

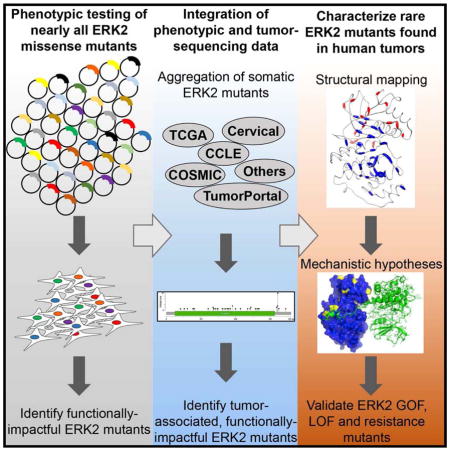
SUMMARY
Tumor-specific genomic information has the potential to guide therapeutic strategies and revolutionize patient treatment. Currently, this approach is limited by an abundance of disease-associated mutants whose biological functions and impacts on therapeutic response are uncharacterized. To begin to address this limitation, we functionally characterized nearly all (99.84%) missense mutants of MAPK1/ERK2, an essential effector of oncogenic RAS and RAF. Using this approach, we discovered rare gain- and loss-of-function ERK2 mutants found in human tumors, revealing that, in the context of this assay, mutational frequency alone cannot identify all functionally impactful mutants. Gain-of-function ERK2 mutants induced variable responses to RAF-, MEK- and ERK-directed therapies, providing a reference for future treatment decisions. Tumor-associated mutations spatially clustered in two ERK2 effector-recruitment domains, yet produced mutants with opposite phenotypes. This approach articulates an allele-characterization framework that can be scaled to meet the goals of genome-guided oncology.
Graphical Abstract
INTRODUCTION
Many thousands of tumor genomes have been sequenced to deepen our understanding of the genetic basis of cancer and to guide therapeutic strategies (Roychowdhury and Chinnaiyan, 2014). Computational approaches that identify significantly mutated genes or detect mutational clusters within individual genes have revealed the complex landscape of genetic alterations in cancer (Cancer Genome Atlas Research et al., 2013; International Cancer Genome et al., 2010; Kamburov et al., 2015; Lawrence et al., 2014; Niu et al., 2016; Porta-Pardo and Godzik, 2014; Tamborero et al., 2013; Vogelstein et al., 2013). As a result, tumor-associated mutations have been detected in nearly all human genes and the number of uncharacterized mutations continues to rise (Lawrence et al., 2014).
Characterizing tumor-associated variants of uncertain significance (VUS) is imperative for druggable therapeutic targets that lack genomic indicators of clinical response. For example, ATP-competitive inhibitors of the Ser/Thr kinase MAPK1/ERK2 are in early stage clinical trials (Infante et al., 2015). Tumor-sequencing studies have identified a single mutational hotspot in ERK2 (ERK2E322K) and many VUS. However, the infeasibility of characterizing mutant proteins at massive scale has precluded the identification of functionally impactful ERK2 mutants. In addition, this limitation has prevented the comprehensive association of mutants with response to RAF-, MEK- and ERK- (MAPK-pathway) directed therapies.
To address the challenge of interpreting mutations in ERK2, we employed saturation mutagenesis to comprehensively characterize the activity and drug responsiveness of 6,810 ERK2 missense mutants. We identify rare ERK2 mutants found in human tumors that have gain- or loss-of-function activity and drive differential responses to MAPK pathway-directed therapies. Collectively, this method provides a scalable framework for mutant characterization that can be used to deepen our understanding of disease-associated mutants.
RESULTS
Generation of an ERK2 mutant library
To enable the functional characterization of all possible missense mutations in cancer-associated genes, we established Mutagenesis by Integrated TilEs (MITE), a DNA-synthesis-based approach optimized for use in mammalian cells (Melnikov et al., 2014). We generated a pooled, virally-delivered, doxycycline-induced, cDNA expression library containing all 19 amino acid substitutions at residues 2-360 of ERK2 (Figure 1A, Figure S1A). Massively-parallel sequencing confirmed the presence of 6,810 of 6,821 possible (99.84%) ERK2 mutants in both plasmid DNA and in genomic DNA from transduced A375 cells (Figure S1B, S1C). MITE produced a library with a 4.7-fold increase in the abundance of ERK2 mutants and improved variant representation relative to bacterial mutagenesis (Figure S1A, S1D).
Figure 1. Functional characterization of 6,810 ERK2 mutants.

(A) A pooled-format proliferation screen in A375 cells to measure the functional impact of 6,810 MAPK1/ERK2 mutants.
(B) Heat map of the enrichment/depletion of ERK2 mutants in a pooled assay in which the proliferation of A375 cells serves as an indirect measure of ERK2 function. Data points represent the mean of 4 biological replicates. Grey boxes identify wild-type residues (see also Table S1, S2).
(C) The aggregate impact of amino acid substitutions on ERK2 function in A375 cells.
(D) Composite metric of ERK2 function when mutated at each amino acid, based on mutant enrichment/depletion in A375 cells.
See also Figure S1, S2 and Table S1, S2 and S3.
