

Abstract
Birth defects are among the leading causes of infant mortality and contribute substantially to illness and long-term disability. Defects in Bone Morphogenetic Protein (BMP) signaling are associated with cleft lip/palate. Many craniofacial syndromes are caused by defects in signaling pathways that pattern the cranial neural crest cells (CNCCs) along the dorsal-ventral axis. For example, auriculocondylar syndrome is caused by impaired Endothelin-1 (Edn1) signaling, and Alagille syndrome is caused by defects in Jagged-Notch signaling. The BMP, Edn1, and Jag1b pathways intersect because BMP signaling is required for ventral edn1 expression that, in turn, restricts jag1b to dorsal CNCC territory. In zebrafish, the scaffolding protein Wdr68 is required for edn1 expression and subsequent formation of the ventral Meckel’s cartilage as well as the dorsal Palatoquadrate. Here we report that wdr68 activity is required between the 17-somites and prim-5 stages, that edn1 functions downstream of wdr68, and that wdr68 activity restricts jag1b, hey1, and grem2 expression from ventral CNCC territory. Expression of dlx1a and dlx2a was also severely reduced in anterior dorsal and ventral 1st arch CNCC territory in wdr68 mutants. We also found that the BMP agonist isoliquiritigenin (ISL) can partially rescue lower jaw formation and edn1 expression in wdr68 mutants. However, we found no significant defects in BMP reporter induction or pSmad1/5 accumulation in wdr68 mutant cells or zebrafish. The Transforming Growth Factor Beta (TGF-β) signaling pathway is also known to be important for craniofacial development and can interfere with BMP signaling. Here we further report that TGF-β interference with BMP signaling was greater in wdr68 mutant cells relative to control cells. To determine whether interference might also act in vivo, we treated wdr68 mutant zebrafish embryos with the TGF-β signaling inhibitor SB431542 and found partial rescue of edn1 expression and craniofacial development. While ISL treatment failed, SB431542 partially rescued dlx2a expression in wdr68 mutants. Together these findings reveal an indirect role for Wdr68 in the BMP-Edn1-Jag1b signaling hierarchy and dorso-anterior expression of dlx1a/2a.
Introduction
Birth defects are among the leading causes of infant mortality and contribute substantially to illness and long-term disability. Craniofacial anomalies, excluding cleft lip and palate, occur in 1 out of every 1600 births in the United States [1]. Defects in Bone Morphogenetic Protein (BMP) signaling are associated with cleft lip/palate [2, 3]. Many craniofacial syndromes are caused by defects in signaling pathways that pattern the cranial neural crest cells (CNCCs) along the dorsal-ventral (D/V) axis. For example, Auriculocondylar syndrome is caused by impaired Endothelin-1 (Edn1) signaling [4–6] and Alagille syndrome is caused by defects in Jagged (Jag)-Notch signaling [7, 8]. The BMP, Edn1, and Jag1b pathways intersect because BMP signaling is required for ventral edn1 expression that, in turn, restricts jag1b to dorsal CNCC territory [9–11].
Craniofacial development in vertebrate organisms is a highly conserved process and involves interactions between multiple signaling pathways. The zebrafish Danio rerio is a model vertebrate organism particularly suited to the study of early developmental events. Craniofacial development begins shortly after the end of gastrulation with the specification and subsequent migration of CNCCs from the dorsal part of the neural tube. The CNCCs migrate ventro-laterally from the dorso-medial neural tube to one of seven pharyngeal arches. Once in the arches, the CNCCs receive and participate in multiple signaling events [12]. Lateral ectoderm and medial endoderm cells together form pouches that cup the CNCCs and deliver various paracrine signals important for both the survival and the patterning of the adjacent CNCCs [13–15]. The zebrafish jaw structures derived from the first arch CNCCs are the dorsal palatoquadrate (PQ) and ventral Meckel’s (M) cartilages [16, 17].
BMP and Jag-Notch signaling regulate ventral and dorsal CNCC patterning, respectively [9–11]. Edn1 signaling is required for ventral CNCC patterning downstream of BMP signaling [10, 18]. The combined action of these signaling pathways sub-define regions of CNCCs along the D/V axis within the first pharyngeal arch. These sub-defined territories are evident in the layered expression patterns of the distal-less (dlx) family of transcription factors [19]. Specifically, loss of Edn1 signaling results in the loss or reduction of ventral and intermediate structures such as Meckel’s cartilage (M) and the jaw joint. Additionally, the loss of Edn1 signaling has been shown to result in the dorsalization of ventral structures [18, 20–22]. In mice, mutants for either edn1-/- or ednra-/- display loss of expression for dlx3-6, and hand2 [21, 22]. Similarly, edn1-/- mutant zebrafish lose ventral and intermediate expression of dlx3-6 and hand2 [18]. Overexpression of Bmp4 expands ventral territory via upregulation of edn1 and the corresponding downstream network of transcription factors including hand2 and several dlx genes [10]. Conversely, disruption of later stage Bmp signaling by overexpressing a dominant negative Bmp receptor after CNCC induction results in the loss of ventral edn1 and dlx6a expression, intermediate-ventral dlx3b expression, and the ventral expansion of the normally dorsal restricted jag1b [9]. In zebrafish, jag2 and jag1b are expressed in the CNCCs and pharyngeal pouch endoderm [11, 23] and antisense knockdown of them results in reductions in dorsal cartilages [24]. Zebrafish jag1b mutants display dorso-posterior defects in PQ formation; antisense morpholino knockdowns of notch2 yield a similar phenotype consistent with a traditional Jag-Notch signaling requirement for dorsal development. Loss of Jagged-Notch signaling results in the dorsal expansion of ventral and intermediate specifying dlx3b and dlx5a markers. Misexpression of jag1b results in the dorsalization of ventral structures, particularly with the transformation of M into a more PQ like structure. Jag1b is normally restricted to the dorsal-most CNCC within the first arch, however ubiquitous overexpression of jag1b results in the loss of ventrally expressed hand2 and edn1 as well as restriction of ventral-intermediate markers dlx3b, dlx5a, and dlx6a to the most ventral territory of the arch [11]. Taken together, these interactions yield a model in which BMP signaling patterns ventral and intermediate territory at least partly through induction of Edn1 signaling, while Jag-Notch signaling patterns dorsal territory, and mutual antagonism stabilizes patterning along the D/V axis [25]. These combined interactions yield spatial restrictions of a nested network of transcription factors into dorsal, intermediate, and ventral territories that prefigure the future lower jaw, joint, and upper jaw cartilages.
BMP2/4/7 ligands are members of the Transforming Growth Factor Beta (TGF-β) superfamily of growth factors. The TGF-β signaling pathway is also important for several patterning events during early embryonic development, including proper craniofacial development [26]. While BMPs act through the phospho-Smad1/5 (pSmad1/5) transcription factors in complex with the common shared Smad4 to regulate downstream target gene expression, the TGF-β1/2/3 ligands instead act through pSmad2/3 also in complex with Smad4 [27, 28]. Interestingly, TGF-β can interfere with BMP signaling via Smad3 displacement of Smad4 from pSmad1/5 complexes [29, 30].
Wdr68/Dcaf7 (hereafter referred to as Wdr68) is a highly conserved 342 amino acid length member of the WD40 repeat domain family of proteins that are generally known to function as scaffolding proteins [31–33]. Vertebrate Wdr68 interacts with several protein kinases including several members of the Dual-specificity tyrosine phosphorylation-regulated kinase (Dyrk) gene family [34–38]. Dyrk1a maps to the Down Syndrome (DS) critical region of human chromosome 21 and overexpression of Dyrk1a is a major contributor to the neurodevelopmental defects present in DS patients [39, 40]. Dyrk1a and Dyrk1b possess RNApII-CTD kinase activity important for the expression of several genes [41]. High-throughput protein interaction assays also suggest that Wdr68 interacts with the pSmad2/3 components of TGF-β signaling [42]. The subcellular localization of Wdr68 is likely regulated. When expressed alone, Wdr68 is found in both the cytoplasm and nucleus. However, co-expression with the RNApII-CTD kinase Dyrk1a induces nuclear translocation of Wdr68 [35, 37, 43]. Wdr68 is expressed widely in early development and regulates ventral cartilage formation in the zebrafish [36, 43]. Specifically, wdr68 is required for expression of edn1 and genes downstream of Edn1 such as bapx1, hand2, and several dlx genes [36]. wdr68 is also required for the expression of the spaw, lft1, and lft2 genes [34] that are downstream of BMP signaling in the zebrafish [44, 45].
Together, these previous findings suggest Wdr68 may modulate TGF-β/BMP signaling to downstream targets such as the Edn1-Jag1b network for D/V patterning. The wdr68hi3812 allele contains a retroviral insertion within the first exon after codon 44 and is therefore considered a null allele [36, 46]. Here we report that the temperature-dependent severity of the cartilage defects present in wdr68hi3812 homozygotes is a consequence of differential perdurance of maternally derived Wdr68. We then exploited the temperature-dependence, along with a Tg(hsp70l:GFP-Wdr68) zebrafish line, to define a temporal window between the 17-somites and prim-5 stages during which wdr68 activity is required for craniofacial development. Notably, this window overlaps substantially with the known onset of edn1 expression and activity during craniofacial development. We also report that edn1 expression can partially rescue dlx6a expression in wdr68 mutants, consistent with it functioning downstream of wdr68. However, edn1 expression was unable to restore hand2 expression in wdr68 mutants. We also found ventral expansion of the expression of the normally dorsal-restricted jag1b, hey1, and grem2 genes in wdr68 mutants. We further report pharmacogenetic interactions between BMP signaling and wdr68. Specifically, the BMP antagonist dorsomorphin (DM) exacerbates the wdr68 cartilage and edn1 expression defects while the BMP agonist isoliquiritigenin (ISL) partially rescues the wdr68 cartilage and edn1 expression defects. Using CRISPR/Cas9 generated mouse C2C12 sublines lacking wdr68, we found no significant changes in pSmad1/5 levels, pYap1 levels, or BRE-Luciferase responsiveness to BMP ligands. We also found little to no defects in pSmad1/5 levels in zebrafish wdr68 mutants. However, co-challenge with TGF-β and BMP ligands revealed significant enhancement of TGF-β interference with BRE-Luciferase responsiveness to BMP ligands in C2C12 sublines lacking wdr68. Suggesting conservation of this mechanism in vivo, we also report that treating wdr68 mutants with the TGF-β inhibitor SB431542 also partially rescued the craniofacial and edn1 expression defects. Further analysis of dorsal CNCC markers revealed that expression of dlx1a and dlx2a was severely reduced in anterior dorsal and ventral 1st arch CNCC territory in wdr68 mutants. While ISL treatment failed to restore dlx2a expression, SB431542 partially rescued dlx2a in wdr68 mutants. Together, the findings reported here reveal an indirect role for Wdr68 in the BMP-Edn1-Jag1b signaling hierarchy and dorso-anterior expression of dlx1a/2a. These findings also suggest interactions between Wdr68 and TGF-β signaling that will require further investigation.
Materials and Methods
Chemicals and reagents
Dorsomorphin (P5499, Sigma-Aldrich), isoliquiritigenin (I3766, Sigma-Aldrich), SB431542 (S4317, Sigma-Aldrich), BMP4 (HZ-1045, Humanzyme), and TGF-β1 (HZ-1011, Humanzyme) were purchased and used according to the manufacturers recommendations. Antibodies used were anti-Wdr68 (HPA022948, Sigma-Aldrich), anti-β-tubulin (sc-55529, Santa Cruz Biotechnology Inc.), anti-pYap1 (13008S, Cell Signaling), anti-Yap1 (4912S, Cell Signaling), anti-pSmad1/5 (9511S, Cell Signaling), goat anti-mouse IgG-HRP (sc-2005, Santa Cruz Biotechnology Inc.), Amersham ECL anti-rabbit IgG, HRP-linked whole antibody (from donkey) (NA934, GE Healthcare).
Zebrafish husbandry
This project was approved and conducted under the approved Cal State LA IACUC protocol (14–2 Renewal 11–3) and the zebrafish animals (TAB14 and AB* backgrounds) used in it were reared in strict accordance with IACUC guidelines. Zebrafish embryos were raised at 24°C, 28.5°C, or 32°C as specified in specific experiments.
Alcian blue staining
Alcian blue staining was performed as previously described [34, 43]. Briefly, after fixation with 4% PFA, embryos were washed in Phosphate Buffered Saline + 0.1% Tween-20 (PBST) and dehydrated through 50% PBST/ 50% Methanol (MeOH) while rocked for 5 minutes. Samples were put into 100% Methanol and stored on ice for 30 minutes. Methanol was then removed and samples stained in 0.1% Alcian Blue overnight while rocking. The next day samples were rinsed twice with 100% Ethanol (EtOH) and then rehydrated through 50% PBST/ 50% MeOH into PBST. Samples were digested in 0.05% Trypsin in saturated Sodium Borate (Na2B4O7) for 3 hours at 37°C. Samples were bleached overnight in a solution of 3% H2O2; 1% KOH at 4°C. The next day samples were washed in PBS + 1.0% Tween-20 for 5 min and then re-suspended in 80% glycerol; 0.1% Tween-20. Animals were scored for the presence of the M and PQ cartilages.
