology. 2015;220(2):236–242. doi: 10.1016/j.imbio.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 7.Yan HH, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70(15):6139–6149. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowanetz M, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010;107(50):21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 11.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457(7225):102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauti LA, et al. Myeloid-derived suppressor cells are implicated in regulating permissiveness for tumor metastasis during mouse gestation. J Clin Invest. 2011;121(7):2794–2807. doi: 10.1172/JCI41936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sceneay J, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012;72(16):3906–3911. doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- 17.Gao D, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72(6):1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pang Y, et al. Transforming growth factor beta signaling in myeloid cells is required for tumor metastasis. Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-12-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6(7):506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 20.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L, et al. Abrogation of TGFbeta Signaling in Mammary Carcinomas Recruits Gr-1+CD11b+ Myeloid Cells that Promote Metastasis. Cancer Cell. 2008;13(1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bierie B, et al. Abrogation of TGF-beta signaling enhances chemokine production and correlates with prognosis in human breast cancer. J Clin Invest. 2009;119(6):1571–1582. doi: 10.1172/JCI37480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ijichi H, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. 2011;121(10):4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kojima Y, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitamura T, et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat Genet. 2007;39(4):467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 26.Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431(7007):405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 27.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9/MIP-1gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. J Cell Biochem. 2002;87(4):386–393. doi: 10.1002/jcb.10319. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, et al. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer's patch CD11b+ dendritic cells. J Immunol. 2003;171(6):2797–2803. doi: 10.4049/jimmunol.171.6.2797. [DOI] [PubMed] [Google Scholar]
- 30.Kitamura T, et al. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci U S A. 2010;107(29):13063–13068. doi: 10.1073/pnas.1002372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara T, et al. Molecular cloning and functional characterization of a novel member of the C-C chemokine family. J Immunol. 1995;155(11):5352–5358. [PubMed] [Google Scholar]
- 32.Okamatsu Y, et al. MIP-1 gamma promotes receptor-activator-of-NF-kappa-B-ligand-induced osteoclast formation and survival. J Immunol. 2004;173(3):2084–2090. doi: 10.4049/jimmunol.173.3.2084. [DOI] [PubMed] [Google Scholar]
- 33.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9(4):285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel VP, et al. Molecular and functional characterization of two novel human C-C chemokines as inhibitors of two distinct classes of myeloid progenitors. Journal of Experimental Medicine. 1997;185(7):1163–1172. doi: 10.1084/jem.185.7.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berahovich RD, et al. Proteolytic activation of alternative CCR1 ligands in inflammation. J Immunol. 2005;174(11):7341–7351. doi: 10.4049/jimmunol.174.11.7341. [DOI] [PubMed] [Google Scholar]
- 38.Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 6(3):e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendoza A, et al. Modeling metastasis biology and therapy in real time in the mouse lung. J Clin Invest. 2010;120(8):2979–2988. doi: 10.1172/JCI40252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 42.Hong SH, Ren L, Mendoza A, Eleswarapu A, Khanna C. Apoptosis resistance and PKC signaling: distinguishing features of high and low metastatic cells. Neoplasia. 2012;14(3):249–258. doi: 10.1593/neo.111498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Owen-Schaub LB, van Golen KL, Hill LL, Price JE. Fas and Fas ligand interactions suppress melanoma lung metastasis. J Exp Med. 1998;188(9):1717–1723. doi: 10.1084/jem.188.9.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medina-Ramirez CM, et al. Apoptosis inhibitor ARC promotes breast tumorigenesis, metastasis, and chemoresistance. Cancer Res. 2011;71(24):7705–7715. doi: 10.1158/0008-5472.CAN-11-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 47.Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends in molecular medicine. 2010;16(3):133–144. doi: 10.1016/j.molmed.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33(10):496–504. doi: 10.1016/j.it.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Acharyya S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15(7):891–903. doi: 10.1093/neuonc/not031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitamura T, Taketo MM. Keeping out the bad guys: gateway to cellular target therapy. Cancer Res. 2007;67(21):10099–10102. doi: 10.1158/0008-5472.CAN-07-2100. [DOI] [PubMed] [Google Scholar]
- 52.Owens P, et al. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc Natl Acad Sci U S A. 2012;109(8):2814–2819. doi: 10.1073/pnas.1101139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010;28(26):4022–4028. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- 54.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cook LM, Hurst DR, Welch DR. Metastasis suppressors and the tumor microenvironment. Semin Cancer Biol. 2011;21(2):113–122. doi: 10.1016/j.semcancer.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sethi N, Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer. 2011;11(10):735–748. doi: 10.1038/nrc3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sleeman JP. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012;31(3-4):429–440. doi: 10.1007/s10555-012-9373-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell. 2013;24(4):410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Gil-Bernabe AM, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119(13):3164–3175. doi: 10.1182/blood-2011-08-376426. [DOI] [PubMed] [Google Scholar]
- 61.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21(2):139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Granot Z, et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20(3):300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Catena R, et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer discovery. 2013;3(5):578–589. doi: 10.1158/2159-8290.CD-12-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Figueroa JD, et al. Expression of TGF-beta signaling factors in invasive breast cancers: relationships with age at diagnosis and tumor characteristics. Breast Cancer Res Treat. 2009;121(3):727–735. doi: 10.1007/s10549-009-0590-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 66.Wilson C, Holen I, Coleman RE. Seed, soil and secreted hormones: potential interactions of breast cancer cells with their endocrine/paracrine microenvironment and implications for treatment with bisphosphonates. Cancer Treat Rev. 2012;38(7):877–889. doi: 10.1016/j.ctrv.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 67.Achyut BR, Yang L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology. 2011;141(4):1167–1178. doi: 10.1053/j.gastro.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravindran C, Cheng YC, Liang SM. CpG-ODNs induces up-regulated expression of chemokine CCL9 in mouse macrophages and microglia. Cell Immunol. 260(2):113–118. doi: 10.1016/j.cellimm.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 70.Iotti G, et al. Expression of CCL9/MIP-1gamma is repressed by BCR/ABL and its restoration suppresses in vivo leukemogenesis of 32D-BCR/ABL cells. Oncogene. 2007;26(24):3482–3491. doi: 10.1038/sj.onc.1210146. [DOI] [PubMed] [Google Scholar]
- 71.Yang YJ, et al. TRAF6 specifically contributes to FcepsilonRI-mediated cytokine production but not mast cell degranulation. J Biol Chem. 2008;283(46):32110–32118. doi: 10.1074/jbc.M802610200. [DOI] [PubMed] [Google Scholar]
- 72.Hembruff SL, Jokar I, Yang L, Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. 2010;12(5):425–433. doi: 10.1593/neo.10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jessen KA, et al. Molecular analysis of metastasis in a polyomavirus middle T mouse model: the role of osteopontin. Breast Cancer Res. 2004;6(3):R157–169. doi: 10.1186/bcr768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ljung BM, et al. Cell dissociation techniques in human breast cancer--variations in tumor cell viability and DNA ploidy. Breast Cancer Res Treat. 1989;13(2):153–159. doi: 10.1007/BF01806527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.