

The title hydrated salt, C10H12N3O3S+·Cl−·H2O, forms centrosymmetric  (20) dimers through intermolecular C—H⋯O hydrogen bonds. These dimers are stacked via N—H⋯O and O—H⋯Cl hydrogen bonds involving the water molecules and chloride anions. Offset π–π interactions are also present.
(20) dimers through intermolecular C—H⋯O hydrogen bonds. These dimers are stacked via N—H⋯O and O—H⋯Cl hydrogen bonds involving the water molecules and chloride anions. Offset π–π interactions are also present.
Keywords: crystal structure, benzimidazole derivative, hydrogen bonding, π–π interactions
Abstract
In the cation of the title hydrated molecular salt, C10H12N3O3S+·Cl−·H2O, the benzimidazolium ring system is almost planar (r.m.s. deviation = 0.006 Å) and the nitro group is inclined at an angle of 4.86 (9)° to this plane. In the crystal, C—H⋯O hydrogen bonds form centrosymmetric R 2 2(20) dimers and these are further aggregated through N—H⋯O and O—H⋯Cl hydrogen bonds involving the water molecules and chloride anions. Aromatic π–π stacking interactions are also found between two parallel benzene rings or the benzene and imidazolium rings, with centroid–centroid distances of 3.5246 (9) and 3.7756 (9) Å, respectively. Analysis of the bond lengths and comparison with related compounds show that the nitro substituent is not involved in conjugation with the adjacent π-system and hence has no effect on the charge distribution of the heterocyclic ring.
Chemical context
Numerous compounds with benzimidazole ring systems display versatile pharmacological activities such as anti-viral, anti-helmintic, spasmolitic, anti-hypertensive and vasodilator properties (Akkurt et al., 2006 ▸). Many benzimidazole derivatives also have anti-microbial and anti-fungal activities (Küçükbay et al., 2003 ▸, 2004 ▸; Puratchikody et al., 2008 ▸; Alasmary et al., 2015 ▸). The synthesis of new benzimidazole derivatives is therefore of considerable current interest. As part of our studies in this area, the title protonated benzimidazole compound (I) has been synthesized and its molecular structure is presented here.
Structural commentary
The molecular structure of the title compound is shown in Fig. 1 ▸. The nine-membered benzimidazolium ring system (N4/C11/N9/C13/C16/C7/C15/C18/C10) is essentially planar, the maximum deviation from planarity being 0.013 (1) Å for atom N4. In addition, atoms N12, C17 and S2 of the nitro, hydroxyethyl and methylsulfanyl substituents lie close to the benzimidazolium ring plane with a maximum deviation of −0.059 (1) Å for atom S2. The least-squares plane of the nitro group (C7/N12/O6/O8) lies close to the benzimidazolium ring system, making a dihedral angle of 4.86 (9)°. In the structure, the bond lengths and angles of the benzimidazolium ring are generally in good agreement with those observed in related structures (Morozov et al., 2004 ▸; Verdan et al., 2009 ▸; Chen et al., 2010 ▸; Yuasa et al., 2010 ▸; Gao et al., 2013 ▸; Samsonov et al., 2013 ▸; Liu et al., 2014 ▸). In addition, the C7—N12 bond length, 1.4667 (19) Å shows that the nitro group is not involved in conjugation with the adjacent π-system and hence has no effect on the charge distribution of the heterocyclic ring. 
Figure 1.

The molecular structure of (I), showing the atomic labelling scheme and displacement ellipsoids drawn at the 50% probability level. H atoms are shown as spheres of arbitrary radius.