Immunofluorescence
Embryos were fixed in 4% PFA in PBST for 2 hours at room temperature (RT), then washed twice with PBST, once with 50% PBST/50% MeOH, once with ice-cold 100% MeOH for 2 min, quickly washed twice with dH2O and then exposed to 100% acetone at -20°C for 4 min followed by three washes with PBST. Embryos were then washed twice with PBSTTD (PBST + 0.1% Triton X-100 + 1% DMSO) for 5 min. Embryos and primary antibodies were pre-blocked for 30 min at RT in Block solution (PBSTTD + 0.5% Boehringer Mannheim Block solution + 10% Lamb serum) and then incubated in primary antibody overnight at 4°C. The next day embryos were washed twice quickly and five times for 10 minutes each in PBSTD (PBST + 1% DMSO) and then re-blocked for 30 minutes before exposure to blocked secondary antibody for 2 hours at RT. Then, embryos were washed twice quickly and five times for 10 minutes each in PBSTD followed by imaging on a Zeiss Apotome microscope.
Confocal imaging
The Tg(sox10:mCherryCAAX) animals were kindly provided by the Crump Lab [47]. Confocal multi-TIFF stacks were captured using an Olympus IX81 confocal microscope on agarose-mounted embryos and analyzed using FIJI volume viewer.
Transgenesis constructs and isolation of transgenic lines
The Tg(hsp70l:GFP-wdr68)csu6 and Tg(hsp70l:GFP-wdr68)csu9 lines were created using the Gateway Tol2kit reagents [48]. Briefly, the GFP-wdr68 fusion fragment from pCS2+GFP-wdr68 [36] was inserted into the pDONR221 plasmid to yield the pME-GFP-wdr68 plasmid that was then combined with p5E-hsp70l, p3E-polyA and pDestTol2CG2 to create pT2-hsp70l:GFP-wdr68-CG2. This plasmid was then co-injected with Tol2 transposase mRNA into zebrafish embryos at the 1-cell stage to create the founders for subsequent isolation of stable transgenic lines.
Microinjections
Zebrafish embryos were harvested and injected with morpholino solution at the one to four cell stages as previously described [34, 43]. The morpholino solutions used were 3812-2/4: 200μM 3812–4 morpholino, 500μM 3812–2 morpholino in 0.1% phenyl red; 0.5x PBS or 3812–1: 700μM 3812–1 morpholino in 0.1% phenyl red; 0.5x PBS [36]. The 3812-2/4 solution blocks endogenous wdr68 mRNA translation, while the 3812–1, which does not block translation, served as a negative control.
Heat Shock Rescues
Tg(hsp70l:GFP-wdr68)csu9 animals were crossed to wildtype TAB14 fish. Embryos were collected at the one cell stage and injected with either 3812-2/4 morpholino, blocking translation of endogenous Wdr68, or with the negative control 3812–1 morpholino. Embryos were then allowed to develop in a 28.5°C incubator. Heat shocks were performed at 39°C for 30min at each of the stages of interest. Heat shocks were conducted at 5-somites, 12-somites, 15-somites, 17-somites, 20-somites, 25-somites, prim-5, prim-12 and prim-20 stages. After 30 minutes of heat shock, animals were returned to 28.5°C, and allowed to develop until the formation of the swim bladder was observed in control animals [approximately 5 days post fertilization (dpf)]. Animals were then fixed for 1 hour at room temperature or overnight at 4°C in 4% PFA in PBST, and cartilages scored via Alcian Blue Staining.
Probe Synthesis and In Situ Hybridization
Probes for jag1b, hey1, and grem2 were previously described [10, 11]. The edn1 plasmid has been previously described [18]. The dlx6a, hand2, dlx1a, and dlx2a plasmids have been previously described [49–52]. All transcription reactions were conducted with the MEGAscript SP6, T7, or T3 Transcription Kits per manufacturer’s instructions (Ambion), with the adaptation of using digoxigenin-labeled-UTP. ISH was performed as previously described [34, 53] with the following modifications. The jag1b, hey1, grem2, and dlx1a probes were used at 0.2ng/μL in hybridization buffer (hyb) overnight at 65°C. The edn1, hand2, dlx6a, and dlx2a probes were used at 0.5ng/μL in hyb buffer overnight at 70°C. Once development was complete, embryos were washed in PBST, bleached in 10% H2O2; 5% formamide in PBST for 20 min. Finally embryos were washed in PBST and cleared in 80% glycerol containing 0.1% Tween-20 in dH2O for imaging.
Embryo Genotyping
Embryos were separated into individual Eppendorf tubes, excess water removed, and then digested in Proteinase K at a final concentration of 100μg/ml for 2 hours at 55°C and were vortexed every 15 minutes during the incubation. Samples were then diluted two-fold with dH2O and vortexed for 1 minute. Proteinase K was then inactivated by heating samples to 96°C for 15 minutes. Samples were then centrifuged at top speed for 1 minute to pellet cellular debris. 2μL of the solution was then used as template in a PCR reaction using the previously described primers 3812-c; 3812–3; and LTR-f1 [36].
Plasmid construction and plasmid rescue assay
The primers edn1-f1 5’-TTCTTCGGATCCACCATGCATTTGAGGATTATTTTCCCAGTTCTG-3’ and edn1-r1 5’-TTCTTCGAATTCCTATGAGTTTTCAGAAATCCACGCTTG-3’ were used to PCR amplify the edn1CDS from a previously described ISH probe plasmid containing edn1 [18]. The insert was TOPO cloned, DNA sequence verified, and subcloned into pCS2+ to yield pCS2+edn1CDS. For rescue, a pCS2+eGFP marker plasmid was co-injected at the 1-cell stage with either pCS2+dsRed or the pCS2+edn1CDS plasmid, each at 50ng/μL.
Drug Treatments of Embryos
Adult wdr68hi3812/+ zebrafish were crossed to obtain groups of embryos that were placed in a 24°C incubator for later dorsomorphin (DM) treatment or a 32°C incubator for later isoliquiritigenin (ISL) or SB431542 (SB) treatment. Dechorionated embryos were exposed to 0.1% DMSO control or DM/ISL/SB starting at the 14- to 15-somites stage and processed at 5 dpf for Alcian staining or at the 20–24 somites stages for ISH analysis. Various drugs were kept on the embryos until fixation.
CRISPR/Cas9-mediated knockouts in C2C12 cells
C2C12 cells were obtained from the ATCC. pLentiCRISPRv2-dcaf7-2 was generated by digesting the pLentiCRISPRv2 plasmid with BsmBI, the oligonucleotides CRISPR-mdcaf7-2f: 5’-CACCGACATCGCCTTCAGCCGCGC-3’ and CRISPR-mdcaf7-2r: 5’-AAACGCGCGGCTGAAGGCGATGTC-3’ were annealed, and then ligated into the vector fragment as previously described [54]. Putative clones were isolated from Stbl3 competent cells and verified by DNA sequencing. Lentiviral particles were generated by co-transfecting 293T cells with the virus packaging plasmids psPAX2 and pCMV-VSV-G along with pLentiCRISPR-dcaf7-2. Cleared virus-containing supernatant was used to transduce C2C12 cells in C2C12 growth medium (GM) (DMEM + 2.5% FBS + 10% calf serum + 7.4 mM L-glutamine + 100μg/mL pen/strep). The transduced C2C12 cells were subjected to 1μg/mL puromycin (GM+puro) selection, and clonal sublines were isolated by serial dilution. Putative knock-out sublines were screened by Western blot to identify functional knock-outs.
Western blots on zebrafish embryo or C2C12 cell extracts
Embryos were ground on ice in a 1.5 mL microcentrifuge tube using a plastic pestle in 50μL ice-cold RIPA buffer (50mM Tris pH 8, 150mM NaCl, 1% Igepal CA630, 0.5% NaDeoxycholate, 0.1% SDS) with 1x Protease Inhibitor Cocktail (PIC). The samples were vortexed for 1 minute and then centrifuged for 1 minute at room temperature at high speed. The samples were maintained on ice for 1 hour and every 10 minutes were vortexed for 1 minute. After the 1-hour period ended, the tubes were centrifuged to pellet debris and supernatants quantified by Bradford assay prior to being subjected to western blot analysis as described further below. C2C12 cell extracts were made from 10cm plates of cells grown in GM or Differentiation Medium (DMEM + 2% Horse serum + 100ug/mL pen/strep), rinsed twice with ice-cold PBS, and then incubated in 0.5mL ice-cold RIPA buffer + 1x PIC for 15 min at 4°C. Cells were then scraped from the plate, incubated on ice for 1 hour, centrifuged at 10,000g at 4°C and supernatants quantified by Bradford assay prior to being subjected to western blot analysis as follows. Equal amounts of protein samples were boiled for 5 minutes at 95°C, ran on 8–16% SDS-PAGE gels, and then transferred onto PVDF membrane (Thermo Scientific). The PVDF membrane was blocked overnight at 4°C with 5% non-fat dry milk in PBST (PBS+0.01% Tween-20) with 0.02% NaN3. The following day the blocking buffer was removed and blocked primary antibody was added. The membrane was then rinsed 3X for 5 minutes with PBST and then blocked secondary antibody was added. After the incubation period the membrane was rinsed 3X for 5 minutes with PBST. In most cases, the same blot was stripped and re-probed for relevant controls (beta-tubulin, total Yap).
Transient transfections and reporter assays
BMP-luciferase (BRE-Luc) reporter plasmid [55] and SV-40 Renilla Luciferase (SV40-Luc) reporter plasmid were co-transfected into the C2C12 sublines using X-tremeGene HP as per the manufacturers recommendations (Roche). After 8 or 16 hours of transfection, DNA-lipid complexes were replaced with fresh medium -/+ BMP4 and/or -/+ TGF-β1. After an additional 8 or 20 hours, cell extracts were harvested for luminometer measurements using the Dual-luciferase reporter (DLR) assay kit reagents (Promega).
Statistical Analysis
Pairwise comparisons were performed using the Student’s T-test. Experiments containing 3 or more conditions were subjected to one-way ANOVA and post-hoc Tukey HSD tests.
Results
wdr68 activity is required during a window between the 17 somites and prim-5 stages
The wdr68hi3812/hi3812 mutant was originally described as presenting a range of jaw defects that varied from only mild joint fusions to near complete losses of the palatoquadrate (PQ) and Meckel’s (M) cartilages [36]. The initial characterization was completed using embryos raised at the standard 28.5°C. We have since found that rearing the animals at 24°C yielded mutant animals that mostly present only the mild M-PQ joint fusion defects (Fig 1B red arrowhead, compare to 1A). Specifically at 24°C, 79% of mutants presented only mild M-PQ fusions versus 21% displaying severe loss of the M and/or PQ cartilage. In contrast, rearing embryos at 32°C yielded mutant animals that mostly present severe losses of the M and PQ cartilages (Fig 1C compare to 1A, 1B). Specifically at 32°C, 6% of mutants presented only mild M-PQ fusions versus 94% displaying severe loss of the M and/or PQ cartilage. Because the wdr68hi3812 allele is a retroviral insertion within exon-1 near a splicing junction [36], and insertional mutations can cause temperature-dependent splicing defects [56], we examined animals reared at 24°C versus 32°C by RT-PCR in an effort to detect potential alternative splicing products but found no evidence to support such a mechanism (data not shown).
Fig 1. Wdr68 is required for craniofacial development between the 17 somites and prim-5 stages.

A) Ventral view of 5dpf alcian blue stained cartilages in wild type zebrafish. M: Meckel’s PQ: Palatoquadrate. B) Mild mutant phenotype resulting from rearing of embryos at 24°C, characterized by joint fusions (arrowhead) between M and PQ. C) Severe mutant phenotype resulting from rearing of embryos at 32°C, characterized by the loss of M and PQ. D) Immunohistochemistry readily detects maternal Wdr68 protein up to the 17 somites stage in wdr68hi3812/hi3812 mutants raised at the permissive 24°C temperature (blue line). Maternal Wdr68 is lost by the 17 somites stage in wdr68hi3812/hi3812 mutants raised at the non-permissive 32°C temperature (red line). E) wdr68-MO injected non-transgenic (Non-Tg) animals display jaw defects regardless of heat shock. wdr68-MO injected Tg(hsp70l:GFP-Wdr68)csu9 animals that are heat shocked by the prim-5 stage show rescue from jaw defects. Error bars indicate standard deviation. Additional abbreviations: Shield (Sh), 15 somites (15s), 17 somites (17s), 20 somites (20s), 25 somites (25s), prim-5 (p5), prim-25 (p25), no heat shock (-HS).