Comprehensive assessment of mutant ERK2 phenotypes
Most cancer-associated mutations are extraordinarily rare. As a result, the functional impact of these mutations are unknown and their contribution to disease biology is poorly understood. As an initial step towards disambiguating functionally impactful ERK2 mutants from inert mutants, we sought to test the function of all possible ERK2 point mutants and use these data to comprehensively identify cancer-associated gain-of-function (GOF) and loss-of-function (LOF) mutants. Testing the function of many thousands of ERK2 variants requires scalable assays that employ simple phenotypes to measure ERK2 function. Towards this goal, the A375 cell line is a useful model for studying ERK2 function. A375 cells harbor a BRAFV600E mutation that leads to constitutive activation of ERK1/2. In this context, ERK activity is homeostatic and is required for sustained proliferation. As a result, increases or decreases in ERK function affect A375 proliferation. For example, treating A375 cells with RAF, MEK or ERK inhibitors reduces ERK signaling and impairs cellular proliferation (Emery et al., 2009). However, increased pathway activity also negatively affects cellular proliferation. For instance, expression of constitutively-active BRAF, MEK1 or wild-type ERK2 decreases cell growth as a function of MAPK-pathway output (Figure 1A) (Goetz et al., 2014). We took advantage of this system to assess the function of all 6,810 ERK2 point mutants. We hypothesized that, in A375, cells expressing GOF ERK2 mutants will proliferate slower than cells expressing wild-type ERK2, whereas cells expressing LOF ERK2 mutants will proliferate faster than cells expressing wild-type ERK2 (Figure 1A). Thus, in a pooled competition assay, we predicted that LOF ERK2 mutants would be enriched relative to those with wild-type activity, whereas GOF mutants would be to be depleted.
We induced mutant library expression with doxycycline in A375 cells and quantified the relative abundance of each mutant after 96 hours via massively-parallel sequencing (See Supplemental Experimental Procedures, Figure S2A). Enrichment and depletion of ERK2 variants was calculated relative to their initial abundance in non-induced A375 (Figure 1B, S2B, S2C, Table S1). We predicted that LOF mutants would be enriched and GOF mutants would be depleted in this assay. Consistent with this notion, mutations expected to disrupt ERK2 protein function, including charged (arginine, aspartate, glutamate and lysine) and rigid (proline) substitutions, were the most widely enriched across all ERK2 mutant proteins (Figure 1C). Additionally, variants encoding substitutions in functionally essential ERK2 protein domains, including a subset of residues in the ATP-binding pocket, regulatory phosphorylation sites, the glycine-rich loop, the catalytic loop, the activation loop and the F-helix were universally enriched and captured a number of previously-reported LOF ERK2 mutants (Goetz et al., 2014; Madhani et al., 1997; McReynolds et al., 2016; Shin et al., 2010) (Figure 1B, Figure S2D, Table S2). We generated a composite, phenotypic score for each residue that summarizes the impact of ERK2 mutation on phenotypic behavior. Briefly, we summed the log(2) enrichment/depletion values for all substitutions across each amino acid and normalized them to that of the most depleted residue. In total, we identify 37 ERK2 residues that, when mutated, lead to mutant enrichment greater than or equal to Thr-185—an essential phosphorylation site in the ERK2 active site (Table S3). Moreover, substitutions at Lys-54, the kinase-essential “active lysine” traditionally mutated to create kinase-impaired ERK2 were also widely enriched (albeit to varying degrees) collectively suggesting that variant enrichment might serve as a proxy for decreased ERK2 activity (Figure 1B, S2E). However, we cannot conclude that all mutants that underwent enrichment are in fact LOF, as depletion may arise as a consequence of additional mechanisms.
To further test the hypothesis that proliferative changes induced by a subset ERK2 mutants may reflect altered ERK2 function, we asked if mutation of highly conserved ERK2 residues were enriched in our assay. To do this, we generated a cumulative phenotypic score for each ERK2 residue by summing the log2 depletion/enrichment of all substitutions at each amino acid and normalizing this value to the residue with the highest cumulative depletion (Figure 1D, Figure S2E, and Table S3). We observed that substitutions in highly-conserved residues of ERK2 homologues have a significantly greater propensity to be enriched, have a low phenotypic score (and therefore may be damaging) compared to substitutions in lowly conserved residues (Figure S2F). Expression of ERK2 mutants found in the germlines of healthy individuals—and therefore expected to be inert—were neither enriched nor depleted (Exome Aggregation Consortium, 2015) (Figure S2G). Moreover, we found that mutations in internally buried residues presumed to maintain ERK2 structure were enriched (Figure S2D). Collectively, these observations suggested that variant enrichment may serve as a proxy for reduced ERK2 activity and can rediscover previously characterized ERK2 functional domains.
A number of catalytically active ERK2 mutants have been previously identified (Bott et al., 1994; Chu et al., 1996; Emrick et al., 2001; Emrick et al., 2006; Goetz et al., 2014; Levin-Salomon et al., 2008; Levin-Salomon et al., 2009; Robinson et al., 1998; Shin et al., 2010; Smorodinsky-Atias et al., 2016) (Table S2). However, the identification of tumor-associated ERK2 mutants with GOF in-cell phenotypes has remained elusive in spite of the prevalence of constitutively active oncogenic missense mutants in upstream pathway members, including Ras-family members, BRAF and MEK1/2 that signal through ERK1/2 (Roskoski, 2012). Only a small subset of GOF ERK2 mutants, including the sevenmaker mutation (human ERK2D321N) and ERK2E322K have been found in human tumors or cancer cell lines (Bott et al., 1994; Chu et al., 1996; Goetz et al., 2014). We observed extensive depletion of previously identified ERK2 GOF mutants in our assay (Table S2). For example, Glu-322 and Asp-321 exhibited high phenotypic scores and were 11th and 19th, respectively, of all 360 ERK2 amino acids ranked from most depleted (#1) to least depleted (#360) (Figure 1D, S2E, Table S3). In addition, we found an additional 19 mutant ERK2 residues that were depleted to a degree equivalent to or greater than that of Glu-322 and Asp-321 (Figure S2E, Table S3). The observation that previously characterized GOF ERK2 mutants are depleted in this assay suggests (but does not prove) that a subset of uncharacterized, highly-depleted ERK2 mutants may also be GOF. However, until a comprehensive validation of all such mutants is complete, we cannot conclude that all such mutants are in fact GOF, nor that they have increased catalytic activity. These data raise the possibility that comprehensive approaches can be employed to discover additional, cancer-associated mutants for deeper validation. In addition, a complete mapping of residues required for normal ERK2 function may ultimately be useful for the design of allosteric ERK2 inhibitors.
