

Abstract
Background
Synthetic riboswitches have been increasingly used to control and tune gene expression in diverse organisms. Although a set of theophylline-responsive riboswitches have been developed for bacteria, fully functional expression elements mediated by synthetic riboswitches in Bacillus subtilis are rarely used because of the host-dependent compatibility between the promoters and riboswitches.
Results
A novel genetic element composed of the promoter P43 and a theophylline-riboswitch was developed and characterized in B. subtilis. When combined with a P43 promoter (P43′-riboE1), the theophylline-riboswitch successfully switched the constitutive expression pattern of P43 to an induced pattern. The expression mediated by the novel element could be activated at the translational level by theophylline with a relatively high induction ratio. The induction ratios for P43′-riboE1 by 4-mM theophylline were elevated during the induction period. The level of induced expression was dependent on the theophylline dose. Correspondingly, the induction ratios gradually increased in parallel with the elevated dose of theophylline. Importantly, the induced expression level was higher than three other strong constitutive promoters including PsrfA, PaprE, and the native P43. It was found that the distance between the SD sequence within the expression element and the start codon significantly influenced both the level of induced expression and the induction ratio. A 9-bp spacer was suitable for producing desirable expression level and induction ratio. Longer spacer reduced the activation efficiency. Importantly, the system successfully overexpressed β-glucuronidase at equal levels, and induction ratio was similar to that of GFP.
Conclusion
The constructed theophylline-inducible gene expression system has broad compatibility and robustness, which has great potential in over-production of pharmaceutical and industrial proteins and utilization in building more complex gene circuits.
Keywords: Controllable gene expression, RNA switch, Theophylline, Synthetic riboswitches, Bacillus subtilis
Background
Controllable gene expression systems are important for determining the timing and strength of heterologous protein expression. The diversity and availability of easy-to-use biological parts enables the development of sophisticated devices based on tuneable gene expression systems. These devices and systems have been successfully utilized for the overproduction of recombinant proteins as well as for rewiring metabolic pathways [1]. A number of biological elements derived from natural operons, such as the IPTG-induced lac operon and the arabinose-activated araC promoter, have been widely used for conditional gene expression in Escherichia coli and Bacillus subtilis, two commonly utilized hosts in synthetic biology [2, 3]. However, there are still obvious drawbacks when employing these genetic tools in diverse hosts. For example, it has been broadly recognised that protein-based ligand-inducible tools that well functioned in E. coli do not work well in other bacteria, such as B. subtilis.
To overcome the limitations of protein-based gene expression tools, RNA-based elements have increasingly been viewed as superior genetic tools for a range of applications in synthetic biology [4, 5]. One such RNA regulator is the riboswitch, which is an RNA-encoded genetic control element that regulates gene expression in a ligand-dependent fashion without the need for proteins [6]. Riboswitches are composed of an aptamer domain, which recognizes the ligand, and an expression platform, whose conformational change is induced by the ligand binding to the aptamer, which directly modulates transcriptional termination or translational initiation [7, 8]. Ligand-inducible expression systems are essential genetic tools for commonly used bacteria such as E. coli and B. subtilis. Recent reports demonstrated an amino acid-activated riboswitch from B. subtilis that contains tandem glycine aptamers. Heterologous gene expression was activated by the glycine-riboswitch when it was bound to the added glycine [9]. Although the glycine-riboswitch system only resulted in a sixfold induction, it is still a useful model for the development of more riboswitch-dependent gene expression systems and other biological devices.
In addition to natural riboswitches that control gene expression in response to endogenous metabolites, a variety of synthetic riboswitches that respond to non-endogenous small molecules have been developed to control heterologous gene expression in different hosts [10, 11]. Using rational design, the Gallivan’s laboratory developed a set of synthetic riboswitches referred to as A-E that are activated by theophylline rather than the native metabolites. When combined with diverse synthetic ribosome binding sites (RBS), these artificial elements are able to function in various Gram-negative and Gram-positive bacteria to regulate translation [12]. Theophylline, which bears structural and pharmacological similarity to theobromine and caffeine, is a cheap and nontoxic chemical, enabling it to be an ideal inducer for triggering heterologous expression in microbial hosts [12]. The universality and availability of these synthetic riboswitches in other bacteria, such as Mycobacteria, Streptomyces, and Cyanobacteria, has been investigated. The performance of these riboswitches, including their activities and induction efficiency, varied according to the host organism, especially, when different promoters were placed upstream [13]. Therefore, the performance of a given riboswitch relies on different determinants, so no one riboswitch is well-suited to all genetic contexts.
Bacillus subtilis, a low G + C%-containing Gram-positive soil bacterium, has been commonly used as a host strain for producing numerous industrial proteins and fine chemicals because of several characteristics, including its status as generally recognized as safe (GRAS), its high cell density, its well-characterized protein secretion mechanism, the existence of well-established methods for its genetic manipulation, and its applicability for large-scale industrial production [14, 15]. Recent studies have concentrated on using B. subtilis to developed a sophisticated synthetic biology system, which depends on complete genome sequencing, reannotation, and the use of various biological tools for gene manipulation [16]. Although conditional control of gene expression by many protein-based elements in B. subtilis has been well-characterized and routinely used, the activation and tuning of gene expression with these elements depends on addition of expensive chemicals, which limit their scalability for industry application [1]. Recently, a set of synthetic theophylline-riboswitches was developed and optimized to function in diverse bacteria. Among these, the riboswitch E used with a constitutive promoter is reportedly suitable for B. subtilis [12]. However, one report showed that the gene expression level induced by the promoter-riboswitch combination did not consistently correlate with the promoter strength in certain bacteria [17]. This suggests that the compatibility of the riboswitch with specific homologous or heterologous promoters will be broad or narrow, respectively, in B. subtilis and other bacteria. The influence of combing riboswitch with other constitutive promoters on protein expression has not been well characterized until now.
To expand the toolbox for conditional gene expression in B. subtilis, a strong constitutive promoter, P43, was placed upstream of the synthetic riboswitch E1 (derived from the reported riboswitch E [12]). By using green fluorescent protein (GFP) as a reporter gene, riboswitch E1 with P43 was able to achieve inducible expression in B. subtilis. The conditional expression pattern was theophylline-dependent with high stringency over a wide range of concentrations. Moreover, the optimal spacing between the SD sequence and the start codon within the synthetic element was also determined by varying the distance. This resulted in the successful engineering a strong constitutive promoter with an RNA regulator-based element, which could be broadly applied in controlling heterologous gene expression and used as an element for input signal in construction of complex gene circuits.