We next used immunofluorescence to determine whether Wdr68 protein might exhibit temperature-dependent differences in perdurance (Fig 1D and S1A–S1D Fig). We expected to find Wdr68 protein in very early stage wdr68hi3812/hi3812 mutants because we previously reported that wdr68 transcripts are maternally supplied [36]. Consistent with that expectation, 100% of shield-stage embryos obtained from wdr68hi3812/+ in-crosses that were analyzed by immunofluorescence were positive for Wdr68 expression when raised at either 24°C or 32°C. Random genotyping identified 2/16 wdr68hi3812/hi3812 mutants confirming that the appropriate breeding cross had been made. However, by the 17-somites stage embryos raised at 32°C were strikingly different than those raised at 24°C (Fig 1D, 17s red versus blue). While no embryos raised at 24°C displayed severe loss of Wdr68 expression, a near-mendelian ratio (20%) of embryos raised at 32°C lacked Wdr68 expression. Genotyping confirmed that 5/5 embryos raised at 32°C and lacking Wdr68 expression by the 17-somites stage were wdr68hi3812/hi3812 mutants. Likewise, 9/9 embryos raised at 32°C and possessing Wdr68 expression were wildtype or wdr68hi3812/+ (hereafter summarized as +/*). Random genotyping of the embryos raised at 24°C that all had Wdr68 expression identified 3/10 wdr68hi3812/hi3812 mutants confirming that the appropriate breeding cross had been made. By the prim-25 stage, both embryos raised at 24°C and 32°C yielded near-mendelian ratios (24.1% and 23.8%, respectively) of embryos lacking Wdr68 expression. Thus, a critical function for M and PQ formation is performed during late somitogenesis by Wdr68 that is cut short in embryos raised at 32°C.
To further delineate the temporal window during which Wdr68 functions to mediate M and PQ formation, we generated Tg(hsp70l:GFP-wdr68) zebrafish lines (S1E–S1G Fig). The lines were first characterized by heat shocking them at 39°C for various periods of time followed by fluorescence imaging (S1E and S1F Fig) and then harvested for western blot analysis using anti-Wdr68 antibody (S1G Fig). We found that a 30-minute heat shock was sufficient to yield fusion protein expression that lasted for at least 7 more hours (S1F and S1G Fig, lane 7). We then injected embryos from outcrosses of the Tg(hsp70l:GFP-wdr68) adults with a previously described antisense wdr68-MO [36] and subjected them to 30-minute heat shocks at various developmental stages to determine how late in development restored Wdr68 activity could still rescue craniofacial development (Fig 1E). Overexpression of GFP-Wdr68 in otherwise wildtype embryos did not yield any observable defects (S2A and S2B Fig). Over 75% of non-transgenic wdr68-MO injected sibling embryos displayed jaw defects in spite of receiving heat-shocks at all developmental time-points examined indicating that heat shock alone was unable of rescue (Fig 1E, red line non-Tg). These embryos were also indistinguishable from both transgenic and non-transgenic wdr68-MO injected embryos that did not receive any heat shock (Fig 1E, -HS data point at the far right for each line; S2C and S2D Fig representative images for mild and severe phenotypes). In contrast, less than 25% of Tg(hsp70l:GFP-wdr68) sibling embryos heat shocked at 39°C between the 17-somites and prim-5 stages displayed jaw defects indicating rescue of craniofacial development as late as the prim-5 stage (Fig 1E, blue line; S2G Fig compare 25s and p5 with p25 and -HS). However, heat shocking the wdr68-MO-injected Tg(hsp70l:GFP-wdr68) embryos at the prim-25 stage failed to rescue craniofacial development (Fig 1E, blue line; S2G Fig). Thus, restoring Wdr68 function as late as the prim-5 stage is sufficient to rescue craniofacial development. Together these findings indicate that Wdr68 function is important for craniofacial development during a window between the 17-somites and prim-5 stages.
edn1 functions downstream of wdr68
We previously showed that edn1 expression depends on wdr68 activity and that expression of downstream targets of Edn1 signaling, such as dlx6a and hand2, are also reduced or absent in wdr68hi3812/hi3812 mutants [36]. The simplest model for these observations is that edn1 functions downstream of wdr68 and that the primary function of wdr68 in lower jaw development is, either directly or indirectly, the induction of edn1 expression. A simple prediction of this model is that restoring edn1 expression in a wdr68hi3812/hi3812 mutant should also restore the expression of edn1 target genes, such as dlx6a and hand2. To test this model, we generated a plasmid construct for expressing edn1, injected embryos from crosses of wdr68hi3812/+ adults with various combinations of mRNAs or plasmids, and then processed the embryos raised at 32°C for either alcian blue staining of cartilages (Fig 2A–2E) or in situ hybridization (ISH) using dlx6a probe to assess potential rescue (Fig 2F–2Q). The negative control mRNA EF1a had no effect on wildtype siblings and failed to rescue wdr68hi3812/hi3812 mutants (Fig 2A and 2B). In contrast a significantly higher fraction of the embryos injected with Edn1 mRNA displayed partially restored M-like cartilages in wdr68hi3812/hi3812 mutants (Fig 2C and 2E; compare Edn1 vs EF1a, p<0.001). However, Edn1-injected wdr68hi3812/hi3812 mutants largely failed to form normal PQ-like cartilages. As expected, wdr68hi3812/hi3812 mutants injected with Flag-Wdr68 mRNA displayed nearly-normal M and PQ cartilages (Fig 2D and 2E; compare FW vs EF1a, p<0.001).
Fig 2. Edn1 functions downstream of wdr68 for craniofacial development.

(A-D) Ventral views of 5dpf Alcian stained craniofacial cartilages of zebrafish raised at 32°C. A) wildtype sibling injected with EF1a mRNA. B) wdr68 mutant injected with EF1a mRNA. C) wdr68hi3812/hi3812 mutant injected with Edn1 mRNA. D) wdr68hi3812/hi3812 mutant injected with Flag-Wdr68 (FW) mRNA. E) Edn1 mRNA-injected mutants have more M cartilage elements than EF1a controls (p<0.001). (F-Q) ISH analysis of dlx6a expression in embryos raised at 32°C with red arrowhead pointing at 1st arch CNCC territory. F, H, J, L) dorsal view of prim-5 stage. G, I, K, M) lateral view of prim-5 stage. F, G) wildtype sibling injected with GFP/dsRed (G/R) plasmid mix showing normal dlx6a expression. H, I) wdr68hi3812/hi3812 mutant injected with G/R plasmid mix showing loss of dlx6a in 1st arch CNCC. J, K) wildtype sibling injected with GFP/Edn1 (G/Edn1) plasmid mix showing near-normal dlx6a expression. L, M) wdr68hi3812/hi3812 mutant injected with G/Edn1 plasmid mix showing partial rescue of dlx6a in 1st arch CNCC. N-Q) lateral view of dlx6a expression in prim-5 stage embryos raised at 28.5°C. N) heat shocked wildtype sibling control with normal dlx6a. O) heat shocked wildtype sibling injected with Et1-MO showing loss of dlx6a. P) heat shock induced Tg(hsp70l:GFP-Wdr68) sibling control with normal dlx6a. Q) heat shock induced Tg(hsp70l:GFP-Wdr68) sibling injected with Et1-MO showing loss of dlx6a.
As control for ISH analysis, wildtype sibling embryos injected with plasmids expressing GFP and dsRed (G/R) displayed normal expression levels of dlx6a in the pharyngeal arches (Fig 2F and 2G, red arrowhead). Likewise, wdr68hi3812/hi3812 mutants injected with the G/R plasmid mix displayed the expected loss of 1st arch dlx6a expression (Fig 2H and 2I, red arrowhead). Wildtype sibling embryos injected with plasmids expressing GFP and Edn1 (G/Edn1) were slightly smaller than normal but displayed near-normal expression levels of dlx6a in the pharyngeal arches (Fig 2J and 2K, red arrowhead). In contrast, wdr68hi3812/hi3812 mutants injected with the G/Edn1 plasmid mix partially restored dlx6a expression (compare Fig 2L and 2M with 2H and 2I). Genotyping confirmed that 0/12 mutants injected with the G/R plasmid mix had restored dlx6a expression (Fig 2H and 2I, 0% rescue), while 9/14 mutants injected with the G/Edn1 plasmid mix had restored dlx6a expression (Fig 2L and 2M, 64% rescue). Notably, the restoration of dlx6a expression appeared largely restricted to the ventral-most region of the 1st arch in G/Edn1-injected wdr68hi3812/hi3812 mutants (compare Fig 2M to 2K). A significant caveat to these findings is that, at least in our hands, plasmid-based Edn1 is very broadly expressed in these experiments and thus the partial rescue may be the result of a non-local ectopic Edn1 source (S3E and S3F Fig).
To further explore the epistatic relationships between edn1 and wdr68, we examined dlx6a expression in Tg(hsp70l:GFP-Wdr68) embryos injected with antisense morpholino knockdown of edn1 (Et1-MO) (Fig 2N–2Q). Wildtype sibling control embryos displayed normal dlx6a expression (Fig 2N, red arrowhead). As expected, wildtype sibling embryos injected with Et1-MO displayed severe loss of dlx6a expression in all pharyngeal arches (Fig 2O, red arrowhead). Heat shock induction of GFP-Wdr68 overexpression did not alter dlx6a expression in control embryos (Fig 2P) and failed to rescue dlx6a expression in Et1-MO animals (Fig 2Q). We also found that GFP-Wdr68 overexpression did not affect edn1 expression (S3A and S3B Fig). Expression of dlx6a in Et1-MO;wdr68hi3812/hi3812 mutants was severely reduced in all pharyngeal arches similar to that observed in embryos only injected with Et1-MO (S3C and S3D Fig; Fig 2O). In contrast, alcian blue stained cartilages readily detected a PQ-like cartilage in Et1-MO animals that was absent in Et1-MO;wdr68hi3812/hi3812 mutants (S3G and S3H Fig). Thus, edn1 appears to function downstream of wdr68 to facilitate ventral cartilage development and dlx6a expression.
To further explore the regulatory relationships between wdr68 and edn1, we also examined hand2 expression (S5I–S5P Fig). Expression of hand2 is both edn1-dependent and BMP-dependent [9]. Wildtype sibling embryos injected with the G/R plasmid mix displayed normal expression levels of hand2 (S3I and S3J Fig, red arrowhead). Likewise, wdr68hi3812/hi3812 mutants injected with the G/R plasmid mix displayed the expected loss of 1st arch hand2 expression (S3K and S3L Fig, red arrowhead). Wildtype sibling embryos injected with the G/Edn1 plasmid mix displayed near-normal expression levels of hand2 (S3M and S3N Fig, red arrowhead). In contrast to that observed for dlx6a, wdr68hi3812/hi3812 mutants injected with the G/Edn1 plasmid mix failed to rescue hand2 expression (compare S3O and S3P Fig with S3K and S3L Fig). Genotyping confirmed that 0/6 mutants injected with the G/Edn1 plasmid mix had restored hand2 expression (S3O and S3P Fig, 0% rescue). Thus, while edn1 is downstream of wdr68, more complex regulatory interactions exist for at least some edn1 target genes, such as hand2, that require other wdr68-dependent functions for expression.
wdr68 restricts jagged-notch signaling to first arch dorsal territory
Signaling interactions between dorsally-restricted jag1b and notch2 are important for the induction of similarly dorsally-restricted hey1 and grem2 expression [11]. Because edn1 expression is known to be important for proper restriction of jag1b, hey1, and grem2 expression from ventral territory, we examined their expression in embryos raised at 32°C from crosses of wdr68hi3812/+ adults. ISH revealed expansion of jag1b into the ventral territory of the first arch structure in wdr68hi3812/hi3812 mutants (compare Fig 3A to arrowhead in Fig 3B). Genotyping confirmed that 5/5 animals displaying the phenotype shown in Fig 3A were wildtype (+/*), while 6/6 animals displaying the phenotype depicted in Fig 3B were wdr68hi3812/hi3812 mutant animals. Expression of the transcription factor hey1 depends on Jag-Notch signaling [11]. We also found expansion of hey1 expression into the ventral territory of the first arch structure (compare Fig 3C to arrowhead in Fig 3D). Genotyping confirmed that 5/5 animals displaying the phenotype shown in Fig 3C were wildtype (+/*), while 5/5 animals displaying the phenotype depicted in Fig 3D were wdr68hi3812/hi3812 mutant animals. Expression of the normally dorsally-restricted BMP signaling antagonist grem2 is also dependent on Jag-Notch signaling [10]. Mirroring our findings with jag1b and hey1, we found expansion of grem2 expression into the ventral territory of the first arch structure (compare Fig 3E to arrowhead in Fig 3F). Genotyping confirmed that 5/5 animals displaying the phenotype shown in Fig 3E were wildtype (+/*), while 4/4 animals displaying the phenotype depicted in Fig 3F were wdr68hi3812/hi3812 mutant animals. Thus, we found that wdr68 is required to restrict Jagged-Notch signaling events from ventral territory.
Fig 3. Wdr68 restricts expression of jag1b, hey1, and grem2 from ventral territory.

ISH analysis on embryos raised at 32°C. A) normal expression of jag1b in dorsal territory at the prim-25 stage, B) expansion of jag1b into ventral territory in wdr68hi3812/hi3812 mutants, C) normal expression of hey1 in dorsal territory at the prim-25 stage, D) expansion of hey1 into ventral territory in wdr68hi3812/hi3812 mutants, E) normal expression of grem2 in dorsal territory at the prim-25 stage, F) expansion of grem2 into ventral territory in wdr68hi3812/hi3812 mutants.