Supramolecular features
In the crystal, C14—H14B⋯O8 hydrogen bonds (Table 1 ▸) link the organic fragments into centrosymmetric dimers with (20) ring motifs along the [100] direction (Fig. 2 ▸). These dimers are further connected along the [100] and [010] directions by N—H⋯O and O—H⋯Cl hydrogen bonds, respectively, generating  (22) rings. In the latter ring motifs, both the water molecule and the oxygen atom of the hydroxyethyl substituent act as donors with the chloride anion as acceptor. The O3 atom of the water molecule serves as acceptor for the H9 atom of the imidazolium NH group (Fig. 3 ▸). The pattern formed by the water molecules connecting the chloride anions, and forming an
(22) rings. In the latter ring motifs, both the water molecule and the oxygen atom of the hydroxyethyl substituent act as donors with the chloride anion as acceptor. The O3 atom of the water molecule serves as acceptor for the H9 atom of the imidazolium NH group (Fig. 3 ▸). The pattern formed by the water molecules connecting the chloride anions, and forming an  (8) ring, is reminiscent of a parallelogram (Fig. 3 ▸). The supramolecular aggregation is completed by π–π stacking interactions between two parallel benzene rings and between the benzene and imidazolium rings: Cg2⋯Cg2(1 − x, −y, −z) = 3.5246 (9), Cg1⋯Cg2(1 − x, −y, −z) = 3.7756 (9) Å, slippage = 1.190 Å Cg1 and Cg2 are the centroids of the imidazolium and benzene rings respectively. The centroid–centroid separations are less than 3.8 Å, the maximum regarded as suitable for an effective π–π interaction (Janiak, 2000 ▸) (Fig. 4 ▸)).
(8) ring, is reminiscent of a parallelogram (Fig. 3 ▸). The supramolecular aggregation is completed by π–π stacking interactions between two parallel benzene rings and between the benzene and imidazolium rings: Cg2⋯Cg2(1 − x, −y, −z) = 3.5246 (9), Cg1⋯Cg2(1 − x, −y, −z) = 3.7756 (9) Å, slippage = 1.190 Å Cg1 and Cg2 are the centroids of the imidazolium and benzene rings respectively. The centroid–centroid separations are less than 3.8 Å, the maximum regarded as suitable for an effective π–π interaction (Janiak, 2000 ▸) (Fig. 4 ▸)).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O5—H5⋯Cl1 | 0.82 | 2.40 | 3.1840 (15) | 161 |
| C17—H17B⋯S2 | 0.97 | 2.68 | 3.1514 (18) | 110 |
| O3—H3B⋯Cl1 | 0.83 (2) | 2.28 (2) | 3.1090 (14) | 178 (2) |
| O3—H3A⋯Cl1i | 0.79 (2) | 2.37 (2) | 3.1561 (14) | 174 (2) |
| C14—H14B⋯O8ii | 0.97 | 2.60 | 3.189 (2) | 119 |
| N9—H9⋯O3iii | 0.86 | 1.85 | 2.6949 (16) | 165 |
Symmetry codes: (i)  ; (ii)
; (ii) 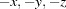 ; (iii)
; (iii)  .
.
Figure 2.

The crystal packing of (I), showing the supramolecular aggregation resulting from the three-dimensional hydrogen-bonded network. Dashed lines indicate hydrogen bonds. H atoms not involved in hydrogen bonding have been omitted for clarity.
Figure 3.

The molecular packing of (I), showing the pattern formed by the water molecules hydrogen bonded to the chloride anions.
Figure 4.

A view of the crystal packing, showing π–π stacking interactions (dashed lines). The brown dots are the centroids of the rings. H atoms have been omitted for clarity.
Database survey
A CSD search (Web CSD version 5.37; August 19, 2016; Groom et al., 2016 ▸) found eight benzimidazolium structures with substituents at the 1 and 2 positions of the imidazolium ring system (Morozov et al., 2004 ▸; Verdan et al., 2009 ▸; Chen et al., 2010 ▸; Yuasa et al., 2010 ▸; Gao et al., 2013 ▸; Samsonov et al., 2013 ▸; Liu et al., 2014 ▸; Kerimov et al., 2012 ▸). In these structures, the imidazolium rings generally show two long (in the range 1.36–1.40 Å) and two short (1.30–1.34 Å) C—N distances. This pattern is clearly repeated here with N4—C11 = 1.3492 (18) and N9—C11 = 1.3390 (17) Å while N4—C10 = 1.3898 (18) Å and N9—C13 = 1.3867 (16)Å. The sole exception to this pattern is the compound, 2-(4-chlorophenyl)-3-[(5-(3,5-dinitrophenyl)-1,3,4-oxadiazol-2-yl]methyl)-1H-benzimidazole (Kerimov et al., 2012 ▸), with an imidazolium ring, which reveals three long (1.37–1.39 Å) and one short ( 1.30 Å) C—N bonds, a pattern that is also displayed in benzimidazole structures (Abou et al., 2007 ▸; Yavo et al., 2007 ▸; Kakou-Yao et al., 2007 ▸; Akonan et al., 2010 ▸; Lokaj et al., 2009 ▸).
