

Abstract
Endometrial carcinoma is one of the most common cancers in women. A limited number of endometrial carcinoma cell lines are available for studies of signal transduction pathways and experimental therapeutics in vitro. However, these cell lines have not been comprehensively characterized. In this study, we used genome-wide microarray-based comparative genomic hybridization (aCGH) technology to characterize five of the more commonly used endometrial cancer cell lines. We detected DNA copy–number gains in chromosomal regions 2q, 3p, 3q, 5q, 7p, 17q, and19q in all five cell lines. Other common sites of copy–number gains, which were detected in four of five cell lines, included segments of chromosomes 1, 6, 8, 9, 11, 12, and 16. In all five cell lines, we found DNA copy–number losses in regions 3p, 10p, 10q, 11q, 11p, 14q, 15q, 18p, and 21q. Other common sites of genetic aberrations included segments of chromosomes 1, 2, 4, 5, 6, 16, 20, and 22. The genes involved in the copy-number alterations included the oncogenes PIK3CA (3q26.3), K-ras (12p12.1), R-ras (19q13.3-qter), Raf-1 (3p25), EGFR (7p12), Akt1 (14q32.32), and Akt2 (19q13.1-q13.2). A pathway analysis showed that genes in the PI3K and Wnt pathways are commonly affected. Our characterization of genomic alterations in these five commonly used endometrial cancer cell lines provides valuable genomic information for research that focuses on these key oncogenic pathways in endometrial cancer.
Keywords: Endometrial cancer, Amplification, Deletion, Pathway, aCGH
Introduction
Endometrial carcinoma (EC) is the leading cancer of the female genital tract in the United States and the fourth most common cancer among women after breast, lung, and colorectal cancer. In the United States, 42,160 new cases and 7,780 deaths from EC are expected in 2009 (1). EC is commonly classified into two categories: type I estrogen-dependent EC and type II nonestrogen-dependent EC. The majority of ECs are type I (approximately 70% to 80%), which generally have low-grade endometrioid histologies, often arising in a background of endometrial hyperplasia. In contrast, the less common type II ECs often arise in a relatively atrophic endometrium and are characterized by nonendometrioid histologies and a more aggressive clinical course (2). On the basis of their distinct clinico-pathological characteristics, the molecular alterations in these two types of EC appear to be different. Chromosomal abnormalities, including DNA copy–number gains and/or losses are hallmarks of cancers (3). In a comparative genomic hybridization (CGH) study of 98 cases of EC, Levan et al., (4) reported frequent amplifications in the chromosomal regions 1q25-42, 19pter-p13.1, 19q13.1-q13.3, 8q21-22, 10q21-q23, and 10p, and frequent losses in the regions 4q22-qter, 16q21-qter, and 18q21-1ter. Another CGH analysis of 43 human primary ECs revealed gains at 1q25-q41, 8q11.1-q21.1, and 8q21.3-qter, whereas the most frequently detected loss was at 16q11.2-q22 (5).
At the gene level, the most frequent genetic alteration of type I ECs is PTEN inactivation (mutation), followed by microsatellite instability (MSI), and mutations of K-ras and ß-catenin; activation of the PI3K pathway is common as well. Mutation of PIK3CA is seen in 36% of type I ECs and is most common in tumors with PTEN mutations (6). Oda et al., (7) reported that PIK3CA mutations coexisted with K-ras and PTEN mutations in EC, and mutant levels of p-AKT (Ser473) induced by mutant Ras or knockdown of PTEN were dramatically increased by addition of mutant PIK3CA. Catasus et al., showed that PIK3CA mutations occurred in 29% (32/109) of the endometrioid adenocarcinomas they studied, and all had myometrial invasion (8). Recently, the authors also found that EC patients with a deregulated PI3K/AKT pathway (exon 20 PIK3CA and/or PTEN mutation) and p53 alterations had worse prognosis than patients with only p53 alterations (9). Conversely, p53 mutation is the most frequent genetic alteration in the more aggressive type II EC. Other frequent events in type II ECs include inactivation of p16, loss of E-cadherin, and amplification of human epidermal growth factor receptor 2 (HER2)/neu (10–14). Identification of gene amplifications has critical implications for the development of targeted therapeutics. The recent discovery of frequent PIK3CA gene mutations in ECs has led to translational investigations of whether blocking the PI3K pathway is a viable approach for the treatment of ECs.
The current paradigm of cancer translational research relies heavily on the use of cancer cell lines derived from patients. A loss-of-function approach, using small interfering RNA (siRNA) or small-molecule inhibitors is commonly used to block the suspected oncogenic targets that are often amplified, mutated, and/or overexpressed in cancer. A gain-of-function approach by transfection of an expression vector is commonly used to investigate the potential tumor-suppressing function of genes that are commonly deleted in cancers. Therefore, characterization of in vitro cell model systems is important for the selection of the appropriate cell lines for future investigations. In this report, we describe the results of a comprehensive analysis of five commonly used EC cell lines by array CGH (aCGH) to identify the most commonly occurring gene copy–number aberrations. We also discuss our findings regarding these gene aberrations in relation to the PI3K/Akt, Wnt/β-catenin, and other important cancer-related pathways, which may be potential candidates for therapeutic interventions.
Materials and Methods
Cell Culture
The EC cell lines AN3CA (metastatic undifferentiated EC), ECC-1 (well-differentiated adenocarcinoma), Ishikawa (well-differentiated adenocarcinoma), HEC1A, and HEC1B (moderately well-differentiated adenocarcinoma) were used in this study. All the cell lines were obtained from ATCC. HEC1A was maintained in McCoy’s 5A medium, while the other four cell lines were maintained in Eagle’s minimum essential medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2.
Array CGH
Genomic DNA was extracted from 5 × 106 cells using the Qiagen DNA/RNA Prep Kit according to the manufacturer’s instructions (Valencia, CA). Labeled genomic DNA was hybridized to a human whole-genome CGH microarray (4 × 44 k; Agilent Technologies, Palo Alto, CA). More than 43,000 coding and noncoding human sequences were represented in these arrays, yielding an average 35-kbp oligonucleotide probe spatial resolution. At least one target sequence was measured for every well-characterized gene, and known cancer genes were measured using at least two probes. The probe design was based on The University of California Santa Cruz hg17 human genome (National Center for Biotechnology Information, NCBI Build 35).
Data Analysis
Data were extracted from microarrays with Agilent’s feature-extraction software (version 9.5; Santa Clara, CA) using the default settings. The intensity values were median-normalized, and ratios of normalized intensity values from EC cell line–derived and normal control genomic DNA were transformed to log2 space. The log ratio data were then subjected to a circular binary segmentation algorithm to reduce the effect of noise (15). A CGH call algorithm was used to label segments of constant copy number as either a gain or a loss (16). As a result of this procedure, each target was given an aberration label of “normal,” “deletion,” or “amplification.”
DNA sequences were classified as recurrent aberrations if the number of aberration labels given to them exceeded a threshold of statistical significance. A permutation test was applied to estimate the value of this threshold. The aberration labels of the targets were permutated, and sums of the gain and loss labels were computed for each target. Gains and losses were considered separately in this procedure; therefore, both genomic states had different statistical significance thresholds. The 99th percentile values for both sums were chosen as thresholds of significance. Contiguous DNA sequences whose true aberration-label counts exceeded the significance threshold formed segments of recurrent aberrations. These segments varied in length from one gene to a whole chromosome. A mean of the aberration counts for sequences in a recurrent aberration segment (probe average recurrence) was computed as a rough measure of the recurrence rate for the segment. All of the aCGH data will be deposited in the GEO (Gene Expression Omnibus) database.
Results
Detection of Common DNA Copy–Number Aberrations in Five EC Cell Lines
Genomic DNA was isolated from the five EC cell lines and used for whole genome aCGH. The global patterns of gene copy– number aberrations of the five EC cell lines are shown in Figure 1. Among the five cell lines, the HEC1A and HEC1B cell lines exhibited more observable alterations than the other three. Because all five EC cell lines were derived from type I ECs, which are known to be mostly diploid, the global aCGH patterns of these cell lines appear to be consistent with what is known about type I primary ECs. Nevertheless, gene copy–number aberrations were clearly observed from the aCGH analysis.