Comprehensive identification of ERK-inhibitor resistant ERK2 mutants
Guiding treatment strategies with tumor-specific genomic data requires an understanding of how each mutant impacts response to cancer therapeutics. Thus, we wished to discover all possible ERK2 point mutants that could confer resistance to ERK-directed small molecule inhibitors. In BRAFV600E-mutant melanoma cell lines, exposure to small-molecule ERK inhibitors suppresses proliferation. However, this phenotype can be rescued by restoring MAPK signaling to baseline levels by expressing drug-resistant ERK2 mutants (Figure 1A) (Goetz et al., 2014). By performing these experiments in the presence of ERK2 inhibitors, and therefore in a background of reduced ERK activity and cell proliferation, expression of drug-resistant ERK2 mutants are sufficient to restore ERK signaling and rescue cell proliferation. To discover ERK2 mutants that are resistant to ERK-inhibitors, we induced expression of mutant ERK2 proteins in BRAFV600E-mutant A375 cells and exposed them to sub-lethal doses of two small molecule ERK1/2 kinase inhibitors, VRT-11E or SCH772984 (Aronov et al., 2009; Morris et al., 2013) (Figure S3A). After 12 days of drug exposure, enrichment of ERK2 mutants was quantified relative to initial populations (Figure S3B, S3C). The majority of ERK2 mutants were depleted from the population when exposed to drug (Figure S3B, S3C). The observation that most ERK2 mutants were depleted is a result of calculating the ratio of ERK2 mutants exposed to drug relative to their initial abundance prior to drug exposure.
We considered ERK2 variants that were enriched 4-fold or greater (p ≤ 0.025) in drug to be candidate resistance mutants. We identified 134 substitutions in 30 residues that conferred resistance to VRT-11E and 105 mutants in 36 residues that conferred resistance to SCH772984 (Figure 2A, Table S4). Among these, 59 substitutions in 18 residues conferred resistance to both VRT-11E and SCH772984 (Figure 2B, Table S4). We recovered all of the ERK2 variants previously discovered using traditional mutagenesis approaches, but increased ERK2 drug-resistant mutant discovery by 15–19 fold (Goetz et al., 2014). Surprisingly, resistance-conferring events were uncorrelated with ERK2 enrichment/depletion in the absence of ERK inhibition (Table S3).
Figure 2. Comprehensive identification of ERK2 mutants that induce resistance to ERK-directed kinase inhibitors.

(A) Heat map displaying the normalized enrichment/depletion of individual ERK2 mutants expressed in A375 cells in the presence of the ERK-inhibitors SCH772984 or VRT-11E. Data points represent the mean of 4–6 biological replicates. Grey boxes identify wild-type residues.
(B) Annotation of ERK2 amino acids that when mutated confer resistance to VRT-11E (VRT, red), SCH772984 (SCH, orange) or both inhibitors (S+V, black) when expressed in A375 cells.
(C) Composite metric of ERK2 function when mutated at each amino acid, based on mutant enrichment/depletion in A375 cells.
We identified resistance mutants that map to residues in the ATP binding pocket (Figure 2A, 3A). However, we also found drug-resistant mutants in other ERK2 protein domains, including substitutions in the αL16-helix (ERK2I347W, ERK2I347Y), the DFG-motif (ERK2G169D) and mutations in uncharacterized regions of ERK2, including ERK2R15P, ERK2D124T, ERK2C127I and 11 substitutions at Pro-58 (Figure 2A, 3A). A single mutation in the MAPK3/ERK1 DFG-motif (ERK1G186D) was recently discovered in a SCH772984-resistant mutagenic cell line (Jha et al., 2016). Encouragingly, we found that a G→D substitution at the orthologous residue in ERK2 (ERK2G169D) was the only alteration at Gly-169 sufficient to induce resistance to ERK inhibitors (Figure 2A, 2B).
Figure 3. Spatial mapping of ERK2 mutants that induce resistance to ERK-directed kinase inhibitors.

(A) Projection of ERK2 mutant enrichment in A375 cells in the presence of VRT-11E or SCH772984 on the structure of drug-bound ERK2 (PDB ID: 4QTE and 4QTA, respectively). All mutants displaying a log(2) fold-change of ≥2 are represented. Blue lines identify intramolecular contacts between ERK2 and inhibitor. Inhibitors are shown in blue.
(B) Venn diagram highlighting the overlap of ERK2 residues that, when mutated, confer resistance to either VRT-11E only, SCH772984 only, or both VRT-11E and SCH772984.
(C) Structural representation of ERK2 residues that, when mutated, confer resistance to VRT-11E (PDB ID: 4QTE). VRT-11E is shown in blue, residues are colored and sized as in (A).
(D) Structural representation of ERK2 residues that, when mutated, confer resistance to VRT-11E and SCH772984 (PDB ID: 4FMQ). ANP is shown in blue, MAPK-docking peptide is not shown, residues are colored and sized as in (A).
(E) Structural representation of ERK2 residues that, when mutated, confer resistance to SCH772984 (PDB ID: 4QTE). SCH772984 is shown in blue, residues are colored and sized as in (A).
(F) Venn diagram highlighting the overlap of VRT-11E-specific residues with residues that form direct contacts with VRT-11E.
(G) Venn diagram highlighting the overlap of VRT-11E/SCH772984-specific residues with residues that form direct contacts with both inhibitors.
(H) Venn diagram highlighting the overlap of VRT-11E-specific residues with residues that form direct contacts with SCH772984.
See also Figure S3, S4 and Table S1, S3, S4 and S5.