Results
The strong constitutive P43 promoter was successfully combined with a theophylline-activated expression element
Previously many diverse theophylline-dependent riboswitches were engineered and shown to function in various Gram-positive and Gram-negative bacteria at the translational level [12]. However, it was unclear if these riboswitches were compatible with most constitutive promoters. Usually, constitutive promoters are paired with theophylline riboswitches to achieve controllable and tuneable transcription [17, 18]. Here, a new riboswitch, theophylline riboswitch riboE1 containing a modified SD sequence AAAGGAGG was constructed by modifying the riboswitch E constructed by Topp et al. [12]. RiboE1 was genetically fused to a strong promoter P43, in which the native downstream SD sequence was deficient (P43′), yielding a novel dual expression element P43′-riboE1. The P43′ retains the full upstream sequence including the core region (−35 and −10), the transcriptional start site (TSS), and the 5′UTR between the TSS and the native SD sequence. The synthetic theophylline riboswitch is composed of an aptamer region, a synthetic SD sequence, and a spacer (Fig. 1a). Theoretically, the transcription of the riboE1 control element is triggered by P43 during the cell growth. However, the synthetic SD located downstream of the aptamer is sequestered via pairing with the bases in the stem of the riboswitch, resulting in translational block. The binding of theophylline to the aptamer domain initiates translation by altering the downstream base pairing, which releases the SD. Subsequently, GFP expression begins after the ribosome binding to the SD (Fig. 1b). We compared the cell growth and the expression pattern of GFP between the constitutive expression system driven by P43 and the inducible expression system driven by P43′-riboE1. The growth curve showed that the increase in cell density was similar for both the recombinant strain BSG11 (B. subtilis harbouring pBSG11, inducible expression of GFP) and the BSG43 (B. subtilis harbouring pP43-gfp, constitutive expression of GFP) strain during the culture period (Fig. 1c). This indicates that the recombinant strains BSG11 and BSG43 grow identically.
Fig. 1.
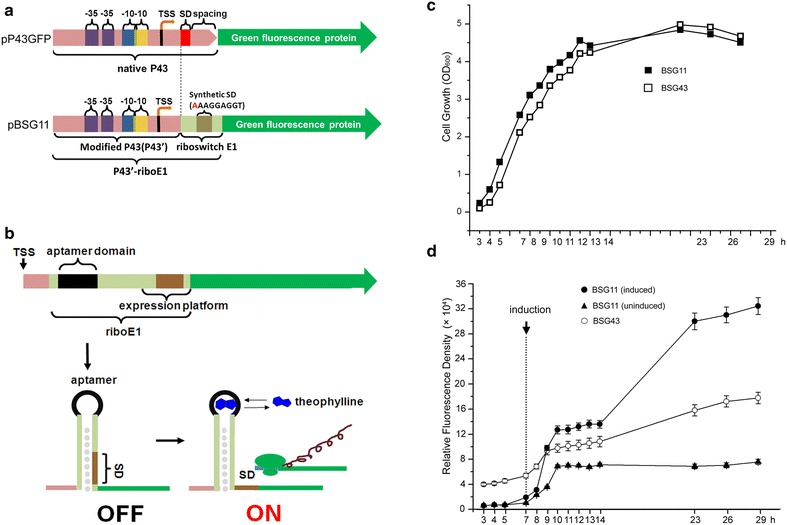
The combination of the novel theophylline-dependent riboswitch E1 and the P43 promoter in Bacillus subtilis. a Diagrams of the expression cassette driven by the native P43 promoter and the novel theophylline-dependent expression element (P43′-riboE1). The green fluorescent protein is represented by the green arrows, and the transcription start site is marked as TSS. The promoter fragment of the novel expression element, P43′, was produced by deletion of the entire sequence downstream of the native P43. The synthetic riboswitch E1 was placed immediately downstream of P43′, resulting in the novel expression element P43′-riboE1. The riboswitch was derived from the riboswitch E (previously reported by Topp et al.), in which a single A was inserted before the 5′ terminus of the original sequence. In the diagram, the inserted nucleotide is coloured red. The dash line denotes the difference between native P43 and the synthetic riboswitch E1 (riboE1). b The schematic diagram of mechanism of the novel genetic element. Theophylline is indicted in blue. c The growth curves of B. subtilis 168 harbouring pP43-gfp (BSG43) and pBSG11 BSG11). The recombinant B. subtilis strains were inoculated by pre-cultures with the initial OD600 of 0.05. The cultures were sampled periodically to measure the cell density until the cell density began to decrease at 27 h. d Expression levels of GFP (y-axis) against time (x-axis) during the culture period. The dashed line denotes the induction by 4 mM theophylline. The GFP fluorescence was measured in triplicates and the data were shown in mean ± SD
Meanwhile, the constitutive expression in BSG43 carrying pP43-gfp and the inducible expression in BSG11 carrying pBSG11 were monitored by measuring the GFP fluorescence intensity in the cultures. The data showed that GFP fluorescence intensity for BSG43 increased gradually during the culture period. After 29 h, the relative fluorescence units reached approximately 180,000 (a.u/OD600). In contrast, the GFP fluorescence in BSG11 displayed very low levels in the absence of theophylline. After culturing for 7 h, 4 mM theophylline was added to the medium, and GFP fluorescence dramatically increased until peaking at approximately 320,000 (a.u/OD600), whereas the BSG11 without induction kept constant low leaky expression levels over the cultural process (Fig. 1d). This indicated that the fusion of the theophylline-dependent riboswitch to the P43 resulted in a controllable expression element.
Characterization of the induction levels and ratios driven by P43′–riboE1 element
To characterize the activated level and efficiency driven by the P43′-riboE1 element, the BSG11 strain was cultivated in LB medium for 7 h prior to treatment with 4 mM theophylline. Figure 2 shows the theophylline-dependent induction of GFP fluorescence after culturing for 24 h. The histograms show that the activated expression levels of GFP and the induction ratios increased gradually during the induction period. After 24 h of induction, the expression level exceeded 350,000. The induction ratios constantly increased during the induction period, and were consistent with the expression levels. Expression due to leakage remained low during the induction period. Finally, the induction ratio peaked at 4.3 after 24 h of induction (Fig. 2).
Fig. 2.