Figure 1.
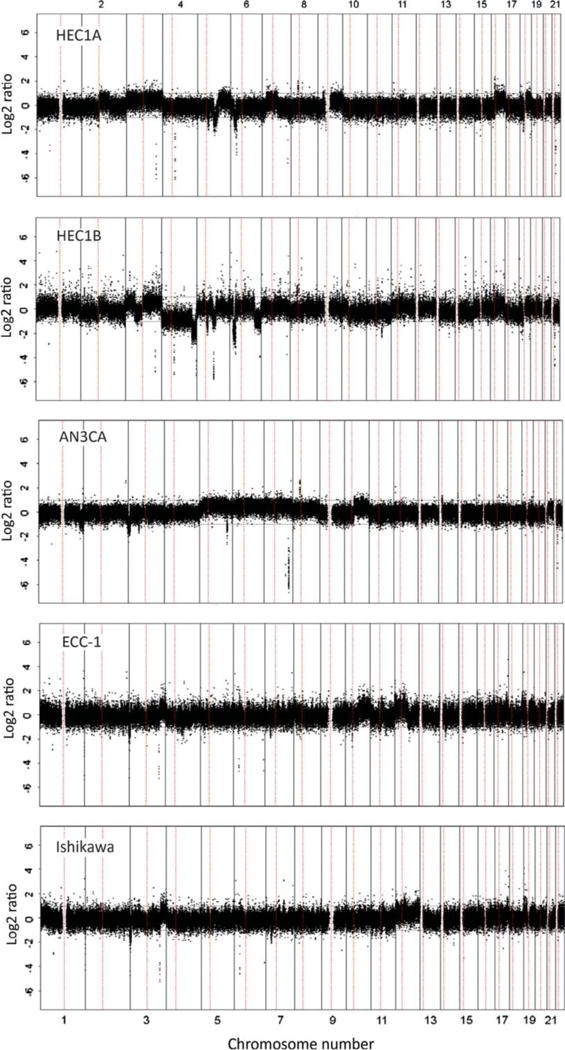
Array-based comparative genomic hybridization profile of five endometrial carcinoma cell lines. The chromosomes are aligned sequentially on the x-axis and are demarcated by the vertical lines. The y-axis shows chromosomal regions of either gain (positive numbers) or loss (negative numbers) relative to normal control genomic DNA isolated from healthy subjects.
We first sought to identify the most frequently altered regions in either all five or in four of the five cell lines. The result of the frequency analysis is illustrated in Figure 2 and summarized in Table I. In all five EC cell lines, chromosomal regions 2q37.1, 3p21, 3q29, 5q23, 5q31, 7p15, 17q12-q25, and 19q13 were significantly amplified, whereas regions 3p12-14, 10p11, 10q11, 11p11, 11q11, 11q23, 14q31, 15q25, 18p11, and 21q11 showed significant deletions. These regions included 67 genes with amplifications and 45 genes with deletions (Table I). Among the amplified genes were R-Ras and PIK3CA (in four of five lines), representing two pathways known to be activated in EC. The Wnt pathway gene Wnt8A was also amplified in all cell lines, as were the cancer stem cell–related ALDH gene family members ALDH7A1 and ALDH16A1.
Figure 2.
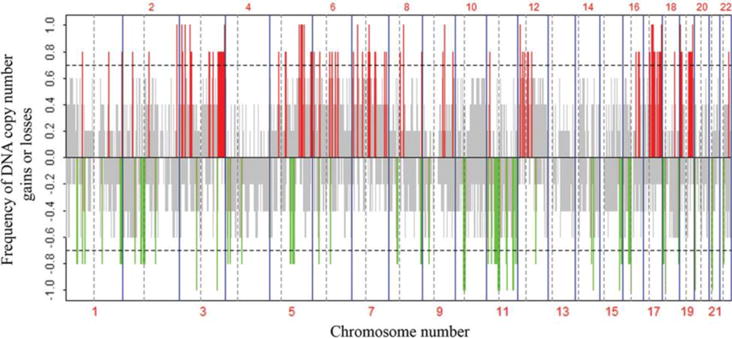
Frequency analysis of DNA copy–number alterations in all five endometrial carcinoma cell lines by array-based comparative genomic hybridization. The y-axis shows the recurrence of gains or losses for each measured sequence, which are aligned evenly in chromosomal order on the x-axis. The horizontal dashed line indicates the threshold for a significant number of aberrations. Red lines indicate a significant frequency of DNA copy–number gains and green lines indicate a significant frequency of DNA copy–number losses. The gray color represents nonsignificant recurrence of aberrations. Borders between chromosomes are indicated by vertical bars, and those between the short arm and long arm of a chromosome are indicated by vertical dashed bars.
Table I.
Consensus regions of genomic imbalance in five EC cell lines.
| Cytogenetic location | Genes |
|---|---|
| Regions of gain | |
| 2q37.1 | NCL |
| 3p21.31 | DHX30 |
| 3p24 | TOP2B |
| 3q29 | ATP13A3, DLG1, GP5 |
| 5q23.2 | C5orf48, RNUXA |
| 5q23.3-q31.1 | LMNB1 |
| 5q31 | ALDH7A1, BRD8, C5orf5, CDC23, CDC25C, FAM53C, KDM3B, KIF20A, NME5, REEP2, TAF7, WNT8A, ERD8 |
| 5q31.1-q31.3 | EGR1, ETF1, HSPA9, GFRA3 |
| 5q32 | LARS |
| 7p15 | HNRNPA2B1 |
| 17q12-q23.2 | CDC27 |
| 17q21-q22 | TOP2A |
| 17q25 | FASN |
| 19pter-q12 | EEF2 |
| 19q13 | SLC17A7 |
| 19q13.1-q13.4 | SLC6A16 |
| 19q13.3 | FCGRT, BCL2L12, CD37, FCGRT, FLT3LG, HRC, KCNA7, LIN7B, PRMT1, RPL13A, RPS11, RUVBL2, SNRP70, TEAD2 |
| 19q13.32 | CGB1, CGB |
| 19q13.33 | PRR12, ALDH16A1, CPT1C, CCDC155, NOSIP, PIH1D1, PPFIA3, PRRG2, RCN3, PTH2, TRPM4, AK097351, DKKL1 |
| 19q13.3-q13. | IRF3 |
| 19q13.3-q13.4 | SCAF1 |
| 19q13.3-qter | RRAS |
| chr19 | AK126060 |
| Regions of loss | |
| 3p12.3 | ZNF717 |
| 3p14.1 | LOC285300 |
| 10p11.1 | HSD17B7P2, CDC10L, LOC100129055, ZNF25 |
| 10p11.2 | ZNF33A, ZNF37A |
| 10p11.21 | ANKRD30A, ZNF248 |
| 10q11.1 | LOC283027 |
| 10q11.2 | ZNF33B, ZNF37B, LOC728064, LOC84856, CCNYL2, BMS1 |
| 11p11.12 | LOC441601, LOC440040, LOC646813 |
| 11p11.12-q12 | TRIM49 |
| 11p11.2 | FOLH1 |
| 11q11 | OR5G5P, OR4A16, TRIM48 |
| 11q12.1 | OR9Q1 |
| 11q23.1-23.2 | NNMT, CADM1, LOC283143 |
| 14q31 | NRXN3 |
| 15q25 | NTRK3, NCRNA00052 |
| 18p11 | ANKRD30B, ANKRD20A5, FLJ44255, CXADRP3, POTEC, ZNF519 |
| 21q11.2 | C21orf99 |
| chr3 | BC019327 |
| chr10 | AL833559, BC035410 |
| chr11 | AB231735, AK096987 |
| chr15 | AL109696 |
The list of commonly deleted genes did not reveal the best-known tumor suppressor genes, such as TP53, p16Ink4a, Rb, and PTEN. Since these cell lines are derived from type I endometrioid tumors, these results were not unexpected. In type I EC, loss of PTEN is most frequently due to mutation. The lack of deletion of these tumor suppressors may indicate that these cell lines, despite being passaged in vitro many times, tend to stay true to their parent tumors.