Given the propensity for resistance mutations to arise in drug targets during the course of treatment, a comprehensive catalogue of resistance mutants to mechanistically-distinct inhibitors may guide the choice of therapies at relapse. To identify inhibitor-specific resistance mutants, we looked for ERK2 mutants that drove resistance to either VRT-11E or SCH772984 (fold change ≥4) but additionally had a differential fold enrichment between inhibitors of 4-fold or greater. We identified 66 mutants that uniquely confer resistance to VRT-11E and 24 mutants that uniquely confer resistance to SCH772984 (Figure 2B, S3D, Table S4). When mutated, only a subset of amino-acids conferred mutually-exclusive drug-resistance to VRT-11E (12 residues) or SCH772984 (9 residues), whereas mutation of 18 residues conferred resistance to both small molecules (Figure 3B, 3C, 3D, 3E, Table S5). While 50% of the ERK2 VRT-11E-specific resistance residues make direct contacts with VRT-11E, none of the SCH772984-specific ERK2 resistance residues make direct contact with SCH772984 (Figure 3F, 3G, 3H, Table S6). Collectively, these observations suggest that drug-specific resistance alterations cannot be fully explained by direct drug-protein interactions alone. In the aggregate, these efforts have revealed an unappreciated diversity of ERK2 drug-resistant mutants that may ultimately guide the choice of therapy at initial diagnosis or relapse.
Massively-parallel characterization of ERK2 mutants found in human tumors
Sequencing of many thousands of human tumors has revealed the landscape of genetic variation across cancer. However, disambiguating inert mutations from mutations that are functionally impactful remains a challenge. Guided by the rediscovery of mutants known to impact ERK2 function, we sought to identify unappreciated, functionally relevant, cancer-associated ERK2 mutants with the ultimate goal of informing initial therapeutic strategies in patients whose tumors harbor these mutants. To do this, we aggregated all tumor-associated ERK2 mutations from The Cancer Genome Atlas, the Catalogue Of Somatic Mutations In Cancer or individual sequencing studies (Figure 4A, Table S7), identifying 83 unique missense mutations or small insertion/deletions in ERK2 (Cerami et al., 2012; Forbes et al., 2015; Gao et al., 2013; Ojesina et al., 2014). We then projected our phenotypic data onto these cancer-associated mutants and identified those that were depleted in our screen (Figure 4A). Using our composite ERK2 phenotypic score, we ranked all 359 ERK2 residues from most depleted (#1) to most enriched (#359). Within the top 20 most depleted ERK2 residues, we found 5 ERK2 residues that contain missense mutations observed in patient tumors or tumor-derived cell lines, including ERK2E322K (n=19), ERK2E322V (n=1), ERK2D321N (n=2), ERK2D321V (n=1), ERK2E81K (n=2), ERK2P58L (n=1) and ERK2S142L (n=1) (Figure 4A, S2E). Collectively, these 5 ERK2 residues contain 32% (27/84) of all observed ERK2 cancer-associated mutants, none of which have yet to be found in the germline of healthy patients (Exome Aggregation Consortium, 2015). Many ERK2 cancer-associated mutants are observed in only a single patient and would be missed by frequency-based methods. Among these somatic events, substitutions at Pro-58 were associated with a drug-resistance phenotype (Figure 2A, 3A, S3E). These data highlight the capacity of comprehensive variant characterization to nominate unappreciated candidate GOF mutants for subsequent validation.
Figure 4. Identification of somatic ERK2 mutants with non-wild-type function.

(A) Bars represent the composite ERK2 function score for each residue of ERK2. Each circle represents an annotated ERK2 mutant found in human tumors. Data bars represent the mean of 4 biological replicates.
(B) Suppression of proliferation in A375 cells expressing ERK2 mutants after treatment with 10 nM dabrafenib (Dab, 96 hours of treatment). Data are represented as a mean (+/− S.D.) of at least 6 replicates.
(C) Suppression of A375 proliferation with 2 nM trametinib (Tra, 96 hours of treatment) and proliferative rescue with GOF ERK2 mutants. Data are represented as a mean (+/− S.D.) of at least 6 replicates.
(D) Suppression of A375 proliferation with 500 nM SCH772984 (SCH, 96 hours of treatment) and proliferative rescue with ERK2 resistance mutants. Data are represented as a mean (+/− S.D.) of at least 6 replicates.
(E) In vitro kinase assay using V5-tagged ERK2 mutants immunoprecipitated from A375 cells treated with DMSO or 4 nM trametinib for 4 hours. Kinase activity is determined by the capacity to phosphorylate purified ELK1 (pERK, phosphorylated ERK; VINC, vinculin; V5 IP KA, immunoprecipitated V5 epitope followed by kinase assay).
See also Figure S2, S4 and Table S1, S2, S3 and S7.
Mutational frequency is often used to nominate oncogenic, activating somatic mutations. However, recurrent ERK2 mutations were not universally depleted in A375. For example, Arg-191 is recurrently mutated to His/Cys in ERK2 (n=3). However, in this phenotypic assay, mutation of Arg-191 was as enriched as mutation of the kinase-essential phosphorylation sites Thr-185/Tyr-187 or the active lysine (Lys-54) (Figure S2E). In fact, somatic variants that encode enriched ERK2 proteins are not uncommon: we identified a number of ERK2 mutants that are enriched disrupt highly-conserved ERK2 residues previously characterized as critical for ATP-binding or ERK2 activation, but have not been observed in the germline of healthy humans, including ERK2T185A (n=1), ERK2D167E/G (n=2), ERK2D149Y (n=1), ERK2R148L/S (n=2) and ERK2H147Y (n=1). These data suggested that, in the contextual constraints of this assay, a subset of enriched, tumor-associated ERK2 mutants may be kinase-impaired, highlighting the need for functional approaches to complement frequency based-computational methods.