Characterization of induction of GFP from pBSG11 harbouring P43′-riboE1 element after induction by 4-mM theophylline. GFP induction is denoted by green histograms at different time points. The solid circles within in the histograms represent the level of induced GFP expression. Similarly, the open circles within the histograms are the corresponding basal expression level. The activation ratio was obtained by dividing the induced expression level by the corresponding basal level
Theophylline-activated gene expression by P43′-riboE1 was dose-dependenct
To examine whether the activation of GFP by theophylline was dose-dependent, the cultivated BSG11 strain was treated with 4% DMSO (0 mM theophylline) or 1, 2, 4 or 8 mM theophylline for 24 h. Then, the fluorescence was measured to determine the dose-dependency. The GFP expression level increased linearly from 0 to 4 mM and continued to increase for the higher concentration of theophylline (up to 8 mM) (Fig. 3a). In particular, the induction ratios were dramatically elevated with the increasing doses of theophylline and reached 6.8 at 8 mM theophylline dose (Fig. 3b). These results indicated that the higher doses of the inducer elicited higher expression levels and a higher induction ratio. Moreover, the overall expression levels of GFP showed dose-dependence under the control of this riboswitch element. Afterwards, SDS-PAGE analysis was used to examine GFP expression under different concentrations of theophylline. The results showed that the accumulation of GFP within the bacterial cells increased at higher theophylline concentrations, which was consistent with the fluorescence assays (Fig. 3c). These results indicated that the expression level of genes mediated by the P43′-riboE1 element can be controlled with different concentrations of the inducer.
Fig. 3.

The activation of GFP expression by the P43′-riboE1 element displayed dose-dependency. a The expression levels of GFP induced by 1, 2, 4, or 8 mM theophylline were determined 24 h after induction, and were linearly correlated. The experiment was repeated independently in triplicates. b The activation ratios were estimated after induction of 1, 2, 4, or 8 mM theophylline. The ratios were elevated in response to the increased theophylline levels. The highest activation ratio was achieved by 8 mM theophylline, which caused an approximately sevenfolds increase. c SDS-PAGE analysis of with increasing amounts of theophylline. The black solid arrow indicates the GFP band. Twenty micrograms of total protein were loaded for each sample. The GFP fluorescence was measured in triplicates and the data were shown in mean ± SD
Comparison of theophylline riboswitch-controlled gene expression levels with expression driven by other constitutive promoters
To comprehensively compare the expression levels between commonly used strong constitutive promoters and P43′-riboE1, three wild-type promoters P43, PaprE and PsrfA, which have been characterized in previous studies [19, 20], were employed to express the GFP reporter. BSG43, BSG04, and BSG03 carrying the three strong promoters respectively, were cultured in LB for 31 h. The BSG11 harbouring pBSG11 was treated with 8 mM theophylline for 24 h. Then, the final expression level was determined by SDS-PAGE analysis. The GFP expression level controlled by P43′-riboE1 was far higher than expression by PaprE and P43 in the presence of 8-mM theophylline and was equivalent to expression by PsrfA. The basal level of P43′-riboE1 was relatively low, which was consistent with the fluorescence intensity shown in Fig. 2. Interestingly, the final expression level mediated by P43′-riboE1 in the presence of 8 mM theophylline was somewhat higher than expression by P43 (Fig. 4a). To quantitatively determine the differences in the expression levels controlled by the four types of gene expression elements, fluorescence intensity was measured. The fluorescence of GFP driven by wild-type promoters and the activated riboswitch element showed a trend similar to the SDS-PAGE analysis of GFP expression (Fig. 4b). This demonstrated that highly controllable and tuneable gene expression can be achieved by using P43′-riboE1 element.
Fig. 4.
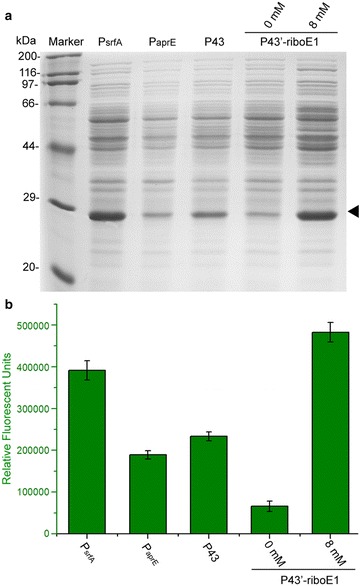
Comparison of the induced expression level with P43′-riboE1 and three strong promoters from B. subtilis. a SDS-PAGE analysis of GFP controlled by the constitutive promoters PsrfA, PaprE, P43 and by the theophylline-induced element P43′-riboE1. The BSG11 strain was activated by 8-mM theophylline for 24 h prior to sampling for SDS-PAGE. b Fluorescence intensity representing the relative expression level was driven by three constitutive promoters as well as by the P43′-riboE1 element after induction by 8-mM theophylline for total 31 and 24-h culture periods, respectively. The GFP fluorescence was measured in triplicates and the data were shown in mean ± SD
A 15-bp spacer between the start codon and the riboswitch element affects induction
To facilitate the cloning of the P43′-riboE1 element into other vectors, it was essential to modularize the element. One method is to insert restriction site either up- or down-stream of the element. Here, we inserted a PstI restriction site immediately downstream of the P43′-riboE1, to produce pBSG10 that contains P43′-riboE1-15 with a 15-bp spacer (Fig. 5a). To compare the effect of the longer spacer on GFP expression, BSG11 and BSG10 were treated with 8 mM theophylline for 12 and 24 h, and the production of GFP was analysed by SDS-PAGE. Obviously, the GFP protein expression was scarcely detected in BSG10 cultures treated with 8 mM theophylline or DMSO treatments in both 12- and 24-h cultures (Fig. 5b). In contrast, GFP expression in BSG11 for 12- and 24-h cultures was substantially higher than in BSG10. Noticeably, the basal expression level in BSG11 also was significantly higher than in BSG10 for both 12- and 24-h cultures (Fig. 5b). Additionally, the GFP fluorescence in BSG10 and BSG11 corresponding to the SDS-PAGE was determined to quantify the expression level. The data were consistent with that measured by SDS-PAGE, which authenticated the difference in heterologous expression level between the two different spacers (Fig. 5c). These results suggested that the failure of P43′-riboE1-15 to induce GFP expression after exposure to theophylline was due to increased spacer sequence length between the SD and the start codon, which probably affects ribosome binding in the translation initiation step [21].
Fig. 5.