Alterations of Genes in Key Pathways Associated with EC Tumorigenesis
Examination of the most commonly altered genes sheds light on the potential key drivers of tumorigenesis; however, cancer is also known for its heterogeneity and complex oncogene interactions in a pathway context. In other words, different genes in a common pathway may be altered in different cancers but the effects can be similar. Therefore, we next performed a pathway-centric analysis and examined the genes involved in the best-characterized oncogenic pathways among the five EC cell lines.
Activation of the PI3K/Akt pathway plays an important role in EC, and PIK3CA is mutated in 24% to 39% of type I ECs (6, 7, 17, 18). Our analysis showed that many genes in this pathway, in addition to PIK3CA, were amplified in EC cell lines (Table II). Interestingly, Akt1 and Akt2 were co-amplified in the ECC-1 and Ishikawa cell lines, whereas Akt3 was deleted in these two cell lines. Akt1 and AKt3 were also deleted in the metastatic AN3CA line, whereas there was no significant aberration in Akt2. Overall, the ECC-1 and Ishikawa lines had amplification of many more PI3K/Akt–pathway genes than the other three cell lines.
Table II.
Genes in the PTEN/P13K/Akt pathway.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| PTEN | 10q23.3 | 0.0431 | −0.2217 | 0.5627 | 0.4458 | −0.0601 | 10 |
| PIK3CA | 3q26.3 | 0.5233 | 0.5533 | 0.0050 | 0.5917 | 0.4001 | 11 |
| PIK3CB | 3q22.3 | 0.5097 | 0.3200 | −0.0328 | 0.1434 | 0.1027 | 10 |
| PIK3CD | 1p36.2 | −0.0513 | −0.0245 | −0.0344 | 0.2615 | 0.4878 | 8 |
| PIK3CG | Tq22.3 | −0.0844 | 0.1829 | 0.5142 | −0.0626 | −0.0638 | 5 |
| PIK3R1 | 5q13.1 | −0.0433 | 0.2323 | 0.6330 | −0.1129 | −0.1190 | 9 |
| PIK3R2 | 19q13.2-q13.4 | −0.1169 | −0.2734 | −0.1508 | 0.3813 | 0.4299 | 3 |
| AKT1 | 14q32.32 | −0.1815 | −0.3197 | −0.3115 | 0.2430 | 0.3230 | 7 |
| AKT2 | 19q13.1-q13.2 | 0.3599 | 0.2232 | −0.1733 | 0.8346 | 0.6278 | 6 |
| AKT3 | 1q43-q44 | −0.2134 | 0.0080 | −0.5262 | −0.1810 | −0.2074 | 36 |
| MTOR | 1p363 | −0.0513 | −0.0245 | 0.0063 | 0.2615 | 0.2784 | 21 |
| p70S6K | 17q23.1 | 0.5455 | 0.3715 | 0.0680 | 0.1566 | 0.2710 | 5 |
| EIF4EBP1 | 8p21 | −0.0444 | 0.0186 | 0.4337 | 0.3302 | 0.3486 | 4 |
| EIF4E | 4q21-q25 | −0.0652 | −0.7867 | −0.0480 | −0.0317 | −0.1648 | 6 |
| EIF4B | 12q13.13 | −0.0595 | 0.1898 | −0.1272 | 0.8488 | 0.7891 | 5 |
Note: Red color indicates amplification: green color indicates deletion.
Another EC-related gene, β-catenin, acts as a downstream transcriptional activator in the Wnt signal-transduction pathway, which has been reported to be activated in EC (19). Gain-of-function mutations in the β-catenin gene are found in 25% to 38% of type I ECs (20). Of the five EC cell lines tested, we found that many Wnt-pathway genes were amplified in the ECC-1 and Ishikawa cell lines (Table III).
Table III.
Genes in the Wnt-β-catenin pathway.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| Wnt1 | 12q13 | −0.0050 | 0.1898 | −0.1965 | 0.8906 | 0.7891 | 1 |
| Wnt2 | 7q31.2 | −0.0844 | 0.2582 | 0.5142 | −0.2159 | −0.1945 | 7 |
| Wnt3 | 17q21 | 0.5451 | 0.2286 | −0.2671 | 0.2182 | 0.2530 | 6 |
| Wnt4 | 1p36.23-p35.1 | −0.0872 | 0.1023 | −0.2132 | 0.2312 | 0.2867 | 3 |
| Wnt5A | 3p21-p14 | 0.5202 | −0.3426 | −0.3303 | −0.1055 | −0.1169 | 4 |
| Wnt5B | 12p13.3 | −0.0508 | 0.0515 | −0.2402 | 0.6476 | 0.5912 | 5 |
| Wnt6 | 2q35 | −0.0757 | −0.2541 | −0.2033 | 0.1608 | 0.2176 | 4 |
| Wnt7A | 3p25 | 0.5202 | 0.4685 | −0.3065 | 0.2961 | 0.0648 | 6 |
| Wnt7B | 22q13 | −0.1082 | −0.3287 | −0.2566 | 0.1980 | 0.2018 | 7 |
| Wnt8A | 5q31 | 0.5752 | 0.2878 | 0.4864 | 0.2412 | 0.3304 | 2 |
| Wnt8B | 10q24 | −0.0501 | −0.2121 | 0.3763 | 0.7915 | 0.2983 | 4 |
| Wnt9A | 1q42 | −0.0503 | 0.0326 | −0.7487 | 0.0367 | 0.0573 | 3 |
| Wnt9B | 17q21 | 0.5451 | 0.2286 | −0.2671 | 0.2182 | 0.2530 | 3 |
| Wnt10A | 2q32 | −0.0757 | −0.2541 | −0.2033 | 0.1608 | 0.2176 | 2 |
| Wnt10B | 12q13 | −0.0050 | 0.1898 | −0.1965 | 0.8906 | 0.7891 | 3 |
| LRP1 | 12q13-q14 | −0.0595 | 0.2042 | −0.1329 | 0.8411 | 0.7952 | 9 |
| CSNK1A1 | 5q32 | 0.6249 | 0.1152 | 0.4036 | 0.1367 | 0.2421 | 6 |
| GSK3A | 19q13.2 | 0.3599 | 0.2232 | −0.2663 | 0.2395 | 0.1462 | 3 |
| GSK3B | 3q13.3 | 0.5097 | 0.5180 | −0.0388 | 0.0270 | −0.0158 | 31 |
| CTNNA11 (α-catenin) | 5q31 | 0.5752 | 0.1587 | 0.4864 | 0.2412 | 0.0955 | 24 |
| CTNNB1 (β-catenin) | 3p21 | 0.5202 | 0.4064 | −0.1461 | −0.1918 | −0.1830 | 5 |
| APC | 5q21-q22 | −0.0955 | 0.2610 | 0.5338 | 0.0394 | 0.0115 | 14 |
Note: Red color indicates amplification: green color indicates deletion.
Receptor tyrosine kinases (RTKs) are often amplified in human cancers and they represent a class of proteins that are considered excellent therapeutic targets. Examination of the five EC cell lines showed that EphB3 and EphB4 were the most commonly amplified RTK genes, and a number of other Eph members were also amplified in these cell lines (Table IV). EGFR was significantly amplified in the AN3CA, HEC1A, and HEC1B cell lines. However, IGF1R, which has been shown to be amplified in some sarcomas, was not amplified in these cell lines. FGFR genes were amplified in four of five cell lines, while the ErbB2 gene was amplified in three of five. MET was only amplified in the HEC1B and metastatic AN3CA cell lines. The ECC-1 and Ishikawa cell lines harbored most of the RTK gene amplifications, whereas the HEC1B cell line had the least.