Validation of candidate GOF or LOF ERK2 mutants
High-throughput assays permit comprehensive characterization of mutant behavior in simple assays (e.g. enrichment/depletion), but are not optimized to the nuances of each mutant and cannot identify more complex phenotypes (e.g. GOF/LOF) a priori. As such, validation of presumed mutant function is critical if we wish to interpret the consequences of mutant function and/or use this information to guide patient treatment strategies. ERK2 mutants with presumed GOF activity should retain function in the context of limited upstream MAPK-pathway activity. Thus, candidate ERK2 GOF mutants should rescue the proliferative effects of RAF or MEK inhibition, whereas mutants with wild-type or LOF activity should not. To test our functional predictions of ERK2 mutants found both in 1) human tumors and 2) displaying depletion/enrichment phenotypes in our screen, we expressed ERK2 mutants in A375 cells and tested their response to inhibitors of RAF or MEK (Figure 4B, 4C). Expression of wild-type ERK2, kinase-impaired ERK2 (ERK2K54R) and candidate LOF mutants (ERK2R191H and ERK2T185A) were insufficient to rescue the anti-proliferative effects of RAF or MEK inhibitors (Figure 4B, 4C). In contrast, putative GOF mutant ERK2 proteins (ERK2E322K, ERK2E322V, ERK2D321N, ERK2D321V, ERK2E81K and ERK2S142L) all restored cell proliferation to levels comparable to cells expressing constitutively active MEK1 (MEK1S218/222D or MEK1DD) or ectopic BRAFV600E (Figure 4B, 4C). Only two ERK2 mutants tested here, ERK2P58L and ERK2P58G, were able to confer ERK-inhibitor resistance (Figure 4D, Figure S3E). ERK2P58L had a modest capacity to rescue RAF inhibition whereas other substitutions at the same Pro-58 residue, including ERK2P58G, had more prominent phenotypes (Figure 4B, 4C). Consistent with these data, immunoprecipitates from candidate GOF ERK2 mutants were able to induce a mobility shift of recombinant ELK1 (a surrogate for ELK1 phosphorylation) in vitro following treatment with trametinib at a concentration that suppressed wild-type ERK2 activity (Figure 4E). In this assay, the candidate LOF mutants, including ERK2R191H and kinase-dead ERK2K54R, were unable to induce ELK1 phosphorylation or mobility shift at baseline or in the presence of trametinib (Figure 4E and S4A). These data suggested that a subset of ERK2 somatic mutants possess GOF activity and that RAF and MEK inhibitors may be contraindicated for patients whose tumors harbor these mutants. Moreover, in spite of their contextual limitations, these studies demonstrate that high-throughput approaches can identify unappreciated GOF and LOF mutants found in human tumors.
Spatially proximal ERK2 mutations encode mutants with opposite phenotypes
Computational approaches have dramatically improved our ability to identify functionally impactful cancer-associated mutants (Kamburov et al., 2015; Lawrence et al., 2014; Porta-Pardo and Godzik, 2014; Tamborero et al., 2013; Vogelstein et al., 2013). These approaches identified a ‘hotspot’ mutation in ERK2 (E322K) that displays GOF properties in validation experiments (Figure 4B, 4C, 4E). Orthogonal computational approaches, including CLUMPS, additionally identified enrichment (p=0.0053) of mutant residues (including E322K) within ERK2 effector recruitment domains (Alexa et al., 2015; Kamburov et al., 2015; Lawrence et al., 2014). This observation was buttressed by the finding that additional ERK2 tumor-associated mutations not found within the CLUMPS mutational search space were are also enriched (p = 4.303 × 10−5, Fishers exact test) in ERK2 effector recruitment domains (Figure 5A) (Lee et al., 2004; Zhang et al., 2003). While these approaches universally nominated ERK2 recruitment domain mutants as functionally important, we were surprised to observe that both depleted and enriched ERK2 mutants, including validated ERK2 GOF and LOF mutants, clustered in ERK2 recruitment domains (Figure 5A). This phenotypic distinction was not random: mutant activity was dictated by their localization within two distinct, well-characterized recruitment regions; alterations in the D-recruitment site (DRS, alternatively known as the common docking domain) contained both depleted and validated GOF proteins whereas alterations in the F-recruitment site (FRS, alternately known as the DEF pocket) encoded enriched and validated LOF proteins (Roskoski, 2012) (Figure 5B, S5A).
Figure 5. Somatic ERK2 mutants co-cluster in effector recruitment domains, but have opposite phenotypes.

(A) ERK2 residues containing tumor-associated mutations are enriched and cluster in ERK2 effector recruitment domains (p = 4.303 × 10−5, Fishers exact test) (PDB ID: 2ERK).
(B) Alteration of residues in the DRS of ERK2 are associated with GOF phenotypes, whereas alterations in FRS residues are associated with LOF phenotypes (p = 2.13 × 107, Students T-test). Data points represent the mean of 4 biological replicates in A375 cells.
(C) Western blot of lysates from A375 cells showing that ERK2 DRS mutants induce DUSP6 expression, whereas FRS mutants behave like kinase impaired ERK2K54R or the phosphorylation site (p-site) mutants ERK2T185A ERK2T187A (pERK, phosphorylated ERK; VINC, vinculin).
(D) ERK2 DRS mutants rescue the anti-proliferative effects of trametinib in A375 cells (Tra, 96 hours of treatment), whereas FRS mutants do not. Data are represented as a mean (+/− S.D.) of at least 6 replicates.
(E) Compound mutation of the FRS (Y233A) in ERK2 DRS mutants abrogate their capacity to rescue the anti-proliferative effects of trametinib (Tra, 96 hours of treatment) in A375 cells. Data are represented as a mean (+/− S.D.) of at least 6 replicates.
(F) Western blot showing that compound mutation of the FRS (Y233A) in DRS ERK2 mutants abrogate their capacity to signal to downstream effectors in A375 cells.
