

Abstract
Kuru was the first human transmissible spongiform encephalopathy (TSE) or prion disease identified, occurring in the Fore linguistic group of Papua New Guinea. Kuru was a uniformly fatal cerebellar ataxic syndrome, usually followed by choreiform and athetoid movements. Kuru imposed a strong balancing selection on the Fore population, with individuals homozygous for the 129 Met allele of the gene (PRNP) encoding for prion protein (PrP) being the most susceptible. The decline in the incidence of kuru in the Fore has been attributed to the exhaustion of the susceptible genotype and ultimately by discontinuation of exposure via cannibalism. Neuropathologically, kuru-affected brains were characterized by widespread degeneration of neurons, astroglial and microglial proliferation, and the presence of amyloid plaques. These early findings have been confirmed and extended by recent immunohistochemical studies for the detection of the TSE-specific PrP (PrPTSE). Confocal laser microscopy also showed the concentration of glial fibrillary acidic protein–positive astrocytic processes at the plaque periphery. The fine structure of plaques corresponds to that described earlier by light microscopy. The successful experimental transmission of kuru led to the awareness of its similarity to Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease and formed a background against which the recent epidemics of iatrogenic and variant Creutzfeldt-Jakob disease could be studied.
Keywords: Kuru, Prion diseases, Transmissible spongiform encephalopathies
INTRODUCTION
Kuru was the first human neurodegenerative (i.e. not inflammatory) disease proven to be transmissible (1). Its discovery led to the transmission of Creutzfeldt-Jakob disease (CJD) 2 years later (2). These studies led to a Nobel prize for D. Carleton Gajdusek in 1976 (3), later to a Nobel prize for Stanley B. Prusiner for the discovery of “prions” (4), and also, indirectly, to a Nobel prize for Kurt Wüthrich for solving its protein structure using nuclear magnetic resonance spectroscopy (5). Currently, these diseases are believed to be caused by prions and are embraced by the term “transmissible spongiform encephalopathies” (TSEs) or “prion diseases.” Prions are an aggregate of a misfolded isoform, TSE-specific prion protein (PrPTSE), of a normal cellular protein (PrPc) that are regarded by the majority, but not all, investigators as the sole cause of this group of diseases (6).
Kuru Etiology—A Discovery of a New Class of Pathogens
Although on epidemiological grounds the etiology of kuru was thought to be infectious even before Gajdusek arrived in Papua New Guinea, all attempts to transmit kuru to small laboratory animals or to isolate a virus using tissue cultures or embryonated hen’s eggs were unsuccessful (7). Gajdusek had initially expressed doubt that cannibalism spread the disease because there was no initial evidence of infection and the fact that cannibalism, widespread in Papua New Guinea, did not lead to any disease in other areas where it was practiced. However, in retrospect, he said, “even a complete drunk could guess that a disease endemic among cannibals must spread through eating corpses” (D. Carleton Gajdusek, personal communication to Pawel Liberski). It must be stressed that Gajdusek and Vin Zigas provided a major early contribution to the epidemiology of the disease, on which later investigators were able to build (8, 9).
On August 6, 1959, in Papua New Guinea, Gajdusek received a letter from William Hadlow, a veterinary neuropathologist (9–11), who, having seen pictures of kuru neuropathology taken by Polish-born neuropathologist Igor Klatzo (see section on neuropathology) at a traveling Welcome Medical Museum exhibition in London, enclosed a copy of a letter to the editor of Lancet that pointed out the similarities between kuru and scrapie (10, 11). Gajdusek found the Hadlow suggestions “much more compelling” (11). A similar observation was made by veterinary neuropathologist James R. M. Innes (D. Carleton Gajdusek, 2008, personal communication) (12). The stage was thus set for Gajdusek to play a leading role, and the first chimpanzee was inoculated on February 17, 1963 (9). In 1965, brain tissue from 3 kuru patients he had inoculated 2 years earlier transmitted the illness to chimpanzees (1, 12) and later to Rhesus monkeys (13), marmosets (14), and Gibbon and Sooty Mangabey monkeys (15). In contrast, sheep, goats, mink, pigs, mongooses, opossums, dogs, ferrets, as well as ducks, geese, and turkeys were totally resistant to infection (16). At some stage, even plants like tomatoes were tried as possible hosts because of a “viroid hypothesis” (17). Overall, 18 cases transmitted kuru with a transmission rate of 95%, which was slightly higher than that for sporadic CJD (87%) (16).