Table IV.
Receptor tyrosine kinases and hormone receptors.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| EGFR | 7p12 | 0.6499 | 0.2086 | 0.3686 | 0.0032 | −0.0616 | 23 |
| FGFR1 | 8p11.2-p11.1 | −0.0444 | 0.0186 | 0.2619 | −0.0819 | 0.1227 | 8 |
| FGFR2 | 10q26 | −0.0945 | −0.2865 | 0.5596 | 0.7241 | 0.1678 | 16 |
| FGFR3 | 4p16.3 | −0.1495 | −0.8308 | −0.3144 | 0.1737 | 0.2016 | 3 |
| FGFR4 | 5q35.1-qter | 0.4573 | 0.1152 | 0.3270 | 0.2664 | 0.2542 | 3 |
| MET | 7q31 | −0.0844 | 0.2582 | 0.5142 | −0.2159 | −0.1945 | 16 |
| KIT | 4q11-q12 | −0.0975 | −1.0117 | −0.2309 | −0.0824 | −0.0936 | 9 |
| PDGFRA | 4q11-q13 | −0.0975 | −1.0117 | −0.2309 | −0.0824 | −0.0936 | 7 |
| PDGFRB | 5q31-q32 | 0.6249 | 0.1152 | 0.1932 | 0.1367 | 0.2421 | 9 |
| EphA1 | 7q34 | −0.1828 | 0.0700 | 0.3990 | 0.1556 | −0.1301 | 4 |
| EphA2 | 1p36 | −0.0590 | −0.0589 | −0.2143 | 0.3379 | 0.2886 | 5 |
| EphB1 | 3q21-q23 | 0.5097 | 0.3200 | −0.3048 | −0.1681 | −0.0909 | 48 |
| EphB2 | 1p36.1-p35 | −0.0872 | 0.1023 | −0.4026 | 0.2312 | 0.2867 | 22 |
| EpRB3 | 3q21-qter | 0.5233 | 0.5533 | −0.2437 | 0.7275 | 0.6853 | 4 |
| EphB4 | 7q22 | −0.0844 | 0.8065 | 0.3805 | 0.3756 | 0.3720 | 4 |
| InsR | 19p13.3-p13.2 | −0.1169 | −0.2921 | −0.1422 | 0.3559 | 0.2705 | 16 |
| IGF1R | 15q26.3 | −0.1227 | −0.3355 | −0.3256 | −0.0229 | −0.0125 | 42 |
| ErbB2 | 17q21.1 | 0.5451 | 0.1954 | −0.3372 | 0.2725 | 0.3018 | 7 |
| ALK | 2p23 | −0.0544 | −0.4034 | −0.2244 | −0.1704 | −0.1032 | 83 |
| ESR1 | 6q25.1 | −0.0496 | −0.8763 | 0.4344 | −0.0944 | −0.1554 | 33 |
| ESR2 | 14q23.2 | −0.0290 | −0.1773 | 0.0876 | 0.1641 | 0.1705 | 14 |
| ESRRA | 11q13 | −0.0968 | −0.2377 | −0.2086 | 0.2758 | 0.3318 | 3 |
| PGR | 11q22-q23 | −0.0903 | −0.1665 | −0.0386 | −0.1330 | −0.2570 | 9 |
| AR | Xq11.2-q12 | −0.1765 | −1.0823 | −0.0950 | −0.1504 | −0.2826 | 20 |
Note: Red color indicates amplification: green color indicates deletion.
Among the common cancer-related oncogenes, R-Ras was amplified in all cell lines and Raf-1 was amplified in four of five cell lines (Table V), consistent with reports of mutations of these oncogenes in EC patients (11, 21). We also found that K-Ras, MDM1, and MDM2 were amplified in three of five cell lines.
Table V.
Other oncogenes.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikiwa | |||
| R-Ras | 19q13.3-qter | 0.3599 | 0.2232 | 0.3676 | 0.3714 | 0.3824 | 3 |
| N-Ras | 1p13.2 | −0.0417 | 0.2089 | 0.0636 | 0.2450 | 0.1566 | 3 |
| K-Ras | 12p12.1 | −0.1126 | 0.2148 | −0.0810 | 0.4107 | 0.3721 | 6 |
| H-Ras | 17p15.5 | −0.1744 | −0.3260 | −0.3291 | 0.4083 | 0.3366 | 1 |
| A-Raf | Xp11.4-p11.2 | −0.2728 | −1.1148 | −0.2564 | −0.8453 | −0.8617 | 3 |
| B-Raf | 7q34 | −0.0844 | 0.2563 | 0.3990 | −0.1669 | 0.0576 | 21 |
| C-Raf (Raf1) | 3p25 | 0.5202 | 0.4685 | −0.0585 | 0.2448 | 0.2516 | 10 |
| MYC | 8q24.21 | −0.0084 | 0.0454 | 0.3018 | 0.1312 | 0.1336 | 3 |
| SMAD4 | 18q21.1 | −0.1019 | −0.3032 | −0.1364 | 0.0450 | 0.0230 | 6 |
| MDM1 | 12q15 | −0.0101 | 0.2394 | −0.0121 | 0.5316 | 0.4902 | 6 |
| MDM2 | 12q14.3-q15 | −0.0101 | 0.2394 | −0.0121 | 0.5194 | 0.4902 | 3 |
| MDM4 | 1q32 | −0.0372 | 0.0843 | −0.2149 | 0.1356 | 0.1940 | 4 |
| SRC | 20q12-q13 | −0.0500 | 0.1290 | −0.2211 | 0.1451 | 0.0904 | 8 |
Note: Red color indicates amplification: green color indicates deletion.
There were no significant alterations in cell cycle–related genes in the EC cell lines, except for Cdk3, which was amplified in four of five cell lines (Table VI). Although no significant alterations were observed in the apoptosis-related genes in these EC cell lines (Table VII), it was interesting that many of these genes were deleted in the HEC1B cell line but amplified in the ECC-1 and Ishikawa cell lines.
Table VI.
Cell cycle–related genes.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| cyclin A1 | 13q123-q13 | −0.0417 | 0.1248 | −0.1107 | −0.1595 | −0.0434 | 3 |
| cyclin A2 | 4q25-q31 | −0.0652 | 0.9613 | 0.0916 | 1.3457 | −0.1648 | 5 |
| cyclin B1 | 5q12 | −0.0433 | 0.2323 | 0.6330 | 0.2964 | 0.2301 | 3 |
| cyclin B2 | 15q22.2 | 0.0309 | −0.2265 | −0.1002 | 0.6990 | 0.2053 | 4 |
| cyclin B3 | Xp11 | −0.0849 | −0.9476 | −0.1311 | −1.0468 | −1.1298 | 8 |
| cyclin D1 | 11q13 | −0.0948 | −0.2486 | −0.3580 | 0.1298 | 0.1793 | 3 |
| cyclin D2 | 12p13 | −0.1817 | −0.0097 | −0.3046 | 0.5258 | 0.5171 | 5 |
| cyclin D3 | 6p21 | −0.0513 | 0.1458 | −0.3446 | 0.2322 | 0.2552 | 3 |
| cyclin E1 | 19q12 | 0.3559 | 0.0855 | −0.2796 | 0.1812 | 0.1281 | 4 |
| cyclin E2 | 8q22.1 | −0.0379 | 0.1257 | 0.5024 | 0.1807 | 0.2668 | 4 |
| Cdk1 (Cdc2) | 10q21.1 | −0.1010 | −0.1904 | 0.4599 | −0.0581 | −0.0295 | 4 |
| Cdk2 | 12q13 | −0.0595 | 0.2042 | −0.1329 | 0.8736 | 0.7952 | 3 |
| Cdk3 | 17q22-qter | 0.4495 | 0.2271 | −0.0969 | 0.9831 | 0.9792 | 1 |
| Cdk4 | 12q14 | −0.0595 | 0.2042 | −0.1329 | 0.8411 | 0.7952 | 3 |
| Cdk5 | 7q36 | −0.1828 | 0.1180 | 0.2972 | 0.1620 | 0.1969 | 1 |
| Cdk6 | 7q21-q22 | −0.0844 | 0.2795 | 0.5650 | −0.1167 | −0.1016 | 27 |
| Aurora A | 20q13.2-q13.3 | −0.1690 | 0.0169 | −0.2292 | 0.0592 | 0.0348 | 5 |
| Aurora B | 17p13.1 | −0.0892 | 0.0619 | −0.1820 | 0.3108 | 0.3234 | 1 |
| Aurora C | 19q13.43 | 0.3599 | 0.1522 | −0.1645 | −0.0504 | −0.1057 | 1 |
| PLK1 | 16p12.2 | −0.1101 | 0.0805 | −0.1403 | 0.1252 | 0.2563 | 3 |
| PLK2 | 5q12.1-q13.2 | −1.0431 | −1.1599 | 0.5580 | −0.1007 | −0.1312 | 1 |
| PLK3 | 1p34.1 | −0.0624 | 0.0996 | −0.0114 | 0.1991 | 0.1655 | 1 |
| PLK4 | 4q28 | −0.0652 | −0.7715 | 0.0916 | 0.0576 | 0.0874 | 3 |
| CHK1 | 11q24-q24 | −0.2516 | −0.3438 | −0.1327 | −0.0822 | −0.0739 | 3 |
| CHK2 | 22q12.1 | −0.0993 | −0.2598 | −0.1119 | 0.1733 | 0.2646 | 7 |
Note: Red color indicates amplification; green color indicates deletion.