To validate the hypothesis that FRS mutants are LOF and that DRS mutants are GOF, we expressed ERK2 domain mutants in A375 cells and exposed them to the MEK inhibitor trametinib. DRS mutants displayed an increased capacity to phosphorylate downstream pathway components (Figure 5C) and could rescue the proliferative decrement caused by MEK inhibition (Figure 5D). In contrast, ERK2 FRS mutants behaved equivalently to kinase-impaired ERK2 (ERK2K54R), were unable to signal to DUSP6, FOS and FRA1 (Figure 5C, S5B) and could not rescue the anti-proliferative effects of MEK inhibition (Figure 5D). Compound mutation of the FRS (ERK2Y233A) in DRS mutants abrogated their capacity to rescue upstream MEK inhibition and curtailed downstream signaling to DUSP6, FOS and FRA1 similar to kinase-impaired ERK2 (ERK2K54R), suggesting that an intact FRS is required for GOF DRS mutants (Figure 5E, 5F). Given the proximity of the FRS to the activation loop and catalytic residues, it is likely that mutations in the FRS would negatively impact ERK2 kinase activity. Consistent with this prediction, FRS LOF ERK2 mutants have no activity in vitro (Figure 4E, S4A). Thus, although ERK2 mutations found in human tumors frequently cluster within ERK2 recruitment sites, they produce proteins with opposing phenotypes.
Tumor-associated ERK2 DRS mutants are refractory to dephosphorylation by DUSP6
High-throughput approaches can identify GOF mutants with equivalent phenotypes. However, determining the mechanism(s) of GOF mutants remains a challenge. ERK2D321N was originally discovered as a GOF mutant that has sustained kinase activity resulting from disrupted negative regulation by the DUSP6 phosphatase—an ERK2 effector that inhibits ERK2 activity through direct dephosphorylation (Bott et al., 1994; Chu et al., 1996; Li et al., 2007). As DUSP6 interacts with ERK2 via the DRS, we hypothesized that disrupted negative regulation by DUSP6 may represent a generalizable mechanistic for ERK2 DRS GOF properties (Farooq et al., 2001). This hypothesis was supported by the observation that ERK2E322, ERK2D321 and ERK2E81 establish the charged surface of the ERK2 DRS and form direct contacts with ERK2 effectors (Figure 6A).
Figure 6. Somatic DRS ERK2 mutants are refractory to de-phosphorylation by the DUSP6 phosphatase.

(A) Somatically mutated residues in the DRS of ERK2 (blue) form direct contacts with ERK2 effectors, including RSK1 (green, PDB ID: 4NIF).
(B) Somatic DRS ERK2 mutants are refractory to dephosphorylation by the DUSP6 phosphatase in transfected HEK293T cells (pERK, phosphorylated ERK; VINC, vinculin).
(C) Quantification of phosphorylated mutant ERK2 when co-expressed with DUSP6, as in (B). Data are represented as the mean (+/− S.D.) from 3 independent experiments in HEK293T cells. Values are normalized to wild-type ERK2.
To test this hypothesis, we co-expressed ERK2 mutants with BRAFV600E and either eGFP or DUSP6 in 293T cells and measured ERK2 phosphorylation. Co-expression of DUSP6 abrogated phosphorylation of wild-type ERK2, kinase-impaired ERK2 (ERK2K54R), ERK2R191H and endogenous ERK2 (Figure 6B, 6C). In contrast, DRS mutants all displayed sustained ERK2 phosphorylation in the presence of ectopic DUSP6. Surprisingly, ERK2S142L showed an equivalent degree of sustained phosphorylation when compared to known DRS ERK2 mutants, suggesting that impaired dephosphorylation by DUSP6 might underlie the GOF properties of DRS mutants and ERK2S142L. Given the physical proximity of Ser-142 to the DRS, these data suggest that Ser-142 may be an unappreciated component of the ERK2 DRS (Figure S5C). Notably, sustained phosphorylation was not a universal property of GOF ERK2 mutants—ERK2P58L exhibited a marked reduction in phosphorylation in the presence of DUSP6 (Figure 6B, 6C). Collectively, these studies suggest that rare cancer mutants, such as ERK2E81K, ERK2S142L and ERK2D321N, can achieve GOF properties through the same mechanisms as recurrent alterations (ERK2E322K). These observations are corroborated by the fact that ERK2 mutants that abrogate DUSP6 dephosphorylation are rarely found in tumors that simultaneously contain alterations leading to frank MAPK pathway activation—in this context there would be little preference for ERK2 mutations that impede ERK2 phosphorylation.
DISCUSSION
Comprehensive saturation mutagenesis has set the stage for mutant protein characterization at scale. We applied these technological advances to characterize mutants of ERK2 and identified a number of functionally consequential ERK2 mutants found in human tumors, including rare GOF ERK2 mutants. We also identified recurrent LOF ERK2 mutants, potentially indicating that these mutants can confer a selection advantage in certain contexts or possess kinase-activity independent functions for ERK2 (Rodriguez and Crespo, 2011). Although it remains to be determined if GOF or LOF alterations impact tumorigenesis or engender a dependency on ERK signaling in tumors, these data raise the possibility that presumed oncogenes can have both GOF or LOF mutations and challenge the gene-centric paradigm in which the classification of cancer genes is driven by the functions of a few deeply studied mutants. Moreover, these results imply that the mutational frequency of individual mutants is not a universal predictor of mutant protein function.
Collectively, these efforts represent an initial step towards comprehensive genome-guided oncology and suggest a general framework for disease-associated mutant classification. As genome sequencing studies multiply, this data remains stable and can continue to serve as a reference for newly discovered mutations. Moreover, as a resource, this library (and future libraries) can be publically deployed in multiple assays and cellular lineages to identify context-specific mutant function not feasible in these initial studies. Moving forward, one could imagine public repositories of aggregated somatic mutations alongside comprehensive phenotypic annotation.