Synthesis and crystallization
2-Chloroethanol (1.3 ml, 19.2 mmol) and potassium carbonate (1.32 g, 9.6 mmol) were added to 2-methylthio-5-nitro-1H-benzimidazole (1.15 g, 4.8 mmol) in dimethyl sulfoxide (DMSO) (10 ml). The reaction mixture was agitated for 5 h at room temperature. 50 ml of water was then added to the reaction mixture, and the products were extracted with dichloromethane (3 × 50 ml). The combined organic extracts were washed with ammonium chloride solution (10 g of ammonium chloride in 100 ml of water), dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (elution: methanol/ethyl acetate, 20:80, v/v). The resulting powder was dissolved in dichloromethane and after three days, yellow crystals suitable for single-crystal X-ray diffraction analysis were obtained in 72% yield with a melting point of 425 K.
1H NMR (DMSO, 300 MHz) δ(p.p.m.): 2.7 (s, 3H, CH3); 3 (s, 2H, H2O); 3.7 (m, 2H, CH2O); 4.3 (m, 2H, CH2N); 5 (t, 1H, OH); 7.5–8.5 (m, 3H, C6H3).
13C (DMSO, 75 MHz) δ (p.p.m.): 114.28 (CH3); 47 (CH2O); 59 (CH2N); 106.56; 110.03; 112.87; 117.13; 136.38; 147.37; 155.52 (C4, C5, C6, C7, C8, C9); 162.23 (C=N).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The water H atoms were located in a difference Fourier map; their positional parameters and U iso(H) were refined with O—H distances restrained to be 0.82 Å with a standard deviation of 0.02 Å. Other H atoms were placed in calculated positions [O—H = 0.82, N—H = 0.86, C—H = 0.93 (aromatic), 0.96 (methyl) or 0.97 Å (methylene)] and refined using a riding-model approximation with U iso(H) constrained to 1.2 (amine, aromatic and methylene group) or 1.5 (hydroxyl, methyl group) times U eq of the respective parent atom.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C10H12N3O3S+·Cl−·H2O |
| M r | 307.75 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 298 |
| a, b, c (Å) | 8.8587 (5), 22.1427 (8), 7.1657 (2) |
| β (°) | 108.497 (3) |
| V (Å3) | 1332.98 (10) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.46 |
| Crystal size (mm) | 0.30 × 0.15 × 0.10 |
| Data collection | |
| Diffractometer | Nonius KappaCCD |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 15850, 3856, 3030 |
| R int | 0.029 |
| (sin θ/λ)max (Å−1) | 0.705 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.037, 0.110, 1.06 |
| No. of reflections | 3856 |
| No. of parameters | 183 |
| No. of restraints | 2 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.29, −0.24 |
Supplementary Material
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989016013657/sj5502sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016013657/sj5502Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989016013657/sj5502Isup3.cml
CCDC reference: 1500918
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors are grateful to the Spectropôle Service of the Faculty of Sciences and Techniques of Saint Jérôme (France) for the use of the diffractometer.