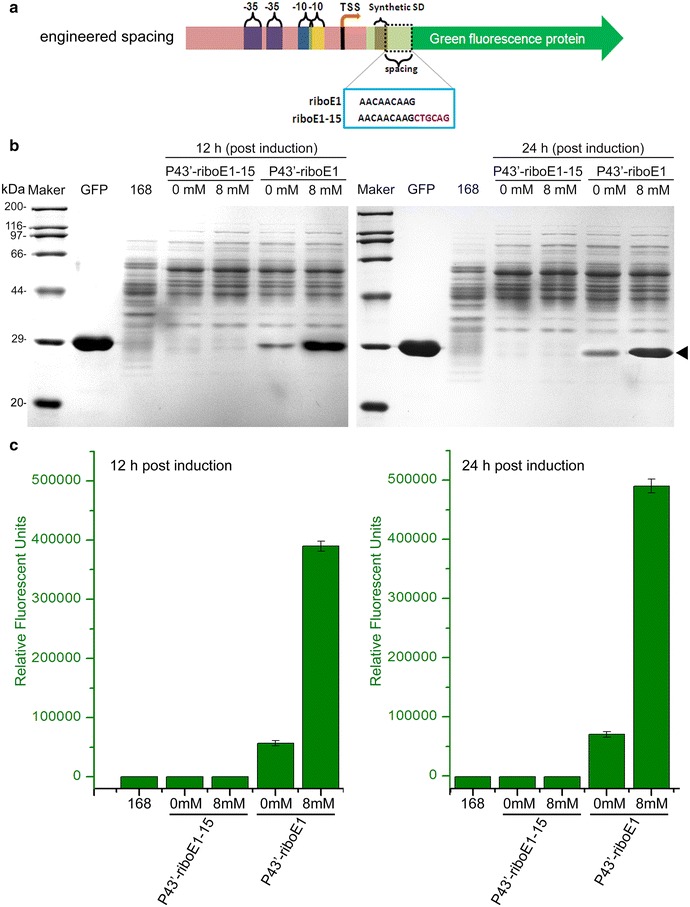
Determination of the effect of length of spacer on the induced level of GFP expression. a Schematic diagram displays the gene structure of riboE1, and riboE1-15 harbouring 15-bp spacer between SD sequence and start codon was produced by insertion of PstI restriction site immediately downstream of the SD sequence. The SD sequences within riboE1 and the modified riboE1-15 are in the azure box. b SDS-PAGE analysis was carried out to detect the expression level of GFP driven by P43′-riboE1 and P43′-riboE1-15 after induction with 8 mM theophylline for both 12 and 24 h. The strains harbouring recombinant plasmids treated with 4% DMSO are designated as 0 mM (controls). Protein extracts from Bacillus subtilis 168, which were collected at 19 and 31-h culture (equal to the induced groups with 12 and 24-h induction, respectively), are used as the negative controls. The GFP stands for the purified GFP protein, which is served as the specific marker to indicate the corresponding position of heterologous GFP. c Measurement of the relative fluorescent units of GFP corresponding to the SDS-PAGE in b
Validation of the novel theophylline-responsive riboswitch element with β-glucuronidase
To test the usefulness and the compatibility of the gene system, the reporter gene gus was used to verify the expression level driven by P43′-riboE1 element. The SDS-PAGE analysis showed some level of gus expression in both the presence and absence of theophylline. The gus was successfully activated at very high level after treatment with 8 mM theophylline for 16 and 20 h. In contrast, the control groups treated with 4% DMSO for the same time showed relatively low GUS expression levels (Fig. 6a).
Fig. 6.
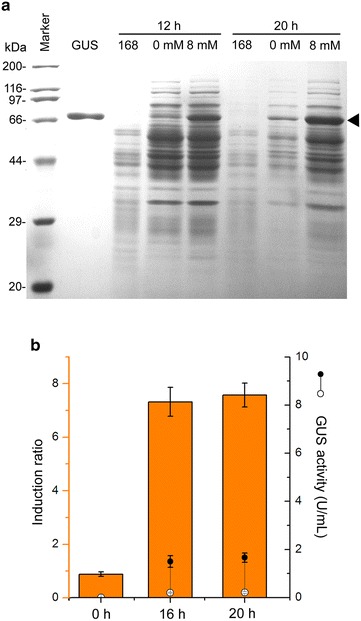
Validation of the theophylline-responsive gene expression system with gus reporter gene. a SDS-PAGE analysis showed the expression level of GUS in the absence and presence of theophylline. The induced expression level of GUS in the recombinant strain, BSGgus, was determined by treatment with 8 mM theophylline for 20 h culture, and the culture treated with 4% DMSO for the same time after induction was designated as controls (0 mM). The solid triangle pointed the overproduced GUS. Protein extracts from B. subtilis 168 sampled simultaneously with the induced recombinant strains was used to be the negative control. GUS, denoted the purified GUS protein, was used to be a specific marker to indicate the bands of GUS on SDS-PAGE. b Enzymatic assay for GUS in BSGgus after induction by 8 mM theophylline for 16 and 20 h. The induced activities of GUS and the levels for leakage are shown by solid and open circles, respectively. The histograms represent the corresponding induction ratios. The enzymatic activity assay was performed in triplicates and the data are presented in mean ± SD
The enzymatic activities and induction ratio of GUS were determined by measuring the GUS activity. The induction ratio in 16- and 20-h induced cultures was 7.3 ± 0.4 and 7.6 ± 0.4 U/mL, and the activated activity of GUS was 1.5 ± 0.2 and 1.7 ± 0.2 U/mL, respectively. Meanwhile, levels for both GUS activity from expression leakage at 16- and 20-h were 0.2 ± 0.03 and 0.3 ± 0.03 U/mL (Fig. 6b), which were rather low compared to that with induction. Approximately equal expression levels in 7-h cultures before the addition of theophylline were observed between the two experimental groups. These results were consistent with those for GFP expression, indicating that the controlled expression system constructed by combining a constitutive promoter and the theophylline-responsive riboswitch, potentially has broad compatibility with numerous heterologous genes and is widely applicable in synthetic biology.
Discussion
Here, a novel controllable expression element composed of an engineered theophylline-dependent riboswitch and the strong constitutive P43 promoter was successfully constructed and characterized in B. subtilis, and inducible heterologous gene expression was efficiently achieved. Compared to the commonly used inducer, IPTG, theophylline is cheaper and nontoxic, which has potential utilization in triggering heterologous gene expression in microbial hosts. The riboswitch E is one of six riboswitches activated by theophylline that has the potential to induce gene expression, but, it has a low induced expression level in B. subtilis [12].
In the Gallivan’s study on fabrication of diverse theophylline riboswitch A–E*, we noticed that the difference in the synthetic SD sequence influenced the induction features in diverse bacteria [12]. Thus, to improve the level of expression in this study, a single adenine base was inserted upstream of the native SD sequence to generate a SD sequence with full complementarity to the ribosome. The highest induction ratio for this novel element was 6.8 after treatment with 8 mM theophylline (Fig. 3b), which was lower than that combined with other promoters in Cyanobacteria [13] and B. subtilis [12]. These unexpected results obviously indicate that the novel riboswitch-mediated element produced higher expression than previously reported, but at the cost of a decreased induction ratio. Riboswitches combined with different promoters display distinct properties in various bacteria [12, 18, 22, 23]. Therefore, it is worthwhile to assess the performance of riboswitch elements in the context of individual genetic backgrounds. Usually, several features such as regulatory efficiency, basal activity, noise, and kinetic parameters are systematically considered under similar experimental settings [24]. The continuous increase in the induction ratios over activation at different times suggested that the induction by P43′-riboE1 functions regularly in the B. subtilis host.