The sevenmaker mutation (ERK2D321N) was discovered in D. melanogaster over 20 years ago and has been extensively characterized as a GOF mutant. However, since its discovery, the sevenmaker mutation has rarely been found in human tumors and, as a consequence, its role in oncology has remained unappreciated. Here, we identify additional cancer-associated GOF ERK2 mutants that mechanistically copy the sevenmaker mechanism of activation, are refractory to negative regulation by DUSP-family phosphatases and lead to sustained ERK2 function in the presence of limiting upstream MEK activation. Thus, while the sevenmaker mutant itself is quite rare in human tumors, the discovery of additional “sevenmaker-like” mutants suggest that an unexpectedly large fraction of all cancer associated ERK2 mutants utilize similar mechanisms of activation, supporting a larger-than-appreciated role for the sevenmaker mutant in cancer. In contrast, we observed that alterations at Pro-58 achieve equivalent GOF phenotypes through mechanistically distinct mechanisms, despite the fact that this mutant has only been observed in a single patient sample.
Our comprehensive, unbiased approach to annotating ERK2 mutants allowed us to uncover unappreciated GOF or LOF somatic missense mutations in a heavily investigated gene and signaling pathway whose activation is long-appreciated in cancer. These studies illustrate the potential of comprehensive mutant-characterization strategies to accelerate GOF mutant discovery, preemptively identify drug resistant mutants, deepen our understanding of gene-regulatory mechanisms and clarify how their disruption contributes to tumorigenesis.
EXPERIMENTAL PROCEDURES
High throughput ERK2 mutant screens
A375 cells were infected in six biological replicates (5.1 × 107 cells/replicate, 19% infection efficiency) with the lentiviral ERK2 mutant library followed by selection with puromycin. Post selection we achieved an allelic representation of ~1171–1482 cells per variant. Each replicate was split into four experimental arms: 1) 24-hour un-induced early time point, 2) 96-hours of 1 μg/ml doxycycline, and 16 hours of 1 μg/ml doxycycline followed by treatment with 3) 2 μM SCH772984 or 4) 3 μM VRT11E for 12 days of continuous culture. Genomic DNA was prepared from cell pellets (2.0 × 107 cells/replicate), treated with RNAse and ERK2 variants amplified with vector-specific primers (forward: TGGGAGGCCTATATAAGCAGAGCTCG, reverse: CGGAACCACGCCCAGAGC). Indexed Nextera libraries were generated from PCR-products and subjected to massively-parallel sequencing on 4 lanes of HighSeq2500, resulting in a minimum of 200 million reads per lane and an average coverage of ~0.3x per sample. Mapping of phenotypic data onto the crystal structure of ERK2 was performed using UCSF Chimera (Pettersen et al., 2004).
Creation of individual ERK2 mutants
Full-length wild-type ERK2 (NM_002745.4) was synthesized (GenScript) and site-directed mutagenesis was performed in pDONOR223 to all reported ERK2 mutants. All mutants were sequence confirmed using Sanger sequencing and transferred by LR-mediated recombination into the lentiviral vectors pLiX403 (AddGene) for inducible gene expression or pLVX307 (AddGene) for constitutive expression in mammalian cells.
Drug-sensitivity measurements
Inducible ERK2-mutant-expressing A375 cells were seeded into 96-well, white-walled, clear bottom plates at a density of 2500 cells/well. Twenty-four hours after seeding, doxycycline was added to the wells at a concentration of 1ug/ml and drugs were added using an HP D300 Digital Dispenser at the following concentrations: Trametinib at 2 nM; SCH722984 at 500 nM; and Dabrafenib at 10 nM, with the final volume of DMSO not exceeding 0.5%. After the addition of drugs, cells were incubated for 96 h and cell viability was measured using CellTiterGlo viability assay (Promega). Viability was calculated as a percentage of control (DMSO treated A375 cells with 1 μg/ml doxycycline). A minimum of six replicates were performed for each independent experiment.
Immunoblots
Adherent cells were washed once with ice-cold PBS and lysed with 1% NP-40 buffer [150 mM NaCl, 50 mM Tris pH 7.5, 2 mM EDTA pH 8, 25 mM NaF and 1% NP-40] containing 2x protease inhibitors (Roche) and 1x Phosphatase Inhibitor Cocktails I and II (CalBioChem). Lysate protein content was quantified (BCA assay) and normalized, then proteins were reduced and denatured (95 °C) and resolved by SDS gel electrophoresis o n 4–20% Tris/Glycine gels (Invitrogen). Resolved protein was transferred to nitrocellulose membranes, blocked in LiCOR blocking buffer and probed with primary antibodies.
Supplementary Material
Acknowledgments
We thank Xiaolan Zhang and Robert Lintner for technical assistance. We thank Amit Majithia, Amanda Walker, Brett Tomson, William G. Kaelin, Jr., Nicole Persky and the Broad Institute Research Associate/Associate Computational Biologist Journal Club for manuscript review. We thank Adam Tracy for assistance in data management. This work was conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Foundation in Mexico. This work was supported by a Career Development Award from the Melanoma Research Foundation (C.M.J.), a Collaborative Research Award from the William Guy Forbeck Research Foundation (C.M.J.), a Broad Institute Scientific Projects to Accelerate Research and Collaboration (T.S.M.), by the Paul C. Zamecnik, MD, Chair in Oncology at Massachusetts General Hospital (G.G.), by the NIH TCGA Genome Data Analysis Center (U24CA143845, G.G.). L.A.G. is a consultant for Foundation Medicine, Novartis, Boehringer Ingelheim, Third Rock; an equity holder in Foundation Medicine; and a member of the Scientific Advisory Board at Warp Drive. L.A.G. receives sponsored research support from Novartis, Astellas, BMS, and Merck.
Footnotes
ACCESSION NUMBERS
ERK2 protein structures were all retrieved from PDB (PDB ID: 4NIF, 2ERK, 4QTE, 4QTA, 4FMQ).