Table VII.
Apoptosis-related genes.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| Bcl-2 | 18q21.3 | −0.1019 | −0.2608 | −0.0680 | 0.0178 | −0.0479 | 23 |
| Bax | 19q13.3-q13.4 | 0.3599 | 0.2232 | −0.1210 | 0.3714 | 0.3824 | 3 |
| Bel-XL | 20q11.21 | −0.0500 | 0.1290 | −0.1827 | 0.6157 | 0.2772 | 7 |
| Bad | 11q13.1 | −0.0968 | −0.2377 | −0.2086 | 0.2758 | 0.3318 | 3 |
| Bim | 2q13 | 0.4906 | 0.1959 | −0.1531 | −0.0458 | −0.0386 | 5 |
| Bid | 22q11.1 | −0.0847 | −0.3390 | −0.1947 | 0.2382 | 0.2477 | 6 |
| Caspase 2 | 7q34-q35 | −0.1828 | 0.0700 | 0.3990 | −0.1583 | −0.1301 | 4 |
| Caspase 3 | 4q34 | −0.0652 | −1.7495 | −0.0425 | 0.2432 | 0.0419 | 7 |
| Caspase 6 | 4q25 | −0.0652 | −0.7584 | −0.0344 | −0.1529 | −0.1648 | 4 |
| Caspase 7 | 10q25 | −0.0467 | −0.2491 | 0.3536 | 0.4831 | 0.0141 | 5 |
| Caspase 8 | 2q33-q34 | −0.0079 | −0.2052 | −0.0094 | 0.0386 | 0.0988 | 8 |
| Caspase 9 | 1p36.3-p36.1 | −0.0590 | −0.0589 | 0.0370 | 0.3379 | 0.2886 | 6 |
| Caspase 10 | 2q33-q34 | −0.0079 | −0.2052 | −0.0094 | 0.2371 | 0.0988 | 6 |
Note: Red color indicates amplification: green color indicates deletion.
Tumor-suppressor gene deletion was not a common event in the five EC cell lines, with the exception of p16, which was deleted in two EC cell lines (Table VIII). Some of the genes, such as VHL, were actually amplified in EC, although it is not clear whether these genes had inactivation mutations or not.
Table VIII.
Other tumor-suppressor genes.
| Target Gene | Cytoband | Log2 ratio
|
Probe number | ||||
|---|---|---|---|---|---|---|---|
| HEC1A | HEC1B | AN3CA | ECC-1 | Ishikawa | |||
| TP53 | 17p13.1 | −0.0892 | 0.0619 | −0.1820 | 0.3108 | 0.3234 | 4 |
| CDKN2A (p16) | 9p21 | −0.1415 | −0.5251 | −0.0593 | −0.2064 | −0.2104 | 5 |
| CDH1(E-cadherin) | 16q22.1 | −0.0577 | 0.1242 | 0.0021 | 0.3063 | 0.2510 | 11 |
| RB1 | 13q14.2 | −0.0417 | 0.1215 | −0.1176 | −0.0601 | −0.1089 | 24 |
| NF1 | 17q11.2 | 0.5670 | 0.3060 | 0.0769 | −0.1205 | −0.1047 | 34 |
| VHL | 3p26-p25 | 0.5202 | 0.4685 | −1.0702 | 0.2749 | 0.3030 | 2 |
Note: Red color indicates amplification: green color indicates deletion.
Discussion
In this study, we used genome-wide aCGH technology to characterize the gene copy–number aberrations in five of the most commonly used EC cell lines. Although limited in scale (the results need to be further compared in a bigger study with a large number of primary EC samples) we have obtained valuable information that provides some insight into the genetic signatures of EC cells. First, the global gene copy–number aberration patterns of the five EC cell lines were less complex than patterns reported for many other cancer cell lines. This suggests that the EC cell lines have not undergone major changes during multiple in vitro passages in the laboratory and, thus, still exhibit the molecular characteristics of the primary type I EC cells, which are known to be more diploid. Therefore, the five cell lines studied rep-resent good model systems for studying EC signaling pathways and, perhaps, therapeutics in vitro and in preclinical animal model experiments. To support this notion, the consistent gains or losses we identified (gains in 2q, 3q, 3p, 5q, 17q, 7p, and 19q and losses in 3p,10p, 10q, 11q, 11p, 14q, 15q, 18p, and 21q) are consistent with those reported for EC patients (4, 5).
Many of the amplifications identified in our study are relatively small in scale. We believe this is also the result of tumor heterogeneity and the co-existence of multiple clones in each cell line. Although this can be further tested by serial dilution and clonal expansion experiments, the results from the HEC1A and HEC1B cell lines, which were derived from the same patient, support this hypothesis. Most of the altered genes are shared between the HEC1A and HEC1B lines, such as amplification on chromosome 3p and deletions on chromosome 1p and 4q. However, there are a few HEC1B-specific gene deletions present, such as InsR, IGF1R, ALK, ESR1, ESRRA, and AR. Thus, these two cell lines may be useful as “isogenic” control lines for the study of EC responses to insulin and IGF.
Our detailed analysis of key oncogenic pathway genes provides additional insights that are relevant to many areas of EC research. O’Toole et al., (22) examined eight EC specimens by aCGH and also found amplification of some of the same oncogenes, including AR, PIK3CA, MET, HRAS, NRAS, 17S1670, FGFR, CTSB, RPS6KB1, LAMC2, MYC, PDGFRA, FGF4/FGF3, PAKI, and FGR. Below, we discuss a few of the major pathways of particular interest in EC in more detail.
PTEN/PI3K/Akt pathway
PTEN, which is located on chromosome 10q23.3, has been reported to be altered (mostly by mutation and less frequently by loss of heterozygosity) in up to 83% of type I ECs (23–25). Inactivation of PTEN through deletion and mutation results in activation of the PI3K/Akt pathway, which is important for cell proliferation, apoptosis, and migration in EC (26–28). Although we only detect PTEN deletion in HEC1B in all of the five EC cell lines we tested, the PIK3CA gene, which encodes the p110α catalytic subunit of PI3K, is amplified in four of the five EC cell lines. We have not performed a sequencing analysis and, therefore, do not yet know whether PIK3CA is mutated. However, it would not be surprising if mutations also occur in this gene, as mutation of the PIK3CA gene has recently been found in 24% to 39% of type I ECs, and these mutations are correlated with poor prognosis (6–9, 17, 18). Amplification of PIK3CA has also been identified in ovarian cancer (29), non-small cell lung cancer (30), squamous cell carcinoma of the oral tongue (31), and cervical cancer (32), as well as in EC (22). Therefore, PIK3CA amplification plays an important role in the tumorigenesis of a wide spectrum of solid tumors.