Although it has been shown that the riboswitch A, E* and the riboswitch F derived from the E combined with different promoters are able to function in Streptomyces [25], Cyanobacteria [13], and Mycobacteria [18], which displayed both high induction ratios and high expression levels, it is still unclear whether a single riboswitch is suitable for fabricating expression elements with diverse constitutive promoters that have a consistent induction ratio and similar dynamics. It is difficult to predict the activation characteristics of specific riboswitch-promoter combinations merely from induction data from the same riboswitch fused to other promoters. As the synthetic theophylline riboswitches function by controlling accessibility to the RBS [12, 26], promoters positioned upstream of the theophylline riboswitch-controlling elements, which share the same functional regions with the corresponding wild-type promoters, ought to output the similar rate of transcription to the corresponding wild-type promoters, and result in comparable translational levels. However, the final expression level of GFP in the P43′-riboE1-harbouring strain (BSG11) was higher than that in the P43-harbouring strain (BSG43) (Fig. 4a). Based on a recent report that described the effect of upstream secondary structure within the 5′UTR on translation efficiency [27], we proposed that the aptamer domain of the theophylline riboswitch naturally forming a stem-loop upstream of the SD sequence after transcription augments the accessibility of the ribosome to the SD, eventually producing higher levels of GFP. These results are consistent with previously reported data in which the induced expression levels altered after combining different promoters with the same riboswitch [22, 23].
The capability to control and tune the expression of proteins is an advantageous feature of synthetic riboswitches. Some theophylline riboswitches have been further improved by their combination with different promoters to achieve dose-dependent induced expression levels, allowing for the precise regulation of a gene [13, 28]. The theophylline riboswitch combined with the P43 promoter that was constructed in this study can be induced to different degree across a wide range of concentrations and at the same time produce corresponding induction ratios (Fig. 3). This gives the P43′-riboE1 broad utility for regulating gene expression. Although the P43′-riboE1 expression element produced lower induction ratios than theophylline riboswitch in other bacteria at same concentrations of theophylline, it produced higher levels of expression. In several Cyanobacterial species, theophylline riboswitches combined with native strong promoter ConII led to high induction ratios mediated that were achieved at 2 or 3 mM inducer concentrations [13]. Even in other Gram-positive bacteria, such as Mycobacteria and Streptomyces, high levels of induced gene expression, controlled by riboswitch E* fused to other constitutive promoters, were obtained with treatment of no more than 5 mM theophylline [18, 22, 25].
The length of the spacer between the SD sequence and the start codon has been shown to strongly influence the efficiency of translation initiation [21]. In the Gram-negative bacteria E. coli, only rare mRNAs have spacer lengths shorter than 7 bp and longer than 15 bp. The optimal length is approximately 9 bp [27]. Although the principle is derived from a systematic analysis of E. coli native genes, it might also be suitable to synthetic genetic elements such as riboswitches. A 15 bp length between the SD sequence and the start codon strikingly impaired the expression of GFP, indicating that a longer spacer length decreases the translation initiation efficiency (Fig. 5b). The optimal spacing length occurring in the native mRNAs is reported to be approximately 10 bp long, which improves the efficiency of translation initiation. However, in this study, the desired expression level and activation efficiency were obtained with 9-bp spacer. This inconsistency suggests that the gene expression controlled by a synthetic riboswitch functions in a more complex way in different bacteria. The range of use for this novel expression element was verified by production of the commonly used reporter, gus. The highest induced level and induction ratio of GUS in this study was 1.6 and 7.5 U/mL, respectively. The induction ratio was similar to that calculated for GFP. The leaky expression level corresponding to the highest expression level was 0.3 U/mL with this element, which was significantly lower than that with induction. Lower leaky expression level produced by riboswitch E was also observed in other theophylline-mediated inducible system in Streptomyces coelicolor, in which the leakiness of GUS level is 0.1 GU. However, the induced expression level of GUS in the Streptomyces system was lower than that in our construct [22].
Conclusion
In this work, we constructed an inducible gene expression element composed of theophylline-dependent riboswitch and a constitutive promoter and characterized the induction properties in B. subtilis. Our data demonstrated that the P43 has good compatibility to the engineered riboswitch, which displayed high activation ratio and expression level in a dose-dependent manner. Combination of P43 to the engineered theophylline riboswitch did not decrease the expression but enhanced the expression level. Two reporters, GFP and GUS, were successfully over-expressed in recombinant B. subtilis by activation with theophylline. These results imply that the element reported here was efficient at tuning gene expression to a wide range of levels in B. subtilis. In summary, the novel theophylline-dependent riboswitch fused with strong constitutive promoter is able to efficiently drive gene expression in B. subtilis. The properties of high induction efficiency and controllable expression level are not only of great interest for constructing and designing more complex genetic circuits in B. subtilis but they also provide a more diverse set of genetic tools for synthetic biology.
Methods
Strains and culture conditions
The bacterial strains and plasmids that were used in this study are listed in Table 1. Escherichia coli JM109 was used for cloning and plasmid propagation, and B. subtilis 168 and its derivatives were used for evaluation of the inducible riboswitch. All of the strains were cultured with aeration in Luria–Bertani (LB) at 37 °C. When appropriate, the B. subtilis growth medium was supplemented with kanamycin (5 μg/mL), and the E. coli growth medium was supplemented with ampicillin (10 μg/mL). Cell density was determined by measuring the OD600 with a UV-1800/PC spectrophotometer (MAPADA Instrument Co., Ltd., Shanghai, China). The plasmid pBSG03 was used as a backbone to construct the plasmids containing various riboswitch elements. Plasmid pUC19A harbouring the synthetic riboswitch E1 (hereafter referred to as riboE1) was used as the cloning template and was provided by the Shanghai Sangon Biotech Co., Ltd. (Shanghai, China).
Table 1.