A BMP antagonist exacerbates the severity of craniofacial defects and loss of edn1 expression in wdr68 mutants
BMP signaling is required for edn1 expression and ectopic BMP signaling can induce ectopic edn1 expression [9, 10]. Thus, edn1 is a downstream target of BMP signaling. We previously showed that edn1 expression is also downstream of wdr68 function [36] (and Fig 2). ISH analysis failed to detect any defects in BMP4 expression in wdr68 mutants (S4 Fig), suggesting wdr68 functions downstream or in parallel to BMP signaling. A simple model combining these observations is that wdr68 facilitates BMP induction of edn1. If true, then treating wdr68hi3812/hi3812 mutants with a BMP antagonist will exacerbate loss of M formation and edn1 expression. To test this, we treated embryos, from matings of wdr68hi3812/+ adult zebrafish, raised at 24°C with 10μM Dorsomorphin (DM) starting at the 14–15 somites stage. We chose to start treatments at this relatively late stage in order to avoid perturbing the very early developmental roles of BMP signaling. When grown at 24°C, wdr68hi3812/hi3812 embryos displayed only mild M-PQ joint fusions (see Fig 1B). We found that DMSO-treated wildtype (+/*) embryos exhibited normal cartilage formation while wdr68hi3812/hi3812 embryos exhibited M-PQ joint fusions (compare Fig 4A to 4B). Genotypic analysis of embryo tails separated from heads prior to Alcian blue staining confirmed that 6/6 embryos like that shown in Fig 4B were wdr68hi3812/hi3812 mutants. DM-treated wildtype (+/*) embryos appeared the same as DMSO-treated wildtypes (compare Fig 4C to 4A). However, DM-treated wdr68hi3812/hi3812 mutants lost M and also had severely reduced PQ cartilages while vehicle treated mutants displayed only joint fusions between M and PQ (compare Fig 4D to 4B). Genotypic analysis confirmed that 6/6 embryos like that shown in Fig 4D were wdr68hi3812/hi3812 mutants. We also quantified the fraction of mutants in each treatment exhibiting the mild M-PQ fusion and found that 23.2% of DMSO-treated animals displayed the M-PQ fusion versus only 4.2% of the DM-treated animals (Fig 4E, blue bars, p<0.046). Conversely, we found that only 3.7% of DMSO-treated animals displayed severe losses of M and PQ versus 20.0% of DM-treated animals (Fig 4E, orange bars, p<0.040).
Fig 4. DM treatment induces loss of M cartilage and edn1 expression in wdr68hi3812/hi3812 zebrafish.
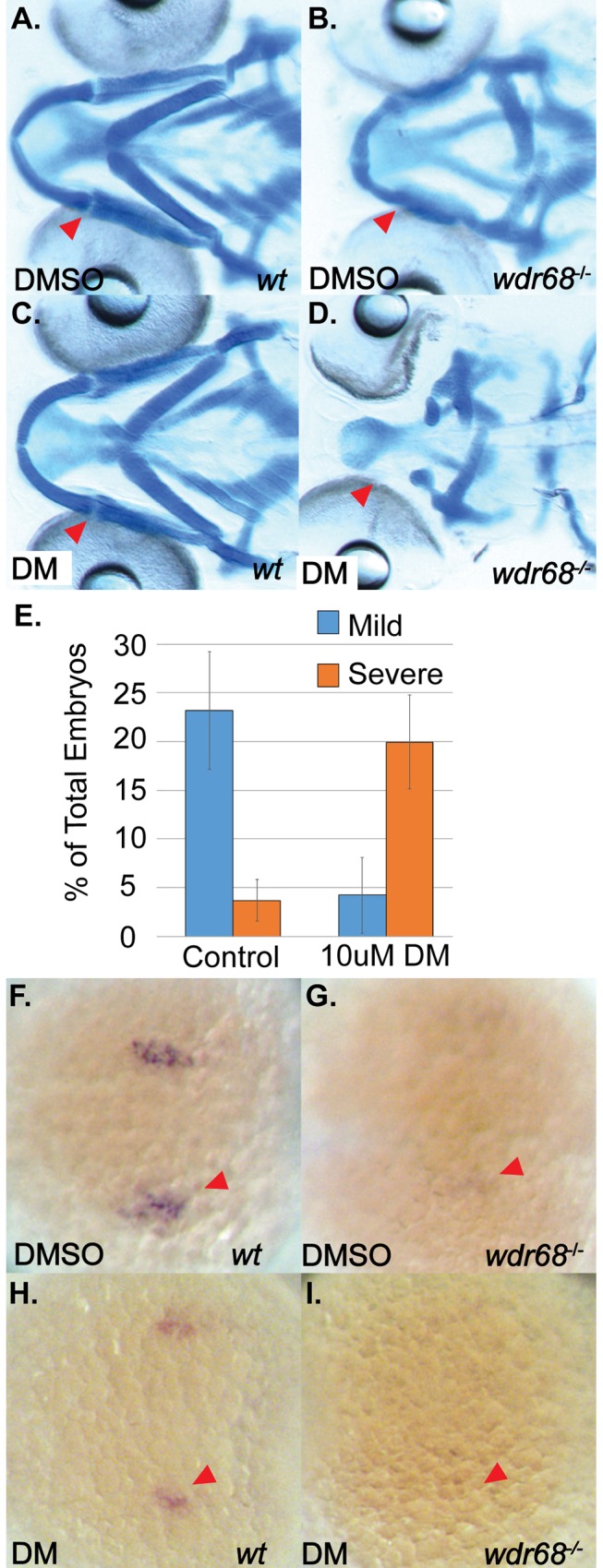
(A-D) Ventral views of 5dpf Alcian stained craniofacial cartilages of zebrafish raised at 24°C and treated with DMSO or 10μM DM at 14–15 somites stage. A) Wildtype embryo treated with DMSO control. B) wdr68hi3812/hi3812 mutant treated with DMSO show joint fusions between M and PQ. C) Wildtype embryo treated with DM reveal no defects in craniofacial cartilages. D) wdr68hi3812/hi3812 mutants treated with DM show severe reduction in PQ and deletion of M. E) DM-treated mutants show significantly more severe defects compared to the control group (p<0.040). (F-I) Dorsal views of edn1 ISH analysis on 20 somites stage embryos treated with DMSO or 10μM DM starting at the 14–15 somites stage. F) Wildtype embryos treated with DMSO control. G) wdr68hi3812/hi3812 mutants treated with DMSO control show reduced edn1 expression. H) Wildtype embryos treated with DM show mildly reduced edn1 expression. I) wdr68hi3812/hi3812 mutants treated with DM lack edn1 expression.
To test whether partial inhibition of BMP signaling in mild wdr68hi3812/hi3812 embryos would yield a synergistic reduction of edn1 expression, we again treated embryos raised at 24°C with DM starting at the 14–15 somites stage. We then used ISH to observe expression of edn1 at the 20 somites stage. In embryos treated with DMSO, we found normal expression of edn1 in wildtype (+/*) siblings (Fig 4F, red arrowhead) and reduced but still barely detectable expression of edn1 in wdr68hi3812/hi3812 mutants (Fig 4G, red arrowhead). Genotypic analysis confirmed that 7/7 embryos in Fig 4F were wildtype (+/*) and 5/5 embryos shown in Fig 4G were wdr68hi3812/hi3812 mutants. In embryos treated with DM, we found mildly reduced expression of edn1 in wildtype (+/*) siblings (Fig 4H, red arrowhead) that is consistent with the known role of DM as a BMP signaling antagonist (Yu et al., 2008) and with the fact that edn1 is a downstream target of BMP signaling (Alexander et al., 2011; Zuniga et al., 2011). In the DM-treated wdr68hi3812/hi3812 mutants, we were unable to detect edn1 expression (Fig 4I, red arrowhead). Genotypic analysis confirmed that 8/8 embryos shown in Fig 4H were wildtype (+/*) and that 5/5 embryos shown in Fig 4I were wdr68hi3812/hi3812 mutants.
A BMP agonist can partially rescue Meckel’s cartilage formation and edn1 expression in wdr68 mutants
If wdr68 facilitates BMP induction of edn1, then treating wdr68 mutants with a BMP agonist should at least partially rescue M cartilage formation and edn1 expression. To test this, we treated embryos raised at 32°C with the BMP signaling agonist isoliquiritigenin (ISL) starting at the 14–15 somites stages [57]. Embryos were raised at 32°C so that mutants would exhibit severe loss of the M cartilage from which potential rescue of M cartilage formation could be most clearly assessed (see Fig 1C). Alcian blue stained zebrafish cartilages were dissected and flat mounted on slides for additional clarity. At 32°C, we found that DMSO-treated wildtype zebrafish developed the M cartilage as expected (Fig 5A, red arrowhead). Also at 32°C, we found that most DMSO-treated mutants exhibited complete loss of M and reduction of PQ as previously described (Fig 5B, red arrowhead). ISL-treated wildtype embryos were indistinguishable from DMSO-treated wildtype (compare Fig 5C to 5A). In contrast, ISL-treated mutants raised at 32°C exhibited a partial restoration of the M cartilage (compare Fig 5D to 5B, red arrowheads). However, ISL treatment did not appear to significantly restore formation of the PQ (compare Fig 5D to 5A and 5C). We quantified the fraction of mutants in each treatment exhibiting a discernible M-like cartilage element and found that 59% of ISL-treated animals displayed an M-like cartilage versus only 17% of DMSO-treated animals (Fig 5E, p<0.006).
Fig 5. ISL treatment partially rescues M cartilage and edn1 expression in wdr68hi3812/hi3812 zebrafish.
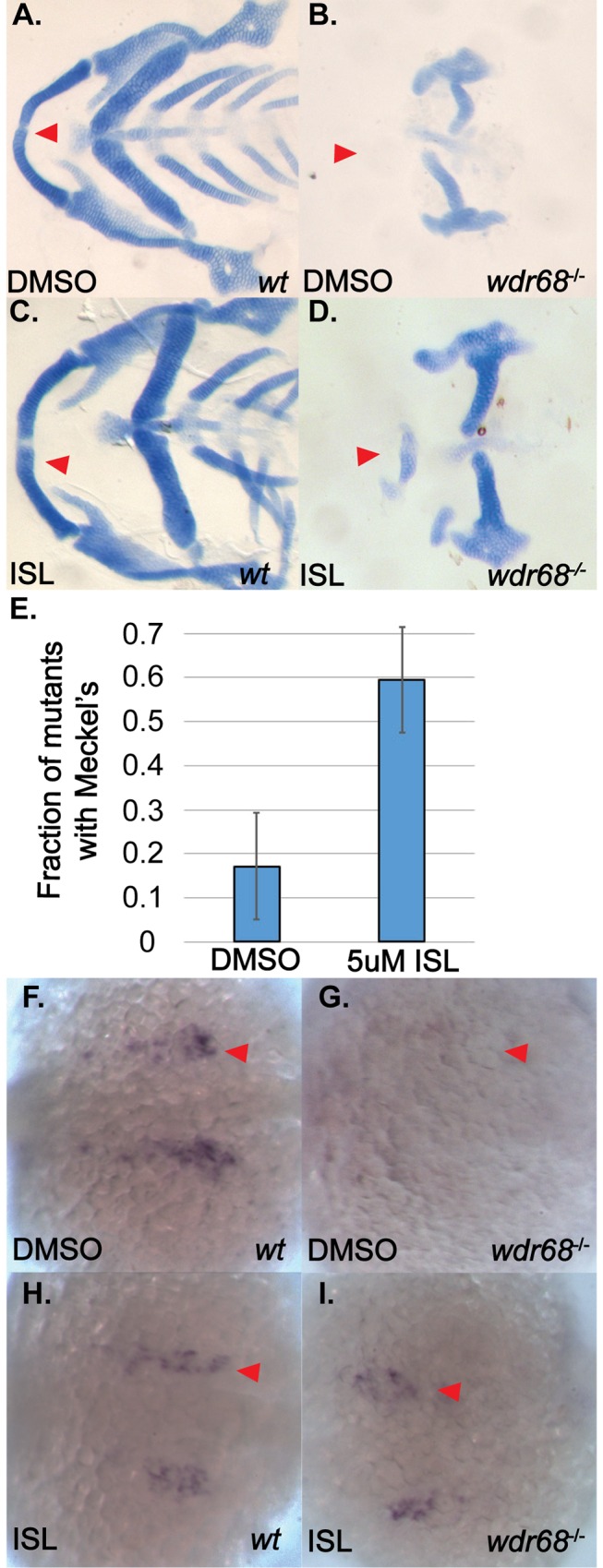
(A-D) Flatmounts of 5dpf ventral cartilages of Alcian stained zebrafish raised at 32°C and treated with DMSO or 5μM ISL starting at the 14- to 15-somites stage. A) Wildtype zebrafish treated with DMSO control. Red arrow indicate M. B) wdr68hi3812/hi3812 mutants treated with DMSO control show a lack of M cartilage. C) Wildtype zebrafish treated with 5μM ISL show normal craniofacial cartilage formation. D) wdr68hi3812/hi3812 mutants treated with 5μM ISL show a partial rescue of M. E) Fraction of mutant embryos with partial M is significantly greater in the ISL treated group (p<0.006). (F-I) Dorsal views of edn1 ISH analysis on 20-somites stage embryos treated with DMSO or 5μM ISL starting at the 14- to 15-somites stage. F) Wildtype embryos treated with DMSO control. G) wdr68hi3812/hi3812 mutants treated with DMSO control show lack of edn1 expression. H) Wildtype embryos treated with ISL show similar expression compared to wild type. I) wdr68hi3812/hi3812 mutants treated with ISL are indistinguishable from that of wildtype.