Transmission of kuru to primates followed by transmission of CJD and, ultimately, other TSEs raised a persistent question of physicochemical structure of the agent. This question remained open until the “protein-only” theory was formulated (18, 19), a protein that copurified with the infectivity was found (20), and S. B. Prusiner received a Nobel Prize. The physicochemical properties of this misfolded protein, PrPTSE, and the results of transmission to transgenic mice define the strains of prion (21). The so-called molecular strains are defined on the basis of immunoblots of PrPTSE following limited proteinase K digestion, which produces fragments of different sizes and in different ratios. In kuru, the pattern of protease-resistant PrPTSE fragments corresponded to the type 2 or type 3 patterns according to the Collinge classification (21, 22). With regard to transmission to transgenic mice, those expressing human PrP 129 valine on the null background lack the transmission barrier to sporadic and iatrogenic CJD. These mice also lack the transmission barrier to kuru inoculum, and the type 3 PrPTSE from kuru inoculum is preserved on transmission. In contrast, inocula characterized by the type 2 PrPTSE pattern change to the type 3 pattern on transmission (21). Collectively, in cited experiments, kuru behaves like sporadic CJD.
Genetics of Kuru
Genetic studies of TSEs during the past 2 decades resulted in the discovery that each hereditary form of TSE, familial CJD, Gerstmann-Sträussler-Scheinker disease (GSS), and fatal familial insomnia is associated with mutations in the PRNP gene coding for the PrPC. The methionine/valine (Met/Val) variation at position 129 does not by itself cause the disease but dramatically influences the phenotype resulting from other PRNP mutations (23) and influences susceptibility/resistance to kuru and iatrogenic and sporadic forms of CJD (24–26). The genotype frequencies determined in the background populations vary from 0.32Met/Met/0.43Met/Val/0.24Val/Val in New Guineans to 0.37/0.51/0.12 in the British to 0.92/0.08/0 in the Japanese.
The results of early clinical, neuropathologic, and epidemiologic studies led investigators to the hypothesis that kuru was a genetically determined or genetically mediated illness based on the following facts: the disease was generally restricted to Fore natives; three quarters of patients were related to someone who had succumbed to kuru; the age and gender distributions indicated a higher susceptibility of certain population groups; and clinical and laboratory findings were not suggestive of an infectious disease (27). Large kuru pedigrees were compiled and analyzed (28). A genetic theory based on family/population analyses proposed that kuru might be controlled by a single autosomal gene that, if mutated, behaves as an autosomal recessive trait in young children but is autosomal dominant in older females (29).
After transmissibility of kuru was discovered (1) the epidemiologic phenomenon was explained by the spread of an infectious agent through the practice of cannibalism, with the pattern of kuru distribution continuing to suggest a role for genetic predisposition (30). A study of erythrocyte antigens and serum proteins in representative groups of local populations was conducted. The extremely polymorphic group-specific component (Gc) system was widely used. The rare Australian Gc variant GcAborigine (GcAb) has a single-nucleotide alteration in the second position of α-2-globulin codon 429 resulting in an electrophoretically distinguishable pattern (31). The GcAb allele occurs with an extremely high frequency in the kuru region, the highest in the North and South Fore (10%–24%), Keiagana (24%), Gimi (10%–21%), and Auyana (16%) groups. The distant second highest frequency of the GcAb allele was observed in Australian aborigines (5%–8%). Kuru patients possess the GcAb allele more often than the general population (32, 33); overall, 38.6% of kuru patients from kuru-affected areas carried at least one GcAb allele, and the calculated risk of contracting kuru for the GcAb-Ab genotype carrier was 6.14:1 (33). The results describing the prevalence of the GcAb-Ab genotype in kuru patients are very difficult to interpret; it is possible that the 4q12 locus harbors an unknown gene whose product may interact with PrP or be indirectly responsible for an increased susceptibility to kuru.