AUTHOR CONTRIBUTIONS
Conceptualization, C.M.J; Methodology, T.S.M., F.P., A.K.; Validation, L.B., A.A.; Formal Analysis, N.S.P., O.C., A.K., C.Z., A.B.B; Investigation, L.B., A.A., S.P., M.B., D.C., E.M.G., N.S.P.; Resources, T.S.M; Writing – Original Draft, C.M.J., Writing – Review & Editing, C.M.J., L.B., A.A., D.E.R, F.P., C.Z., O.C., A.K., E.M.G., N.S.P. Supervision, C.M.J., L.A.G., G.G., D.E.R., F.P.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexa A, Gogl G, Glatz G, Garai A, Zeke A, Varga J, Dudas E, Jeszenoi N, Bodor A, Hetenyi C, Remenyi A. Structural assembly of the signaling competent ERK2-RSK1 heterodimeric protein kinase complex. Proc Natl Acad Sci U S A. 2015;112:2711–2716. doi: 10.1073/pnas.1417571112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronov AM, Tang Q, Martinez-Botella G, Bemis GW, Cao J, Chen G, Ewing NP, Ford PJ, Germann UA, Green J, et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J Med Chem. 2009;52:6362–6368. doi: 10.1021/jm900630q. [DOI] [PubMed] [Google Scholar]
- Berger Alice H, Brooks Angela N, Wu X, Shrestha Y, Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M, Hogstrom L, et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. doi: 10.1016/j.ccell.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott CM, Thorneycroft SG, Marshall CJ. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 1994;352:201–205. doi: 10.1016/0014-5793(94)00958-9. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- Dogruluk T, Tsang YH, Espitia M, Chen F, Chen T, Chong Z, Appadurai V, Dogruluk A, Eterovic AK, Bonnen PE, et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015;75:5341–5354. doi: 10.1158/0008-5472.CAN-15-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrick MA, Hoofnagle AN, Miller AS, Ten Eyck LF, Ahn NG. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J Biol Chem. 2001;276:46469–46479. doi: 10.1074/jbc.M107708200. [DOI] [PubMed] [Google Scholar]
- Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc Natl Acad Sci U S A. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Aggregation Consortium e. a. Analysis of protein-coding genetic variation in 60,706 humans. 2015 doi: 10.1038/nature19057. biorxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, Dhalluin C, Ashton R, Zhou MM. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell. 2001;7:387–399. doi: 10.1016/s1097-2765(01)00186-1. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74:7079–7089. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JR, Janku F, Tolcher AW, Patel MR, Sullivan RJ, Flaherty KT, Carvajal RD, Varghese AM, Wong DJL, Sznol M, et al. Dose escalation stage of a first-in-class phase I study of the novel oral ERK 1/2 kinase inhibitor BVD-523 (ulixertinib) in patients with advanced solid tumors. J Clin Oncol. 2015;33:33. [Google Scholar]
- International Cancer Genome C. Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Morris EJ, Hruza A, Mansueto MS, Schroeder G, Arbanas J, McMasters D, Restaino CR, Dayananth P, Black S, et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol Cancer Ther. 2016 doi: 10.1158/1535-7163.MCT-15-0172. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, Garraway LA. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Lawrence MS, Polak P, Leshchiner I, Lage K, Golub TR, Lander ES, Getz G. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc Natl Acad Sci U S A. 2015;112:E5486–5495. doi: 10.1073/pnas.1516373112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C, Yang X, Lubonja R, Tran N, Nguyen C, et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov. 2016;6:714–726. doi: 10.1158/2159-8290.CD-16-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- Levin-Salomon V, Kogan K, Ahn NG, Livnah O, Engelberg D. Isolation of intrinsically active (MEK-independent) variants of the ERK family of mitogen-activated protein (MAP) kinases. J Biol Chem. 2008;283:34500–34510. doi: 10.1074/jbc.M806443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Salomon V, Maayan I, Avrahami-Moyal L, Marbach I, Livnah O, Engelberg D. When expressed in yeast, mammalian mitogen-activated protein kinases lose proper regulation and become spontaneously phosphorylated. Biochem J. 2009;417:331–340. doi: 10.1042/BJ20081335. [DOI] [PubMed] [Google Scholar]
- Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007;134:167–176. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Styles CA, Fink GR. MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell. 1997;91:673–684. doi: 10.1016/s0092-8674(00)80454-7. [DOI] [PubMed] [Google Scholar]
- McReynolds AC, Karra AS, Li Y, Lopez ED, Turjanski AG, Dioum E, Lorenz K, Zaganjor E, Stippec S, McGlynn K, et al. Phosphorylation or Mutation of the ERK2 Activation Loop Alters Oligonucleotide Binding. Biochemistry. 2016;55:1909–1917. doi: 10.1021/acs.biochem.6b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014;42:e112. doi: 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang WW, Zhang Q, McLellan MD, et al. Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat Genet. 2016 doi: 10.1038/ng.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Porta-Pardo E, Godzik A. e-Driver: a novel method to identify protein regions driving cancer. Bioinformatics. 2014;30:3109–3114. doi: 10.1093/bioinformatics/btu499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;8:1141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Crespo P. Working without kinase activity: phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci Signal. 2011;4:re3. doi: 10.1126/scisignal.2002324. [DOI] [PubMed] [Google Scholar]
- Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S, Chinnaiyan AM. Translating genomics for precision cancer medicine. Annu Rev Genomics Hum Genet. 2014;15:395–415. doi: 10.1146/annurev-genom-090413-025552. [DOI] [PubMed] [Google Scholar]
- Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010;38:114–127. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smorodinsky-Atias K, Goshen-Lago T, Goldberg-Carp A, Melamed D, Shir A, Mooshayef N, Beenstock J, Karamansha Y, Darlyuk-Saadon I, Livnah O, et al. Intrinsically active variants of Erk oncogenically transform cells and disclose unexpected autophosphorylation capability that is independent of TEY phosphorylation. Mol Biol Cell. 2016;27:1026–1039. doi: 10.1091/mbc.E15-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29:2238–2244. doi: 10.1093/bioinformatics/btt395. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou B, Zheng CF, Zhang ZY. A bipartite mechanism for ERK2 recognition by its cognate regulators and substrates. J Biol Chem. 2003;278:29901–29912. doi: 10.1074/jbc.M303909200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.