Akt, one of the downstream effectors of PI3K, is an evolutionarily conserved serine/threonine kinase that has three isoforms: Akt1, Akt2, and Akt3. Once activated, Akt regulates multiple cellular functions such as cell proliferation, survival, apoptosis, glucose metabolism, ribosomal function, transcription, and cell migration (33, 34). The expression of Akt1 and Akt2 is ubiquitous, whereas Akt3 is expressed predominantly in the brain, heart, and kidney (35). The emerging picture from recent studies is that the three Akt isoforms play different roles in different cancers. For example, in breast cancer, Akt3 is upregulated in ER-negative breast carcinomas (36), and overexpression of Akt2 could stimulate tumor cell invasion both in vitro and in vivo (37). A recent report by Irie et al., (38) showed that silencing Akt1 expression with shRNA actually increased cell migration, whereas silencing Akt2 had no effect on nontransformed MCF10A breast epithelial cells. Gagnon et al., (39) showed that Akt1 mRNA and protein were present in both HEC1A and KLE EC cells, whereas Akt2 and Akt3 mRNAs and proteins were strongly expressed in KLE cells. Knockout of Akt isoforms increased the sensitivity of KLE cells toward cisplatin and caused a significant induction of cell death. In this study, we found that Akt2 was significantly amplified in four of five EC cell lines, and Akt1 was amplified in two of five cell lines. In contrast, we found that Akt3 was deleted in the majority of EC cell lines. Thus, Akt3 may function as a negative regulator in EC cells. More detailed functional studies are warranted to delineate the role of different Akt isoforms in EC.
Wnt/β-catenin pathway
The Wnt family consists of highly conserved genes associated with oncogenesis. The Wnt pathway is activated in a wide variety of tumors such as prostate cancer (40), renal cancer (41), ovarian cancer (42), and EC (19). Wang et al.,(43) reported that progesterone inhibits Wnt signaling in the human endometrium through induction of DKK1 and FOXO1. The inhibitory effect of progesterone on Wnt signaling may, in part, play a role in the maintenance of endometrial homeostasis. Wagner et al., (44) found that the estrogen receptor is involved in the regulation of Wnt7a in Ishikawa cells and the modulation of Wnt gene expression by estrogen might be a novel mechanism for EC tumorigenesis. The aCGH results of our study show that there were significant alterations in the genes in the Wnt/b-catenin pathway. Wnt8A, Wnt10B, and CSNK1A1 were amplified in all five cell lines, while Wnt7A, Wnt9B, and GSK3B were amplified in four of five cell lines. Most genes in this pathway were amplified in the ECC-1 and Ishikawa cells, which predominantly express estrogen receptor (45, 46). Thus, these two cell lines may represent ideal model systems for studying the relationship between the Wnt pathway and the hormone response and their roles in EC tumorigenesis.
The Ras-Raf pathway
Our study showed that R-Ras was amplified in all cell lines and that K-Ras and C-Raf were amplified in four of five cell lines. Ras mutation is a well-recognized event in EC. The Raf family of proto-oncogenes encodes cytoplasmic serine/threonine protein kinases, which play a pivotal role in cell growth and oncogenesis. B-Raf has been found to be activated by mutations in a multitude of human cancers (47–49). However, most of the analyses have shown a low prevalence of B-Raf mutations in EC (50–52). More interestingly, B-Raf mutation was more frequently found in hMLH1-negative than hMLH1-positive EC cases (53). Alterations in C-Raf expression have been suggested to play a role in melanoma and lung cancer (54, 55). In this study, we found that C-Raf was amplified in four of five EC cell lines, suggesting that C-Raf is a key isoform for EC oncogenesis. To date, the role of A-Raf in tumorigenesis has not been published. However, recently, Hagemann et al., (56) found A-Raf expression did not have any influence on the proliferation or migration of glioblastoma cells, and A-Raf expression was negatively associated with prognosis in patients with glioblastomas. Therefore, A-Raf may be a negative regulator of cell metabolism (57, 58). Supporting this hypothesis, we found that A-Raf was deleted in almost all EC cell lines. Thus, it appears that members of the Raf family may control cell growth both positively and negatively. Raf family members are likely regulated by each other, because in A-Raf–deficient mouse embryonic fibroblasts, both B-Raf and C-Raf activities towards MEK are significantly increased (59). Thus, it will be worthwhile to investigate how the Raf family members together control EC tumorigenesis.
Eph Receptor Pathway
Eph receptors constitute the largest family of RTKs and are involved in a wide range of processes directly related to tumorigenesis and metastasis (60). Our aCGH data showed that EphB3 (3q21-qter) and EphB4 (7q22) were amplified in four of five EC cell lines and EphA2, which has been reported to be overexpressed in EC(61), was amplified in two of the cell lines. Takai et al.,(62) analyzed 20 cases of EC and 20 normal endometrial cases, and found that EphB4 expression was significantly associated with histological grade and certain clinical stages, while Ephrin-B2 expression was significantly associated with the presence of deeper invasion. In addition, Berclaz et al., (63) showed that the EphB4 protein was not detected in normal endometrial tissue, but increased drastically in the majority of hyperplasias and carcinomas. These studies together provide strong evidence that EphB4 activation is likely an early oncogenic event in EC development and may represent an important diagnostic/prognostic marker and a target for therapeutic intervention. At the present time, little is known about other Eph receptors’ roles in EC, although, EphB3 was reported to be amplified in lung and colon cancers and in rhabdomyosarcoma (64–66).
In summary, our genomic characterization of commonly used endometrial cancer cell lines, although limited in scale and needing further validation studies using primary tumor tissues, provides potentially important insights into the genetic events that underly the tumorigenesis of this common type of cancer. Further investigations using primary EC cells, as well as functional studies with these in vitro EC cell models coupled with preclinical mouse model experiments, should allow the identification of clinical markers for diagnosis/prognosis and potential molecular targets for therapeutic intervention.
Acknowledgments
We would like to thank Kate J. Newberry at the Department of Scientific Publications of M. D. Anderson Cancer Center for her editorial assistance. Yingmei Wang was partially supported by a China Scholarship Council Education Fellowship.