Strains and plasmids used in this study
| Plasmids | Properties and characteristics | Reference |
|---|---|---|
| pBSG03 | PsrfA, gfp, bla, Km r, shuttle vector | [20] |
| pBSG26 | PaprE-HpaII, promoter, BSAP(aminopeptidase), bla, Km r, template for aprE Promoter (PaprE) | [19] |
| pUC57B | pUC19 derivative, PaprE fused with modified theophylline riboswitch E (riboE1, 9-bp spacer), bla | This study |
| pP43-gfp | pBSG03 derivative, ΔPsrfA::P43(from Bacillus subtilis 168), gfp, bla, Km r | This study |
| pBSG04 | pBSG03 derivative, ΔPsrfA::PaprE, gfp, bla, Km r | This study |
| pBSG61 | pBSG03 derivative, ΔPsrfA::PaprE-riboE1 | This study |
| pBSG11 | pBSG61 derivative, ΔPaprE::P43, riboE1, gfp, bla, Km r | This study |
| pBSG10 | pBSG11 derivative, insertion of PstI endonuclease immediately downstream of the 9-bp spacer (15-bp spacer), riboE1-15, gfp, bla, Km r | This study |
| p43E-gus | pBSG11 derivative, Δgfp::gus, riboE1, bla, Km r | This study |
| Strains | ||
| E. coli JM109 | Lab stock | |
| B. subtilis 168 | trpC2 | Lab stock |
| BSG11 | B. subtilis 168 derivative, harbouring pBSG11 | This study |
| BSG10 | B. subtilis 168 derivative, harbouring pBSG10 | This study |
| BSG61 | B. subtilis 168 derivative, harbouring pBSG61 | This study |
| BSG04 | B. subtilis 168 derivative, harbouring pBSG04 | This study |
| BSG43 | B. subtilis 168 derivative, harbouring pP43-gfp | This study |
| BSGgus | B. subtilis 168 derivative, harbouring p43E-gus | This study |
Gene manipulation
Escherichia coli JM109 was used for propagating all plasmids. Inverse-PCR was used to insert or substitute target genes into the plasmid as previously described [20]. DpnI (New England BioLabs) was used to digest the template plasmid after the inverse-PCR reactions. PrimeSTAR® DNA Polymerase (TaKaRa Bio Company, Dalian, China) was used for the PCR reactions. A DNA extraction kit was purchased from TIANGEN Biotech (Beijing) Co., Ltd. (Beijing, China) and was used for plasmid DNA isolation.
Plasmids construction
All of the plasmids used in this study are listed in Table 1. All the primers used in this study were shown in Table 2. The synthetic theophylline riboswitch E [12] (riboE for short) combined with the truncated aprE promoter lacking the downstream sequence of the native SD (termed PaprE’), was previously synthesized and then cloned into pUC19 by Shanghai Sangon Biotech Co., Ltd, the resulting plasmid was called pUC57A. pUC57B, carrying the novel riboswitch element riboE1 (Table 3) with a modified SD sequence containing a single A upstream of the SD sequence from pUC57A, was constructed by whole-plasmid inverse PCR with primers p57F and p57R and used as the template to build the following E. coli–B. subtilis shuttle vector containing the theophylline riboE1. To sub-clone the riboE1-mediated expression element into the E. coli–B. subtilis shuttle vector, the PaprE’-riboE1 fragment and 36-bp flanking sequences with homology to the upstream and downstream regions of srfA promoter (PsrfA) on plasmid pBSG03 were amplified using primers G61F and G61R. This fragment was used as a mega-primer for whole-plasmid inverse PCR to insert the template into plasmid pBSG03 in place of PsrfA [19], yielding shuttle vector pBSG61. The expression level from the native aprE promoter in pBSG04 was compared to expression from PsrfA and P43. The pBSG04 plasmid was constructed in two steps. First, the native PaprE was amplified from pBSG26 using primers G04F and G04R, resulting in a 573-bp fragment and 36-bp flanking sequences homologous to the up- and down-stream sequences of PsrfA from pBSG03. The fragment was inserted into pBSG03 using aforementioned inverse-PCR method. This resulted in the substitution of the PsrfA with PaprE and yielding pBSG04. Similarly, the plasmid pBSG11 harbouring the P43 promoter combined with riboE1 was constructed in two steps. In the first step the P43′ (lacking the downstream SD sequence) and 36-bp flanking sequences homologous to PaprE’ from pBSG61 were amplified. Next, inverse PCR was used to substitute the PaprE’ upstream of the riboE1 with the P43 fragment. Plasmid pBSG10, which contains a longer (15-bp) spacer between the synthetic SD and the start codon relative to pBSG11 (Table 3), was constructed by inserting of a PstI restriction site upstream of the start codon in pBSG11 by inverse PCR with the G10F and G10R primers. To compare the induced systems with constitutive system, pP43-gfp was constructed by substituting the PsrfA on pBSG03 by two steps. Firstly, native P43 from Bacillus subtilis 168 flanking 20-bp homologous sequence up- and down-stream of PsrfA on pBSG03 was amplified with primers pP43F and pP43R. Next, PsrfA on pBSG03 was replaced by P43 by inverse PCR using the purified amplified product in first step. To test the ability of the riboswitch element P43′-riboE1 to regulate the production of target proteins, the gene for the β-glucuronidase (GUS, EC: 3.2.1.31) reporter from E. coli JM109 was amplified using the primers E-gus F and E-gus R that contained the flanking sequences homologous to the upstream and downstream of gfp sequences in plasmid pBSG11. Then, the PCR product was purified and was used to replace the GFP sequence by inverse PCR, to produce p43E-gus.
Table 2.
Primers used in this study
| Primer name | Sequence (5′–3′) |
|---|---|
| G61F | CAAAACCCCCCTTTGCTGAGGTGGCAGAGGGCAGGTTGCCGAATTCCATGAACGAGACTTAAAAa |
| G61R | GACAACTCCAGTGAAAAGTTCTTCTCCTTTACTCATCTGAGCAGGGTGCTTGTTGTTACCTCCTTa |
| G04F | CAAAACCCCCCTTTGCTGAGGTGGCAGAGGGCAGGTTGCCGAATTCCATGAACGAGACTTAa |
| G04R | GACAACTCCAGTGAAAAGTTCTTCTCCTTTACTCATTCGGTTCCCTCCTCATTTTTATACCa |
| G10F | CCCTGCTAAAGGAGGTAACAACAAGCTGCAGATGAGTAAAGGAGAAGAACTTTTCAb |
| G10R | TGAAAAGTTCTTCTCCTTTACTCATCTGCAGCTTGTTGTTACCTCCTTTAGCAGGGb |
| E-gus F | CCCTGCTAAAGGAGGTAACAACAAGATGTTACGTCCTGTAGAAACCCCAACCCG |
| E-gus R | CAGATGCGTAAGGAGAAAATACCGCCTACCGGCCGCATAGGCCTTGTTTGCCTCCCTGCT |
| p57F | TGGCAGCACCCTGCTAAAGGAGGTAACAACAAGATG |
| p57R | CATCTTGTTGTTACCTCCTTTAGCAGGGTGCTGCCA |
| pP43F | TGAGGTGGCAGAGGGCAGGTTGATAGGTGGTATGTTTTCGCTTGAAC |
| pP43R | AGTTCTTCTCCTTTACTCATGTGTACATTCCTCTCTTACCTATAATGG |
aThe underlined bold characters indicate the flanking sequence homologous to their respective template plasmid
bThe underlined italic characters denote the inserted PstI restriction site
Table 3.