To determine whether enhanced BMP signaling in severe wdr68hi3812/hi3812 embryos would partially rescue edn1 expression, we again treated embryos raised at 32°C with 5μM ISL starting at the 14–15 somites stages. We then used ISH to observe expression of edn1 at the 20 somites stage. In embryos treated with DMSO, we found normal expression of edn1 in wildtype siblings (Fig 5F) but were unable to detect edn1 expression in wdr68hi3812/hi3812 mutants (Fig 5G). Genotypic analysis confirmed that 10/10 embryos phenotypically like that shown in Fig 5F were wildtype (+/*) and 5/5 embryos phenotypically like that shown in Fig 5G were wdr68hi3812/hi3812 mutants. Strikingly in embryos treated with ISL, no embryos displayed a severe reduction of edn1 expression. Genotyping was performed to unequivocally identify wildtypes and mutants. Wildtype siblings displayed normal expression of edn1 (compare Fig 5H to 5F). The known BMP agonist role for ISL suggested we might see overexpression of edn1 in these animals but that was not observed (Vrijens et al., 2013). Instead, the ISL-treated embryos appeared to yield no phenotypic mutants with respect to edn1 expression level (compare Fig 5I to 5G). Consistent with that notion, random genotypic analysis revealed that 2/9 embryos with normal edn1 expression levels were indeed wdr68hi3812/hi3812 mutants (Fig 5I). Thus, ISL treatment rescued edn1 expression in wdr68hi3812/hi3812 mutants to near-normal levels.
Live confocal imaging of Tg(sox10:mCherryCAAX);wdr68hi3812/hi3812 mutants revealed consistent defects (6/6 mutants genotyped) within the 1st arch CNCC region at the prim-5 and later stages (S5D, S5F and S5H Fig, blue dotted outline). At the prim-5 stage, reduced mCherryCAAX signal was observed in both dorsal and ventral regions of the 1st arch (S5D Fig, compare blue outline to S5C Fig). By the prim-12 stage, the dorsal defect was less apparent but the ventral reduction was still evident (S5F Fig, compare blue outline to S5E Fig) and persisted through at least the prim-25 stage (S5H Fig, compare blue outline to S5G Fig). The mCherryCAAX signal remained detectable through to 4dpf (S5I–S5L Fig). ISL treatment yielded a modest but consistent (4/4 mutants genotyped) rescue of mCherryCAAX signal in the ventral region of the 1st arch (S5N Fig, compare blue outline to S5H Fig).
Canonical BMP signaling is not impaired in cells lacking Wdr68 expression
To further examine whether Wdr68 might directly impact BMP signaling, we used CRISPR/Cas9 gene targeting technology to generate loss-of-function deletions in the Wdr68/Dcaf7 locus in mouse C2C12 cells. Western blot analysis and DNA sequencing confirmed the generation of two independently isolated mutant sublines, Δwdr68-5 and Δwdr68-9 (Fig 6A and S4B Fig) as well as a non-target control (NT1) subline. The transcriptional co-activator Yap is reported to be important for BMP signaling in mammalian cells [58, 59]. In flies, the ortholog of Wdr68, Riquiqui (Riq), is also reported to positively regulate the Yap ortholog yorkie through its interaction with the kinase Minibrain (Mnb) that negatively regulates the Hippo signaling pathway kinase Warts (Wts) [60]. Therefore, we examined both total Yap and pYap levels in the control, Δwdr68-5, and Δwdr68-9 sublines but found no significant differences between them (Fig 6A). To further characterize the sublines, we examined the levels of pSmad1/5 after a one-hour treatment of the cells with 0, 1, 10, or 100ng/mL BMP4 and found no significant reproducible differences between the sublines (Fig 6B). To determine whether Wdr68/Dcaf7 might generally facilitate BMP signaling in a functional assay, we transfected the control and deletion sublines with the BRE-Luc reporter plasmid [55] along with a SV40-Renilla plasmid, and generated a dose-response curve to BMP4 ligand (Fig 6C). All relative light unit responses were normalized to the vehicle-treated control subline (Fig 6C, leftmost column). Overall, no significant decreases in fold-induction were found in the Δwdr68-5 and Δwdr68-9 sublines relative to the control subline (Fig 6C grey bars versus black bars). Immunofluorescence analysis also revealed little consistent difference in pSmad1/5 levels between wildtype and wdr68hi3812/hi3812 mutant zebrafish embryos (Fig 6E and 6F). As expected, ISL treatment did consistently increase pSmad1/5 signal (compare Fig 6H to 6G). Thus, Wdr68/Dcaf7 does not appear to directly modulate BMP signaling pathway activity.
Fig 6. TGF-β interference with BMP signaling is enhanced in cells lacking Wdr68 expression.

A) Isolation of Wdr68/Dcaf7 knock-out C2C12 cell sublines and expression levels in growth medium (GM) versus differentiation medium (Diff). Panel A1) Lanes 1 and 4, Wdr68 protein was detected in the control NT1 cells. Lanes 2 and 5, Δwdr68-5 lacks wildtype Wdr68 protein expression. Lanes 3 and 6, Δwdr68-9 lacks wildtype Wdr68 protein expression. Panel A2) β-tubulin expression was used as a loading control and did not differ substantially between lanes. Panel A3) pYap1 levels did not differ substantially between lanes. Panel A4) Total Yap1 levels did not differ substantially between lanes. B) pSmad1/5 induction was not substantially altered in Δwdr68-5 or Δwdr68-9 sublines. Panel B1) pSmad1/5 levels in control (NT1) or Δwdr68-5 (5) cells after 1 hour of exposure to 0, 1, 10, or 100ng/mL BMP4 in DM. Panel B2) β-tubulin expression was used as a loading control and did not differ substantially between lanes. Panel B3) pSmad1/5 levels in control (NT1) or Δwdr68-9 (5) cells after 1 hour of exposure to 0, 1, 10, or 100ng/mL BMP4 in DM. Panel B4) β-tubulin expression was used as a loading control and did not differ substantially between lanes. C) Transient transfection of NT1, Δwdr68-5, and Δwdr68-9 sublines with BRE-Luc and SV40-Renilla plasmids and induced with 0, 1, 10, or 100ng/mL BMP4 in GM. No significant differences were found between control and deletion sublines. Representative experiment shown from at least 3 independent trials. D) Transient transfection of NT1, Δwdr68-5, and Δwdr68-9 sublines with BRE-Luc and SV40-Renilla plasmids, induced with 10ng/mL BMP4, and then challenged with 0, 0.1, 1.0, or 10ng/mL TGF-®1. At 10ng/mL TGF-®1 interference with BRE-Luc activity was significantly greater in the Δwdr68-5 and Δwdr68-9 sublines relative to NT1 controls (* = p < 0.002). Representative experiment shown from at least 3 independent trials. E-H) Immunofluorescence detection of pSmad1/5 in prim-12 stage zebrafish embryos raised at 32°C. E) wildtype sibling embryo. F) wdr68hi3812/hi3812 mutant embryo. G) DMSO-treated wildtype sibling. H) ISL-treated wildtype sibling.
TGF-β interference with BMP signaling is enhanced in cells lacking Wdr68 expression
TGF-β signaling is important for craniofacial development [26], and can interfere with BMP signaling via Smad3-mediated displacement of Smad4 from pSmad1/5 complexes [29, 30]. Therefore we examined the BRE-Luc responsiveness of the NT1 control, Δwdr68-5 and Δwdr68-9 sublines when co-treated with 10ng/mL BMP4 and 0, 0.1, 1, or 10ng/mL TGF-β1 ligand. As previously reported, we found that TGF-β1 can interfere with BRE-Luc activity in the NT1 control cells (Fig 6D, black bars). We also found a consistent statistically significant further decline of BRE-Luc activity in the Δwdr68-5 and Δwdr68-9 sublines at 10ng/mL TGF-β1 (Fig 6D, grey bars versus black bars, p<0.001).
The TGF-β inhibitor SB431542 can partially rescue the M cartilage defects and edn1 expression in wdr68 mutants
If enhanced TGF-β interference with BMP signaling contributes to the wdr68 mutant phenotype in zebrafish, then inhibiting TGF-β pathway function should at least partially restore cartilage development in wdr68 mutants. SB431542 is a well-characterized inhibitor of the TGF-β family receptor kinases Alk4/5 [61], that is also known to inhibit TGF-β signaling activity in the zebrafish [62–64]. To avoid perturbing earlier developmental roles of TGF-β family signaling, embryos were treated with 10μM SB431542 starting at the 14- to 15-somites stages. Embryos were raised at 32°C so that mutants would exhibit severe loss of the M cartilage from which potential rescue of M cartilage formation could be most clearly assessed (Fig 1C). At 32°C, we found that DMSO-treated wildtype zebrafish developed the M cartilage as expected (Fig 7A, red arrowhead). Also at 32°C, we found that most DMSO-treated mutants exhibited complete loss of M and reduction of PQ as previously described (Fig 7B, red arrowhead). 10μM SB431542-treated wildtype embryos were indistinguishable from DMSO-treated wildtypes (compare Fig 7C to 7A). In contrast, SB431542-treated mutants raised at 32°C exhibited a partial restoration of the M and PQ cartilages (compare Fig 7D to 7B, red arrowheads). Quantitation of the changes in phenotypic severity revealed a significant shift in the number of mutants displaying severe versus mild phenotypes. Under DMSO treatment, mild wdr68hi3812/hi3812 embryos were 8.6% of the total sample population whereas severe wdr68hi3812/hi3812 embryos were 14.3% of the total population (Fig 7E, left). Under SB431542 treatment, mild wdr68hi3812/hi3812 embryos were 19.2% of the total population whereas severe mutants were 8.0% of the total population (Fig 7E, right). Thus, SB431542 treatment decreased the fraction of severe mutants relative to the DMSO control (Fig 7E, red bars, p < 0.012).
Fig 7. Inhibition of TGF-β signaling partially rescues M cartilage and edn1 expression in wdr68hi3812/hi3812 zebrafish.
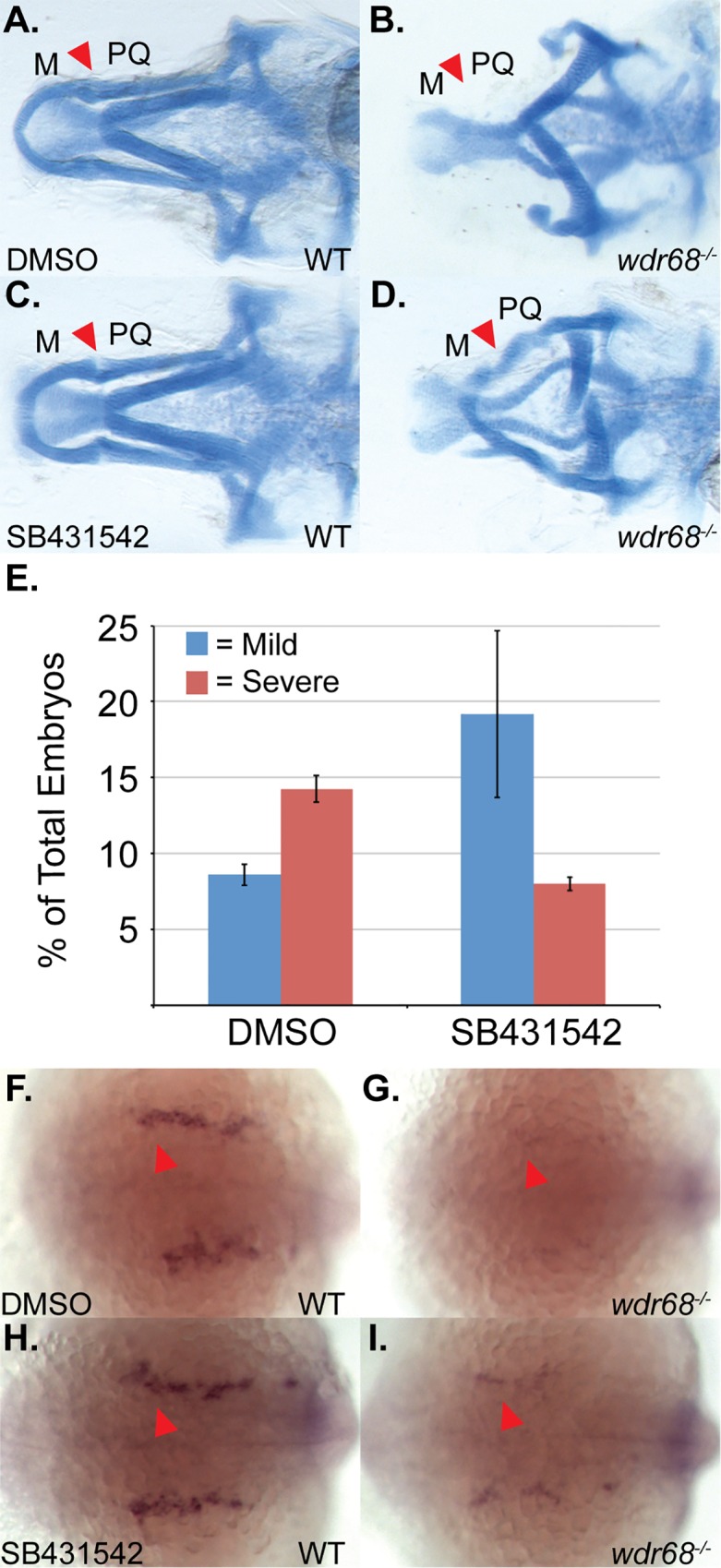
(A-D) Ventral views of 5dpf Alcian stained craniofacial cartilages of zebrafish raised at 32°C and treated with DMSO or 10μM SB431542 at 14- to 15-somites stage. A) Wildtype zebrafish treated with DMSO control. Red arrow indicates M-PQ joint region. B) wdr68hi3812/hi3812 mutants treated with DMSO control show a lack of M cartilage. C) Wildtype zebrafish treated with 10μM SB431542 show normal craniofacial cartilage formation. D) wdr68hi3812/hi3812 mutants treated with 10μM SB431542 show a partial rescue of M. E) SB431542-treated mutants show a significantly reduced fraction of severe defects compared to the control group (p<0.012). (F-I) Dorsal views of edn1 ISH analysis on 22-somites stage embryos treated with DMSO or 10μM SB431542 starting at the 14- to 15-somites stage. F) Wildtype embryos treated with DMSO control. G) wdr68hi3812/hi3812 mutants treated with DMSO control show lack of edn1 expression. H) Wildtype embryos treated with SB431542 show similar expression compared to wild type. I) wdr68hi3812/hi3812 mutants treated with SB431542 show partial restoration of edn1 expression.