Abbreviations
- aCGH
Array-based comparative genomic hybridization
- EC
Endometrial carcinoma
- FBS
Fetal bovine serum
- MSI
Microsatellite instability
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Sherman ME, Bur ME, Kurman RJ. p53 in endometrial cancer and its putative precursors: evidence for diverse pathways of tumorigenesis. Hum Pathol. 1995;26:1268–1274. doi: 10.1016/0046-8177(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 3.Weber BL. Cancer genomics. Cancer Cell. 2002;1:37–47. doi: 10.1016/s1535-6108(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 4.Levan K, Partheen K, Osterberg L, Helou K, Horvath G. Chromosomal alterations in 98 endometrioid adenocarcinomas analyzed with comparative genomic hybridization. Cytogenet Genome Res. 2006;115:16–22. doi: 10.1159/000094796. [DOI] [PubMed] [Google Scholar]
- 5.Hirasawa A, Aoki D, Inoue J, Imoto I, Susumu N, Sugano K, Nozawa S, Inazawa J. Unfavorable prognostic factors associated with high frequency of microsatellite instability and comparative genomic hybridization analysis in endometrial cancer. Clin Cancer Res. 2003;9:5675–5682. [PubMed] [Google Scholar]
- 6.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–10673. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 7.Oda K, Okada J, Timmerman L, Rodriguez-Viciana P, Stokoe D, Shoji K, Taketani Y, Kuramoto H, Knight ZA, Shokat KM, McCormick F. PIK3CA cooperates with other phosphatidylinositol 3′-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008;68:8127–8136. doi: 10.1158/0008-5472.CAN-08-0755. [DOI] [PubMed] [Google Scholar]
- 8.Catasus L, Gallardo A, Cuatrecasas M, Prat J. PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod Pathol. 2008;21:131–139. doi: 10.1038/modpathol.3800992. [DOI] [PubMed] [Google Scholar]
- 9.Catasus L, Gallardo A, Cuatrecasas M, Prat J. Concomitant PI3K-AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod Pathol. 2009;22:522–529. doi: 10.1038/modpathol.2009.5. [DOI] [PubMed] [Google Scholar]
- 10.Konecny GE, Santos L, Winterhoff B, Hatmal M, Keeney GL, Mariani A, Jones M, Neuper C, Thomas B, Muderspach L, Riehle D, Wang HJ, Dowdy S, Podratz KC, Press MF. HER2 gene amplification and EGFR expression in a large cohort of surgically staged patients with nonendometrioid (type II) endometrial cancer. Br J Cancer. 2009;100:89–95. doi: 10.1038/sj.bjc.6604814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bansal N, Yendluri V, Wenham RM. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control. 2009;16:8–13. doi: 10.1177/107327480901600102. [DOI] [PubMed] [Google Scholar]
- 12.Ignatov A, Bischoff J, Schwarzenau C, Krebs T, Kuester D, Herrmann K, Costa SD, Roessner A, Semczuk A, Schneider-Stock R. P16 alterations increase the metastatic potential of endometrial carcinoma. Gynecol Oncol. 2008;111:365–371. doi: 10.1016/j.ygyno.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 13.Yalta T, Atay L, Atalay F, Caydere M, Gonultas M, Ustun H. E-cadherin expression in endometrial malignancies: comparison between endometrioid and non-endometrioid carcinomas. J Int Med Res. 2009;37:163–168. doi: 10.1177/147323000903700119. [DOI] [PubMed] [Google Scholar]
- 14.Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Induction of p16INK4A mediated by beta-catenin in a TCF4-independent manner: implications for alterations in p16INK4A and pRb expression during trans-differentiation of endometrial carcinoma cells. Int J Cancer. 2006;119:2294–2303. doi: 10.1002/ijc.22112. [DOI] [PubMed] [Google Scholar]
- 15.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 16.van de Wiel MA, Kim KI, Vosse SJ, van Wieringen WN, Wilting SM, Ylstra B. CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics. 2007;23:892–894. doi: 10.1093/bioinformatics/btm030. [DOI] [PubMed] [Google Scholar]
- 17.Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, Ellenson LH. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932–5935. doi: 10.1158/1078-0432.CCR-06-1375. [DOI] [PubMed] [Google Scholar]
- 18.Velasco A, Bussaglia E, Pallares J, Dolcet X, Llobet D, Encinas M, Llecha N, Palacios J, Prat J, Matias-Guiu X. PIK3CA gene mutations in endometrial carcinoma: correlation with PTEN and K-RAS alterations. Hum Pathol. 2006;37:1465–1472. doi: 10.1016/j.humpath.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Carmon KS, Loose DS. Secreted frizzled-related protein 4 regulates two Wnt7a signaling pathways and inhibits proliferation in endometrial cancer cells. Mol Cancer Res. 2008;6:1017–1028. doi: 10.1158/1541-7786.MCR-08-0039. [DOI] [PubMed] [Google Scholar]
- 20.Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer. 2000;88:814–824. [PubMed] [Google Scholar]
- 21.Tu Z, Gui L, Wang J, Li X, Sun P, Wei L. Tumorigenesis of K-ras mutation in human endometrial carcinoma via upregulation of estrogen receptor. Gynecol Oncol. 2006;101:274–279. doi: 10.1016/j.ygyno.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 22.O’Toole SA, Dunn E, Sheppard BL, Klocker H, Bektic J, Smyth P, Martin C, Sheils O, O’Leary JJ. Genome-wide analysis of deoxyribonucleic acid in endometrial cancer using comparative genomic hybridization microarrays. Int J Gynecol Cancer. 2006;16:834–842. doi: 10.1111/j.1525-1438.2006.00530.x. [DOI] [PubMed] [Google Scholar]
- 23.Mutter GL. Pten, a protean tumor suppressor. Am J Pathol. 2001;158:1895–1898. doi: 10.1016/S0002-9440(10)64656-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000;92:924–930. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 25.Maurice-Duelli A, Ndoye A, Bouali S, Leroux A, Merlin JL. Enhanced cell growth inhibition following PTEN nonviral gene transfer using polyethylenimine and photochemical internalization in endometrial cancer cells. Technol Cancer Res Treat. 2004;3:459–465. doi: 10.1177/153303460400300507. [DOI] [PubMed] [Google Scholar]
- 26.Sexton E, Van Themsche C, LeBlanc K, Parent S, Lemoine P, Asselin E. Resveratrol interferes with AKT activity and triggers apoptosis in human uterine cancer cells. Mol Cancer. 2006;5:45. doi: 10.1186/1476-4598-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J, Tian J, Lv Y, Shi F, Kong F, Shi H, Zhao L. Leptin induces functional activation of cyclooxygenase-2 through JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways in human endometrial cancer cells. Cancer Sci. 2009;100:389–395. doi: 10.1111/j.1349-7006.2008.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheng S, Qiao M, Pardee AB. Metastasis and AKT activation. J Cell Physiol. 2009;218:451–454. doi: 10.1002/jcp.21616. [DOI] [PubMed] [Google Scholar]
- 29.Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 30.Massion PP, Kuo WL, Stokoe D, Olshen AB, Treseler PA, Chin K, Chen C, Polikoff D, Jain AN, Pinkel D, Albertson DG, Jablons DM, Gray JW. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: implications of the phosphatidylinositol 3-kinase pathway. Cancer Res. 2002;62:3636–3640. [PubMed] [Google Scholar]
- 31.Estilo CL, P OC, Ngai I, Patel SG, Reddy PG, Dao S, Shaha AR, Kraus DH, Boyle JO, Wong RJ, Pfister DG, Huryn JM, Zlotolow IM, Shah JP, Singh B. The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110alpha in squamous cell carcinoma of the oral tongue. Clin Cancer Res. 2003;9:2300–2306. [PubMed] [Google Scholar]
- 32.Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739–2744. doi: 10.1038/sj.onc.1203597. [DOI] [PubMed] [Google Scholar]
- 33.Crowell JA, Steele VE, Fay JR. Targeting the AKT protein kinase for cancer chemoprevention. Mol Cancer Ther. 2007;6:2139–2148. doi: 10.1158/1535-7163.MCT-07-0120. [DOI] [PubMed] [Google Scholar]