The sequences of constructed theophylline riboswitches
| Theophylline riboswitches | Sequencesa |
|---|---|
| riboE1 | ATACGACTCACTATA GGTGATACCAGCATCGTCTTGATGCCCTTGGCAGCACCCTGCTAAAGGAGGTAACAACAAG |
| riboE1-15 | ATACGACTCACTATA GGTGATACCAGCATCGTCTTGATGCCCTTGGCAGCACCCTGCTAAAGGAGGTAACAACAAGCTGCAG b |
aItalic constant sequences; bolditalic theophylline riboswitch
bUnderline inserted PstI site
GFP expression by an engineered theophylline riboswitch E combined with P43
A single clone of B. subtilis 168 harbouring recombinant plasmids was plated on agar plates and individual colonies were inoculated into the test tube containing LB medium and cultured overnight at 37 °C with 200-rpm shaking. Then, the fresh culture was inoculated into 250-mL conical flasks containing 30-mL LB media and kanamycin (50 μg/mL), the initial OD600 was adjusted to 0.05, and the 250-mL flasks were incubated at 37 °C in the shaker at 200 rpm. To monitor the cell growth and reporter gene activation, cultures of B. subtilis harbouring pP43-gfp and pBSG11 were collected periodically before and after activation with 4 mM theophylline. To characterize the induction of P43′-riboE1, recombinant B. subtilis BSG11 harbouring pBSG11 were treated with 4 mM theophylline after 7 h in culture. Non-induced BSG11 was used as control to determine the expression level of leakage. Afterwards, the cultures were sampled periodically. To characterize the dose-dependency of BSG11, different concentrations of theophylline (1, 2, 4 or 8 mM) were added to the medium and the cultures were incubated for 24 h. Then, the recombinant bacterial cells were collected and the relative fluorescence intensities were measured with a microplate reader. To compare the expression levels from pBSG11 and pBSG10, the corresponding recombinant strains were treated with 8 mM theophylline for 12 and 24 h. Cultures treated with 4% DMSO were designated as controls.
Reporter gene assay
The fluorescence measurements were conducted in accordance with a previously reported procedure, with some modifications [20]. In brief, the cultured recombinant B. subtilis cells were collected by centrifugation at 4000 rpm for 5 min and then the supernatant was discarded. The cell pellets were washed with potassium phosphate buffer (PBS, 137 mM NaCl, 2.7 mM KCl, 8 mM Na2PO4 2H2O, 1.76 mM KH2PO4, adjust to pH 7.4 with 1 N HCl) three times. Then, the cell pellets were re-suspended in PBS with appropriate dilution, after which 200-μL aliquot of each sample was transferred to the wells of a black-walled transparent bottomed 96-well plate. The fluorescence intensity (a.u) and cell density (OD600) were simultaneously detected using an Infinite M200 microplate reader (Tecan Group Ltd, Männedorf, Switzerland), with excitation at 495/9 and emission at 535/9. Relative fluorescence units (a.u/OD600) were obtained by dividing the measured fluorescence intensity by the corresponding OD600 value, and were used to represent the level of GFP expression.
SDS-PAGE analysis
A volume of 1 mL of the recombinant B. subtilis cells were harvested by centrifugation at 5000 rpm for 3 min. Then, equal volumes of PBS were added into the 1.5-mL tube and vortexed for 30 s prior to discarding the supernatant after centrifugation again under the same setting. Subsequently, the pellet was thoroughly suspended in 200-μL lysis buffer (100 mM PBS with 5 μg/mL lysozyme), then incubated at 37 °C for 10–15 min. Afterwards, the cells were destroyed completely by ultra-sonication. Then, the lysates were subjected to centrifugation at 14,000 rpm and 4 °C for 25 min. Then, 120-μL supernatant was pipetted and mixed with 30-μL 5× loading buffer. After the determination of the concentration of the extracted proteins by the Bradford method, equal amounts of total proteins were loaded onto the SDS-PAGE to analyse the expression levels.
β-glucuronidase (GUS) enzymatic activity assay
To test the stability and compatibility of expression from the constructs, recombinant B. subtilis BSGgus was used for the determination of the expression level and enzymatic activity of GUS. BSGgus was cultured as described above for the GFP assays. The cultures were sampled periodically after induction by 8 mM theophylline. Samples were washed with PBS buffer three times and then lysed by the addition of equal volumes of lysis buffer at 37 °C for 30 min. After centrifugation at 4 °C for 20 min, the supernatants were collected and diluted to the appropriate concentration for the enzymatic activity assay. For quantification of the product, the standard curve was firstly plotted by measuring the absorbance of a concentration gradient (1, 2, 3, 4, 5, 6, 7 and 8 μg) of p-nitrophenol (Sigma-Aldrich, Shanghai, China) at 405 nm. Afterwards, the reactions were carried out in a 96-well plate and each reaction contained the following components: 50 μL of sample; 50 μL of the substrate 4-nitrophenyl β-d-glucuronide (PNPG, 0.5 mg/mL, Sigma-Aldrich, Shanghai, China). The reactions were incubated at 37 °C for 5 min, followed by the addition of 100 μL Na2CO3 (1 M) to stop the reaction. Then, the absorbance was measured at 405 nm using an Infinite M200 microplate reader. One unit is defined as the amount of enzyme that catalyses the production of 1 μmol of p-nitrophenol within 1 min. The enzymatic assay was independently performed in triplicates and the data are presented as the mean ± SD.
Authors’ contributions
WJC conceived the idea, planned the project, performed the vast majority of experiments analysed the data and wrote the manuscript. LCH and JTC performed the part of reporter gene assay and processing the data. ZML, JLG and LZ performed the part of plasmid clone and analysed the results. ZMZ analysed part of the protein determination data and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Funding
This study was supported by National High-tech Research and Development Project (863 Project, 2014AA021304), the National Natural Science Foundation of China (31400058, 31300087, 31400078), the Natural Science Foundation of Jiangsu Province (BK20130139, BK20130131, BK20140151).
Abbreviations
- SD
Shine–Dalgarno
- riboE1
riboswitch E1
- PsrfA
srfA promoter
- PaprE
aprE promoter
- PaprE’
aprE promoter lacking native SD sequence
- P43′
P43 promoter lacking native SD sequence
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LB
Luria–Bertani medium
- GUS
β-Glucuronidase
- GFP
green fluorescent protein
- DMSO
dimethyl sulfoxide
- PNPG
β-d-glucuronide
- bla
β-Lactamases
- Kmr
kanamycin resistance gene
Contributor Information
Wenjing Cui, Email: wjcui@jiangnan.edu.cn.