To determine whether inhibiting TGF-β signaling in severe wdr68hi3812/hi3812 embryos would partially rescue edn1 expression, we again treated embryos raised at 32°C with 10μM SB431542 starting at the 14- to 15-somites stages. We then used ISH to observe expression of edn1 at the 22-somites stage. In embryos treated with DMSO, we found normal expression of edn1 in wildtype siblings (Fig 7F) but were unable to detect edn1 expression in wdr68hi3812/hi3812 mutants (Fig 7G). Genotypic analysis confirmed that 4/4 embryos phenotypically like that shown in Fig 7F were wildtype (+/*) and 5/5 embryos phenotypically like that shown in Fig 7G were wdr68hi3812/hi3812 mutants. In embryos treated with SB431542, we also found normal expression of edn1 in wildtype siblings (Fig 7H). Importantly, SB431542-treated mutants displayed greater edn1 expression levels than could be observed in DMSO-treated mutants (Fig 7I, compare to Fig 7G), albeit at levels clearly still lower than wildtype siblings (Fig 7I, compare to Fig 7H). Genotyping confirmed that 5/5 embryos phenotypically like that shown in Fig 7H were wildtype (+/*) and 5/5 embryos phenotypically like that shown in Fig 7I were wdr68hi3812/hi3812 mutants. Thus, SB431542-treatment partially rescued edn1 expression in wdr68hi3812/hi3812 mutants.
The TGF-β inhibitor SB431542 can partially rescue dorsal dlx2a expression in wdr68 mutants
A molecular explanation for the absence of the PQ in wdr68hi3812/hi3812 mutants is still lacking. Interestingly, SB431542-treated wdr68hi3812/hi3812 mutants not only displayed partial rescue of the ventral M cartilage, but also a consistent partial rescue of the dorsal PQ cartilage (Fig 7D). This was in contrast to either ectopic expression of edn1 (Fig 2C), or ISL-treatment (Fig 5D), both of which failed to restore the dorsal PQ cartilage in wdr68hi3812/hi3812 mutants. It has been previously reported that simultaneous antisense knockdown of dlx1a and dlx2a causes loss of the PQ while retaining the M and PTP [65]. The reported dlx1a/2a-MO phenotype bears similarities to the residual defects we observed for both ectopic edn1 (Fig 2C) and ISL-treatment (Fig 5D) in wdr68hi3812/hi3812 mutants. Therefore, we re-examined the expression of the dlx1a and dlx2a genes at the prim-12 stage in wdr68hi3812/hi3812 mutant embryos raised at 32°C (Fig 8). In wildtype sibling embryos, we found robust expression of dlx2a in both dorsal and ventral CNCCs of the 1st and 2nd arches (Fig 8A and 8B). In contrast, we found severely reduced dlx2a expression in the anterior portion of the 1st arch in wdr68hi3812/hi3812 mutants (Fig 8C and 8D, compare red underline between 8D and 8B). Consistent with the relatively minor 2nd arch-derived cartilage defects in wdr68hi3812/hi3812 mutants (Figs 1C, 2B, 5B and 7B), expression of dlx2a in the 2nd arch was relatively unchanged in wdr68hi3812/hi3812 mutants (Fig 8D compare blue underline to 8B). To determine whether inhibiting TGF-β signaling in severe wdr68hi3812/hi3812 embryos would partially rescue dlx2a expression, we again treated embryos raised at 32°C with 10μM SB431542 starting at the 14- to 15-somites stages. While SB431542 treatment yielded no discernible changes in wildtype siblings, we found partial rescue of anterior dorsal and ventral 1st arch expression of dlx2a in wdr68hi3812/hi3812 mutants (Fig 8E and 8F, compare red underline to Fig 8C and 8D). Analysis of dlx1a expression revealed a similar loss of anterior dorsal and ventral 1st arch expression in wdr68hi3812/hi3812 mutants (Fig 8H, compare red underline to Fig 8G).
Fig 8. dlx1a and dlx2a expression is wdr68-dependent and responsive to inhibition of TGF-β signaling in wdr68hi3812/hi3812 zebrafish.

(A-H) ISH analysis of prim-12 stage embryos raised at 32°C. A-F) dlx2a expression with red underline for anterior portion of 1st arch and blue underline for 2nd arch. A, C, E) lateral view. B, D, F) dorso-lateral view. A, B) DMSO-treated wildtype sibling showing normal dlx2a. C, D) DMSO-treated wdr68hi3812/hi3812 mutant showing loss of anterior 1st arch dlx2a. E, F) SB431542-treated wdr68hi3812/hi3812 mutant showing partial rescue of anterior 1st arch dlx2a. G, H) dlx1a expression. G) wildtype sibling showing normal dlx1a. H) wdr68hi3812/hi3812 mutant showing loss of anterior 1st arch dlx1a.
Discussion
Temperature dependence of the wdr68 mutant phenotype
Here we report that modest differences in maternally-derived protein stability, caused by incubation at different temperatures, can yield dramatic shifts in phenotypic severity even for mutants that are otherwise expected to be null mutations (Fig 1A–1D). This finding suggests that conducting forward genetic screens at temperatures other than the field standard of 28.5°C may reveal additional mutable loci that have as yet gone undetected.
wdr68 functions upstream of edn1 during a period between the 17-somites and prim-5 stages to induce and restrict dorsal-specifying genes from ventral territory
We identified a developmental period between the 17-somites and prim-5 stages during which wdr68 function is particularly important for craniofacial development (Fig 1D and 1E). Notably, this window contains the point at which edn1 expression begins, around the 18-somites stage [18], and overlaps substantially with the period during which Edn1 signaling is important for craniofacial patterning [9, 10]. Specifically, Alexander et al., 2011 found that restoring Edn1 expression as late as the prim-5 stage was able to partially rescue the defects caused by dnBmpr1 expression. In contrast, restoring Edn1 expression around the prim-25 stage was too late, similar to our findings with Wdr68. We found that ectopic expression of edn1 in wdr68 mutants was able to partially restore the 1st arch expression of a canonical edn1-dependent target gene, dlx6a, consistent with the model that wdr68 functions upstream of edn1 (Fig 2). However, we also found that ectopic edn1 expression was insufficient to restore 1st arch hand2 expression in the absence of wdr68 (S3 Fig). The expression of hand2 depends on both edn1→dlx6a and BMP signaling [9]. Thus, interference with BMP signaling to hand2 in a wdr68 mutant might prevent edn1→dlx6a induction of hand2. As an attempt to test this model, we treated wdr68 mutants with ISL to enhance BMP signaling and edn1 expression simultaneously, but the treatment failed to restore hand2 expression (S4C and S4D Fig). While that result may simply reflect a shortcoming of the experimental procedure (e.g. insufficient levels of BMP signaling, edn1→dlx6a expression, or both), it may also indicate a distinct wdr68-dependent requirement for hand2 expression. Nonetheless, and consistent with the known role for edn1 expression, we found that wdr68 is required to properly restrict jag1b, hey1, and grem2 from ventral territory (Fig 3). The simplest explanation for these ventral expansions of dorsal territory markers is the failure of edn1 expression in wdr68 mutants.
The expansion of dorsal-specifying markers into ventral territory (Fig 3) is often accompanied by transformation of ventral cartilages into dorsal cartilages [9, 10]. Curiously, such transformations are not observed in wdr68 mutants, which instead display losses of both dorsal and ventral cartilage elements. Our findings on the wdr68-dependence of dlx1a and dlx2a expression suggests that jag1b-mediated transformation of CNCCs may require the dlx1a/2a expression program.
The losses of dlx1a/2a expression (Fig 8) combined with the observed reductions of Tg(sox10:mCherryCAAX) label in wdr68 mutants (S5 Fig) raises the possibility that a subset of the CNCCs in wdr68 mutants may be lost due to apoptosis. This is an intriguing possibility that will require further investigation. However, the detection of dorsal-specifying markers such as jag1b and hey1 in both dorsal and ventral territory (Fig 3) indicates that a population of CNCCs remains within the 1st arch of wdr68 mutants at least until about the prim-25 stage. The ultimate fate of the mis-patterned cells remains unclear.
wdr68 function does not directly modulate BMP signaling but may interfere with TGF-β signaling
Although a BMP antagonist and agonist yielded expected exacerbations and rescues of the defects in wdr68 mutants, respectively (Figs 4 and 5), we found no strong evidence of a direct impact on BMP signaling pathway function (Fig 6). Notably, while analysis of the edn1 promoter sequence revealed the presence of DNA sequence elements associated with BMP responsiveness (unpublished observations), it is unclear whether Smad1/5 complexes directly regulate the edn1 promoter.
Interestingly, we found that TGF-β interference with BMP signaling was enhanced in mouse C2C12 cells lacking Wdr68 and that inhibition of TGF-β signaling partially restored jaw cartilage development, edn1, and dlx2a expression in zebrafish wdr68 mutants (Figs 6D, 7 and 8). Nonetheless, is remains unclear whether these wdr68—TGF-β pathway interactions are direct or indirect.
The ISL-mediated rescue of edn1 expression (Fig 5I) was more robust than the SB431542-mediated rescue of edn1 expression (Fig 7I). Yet, ISL-mediated rescue of the jaw cartilages was less complete (Fig 5D) than the SB431542-mediated rescue of the jaw cartilages (Fig 7D). This discrepancy can be explained by the potency of edn1 plus the absence of dlx1a and dlx2a expression in wdr68 mutants. First, fairly low levels of edn1 expression can still mediate M formation (Fig 4G). Second, SB431542 treatment was able to also rescue dlx2a (Fig 8E and 8F) while ISL treatment did not (S4E and S4F Fig). Thus, the combined actions of a relatively potent edn1 signaling molecule and simultaneous restoration of edn1 and dlx2a in SB431521-treated embryos likely underlies the relatively well formed jaw cartilages. That said, the mechanism by which SB431542 restores jaw development in wdr68 mutants is still poorly understood and will require further investigation.
Wdr68 is a scaffolding protein and a large number of interacting factors have been identified by various high- and low-throughput approaches. Interestingly, Wdr68 has been reported to physically interact with Smad2, Smad3, and Ski [42, 66]. Wdr68 also interacts with the kinases Dyrk1a and Dyrk1b [38], that are reported to interact with Smad3 [42] and Ski [67], respectively. Thus, Wdr68 may organize a multi-subunit complex capable of modulating TGF-β pathway function. Together, the interactions of these factors potentially impact numerous disease states including cleft lip/palate, auriculocondylar syndrome, Alagille syndrome, and Down syndrome. Future investigations aimed at testing these potential roles are needed.