The role of genetically determined susceptibility to kuru was reevaluated after the human gene coding for PrP PRNP was cloned and characterized. Multiple mutations in the PRNP gene have been linked to various phenotypes of hereditary, sporadic, and iatrogenic CJD (25, 26). An incidental finding indicating that 2 kuru patients were homozygous at position 129 of the PRNP gene was reported (34) and subsequently confirmed (35), prompting further research on the susceptibility to kuru.
A population-based study in kuru-affected areas of New Guinea was carried out using specimens collected at several points of the kuru epidemic. DNA was recovered from frozen brain tissue, brain suspensions previously used in transmission studies, blood clots, serum samples, and other tissues stored in tissue banks (24, 36). The Kuru Registry book containing information on each kuru patient and the general population of the kuru-affected areas that was started in 1957 (D. Carleton Gajdusek) and continued into the 1990s provided precious medical information about individuals who did and did not develop kuru throughout the epidemic. Kuru patients identified and examined in Fore villages in 1957 through 1959, at the time of the highest kuru incidence, and patients diagnosed during the later stage (1964–1988) were separately studied (37) and compared with controls from within and outside kuru-affected villages.
The PRNP codon 129 genotype frequency in kuru patients at the height of the epidemic was found to deviate significantly from Fore survivors and non-Fore controls. Based on representative statistics, it was concluded that individuals homozygous for the 129 Met allele had an earlier age of disease onset and a shorter incubation time, whereas 129 Met/Val and Val/Val carriers developed kuru at a later age, after an incubation period of 20 years or longer, and many survived the epidemic (24). A group of individuals who lived through the kuru epidemic and never developed the disease was found to deviate from Hardy-Weinberg equilibrium because of almost complete absence of the Met/Met genotype at position 129 (37). The Met/Met genotype depletion in the group of survivors was significant as compared with the healthy population of non-Fore villages sampled during the same 1957–1959 period. This phenomenon was associated with increased susceptibility of the 129 Met/Met carriers who became early victims of the epidemic, disproportionally dying of the disease after the shortest incubation time (24, 37).
To investigate this hypothesis further, genotype frequencies in Fore male patients who (unlike the females) had a single age-related peak of exposure to kuru between 1 and 10 years of age were separately analyzed (37). Most of the young Fore males who developed kuru in 1957–1959 were Met/Met homozygous, whereas survivors of the kuru epidemic who were exposed to the infection at the same age completely lacked the Met/Met genotype. The results of this analysis demonstrate that in the group of young Fore males just entering the age of risk, the Met/Met individuals were preferentially affected with kuru (37). Deficit of codon 129 homozygosity was also found in analysis of elderly females who lived through the epidemic in the high-exposure areas and attended multiple mortuary feasts (38). Having “used up” the more susceptible Met/Met genotype, the kuru epidemic eventually began absorbing the less susceptible Met/Val and Val/Val genotypes prevailing in the region. Indeed, 11 of 16 kuru cases that occurred in 1964–1988 had a 129 Val/Val genotype (and unpublished data) (40–42). Many of the Met/Val and Val/Val genotype carriers survived the epidemic altogether, demonstrating a lower susceptibility to kuru. Lack of the 129 Met/Met individuals in the postepidemic population control may be explained by the massive loss of this genotype to kuru in a small population (37). The last 11 known kuru patients studied at the very end of the epidemic (1996–2004) had an estimated minimum incubation period of 34 to 41 years; 8 of 10 were heterozygous at the PRNP codon 129 (39). The loss of Met/Met genotype and accumulation of alternative alleles in the postepidemic Fore population was also indicated by the finding that the PRNP V127V synonymous substitution that showed the highest frequency in the surviving elderly women is part of the 127GG–129VV haplotype (38).
Kuru imposed a strong balancing selection on the Fore population (40). Apparently, the decline of kuru incidence in the Fore was determined by the discontinuation of exposure and additionally by the exhaustion of the susceptible genotype. However, the exposure disappeared because of an Australian “carrot-and-stick” approach to the practice of cannibalism rather than by any biological or medical reason!
Surprisingly, a balancing selection at the PRNP locus somewhat similar to what occurred in the Fore was discovered in some other world populations and has been interpreted as evidence of selection pressure from possible epidemics of prion diseases in prehistoric humans (40, 41).