supplementary crystallographic information
Crystal data
| C10H12N3O3S+·Cl−·H2O | F(000) = 640 |
| Mr = 307.75 | Dx = 1.534 Mg m−3 |
| Monoclinic, P21/c | Melting point: 425 K |
| Hall symbol: -P 2ybc | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.8587 (5) Å | Cell parameters from 15850 reflections |
| b = 22.1427 (8) Å | θ = 4.1–30.1° |
| c = 7.1657 (2) Å | µ = 0.46 mm−1 |
| β = 108.497 (3)° | T = 298 K |
| V = 1332.98 (10) Å3 | Block, yellow |
| Z = 4 | 0.30 × 0.15 × 0.10 mm |
Data collection
| Nonius KappaCCD diffractometer | 3030 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.029 |
| Graphite monochromator | θmax = 30.1°, θmin = 4.1° |
| f and ω scans | h = −12→12 |
| 15850 measured reflections | k = −31→31 |
| 3856 independent reflections | l = −9→9 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.037 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.110 | w = 1/[σ2(Fo2) + (0.0512P)2 + 0.4116P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max < 0.001 |
| 3856 reflections | Δρmax = 0.29 e Å−3 |
| 183 parameters | Δρmin = −0.24 e Å−3 |
| 2 restraints | Extinction correction: SHELXL2014 (Sheldrick 2015, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 48 constraints | Extinction coefficient: 0.010 (3) |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.13738 (5) | 0.08560 (2) | 0.42268 (6) | 0.05068 (14) | |
| S2 | 0.57871 (5) | 0.21143 (2) | 0.26622 (7) | 0.04747 (14) | |
| O3 | −0.15176 (14) | 0.05681 (6) | 0.56505 (18) | 0.0472 (3) | |
| N4 | 0.36705 (14) | 0.12789 (5) | 0.06943 (17) | 0.0345 (3) | |
| O5 | 0.1477 (2) | 0.20574 (6) | 0.1828 (3) | 0.0718 (4) | |
| H5 | 0.1668 | 0.1773 | 0.2603 | 0.108* | |
| O6 | 0.45778 (19) | −0.14042 (6) | 0.3012 (2) | 0.0634 (4) | |
| C7 | 0.34801 (17) | −0.05387 (6) | 0.1321 (2) | 0.0342 (3) | |
| O8 | 0.24519 (18) | −0.14988 (6) | 0.0528 (2) | 0.0679 (4) | |
| N9 | 0.56755 (14) | 0.08753 (5) | 0.29958 (17) | 0.0314 (2) | |
| H9 | 0.6563 | 0.0838 | 0.3931 | 0.038* | |
| C10 | 0.33709 (16) | 0.06625 (6) | 0.06760 (19) | 0.0313 (3) | |
| C11 | 0.50505 (17) | 0.13941 (6) | 0.2131 (2) | 0.0333 (3) | |
| N12 | 0.35085 (17) | −0.11931 (6) | 0.1646 (2) | 0.0430 (3) | |
| C13 | 0.46514 (15) | 0.04077 (6) | 0.21301 (19) | 0.0288 (3) | |
| C14 | 0.1141 (2) | 0.18374 (9) | −0.0101 (3) | 0.0561 (5) | |
| H14A | 0.0477 | 0.2127 | −0.1014 | 0.067* | |
| H14B | 0.0548 | 0.1463 | −0.0229 | 0.067* | |
| C15 | 0.21856 (18) | −0.02957 (7) | −0.0142 (2) | 0.0395 (3) | |
| H15 | 0.1372 | −0.0546 | −0.0881 | 0.047* | |
| C16 | 0.47534 (16) | −0.02074 (6) | 0.2500 (2) | 0.0310 (3) | |
| H16 | 0.5606 | −0.0383 | 0.3459 | 0.037* | |
| C17 | 0.26321 (19) | 0.17270 (8) | −0.0631 (2) | 0.0443 (4) | |
| H17A | 0.2347 | 0.1583 | −0.1977 | 0.053* | |
| H17B | 0.3207 | 0.2104 | −0.0550 | 0.053* | |