Supporting Information
A-D) Immunofluorescence detection of endogenous Wdr68. A) Wdr68 expression in a wildtype 15-somites stage embryo raised at 32°C. B) a wdr68 mutant lacking Wdr68 protein. C) Wdr68 expression in a wildtype prim-5 stage embryo raised at 32°C. D) a wdr68 mutant lacking Wdr68 protein. E-F) live imaging of GFP fluorescence in Tg(hsp70l:GFP-wdr68)csu9 animals. E) transgenic animal with the only detected GFP expression coming from the cmlc:eGFP marker for transgenesis. F) same animal as in E but after a 0.5 hour heat shock (HS) at 39°C followed by 7 hours of recovery at 28.5°C. G) western blot analysis of Tg(hsp70l:GFP-wdr68)csu9 animals after various lengths of heat shock exposure. Panel G1) GFP-wdr68 expression is induced by heat shock. Panel G2) β-tubulin expression was used as a loading control and did not differ substantially between lanes.
(TIF)
A-F) ventral views of 5dpf alcian blue stained cartilages from embryos raised at 28.5°C. A) heat shocked wildtype sibling displaying normal cartilages. B) heat shock induced Tg(hsp70l:GFP-wdr68) overexpression yielded no discernible cartilage phenotype. C) wdr68-MO injected animal showing the mild M-PQ joint fusion phenotype. D) wdr68-MO injected animal showing the severe loss of M and PQ phenotype. E) wdr68-MO injected heat shock induced Tg(hsp70l:GFP-wdr68) animal showing rescued normal M and PQ cartilages. F) wdr68-MO injected heat shock induced Tg(hsp70l:GFP-wdr68) animal showing rescued mild M-PQ joint fusions. G) plot of the distribution of phenotypes observed in a representative experiment on wdr68-MO injected Tg(hsp70l:GFP-wdr68) animals.
(TIF)
A-B) ISH analysis for edn1 expression on 25-somites stage animals raised at 28.5C that were heat shocked at the bud and 20-somites stages. A) wildtype sibling with normal edn1 expression. B) Tg(hsp70l:GFP-wdr68) embryo overexpressing GFP-Wdr68 with near-normal edn1 expression. C-D) ISH analysis on prim-12 stage embryos. C) wildtype control sibling with normal dlx6a expression. D) Et1-MO;wdr68hi3812/hi3812 mutant lacking dlx6a expression in all arches. E-F) merged green-red channel fluorescence on prim-5 stage embryos injected with either GFP/dsRed (G/R) or GFP/Edn1 (G/Edn1) plasmid mixtures. E) broad GFP/dsRed expression in a G/R embryo. F) broad GFP expression in a G/Edn1 embryo. G-H) lateral view of 5dpf alcian blue stained embryo. G) Et1-MO injected animal showing loss of M and CH but retention of PQ. H) Et1-MO;wdr68hi3812/hi3812 mutant showing loss of M, CH, and PQ. I-P) ISH analysis for hand2 expression on prim-12 stage animals raised at 32°C. Red arrowhead points at 1st arch expression of hand2. I, K, M, O) dorsal view. J, L, N, P) lateral view. I, J) wildtype sibling injected with G/R mix showing normal hand2. K, L) wdr68hi3812/hi3812 mutant injected with G/R mix showing loss of 1st arch hand2. M, N) wildtype sibling injected with G/Edn1 mix showing normal hand2. O,P) wdr68hi3812/hi3812 mutant injected with G/Edn1 mix showing loss of 1st arch hand2.
(TIF)
A) ISH analysis of prim-25 stage wildtype and wdr68 mutant animals revealed no differences in expression of bmp4. B) C2C12 wildtype and deletion subline sequences at the targeted locus in exon-5. The yellow highlight indicates the guide RNA target sequence followed by the TGG PAM sequence. C and D) ISH analysis for hand2 expression. C) ISL-treated wildtype sibling. D) ISL-treated mutant sibling lacking rescue. E and F) ISH analysis for dlx2 expression. E) ISL-treated wildtype sibling. F) ISL-treated mutant sibling lacking rescue.
(TIF)
A-N) Confocal images of the pharyngeal arch regions of embryos live-mounted in 0.7% agarose containing 0.0167% Tricaine. A) 25-somites stage wildtype sibling. B) 25-somites stage wdr68hi3812/hi3812 mutant. C) prim-5 stage wildtype sibling with 1st arch region outlined in blue. D) prim-5 stage wdr68hi3812/hi3812 mutant with same outline as in C to indicate regions of reduced mCherryCAAX signal. E) prim-12 stage wildtype sibling with ventral 1st arch region outlined in blue. F) prim-12 stage wdr68hi3812/hi3812 mutant with same outline as in E to indicate reduced ventral mCherryCAAX signal. G) prim-25 stage wildtype sibling with ventral 1st arch region outlined in blue. H) prim-25 stage wdr68hi3812/hi3812 mutant with same outline as in G to indicate reduced ventral mCherryCAAX signal. I) lateral view of 4-dpf wildtype sibling cartilages. J) lateral view of 4-dpf wdr68hi3812/hi3812 mutant severely reduced M and PQ cartilages. K) ventral view of animal in I. L) ventral view of animal in J. M) ISL-treated prim-25 stage wildtype sibling with ventral 1st arch region outlined in blue. N) ISL-treated prim-25 stage wdr68hi3812/hi3812 mutant with same outline as in M, H, G to indicate modest rescue of ventral 1st arch mCherryCAAX signal.
(TIF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the United States National Institute of Dental and Craniofacial Research (http://www.nidcr.nih.gov/) grant R15DE022902-01A1 to Robert M Nissen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO. Global strategies to reduce the health-care burden of craniofacial anomalies: World Health Organization; 2002. [Google Scholar]
- 2.Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, et al. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132(6):1453–61. Epub 2005/02/18. doi: dev.01676 [pii] 10.1242/dev.01676 . [DOI] [PubMed] [Google Scholar]
- 3.Suzuki S, Marazita ML, Cooper ME, Miwa N, Hing A, Jugessur A, et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet. 2009;84(3):406–11. Epub 2009/03/03. doi: S0002-9297(09)00063-9 [pii] 10.1016/j.ajhg.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder MJ, Green GE, Park SS, Stamper BD, Gordon CT, Johnson JM, et al. A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am J Hum Genet. 2012;90(5):907–14. Epub 2012/05/09. doi: S0002-9297(12)00202-9 [pii] 10.1016/j.ajhg.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon CT, Petit F, Kroisel PM, Jakobsen L, Zechi-Ceide RM, Oufadem M, et al. Mutations in endothelin 1 cause recessive auriculocondylar syndrome and dominant isolated question-mark ears. Am J Hum Genet. 2013;93(6):1118–25. 10.1016/j.ajhg.2013.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romanelli Tavares VL, Gordon CT, Zechi-Ceide RM, Kokitsu-Nakata NM, Voisin N, Tan TY, et al. Novel variants in GNAI3 associated with auriculocondylar syndrome strengthen a common dominant negative effect. Eur J Hum Genet. 2015;23(4):481–5. 10.1038/ejhg.2014.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243–51. 10.1038/ng0797-243 . [DOI] [PubMed] [Google Scholar]
- 8.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235–42. 10.1038/ng0797-235 . [DOI] [PubMed] [Google Scholar]
- 9.Alexander C, Zuniga E, Blitz IL, Wada N, Le Pabic P, Javidan Y, et al. Combinatorial roles for BMPs and Endothelin 1 in patterning the dorsal-ventral axis of the craniofacial skeleton. Development. 2011;138(23):5135–46. Epub 2011/10/28. dev.067801 [pii] 10.1242/dev.067801. 10.1242/dev.067801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuniga E, Rippen M, Alexander C, Schilling TF, Crump JG. Gremlin 2 regulates distinct roles of BMP and Endothelin 1 signaling in dorsoventral patterning of the facial skeleton. Development. 2011;138(23):5147–56. Epub 2011/10/28. dev.067785 [pii] 10.1242/dev.067785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuniga E, Stellabotte F, Crump JG. Jagged-Notch signaling ensures dorsal skeletal identity in the vertebrate face. Development. 2010;137(11):1843–52. Epub 2010/05/01. dev.049056 [pii] 10.1242/dev.049056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clouthier DE, Schilling TF. Understanding endothelin-1 function during craniofacial development in the mouse and zebrafish. Birth Defects Res C Embryo Today. 2004;72(2):190–9. 10.1002/bdrc.20007 [DOI] [PubMed] [Google Scholar]
- 13.Crump JG, Maves L, Lawson ND, Weinstein BM, Kimmel CB. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131(22):5703–16. 10.1242/dev.01444 [DOI] [PubMed] [Google Scholar]
- 14.David NB, Saint-Etienne L, Tsang M, Schilling TF, Rosa FM. Requirement for endoderm and FGF3 in ventral head skeleton formation. Development. 2002;129(19):4457–68. . [DOI] [PubMed] [Google Scholar]
- 15.Nissen RM, Yan J, Amsterdam A, Hopkins N, Burgess SM. Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development. 2003;130(11):2543–54. . [DOI] [PubMed] [Google Scholar]
- 16.Kimmel CB, Miller CT, Kruze G, Ullmann B, BreMiller RA, Larison KD, et al. The shaping of pharyngeal cartilages during early development of the zebrafish. Dev Biol. 1998;203(2):245–63. 10.1006/dbio.1998.9016 [DOI] [PubMed] [Google Scholar]
- 17.Kimmel CB, Miller CT, Moens CB. Specification and morphogenesis of the zebrafish larval head skeleton. Dev Biol. 2001;233(2):239–57. 10.1006/dbio.2001.0201 [DOI] [PubMed] [Google Scholar]
- 18.Miller CT, Schilling TF, Lee K, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127(17):3815–28. [DOI] [PubMed] [Google Scholar]
- 19.Depew MJ, Simpson CA, Morasso M, Rubenstein JL. Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. J Anat. 2005;207(5):501–61. Epub 2005/11/30. JOA487 [pii] 10.1111/j.1469-7580.2005.00487.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nair S, Li W, Cornell R, Schilling TF. Requirements for Endothelin type-A receptors and Endothelin-1 signaling in the facial ectoderm for the patterning of skeletogenic neural crest cells in zebrafish. Development. 2007;134(2):335–45. Epub 2006/12/15. dev.02704 [pii] 10.1242/dev.02704 . [DOI] [PubMed] [Google Scholar]
- 21.Ozeki H, Kurihara Y, Tonami K, Watatani S, Kurihara H. Endothelin-1 regulates the dorsoventral branchial arch patterning in mice. Mech Dev. 2004;121(4):387–95. 10.1016/j.mod.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 22.Ruest LB, Xiang X, Lim KC, Levi G, Clouthier DE. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131(18):4413–23. 10.1242/dev.01291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zecchin E, Conigliaro A, Tiso N, Argenton F, Bortolussi M. Expression analysis of jagged genes in zebrafish embryos. Dev Dyn. 2005;233(2):638–45. 10.1002/dvdy.20366 . [DOI] [PubMed] [Google Scholar]
- 24.Lorent K, Yeo SY, Oda T, Chandrasekharappa S, Chitnis A, Matthews RP, et al. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131(22):5753–66. 10.1242/dev.01411 . [DOI] [PubMed] [Google Scholar]
- 25.Medeiros DM, Crump JG. New perspectives on pharyngeal dorsoventral patterning in development and evolution of the vertebrate jaw. Dev Biol. 2012;371(2):121–35. 10.1016/j.ydbio.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chai Y, Ito Y, Han J. TGF-beta signaling and its functional significance in regulating the fate of cranial neural crest cells. Crit Rev Oral Biol Med. 2003;14(2):78–88. . [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. Epub 2003/06/18. S009286740300432X [pii]. . [DOI] [PubMed] [Google Scholar]
- 28.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95(6):737–40. Epub 1998/12/29. S0092-8674(00)81696-7 [pii]. . [DOI] [PubMed] [Google Scholar]
- 29.Gronroos E, Kingston IJ, Ramachandran A, Randall RA, Vizan P, Hill CS. Transforming growth factor beta inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol Cell Biol. 2012;32(14):2904–16. 10.1128/MCB.00231-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Upton PD, Davies RJ, Tajsic T, Morrell NW. Transforming growth factor-beta(1) represses bone morphogenetic protein-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3. Am J Respir Cell Mol Biol. 2013;49(6):1135–45. 10.1165/rcmb.2012-0470OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Vetten N, Quattrocchio F, Mol J, Koes R. The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes Dev. 1997;11(11):1422–34. . [DOI] [PubMed] [Google Scholar]
- 32.Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci. 2010;35(10):565–74. Epub 2010/05/11. S0968-0004(10)00072-1 [pii] 10.1016/j.tibs.2010.04.003 . [DOI] [PubMed] [Google Scholar]