Laichuang Han, Email: hanlaichuang@163.com.
Jintao Cheng, Email: 6140203011@vip.jiangnan.edu.cn.
Zhongmei Liu, Email: zliu@jiangnan.edu.cn.
Li Zhou, Email: lizhou@jiangnan.edu.cn.
Junling Guo, Email: guojunling@jiangnan.edu.cn.
Zhemin Zhou, Phone: +86 510 8532 5210, Email: zhmzhou@jiangnan.edu.cn.
References
- 1.Topp S, Gallivan JP. Emerging applications of riboswitches in chemical biology. ACS Chem Biol. 2010;5(1):139–148. doi: 10.1021/cb900278x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen PT, Shaw JF, Chao YP, David Ho TH, Yu SM. Construction of chromosomally located T7 expression system for production of heterologous secreted proteins in Bacillus subtilis. J Agric Food Chem. 2010;58(9):5392–5399. doi: 10.1021/jf100445a. [DOI] [PubMed] [Google Scholar]
- 3.Khlebnikov A, Risa O, Skaug T, Carrier TA, Keasling JD. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J Bacteriol. 2000;182(24):7029–7034. doi: 10.1128/JB.182.24.7029-7034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isaacs FJ, Dwyer DJ, Collins JJ. RNA synthetic biology. Nat Biotechnol. 2006;24(5):545–554. doi: 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- 5.Benenson Y. Synthetic biology with RNA: progress report. Curr Opin Chem Biol. 2012;16(3–4):278–284. doi: 10.1016/j.cbpa.2012.05.192. [DOI] [PubMed] [Google Scholar]
- 6.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 7.Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15(3):342–348. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Berens C, Suess B. Riboswitch engineering—making the all-important second and third steps. Curr Opin Biotechnol. 2015;31:10–15. doi: 10.1016/j.copbio.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 9.Phan TT, Schumann W. Development of a glycine-inducible expression system for Bacillus subtilis. J Biotechnol. 2007;128(3):486–499. doi: 10.1016/j.jbiotec.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Desai SK, Gallivan JP. Genetic screens and selections for small molecules based on a synthetic riboswitch that activates protein translation. J Am Chem Soc. 2004;126(41):13247–13254. doi: 10.1021/ja048634j. [DOI] [PubMed] [Google Scholar]
- 11.Suess B, Fink B, Berens C, Stentz R, Hillen W. A theophylline responsive riboswitch based on helix slipping controls gene expression in vivo. Nucleic Acids Res. 2004;32(3):1610–1614. doi: 10.1093/nar/gkh321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topp S, Reynoso CM, Seeliger JC, Goldlust IS, Desai SK, Murat D, et al. Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl Environ Microbiol. 2010;76(23):7881–7884. doi: 10.1128/AEM.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma AT, Schmidt CM, Golden JW. Regulation of gene expression in diverse cyanobacterial species by using theophylline-responsive riboswitches. Appl Environ Microbiol. 2014;80(21):6704–6713. doi: 10.1128/AEM.01697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189. doi: 10.1016/S0065-2164(07)62006-1. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Liu YF, Shin HD, Chen RR, Wang NS, Li JH, et al. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Appl Microbiol Biotechnol. 2013;97(14):6113–6127. doi: 10.1007/s00253-013-4960-4. [DOI] [PubMed] [Google Scholar]
- 16.Harwood CR, Pohl S, Smith W, Wipat A. Bacillus subtilis model Gram-positive synthetic biology chassis. Methods in microbiology. Microbial synthetic biology. Oxford: Academic Press; 2013. pp. 87–117. [Google Scholar]
- 17.Van Vlack ER, Seeliger JC. Using riboswitches to regulate gene expression and define gene function in Mycobacteria. Methods Enzymol. 2015;550:251–265. doi: 10.1016/bs.mie.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seeliger JC, Topp S, Sogi KM, Previti ML, Gallivan JP, Bertozzi CR. A riboswitch-based inducible gene expression system for Mycobacteria. PLoS ONE. 2012;7(1):e29266. doi: 10.1371/journal.pone.0029266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan C, Cui W, Cheng J, Liu R, Liu Z, Zhou L, et al. Construction of a highly active secretory expression system via an engineered dual promoter and a highly efficient signal peptide in Bacillus subtilis. New Biotechnol. 2016;33(3):372–379. doi: 10.1016/j.nbt.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Guan C, Cui W, Cheng J, Zhou L, Guo J, Hu X, et al. Construction and development of an auto-regulatory gene expression system in Bacillus subtilis. Microb Cell Fact. 2015;14(1):150. doi: 10.1186/s12934-015-0341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vellanoweth RL, Rabinowitz JC. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol. 1992;6(9):1105–1114. doi: 10.1111/j.1365-2958.1992.tb01548.x. [DOI] [PubMed] [Google Scholar]
- 22.Rudolph MM, Vockenhuber MP, Suess B. Conditional control of gene expression by synthetic riboswitches in Streptomyces coelicolor. Methods Enzymol. 2015;550:283–299. doi: 10.1016/bs.mie.2014.10.036. [DOI] [PubMed] [Google Scholar]
- 23.Nakahira Y, Ogawa A, Asano H, Oyama T, Tozawa Y. Theophylline-dependent riboswitch as a novel genetic tool for strict regulation of protein expression in cyanobacterium Synechococcus elongatus PCC 7942. Plant Cell Physiol. 2013;54(10):1724–1735. doi: 10.1093/pcp/pct115. [DOI] [PubMed] [Google Scholar]
- 24.Berens C, Groher F, Suess B. RNA aptamers as genetic control devices: the potential of riboswitches as synthetic elements for regulating gene expression. Biotechnol J. 2015;10(2):246–257. doi: 10.1002/biot.201300498. [DOI] [PubMed] [Google Scholar]
- 25.Rudolph MM, Vockenhuber MP, Suess B. Synthetic riboswitches for the conditional control of gene expression in Streptomyces coelicolor. Microbiology. 2013;159(7):1416–1422. doi: 10.1099/mic.0.067322-0. [DOI] [PubMed] [Google Scholar]
- 26.Lynch SA, Desai SK, Sajja HK, Gallivan JP. A high-throughput screen for synthetic riboswitches reveals mechanistic insights into their function. Chem Biol. 2007;14(2):173–184. doi: 10.1016/j.chembiol.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osterman IA, Evfratov SA, Sergiev PV, Dontsova OA. Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 2013;41(1):474–486. doi: 10.1093/nar/gks989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynoso CM, Miller MA, Bina JE, Gallivan JP, Weiss DS. Riboswitches for intracellular study of genes involved in Francisella pathogenesis. MBio. 2012;3(6):e00253. doi: 10.1128/mBio.00253-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.