| C18 | 0.21161 (17) | 0.03179 (7) | −0.0491 (2) | 0.0390 (3) | |
| H18 | 0.1269 | 0.0493 | −0.1461 | 0.047* | |
| C19 | 0.7761 (2) | 0.19682 (8) | 0.4278 (3) | 0.0560 (5) | |
| H19A | 0.7703 | 0.1756 | 0.5421 | 0.084* | |
| H19B | 0.8311 | 0.2344 | 0.4673 | 0.084* | |
| H19C | 0.8327 | 0.1727 | 0.3606 | 0.084* | |
| H3B | −0.076 (2) | 0.0647 (10) | 0.524 (3) | 0.060 (6)* | |
| H3A | −0.152 (3) | 0.0212 (7) | 0.573 (4) | 0.070 (8)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0492 (2) | 0.0521 (3) | 0.0514 (2) | −0.00027 (17) | 0.01683 (18) | 0.00853 (17) |
| S2 | 0.0580 (3) | 0.02568 (18) | 0.0586 (3) | −0.00229 (15) | 0.0185 (2) | −0.00067 (15) |
| O3 | 0.0382 (6) | 0.0495 (7) | 0.0520 (7) | 0.0023 (5) | 0.0116 (5) | 0.0069 (5) |
| N4 | 0.0362 (6) | 0.0320 (6) | 0.0354 (6) | 0.0051 (4) | 0.0116 (5) | 0.0048 (4) |
| O5 | 0.1054 (13) | 0.0430 (7) | 0.0891 (11) | 0.0073 (7) | 0.0620 (10) | 0.0007 (7) |
| O6 | 0.0788 (10) | 0.0336 (6) | 0.0715 (9) | −0.0019 (6) | 0.0152 (7) | 0.0062 (6) |
| C7 | 0.0393 (7) | 0.0307 (6) | 0.0376 (7) | −0.0053 (5) | 0.0193 (6) | −0.0049 (5) |
| O8 | 0.0661 (9) | 0.0436 (7) | 0.0917 (11) | −0.0218 (6) | 0.0217 (8) | −0.0198 (7) |
| N9 | 0.0322 (6) | 0.0264 (5) | 0.0335 (6) | −0.0009 (4) | 0.0075 (4) | −0.0016 (4) |
| C10 | 0.0325 (6) | 0.0329 (6) | 0.0299 (6) | 0.0013 (5) | 0.0119 (5) | 0.0003 (5) |
| C11 | 0.0380 (7) | 0.0283 (6) | 0.0360 (7) | 0.0018 (5) | 0.0150 (5) | 0.0011 (5) |
| N12 | 0.0507 (8) | 0.0323 (6) | 0.0540 (8) | −0.0091 (5) | 0.0278 (6) | −0.0085 (6) |
| C13 | 0.0276 (6) | 0.0301 (6) | 0.0296 (6) | −0.0013 (5) | 0.0103 (5) | −0.0019 (5) |
| C14 | 0.0517 (10) | 0.0490 (10) | 0.0725 (12) | 0.0175 (8) | 0.0269 (9) | 0.0167 (9) |
| C15 | 0.0352 (7) | 0.0463 (8) | 0.0379 (7) | −0.0097 (6) | 0.0129 (6) | −0.0094 (6) |
| C16 | 0.0330 (6) | 0.0297 (6) | 0.0322 (6) | −0.0002 (5) | 0.0128 (5) | −0.0003 (5) |
| C17 | 0.0451 (8) | 0.0437 (8) | 0.0447 (8) | 0.0122 (7) | 0.0151 (7) | 0.0151 (7) |
| C18 | 0.0317 (7) | 0.0491 (8) | 0.0335 (7) | 0.0004 (6) | 0.0066 (5) | −0.0011 (6) |
| C19 | 0.0531 (10) | 0.0394 (8) | 0.0723 (12) | −0.0149 (7) | 0.0153 (9) | −0.0057 (8) |
Geometric parameters (Å, º)
| S2—C11 | 1.7194 (14) | N9—H9 | 0.8600 |
| S2—C19 | 1.794 (2) | C10—C18 | 1.388 (2) |
| O3—H3B | 0.833 (16) | C10—C13 | 1.3932 (18) |
| O3—H3A | 0.792 (16) | C13—C16 | 1.3849 (18) |
| N4—C11 | 1.3492 (18) | C14—C17 | 1.506 (2) |
| N4—C10 | 1.3898 (18) | C14—H14A | 0.9700 |
| N4—C17 | 1.4758 (18) | C14—H14B | 0.9700 |
| O5—C14 | 1.405 (3) | C15—C18 | 1.379 (2) |
| O5—H5 | 0.8200 | C15—H15 | 0.9300 |
| O6—N12 | 1.219 (2) | C16—H16 | 0.9300 |
| C7—C16 | 1.3853 (19) | C17—H17A | 0.9700 |
| C7—C15 | 1.393 (2) | C17—H17B | 0.9700 |
| C7—N12 | 1.4667 (19) | C18—H18 | 0.9300 |
| O8—N12 | 1.2251 (18) | C19—H19A | 0.9600 |
| N9—C11 | 1.3390 (17) | C19—H19B | 0.9600 |