- 34.Shtilbans V, Wu M, Burstein DE. Current overview of the role of Akt in cancer studies via applied immunohistochemistry. Ann Diagn Pathol. 2008;12:153–160. doi: 10.1016/j.anndiagpath.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Yang ZZ, Tschopp O, Baudry A, Dummler B, Hynx D, Hemmings BA. Physiological functions of protein kinase B/Akt. Biochem Soc Trans. 2004;32:350–354. doi: 10.1042/bst0320350. [DOI] [PubMed] [Google Scholar]
- 36.Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, Roth RA. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem. 1999;274:21528–21532. doi: 10.1074/jbc.274.31.21528. [DOI] [PubMed] [Google Scholar]
- 37.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 38.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagnon V, Mathieu I, Sexton E, Leblanc K, Asselin E. AKT involvement in cisplatin chemoresistance of human uterine cancer cells. Gynecol Oncol. 2004;94:785–795. doi: 10.1016/j.ygyno.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 40.Robinson DR, Zylstra CR, Williams BO. Wnt signaling and prostate cancer. Curr Drug Targets. 2008;9:571–580. doi: 10.2174/138945008784911831. [DOI] [PubMed] [Google Scholar]
- 41.Guillen-Ahlers H. Wnt signaling in renal cancer. Curr Drug Targets. 2008;9:591–600. doi: 10.2174/138945008784911813. [DOI] [PubMed] [Google Scholar]
- 42.Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO, Fearon ER, Cho KR. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Hanifi-Moghaddam P, Hanekamp EE, Kloosterboer HJ, Franken P, Veldscholte J, van Doorn HC, Ewing PC, Kim JJ, Grootegoed JA, Burger CW, Fodde R, Blok LJ. Progesterone inhibition of Wnt/beta-catenin signaling in normal endometrium and endometrial cancer. Clin Cancer Res. 2009;15:5784–5793. doi: 10.1158/1078-0432.CCR-09-0814. [DOI] [PubMed] [Google Scholar]
- 44.Wagner J, Lehmann L. Estrogens modulate the gene expression of Wnt-7a in cultured endometrial adenocarcinoma cells. Mol Nutr Food Res. 2006;50:368–372. doi: 10.1002/mnfr.200500215. [DOI] [PubMed] [Google Scholar]
- 45.Mo B, Vendrov AE, Palomino WA, DuPont BR, Apparao KB, Lessey BA. ECC-1 cells: a well-differentiated steroid-responsive endometrial cell line with characteristics of luminal epithelium. Biol Reprod. 2006;75:387–394. doi: 10.1095/biolreprod.106.051870. [DOI] [PubMed] [Google Scholar]
- 46.Wormke M, Castro-Rivera E, Chen I, Safe S. Estrogen and aryl hydrocarbon receptor expression and crosstalk in human Ishikawa endometrial cancer cells. J Steroid Biochem Mol Biol. 2000;72:197–207. doi: 10.1016/s0960-0760(00)00030-3. [DOI] [PubMed] [Google Scholar]
- 47.Ueda M, Toji E, Nunobiki O, Izuma S, Okamoto Y, Torii K, Noda S. Mutational analysis of the BRAF gene in human tumor cells. Hum Cell. 2008;21:13–17. doi: 10.1111/j.1749-0774.2008.00046.x. [DOI] [PubMed] [Google Scholar]
- 48.Jovanovic B, Egyhazi S, Eskandarpour M, Ghiorzo P, Palmer JM, Bianchi Scarra G, Hayward NK, Hansson J. Coexisting NRAS and BRAF Mutations in Primary Familial Melanomas with Specific CDKN2A Germline Alterations. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oikonomou E, Kosmidou V, Katseli A, Kothonidis K, Mourtzoukou D, Kontogeorgos G, Andera L, Zografos G, Pintzas A. TRAIL receptor upregulation and the implication of KRAS/BRAF mutations in human colon cancer tumors. Int J Cancer. 2009;125:2127–2135. doi: 10.1002/ijc.24613. [DOI] [PubMed] [Google Scholar]
- 50.Pijnenborg JM, Dam-de Veen GC, Kisters N, Delvoux B, van Engeland M, Herman JG, Groothuis PG. RASSF1A methylation and K-ras and B-raf mutations and recurrent endometrial cancer. Ann Oncol. 2007;18:491–497. doi: 10.1093/annonc/mdl455. [DOI] [PubMed] [Google Scholar]
- 51.Kawaguchi M, Yanokura M, Banno K, Kobayashi Y, Kuwabara Y, Kobayashi M, Nomura H, Hirasawa A, Susumu N, Aoki D. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int J Oncol. 2009;34:1541–1547. doi: 10.3892/ijo_00000283. [DOI] [PubMed] [Google Scholar]
- 52.Salvesen HB, Kumar R, Stefansson I, Angelini S, MacDonald N, Smeds J, Jacobs IJ, Hemminki K, Das S, Akslen LA. Low frequency of BRAF and CDKN2A mutations in endometrial cancer. Int J Cancer. 2005;115:930–934. doi: 10.1002/ijc.20702. [DOI] [PubMed] [Google Scholar]
- 53.Feng YZ, Shiozawa T, Miyamoto T, Kashima H, Kurai M, Suzuki A, Konishi I. BRAF mutation in endometrial carcinoma and hyperplasia: correlation with KRAS and p53 mutations and mismatch repair protein expression. Clin Cancer Res. 2005;11:6133–6138. doi: 10.1158/1078-0432.CCR-04-2670. [DOI] [PubMed] [Google Scholar]
- 54.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fedorov LM, Papadopoulos T, Tyrsin OY, Twardzik T, Gotz R, Rapp UR. Loss of p53 in craf-induced transgenic lung adenoma leads to tumor acceleration and phenotypic switch. Cancer Res. 2003;63:2268–2277. [PubMed] [Google Scholar]
- 56.Hagemann C, Gloger J, Anacker J, Said HM, Gerngras S, Kuhnel S, Meyer C, Rapp UR, Kammerer U, Vordermark D, Flentje M, Roosen K, Vince GH. RAF expression in human astrocytic tumors. Int J Mol Med. 2009;23:17–31. [PubMed] [Google Scholar]
- 57.Le Mellay V, Houben R, Troppmair J, Hagemann C, Mazurek S, Frey U, Beigel J, Weber C, Benz R, Eigenbrodt E, Rapp UR. Regulation of glycolysis by Raf protein serine/threonine kinases. Adv Enzyme Regul. 2002;42:317–332. doi: 10.1016/s0065-2571(01)00036-x. [DOI] [PubMed] [Google Scholar]
- 58.Mazurek S, Drexler HC, Troppmair J, Eigenbrodt E, Rapp UR. Regulation of pyruvate kinase type M2 by A-Raf: a possible glycolytic stop or go mechanism. Anticancer Res. 2007;27:3963–3971. [PubMed] [Google Scholar]
- 59.Mercer K, Chiloeches A, Huser M, Kiernan M, Marais R, Pritchard C. ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene. 2002;21:347–355. doi: 10.1038/sj.onc.1205101. [DOI] [PubMed] [Google Scholar]
- 60.Castano J, Davalos V, Schwartz S, Jr, Arango D. EPH receptors in cancer. Histol Histopathol. 2008;23:1011–1023. doi: 10.14670/HH-23.1011. [DOI] [PubMed] [Google Scholar]
- 61.Kamat AA, Coffey D, Merritt WM, Nugent E, Urbauer D, Lin YG, Edwards C, Broaddus R, Coleman RL, Sood AK. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer. 2009;115:2684–2692. doi: 10.1002/cncr.24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takai N, Miyazaki T, Fujisawa K, Nasu K, Miyakawa I. Expression of receptor tyrosine kinase EphB4 and its ligand ephrin-B2 is associated with malignant potential in endometrial cancer. Oncol Rep. 2001;8:567–573. doi: 10.3892/or.8.3.567. [DOI] [PubMed] [Google Scholar]
- 63.Berclaz G, Karamitopoulou E, Mazzucchelli L, Rohrbach V, Dreher E, Ziemiecki A, Andres AC. Activation of the receptor protein tyrosine kinase EphB4 in endometrial hyperplasia and endometrial carcinoma. Ann Oncol. 2003;14:220–226. doi: 10.1093/annonc/mdg072. [DOI] [PubMed] [Google Scholar]
- 64.Chiu ST, Chang KJ, Ting CH, Shen HC, Li H, Hsieh FJ. Over-expression of EphB3 enhances cell-cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis. 2009;30:1475–1486. doi: 10.1093/carcin/bgp133. [DOI] [PubMed] [Google Scholar]
- 65.Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Identification of novel candidate target genes, including EPHB3, MASP1 and SST at 3q26.2-q29 in squamous cell carcinoma of the lung. BMC Cancer. 2009;9:237. doi: 10.1186/1471-2407-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berardi AC, Marsilio S, Rofani C, Salvucci O, Altavista P, Perla FM, Diomedi-Camassei F, Uccini S, Kokai G, Landuzzi L, McDowell HP, Dominici C. Up-regulation of EphB and ephrin-B expression in rhabdomyosarcoma. Anticancer Res. 2008;28:763–769. [PubMed] [Google Scholar]