- 33.Walker AR, Davison PA, Bolognesi-Winfield AC, James CM, Srinivasan N, Blundell TL, et al. The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell. 1999;11(7):1337–50. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazmanian G, Kovshilovsky M, Yen D, Mohanty A, Mohanty S, Nee A, et al. The zebrafish dyrk1b gene is important for endoderm formation. Genesis. 2010;48(1):20–30. Epub 2009/12/17. 10.1002/dvg.20578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyata Y, Nishida E. DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim Biophys Acta. 2011;1813(10):1728–39. Epub 2011/07/23. S0167-4889(11)00189-3 [pii] 10.1016/j.bbamcr.2011.06.023 . [DOI] [PubMed] [Google Scholar]
- 36.Nissen RM, Amsterdam A, Hopkins N. A zebrafish screen for craniofacial mutants identifies wdr68 as a highly conserved gene required for Endothelin-1 expression. BMC Dev Biol. 2006;6(1):28 10.1186/1471-213X-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritterhoff S, Farah CM, Grabitzki J, Lochnit G, Skurat AV, Schmitz ML. The WD40-repeat protein Han11 functions as a scaffold protein to control HIPK2 and MEKK1 kinase functions. Embo J. 2010;29(22):3750–61. Epub 2010/10/14. emboj2010251 [pii] 10.1038/emboj.2010.251 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skurat AV, Dietrich AD. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem. 2004;279(4):2490–8. 10.1074/jbc.M301769200 [DOI] [PubMed] [Google Scholar]
- 39.Altafaj X, Dierssen M, Baamonde C, Marti E, Visa J, Guimera J, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet. 2001;10(18):1915–23. . [DOI] [PubMed] [Google Scholar]
- 40.Smith DJ, Stevens ME, Sudanagunta SP, Bronson RT, Makhinson M, Watabe AM, et al. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat Genet. 1997;16(1):28–36. 10.1038/ng0597-28 . [DOI] [PubMed] [Google Scholar]
- 41.Di Vona C, Bezdan D, Islam AB, Salichs E, Lopez-Bigas N, Ossowski S, et al. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol Cell. 2015;57(3):506–20. 10.1016/j.molcel.2014.12.026 . [DOI] [PubMed] [Google Scholar]
- 42.Brown KA, Ham AJ, Clark CN, Meller N, Law BK, Chytil A, et al. Identification of novel Smad2 and Smad3 associated proteins in response to TGF-beta1. J Cell Biochem. 2008;105(2):596–611. Epub 2008/08/30. 10.1002/jcb.21860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Doan D, Roman Petersen Y, Alvarado E, Alvarado G, Bhandari A, et al. Wdr68 requires nuclear access for craniofacial development. PLoS One. 2013;8(1):e54363 Epub 2013/01/26. 10.1371/journal.pone.0054363 PONE-D-12-25170 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chocron S, Verhoeven MC, Rentzsch F, Hammerschmidt M, Bakkers J. Zebrafish Bmp4 regulates left-right asymmetry at two distinct developmental time points. Dev Biol. 2007;305(2):577–88. Epub 2007/03/31. S0012-1606(07)00179-0 [pii] 10.1016/j.ydbio.2007.03.001 . [DOI] [PubMed] [Google Scholar]
- 45.Monteiro R, van Dinther M, Bakkers J, Wilkinson R, Patient R, ten Dijke P, et al. Two novel type II receptors mediate BMP signalling and are required to establish left-right asymmetry in zebrafish. Dev Biol. 2008;315(1):55–71. Epub 2008/01/29. S0012-1606(07)01565-5 [pii] 10.1016/j.ydbio.2007.11.038 . [DOI] [PubMed] [Google Scholar]
- 46.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101(35):12792–7. 10.1073/pnas.0403929101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Askary A, Mork L, Paul S, He X, Izuhara AK, Gopalakrishnan S, et al. Iroquois Proteins Promote Skeletal Joint Formation by Maintaining Chondrocytes in an Immature State. Dev Cell. 2015;35(3):358–65. 10.1016/j.devcel.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236(11):3088–99. Epub 2007/10/17. 10.1002/dvdy.21343 . [DOI] [PubMed] [Google Scholar]
- 49.Walker MB, Miller CT, Coffin Talbot J, Stock DW, Kimmel CB. Zebrafish furin mutants reveal intricacies in regulating Endothelin1 signaling in craniofacial patterning. Dev Biol. 2006;295(1):194–205. Epub 2006/05/09. S0012-1606(06)00224-7 [pii] 10.1016/j.ydbio.2006.03.028 . [DOI] [PubMed] [Google Scholar]
- 50.Angelo S, Lohr J, Lee KH, Ticho BS, Breitbart RE, Hill S, et al. Conservation of sequence and expression of xenopus and zebrafish dHAND during cardiac, branchial arch and lateral mesoderm development [In Process Citation]. Mech Dev. 2000;95(1–2):231–7. [DOI] [PubMed] [Google Scholar]
- 51.Akimenko MA, Ekker M, Wegner J, Lin W, Westerfield M. Combinatorial expression of three zebrafish genes related to distal- less: part of a homeobox gene code for the head. J Neurosci. 1994;14(6):3475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellies DL, Stock DW, Hatch G, Giroux G, Weiss KM, Ekker M. Relationship between the genomic organization and the overlapping embryonic expression patterns of the zebrafish dlx genes. Genomics. 1997;45(3):580–90. 10.1006/geno.1997.4978 [DOI] [PubMed] [Google Scholar]
- 53.Thisse B, Heyer V, Lux A, Alunni V, Degrave A, Seiliez I, et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004;77:505–19. . [DOI] [PubMed] [Google Scholar]
- 54.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277(7):4883–91. 10.1074/jbc.M111023200 . [DOI] [PubMed] [Google Scholar]
- 56.Bingham PM, Chapman CH. Evidence that white-blood is a novel type of temperature-sensitive mutation resulting from temperature-dependent effects of a transposon insertion on formation of white transcripts. Embo J. 1986;5(12):3343–51. Epub 1986/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vrijens K, Lin W, Cui J, Farmer D, Low J, Pronier E, et al. Identification of small molecule activators of BMP signaling. PLoS One. 2013;8(3):e59045 10.1371/journal.pone.0059045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139(4):757–69. 10.1016/j.cell.2009.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, et al. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011;25(12):1275–88. 10.1101/gad.2060811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Degoutin JL, Milton CC, Yu E, Tipping M, Bosveld F, Yang L, et al. Riquiqui and Minibrain are regulators of the Hippo pathway downstream of Dachsous. Nat Cell Biol. 2013. Epub 2013/08/21. ncb2829 [pii] 10.1038/ncb2829 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62(1):65–74. . [DOI] [PubMed] [Google Scholar]
- 62.Casari A, Schiavone M, Facchinello N, Vettori A, Meyer D, Tiso N, et al. A Smad3 transgenic reporter reveals TGF-beta control of zebrafish spinal cord development. Dev Biol. 2014;396(1):81–93. 10.1016/j.ydbio.2014.09.025 . [DOI] [PubMed] [Google Scholar]
- 63.Chablais F, Jazwinska A. The regenerative capacity of the zebrafish heart is dependent on TGFbeta signaling. Development. 2012;139(11):1921–30. 10.1242/dev.078543 . [DOI] [PubMed] [Google Scholar]
- 64.Ho DM, Chan J, Bayliss P, Whitman M. Inhibitor-resistant type I receptors reveal specific requirements for TGF-beta signaling in vivo. Dev Biol. 2006;295(2):730–42. 10.1016/j.ydbio.2006.03.050 . [DOI] [PubMed] [Google Scholar]
- 65.Talbot JC, Johnson SL, Kimmel CB. hand2 and Dlx genes specify dorsal, intermediate and ventral domains within zebrafish pharyngeal arches. Development. 2010;137:2507–17. Epub 2010/06/25. dev.049700 [pii] 10.1242/dev.049700 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rashidian J, Le Scolan E, Ji X, Zhu Q, Mulvihill MM, Nomura D, et al. Ski regulates Hippo and TAZ signaling to suppress breast cancer progression. Sci Signal. 2015;8(363):ra14 10.1126/scisignal.2005735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R, et al. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3(4):1306–20. 10.1016/j.celrep.2013.03.027 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A-D) Immunofluorescence detection of endogenous Wdr68. A) Wdr68 expression in a wildtype 15-somites stage embryo raised at 32°C. B) a wdr68 mutant lacking Wdr68 protein. C) Wdr68 expression in a wildtype prim-5 stage embryo raised at 32°C. D) a wdr68 mutant lacking Wdr68 protein. E-F) live imaging of GFP fluorescence in Tg(hsp70l:GFP-wdr68)csu9 animals. E) transgenic animal with the only detected GFP expression coming from the cmlc:eGFP marker for transgenesis. F) same animal as in E but after a 0.5 hour heat shock (HS) at 39°C followed by 7 hours of recovery at 28.5°C. G) western blot analysis of Tg(hsp70l:GFP-wdr68)csu9 animals after various lengths of heat shock exposure. Panel G1) GFP-wdr68 expression is induced by heat shock. Panel G2) β-tubulin expression was used as a loading control and did not differ substantially between lanes.
(TIF)
A-F) ventral views of 5dpf alcian blue stained cartilages from embryos raised at 28.5°C. A) heat shocked wildtype sibling displaying normal cartilages. B) heat shock induced Tg(hsp70l:GFP-wdr68) overexpression yielded no discernible cartilage phenotype. C) wdr68-MO injected animal showing the mild M-PQ joint fusion phenotype. D) wdr68-MO injected animal showing the severe loss of M and PQ phenotype. E) wdr68-MO injected heat shock induced Tg(hsp70l:GFP-wdr68) animal showing rescued normal M and PQ cartilages. F) wdr68-MO injected heat shock induced Tg(hsp70l:GFP-wdr68) animal showing rescued mild M-PQ joint fusions. G) plot of the distribution of phenotypes observed in a representative experiment on wdr68-MO injected Tg(hsp70l:GFP-wdr68) animals.
(TIF)
A-B) ISH analysis for edn1 expression on 25-somites stage animals raised at 28.5C that were heat shocked at the bud and 20-somites stages. A) wildtype sibling with normal edn1 expression. B) Tg(hsp70l:GFP-wdr68) embryo overexpressing GFP-Wdr68 with near-normal edn1 expression. C-D) ISH analysis on prim-12 stage embryos. C) wildtype control sibling with normal dlx6a expression. D) Et1-MO;wdr68hi3812/hi3812 mutant lacking dlx6a expression in all arches. E-F) merged green-red channel fluorescence on prim-5 stage embryos injected with either GFP/dsRed (G/R) or GFP/Edn1 (G/Edn1) plasmid mixtures. E) broad GFP/dsRed expression in a G/R embryo. F) broad GFP expression in a G/Edn1 embryo. G-H) lateral view of 5dpf alcian blue stained embryo. G) Et1-MO injected animal showing loss of M and CH but retention of PQ. H) Et1-MO;wdr68hi3812/hi3812 mutant showing loss of M, CH, and PQ. I-P) ISH analysis for hand2 expression on prim-12 stage animals raised at 32°C. Red arrowhead points at 1st arch expression of hand2. I, K, M, O) dorsal view. J, L, N, P) lateral view. I, J) wildtype sibling injected with G/R mix showing normal hand2. K, L) wdr68hi3812/hi3812 mutant injected with G/R mix showing loss of 1st arch hand2. M, N) wildtype sibling injected with G/Edn1 mix showing normal hand2. O,P) wdr68hi3812/hi3812 mutant injected with G/Edn1 mix showing loss of 1st arch hand2.
(TIF)
A) ISH analysis of prim-25 stage wildtype and wdr68 mutant animals revealed no differences in expression of bmp4. B) C2C12 wildtype and deletion subline sequences at the targeted locus in exon-5. The yellow highlight indicates the guide RNA target sequence followed by the TGG PAM sequence. C and D) ISH analysis for hand2 expression. C) ISL-treated wildtype sibling. D) ISL-treated mutant sibling lacking rescue. E and F) ISH analysis for dlx2 expression. E) ISL-treated wildtype sibling. F) ISL-treated mutant sibling lacking rescue.
(TIF)
A-N) Confocal images of the pharyngeal arch regions of embryos live-mounted in 0.7% agarose containing 0.0167% Tricaine. A) 25-somites stage wildtype sibling. B) 25-somites stage wdr68hi3812/hi3812 mutant. C) prim-5 stage wildtype sibling with 1st arch region outlined in blue. D) prim-5 stage wdr68hi3812/hi3812 mutant with same outline as in C to indicate regions of reduced mCherryCAAX signal. E) prim-12 stage wildtype sibling with ventral 1st arch region outlined in blue. F) prim-12 stage wdr68hi3812/hi3812 mutant with same outline as in E to indicate reduced ventral mCherryCAAX signal. G) prim-25 stage wildtype sibling with ventral 1st arch region outlined in blue. H) prim-25 stage wdr68hi3812/hi3812 mutant with same outline as in G to indicate reduced ventral mCherryCAAX signal. I) lateral view of 4-dpf wildtype sibling cartilages. J) lateral view of 4-dpf wdr68hi3812/hi3812 mutant severely reduced M and PQ cartilages. K) ventral view of animal in I. L) ventral view of animal in J. M) ISL-treated prim-25 stage wildtype sibling with ventral 1st arch region outlined in blue. N) ISL-treated prim-25 stage wdr68hi3812/hi3812 mutant with same outline as in M, H, G to indicate modest rescue of ventral 1st arch mCherryCAAX signal.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.