| N9—C13 | 1.3867 (16) | C19—H19C | 0.9600 |
| C11—S2—C19 | 101.51 (8) | C17—C14—H14A | 109.2 |
| H3B—O3—H3A | 105 (2) | O5—C14—H14B | 109.2 |
| C11—N4—C10 | 108.48 (11) | C17—C14—H14B | 109.2 |
| C11—N4—C17 | 126.35 (13) | H14A—C14—H14B | 107.9 |
| C10—N4—C17 | 125.16 (12) | C18—C15—C7 | 119.69 (13) |
| C14—O5—H5 | 109.5 | C18—C15—H15 | 120.2 |
| C16—C7—C15 | 124.82 (13) | C7—C15—H15 | 120.2 |
| C16—C7—N12 | 117.17 (13) | C13—C16—C7 | 114.40 (12) |
| C15—C7—N12 | 118.01 (13) | C13—C16—H16 | 122.8 |
| C11—N9—C13 | 108.53 (11) | C7—C16—H16 | 122.8 |
| C11—N9—H9 | 125.7 | N4—C17—C14 | 111.36 (13) |
| C13—N9—H9 | 125.7 | N4—C17—H17A | 109.4 |
| C18—C10—N4 | 131.27 (13) | C14—C17—H17A | 109.4 |
| C18—C10—C13 | 122.29 (13) | N4—C17—H17B | 109.4 |
| N4—C10—C13 | 106.44 (11) | C14—C17—H17B | 109.4 |
| N9—C11—N4 | 109.42 (12) | H17A—C17—H17B | 108.0 |
| N9—C11—S2 | 128.39 (11) | C15—C18—C10 | 116.82 (13) |
| N4—C11—S2 | 122.18 (11) | C15—C18—H18 | 121.6 |
| O6—N12—O8 | 123.47 (15) | C10—C18—H18 | 121.6 |
| O6—N12—C7 | 118.44 (13) | S2—C19—H19A | 109.5 |
| O8—N12—C7 | 118.09 (15) | S2—C19—H19B | 109.5 |
| C16—C13—N9 | 130.92 (12) | H19A—C19—H19B | 109.5 |
| C16—C13—C10 | 121.98 (12) | S2—C19—H19C | 109.5 |
| N9—C13—C10 | 107.10 (11) | H19A—C19—H19C | 109.5 |
| O5—C14—C17 | 112.04 (17) | H19B—C19—H19C | 109.5 |
| O5—C14—H14A | 109.2 | ||
| C11—N4—C10—C18 | 179.32 (14) | C11—N9—C13—C10 | 0.53 (15) |
| C17—N4—C10—C18 | −0.1 (2) | C18—C10—C13—C16 | 0.1 (2) |
| C11—N4—C10—C13 | −1.53 (14) | N4—C10—C13—C16 | −179.10 (12) |
| C17—N4—C10—C13 | 179.07 (13) | C18—C10—C13—N9 | 179.86 (12) |
| C13—N9—C11—N4 | −1.51 (15) | N4—C10—C13—N9 | 0.61 (14) |
| C13—N9—C11—S2 | 178.15 (11) | C16—C7—C15—C18 | −0.1 (2) |
| C10—N4—C11—N9 | 1.90 (15) | N12—C7—C15—C18 | 179.95 (13) |
| C17—N4—C11—N9 | −178.70 (13) | N9—C13—C16—C7 | 179.86 (13) |
| C10—N4—C11—S2 | −177.78 (10) | C10—C13—C16—C7 | −0.51 (19) |
| C17—N4—C11—S2 | 1.6 (2) | C15—C7—C16—C13 | 0.5 (2) |
| C19—S2—C11—N9 | 11.50 (16) | N12—C7—C16—C13 | −179.58 (12) |
| C19—S2—C11—N4 | −168.88 (13) | C11—N4—C17—C14 | −106.73 (18) |
| C16—C7—N12—O6 | 5.1 (2) | C10—N4—C17—C14 | 72.56 (19) |
| C15—C7—N12—O6 | −174.94 (15) | O5—C14—C17—N4 | 60.0 (2) |
| C16—C7—N12—O8 | −174.90 (14) | C7—C15—C18—C10 | −0.2 (2) |
| C15—C7—N12—O8 | 5.0 (2) | N4—C10—C18—C15 | 179.29 (14) |
| C11—N9—C13—C16 | −179.79 (14) | C13—C10—C18—C15 | 0.3 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O5—H5···Cl1 | 0.82 | 2.40 | 3.1840 (15) | 161 |
| C17—H17B···S2 | 0.97 | 2.68 | 3.1514 (18) | 110 |
| O3—H3B···Cl1 | 0.83 (2) | 2.28 (2) | 3.1090 (14) | 178 (2) |
| O3—H3A···Cl1i | 0.79 (2) | 2.37 (2) | 3.1561 (14) | 174 (2) |
| C14—H14B···O8ii | 0.97 | 2.60 | 3.189 (2) | 119 |
| N9—H9···O3iii | 0.86 | 1.85 | 2.6949 (16) | 165 |
Symmetry codes: (i) −x, −y, −z+1; (ii) −x, −y, −z; (iii) x+1, y, z.
References
- Abou, A., Bany, G. E., Kakou-Yao, R., Seikou, T. & Ebby, N. D. (2007). Acta Cryst. E63, o4218.
- Akkurt, M., Türktekin, S., Şireci, N., Küçükbay, H. & Büyükgüngör, O. (2006). Acta Cryst. E62, o185–o187.
- Akonan, L., Molou, K. Y. G., Adohi-Krou, A., Abou, A. & Tenon, A. J. (2010). Acta Cryst. E66, o442. [DOI] [PMC free article] [PubMed]
- Alasmary, F. A. S., Snelling, A. M., Zain, M. E., Alafeefy, A. M., Awaad, A. S. & Karodia, N. K. (2015). Molecules, 20, 15206–15223. [DOI] [PMC free article] [PubMed]
- Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., Giacovazzo, C., Polidori, G. & Spagna, R. (2005). J. Appl. Cryst. 38, 381–388.
- Chen, S. H., Yang, F. R., Wang, M. T. & Wang, N. N. (2010). C. R. Chim. 13, 1391–1396.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Gao, X. J., Jin, S., Liang, S., Chen, W. & Wang, D. (2013). J. Mol. Struct. 1039, 144–152.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hooft, R. (1998). COLLECT. Nonius BV, Delft, The Netherlands.
- Janiak, J. (2000). J. Chem. Soc. Dalton Trans. pp. 3885–3896.
- Kakou-Yao, R., Abou, A., Adjou, A., Bany, G. E. & Ebby, N. D. (2007). Acta Cryst. E63, o4463.
- Kerimov, I., İlgar, , Ayhan-Kılcıgil, G., Özdamar, E. D., Can-Eke, B., Çoban, T., Özbey, S. & Kazak, C. (2012). Arch. Pharm. Pharm. Med. Chem. 345, 549–556. [DOI] [PubMed]
- Küçükbay, H., Durmaz, R., Okuyucu, N., Günal, S. & Kazaz, C. (2004). Arzneim.-Forsch. Drug. Res. 54, 64–68. [DOI] [PubMed]
- Küçükbay, H., Durmaz, R., Orhan, E. & Günal, S. (2003). Farmaco, 58, 431–437. [DOI] [PubMed]
- Liu, J. & Pan, Q. (2014). Z. Kristallogr. New Cryst. Struct. 229, 111–112.
- Lokaj, J., Kettmann, V., Milata, V. & Solčan, T. (2009). Acta Cryst. E65, o1788. [DOI] [PMC free article] [PubMed]
- Morozov, P. J., Kurbatov, S. V., Dolgushin, F. M., Antipin, M. Y. & Olekhnovich, L. P. (2004). Russ. Chem. Bull. 9, 1990–1994.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Puratchikody, A., Nagalakshmi, G. & Doble, M. (2008). Chem. Pharm. Bull. 56, 273–281. [DOI] [PubMed]
- Samsonov, V. A., Gatilov, Y. V. & Savel’ev, V. A. (2013). Russ. J. Org. Chem. 49, 1208–1214.
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Verdan, S., Melich, X., Bernardinelli, G. & Williams, A. F. (2009). CrystEngComm, 11, 1416–1426.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yavo, E. A., Kakou-Yao, R., Coulibaly, S., Abou, A. & Tenon, A. J. (2007). Acta Cryst. E63, o4551.
- Yuasa, J., Ogawa, T. & Kawai, T. (2010). Chem. Commun. 46, 3693–3695. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989016013657/sj5502sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016013657/sj5502Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989016013657/sj5502Isup3.cml
CCDC reference: 1500918
Additional supporting information: crystallographic information; 3D view; checkCIF report