Kuru and Cannibalism
When kuru was first reported as occurring in certain families and hamlets, confined to the Fore and adjacent people with whom they intermarried, and showing a predilection for young children and adult women (27), a genetic or hormonal explanation seemed to provide a key. It was thought to be a hereditary disorder determined by a single autosomal gene, dominant in females but recessive in males (7, 8, 29). Further investigation was hampered, however, by a lack of information about Fore kinship and social life. With a grant from the Genetics Department at Adelaide University and a request to focus on kinship, anthropological investigation began in 1961.
Anthropological studies by Robert Glasse and Shirley Lindenbaum soon indicated that the genetic hypothesis was not tenable. Many kuru victims were not closely related biologically but were considered by the Fore to be kin in what we would call a “social sense.” Nearby or distant Fore immigrant groups were welcomed and, in time, were said to possess “one blood” and to stem from a common ancestor. Fore genealogies gave legitimacy to ties based on culturally defined notions of kinship, providing a moral guide for living but were not reliable statements of genetic proximity (42).
Further doubts about the genetic hypothesis arose when the Fore reported that kuru had spread through their territory within living memory and that its path followed a specific route, entering from the northwest around the turn of the century and arriving in the North Fore about 1920 (43). From here, it traveled down their southeastern border, arriving in the central South Fore and further south in 1930. In some southwestern and southeastern locations, it appeared as late as the 1940s. The genetic model had implied that kuru must have been of remote evolutionary origin and in epidemiological equilibrium. However, the disease was judged to be too common and too fatal to be a purely genetic disorder, unless the hypothetical kuru gene was maintained at high frequency by a mechanism of balanced polymorphism for which, at that time, there was no evidence (44). Moreover, the Fore could name those who had died of kuru and those who had participated in the consumption of the deceased person, which allowed for an account of the appearance of the disease in particular hamlets some 4 to 20 years after ingestion. This information redirected the anthropological investigation to inquire further about the practice of cannibalism.
The relationship between kuru and cannibalism seemed to be confirmed by the rules for the consumption of human flesh, which tallied with the epidemiological evidence. No longer present in the 1960s, cannibalism had been suppressed by the government and missions, but the Fore spoke openly about their practice of consuming deceased relatives. The Lutheran missionaries in the North Fore were the first to discourage the practice in the early 1950s. The government also played a part. Patrol officers who arrived with firearms demonstrated their power by purchasing a local pig and shooting it at a distance. In addition to banning cannibalism (enforcing an Australian law), they told people to stop fighting, to pull down the barricades around hamlets, and to move their houses into open ground where they could be counted for a census. The first government patrols in the late 1940s had also reported cannibalism to be customary throughout much of the region. Beyond the Fore, the practice was to consume enemies (exocannibalism), not deceased kin (endocannibalism), the Fore pattern. By the end of the 1950s, a government road was constructed through the region, bringing the Fore into contact with people who did not consume the dead. Nevertheless, the South Fore reported that some people had continued to hide and eat deceased kin until the road reached them. Cannibalism thus continued longer in the south, the area with the highest incidence of kuru in the 1960s.
Not all deceased persons were eaten. Those who died of dysentery, leprosy, and possibly yaws were not consumed, but kuru victims were viewed favorably, especially if the body was not emaciated. All body parts were eaten except the gallbladder, which was considered too bitter. Significantly, not all the Fore were cannibals. Adult men in the North Fore consumed the dead more frequently than in the South, where adult men rarely ate human flesh, and those who did avoided eating the bodies of women, the main kuru victims. South Fore children, living in houses with their mothers, ate what their mothers gave them. Rules for consumption were very specific. Body parts were allocated according to kinship rights and gender. Small children were never given meat and were also kept away from the ceremony. Female relatives by marriage, who were the main consumers, could also request certain body parts. For example, the daughters-in-law of a deceased man or woman usually requested the head. Brain tissue mixed with wild green vegetables was cooked in bamboo tubes, which the daughters-in-law shared with other women (45). Initiated youths moved to the men’s house at approximately age 12 years, leaving behind the world of immaturity, femininity, and cannibalism. Male children thus had less exposure to the kuru agent than their sisters who continued to live with their mothers. Consumption of human flesh and exposure to the infectious agent was thus limited to adult women, children of both sexes, and a few adult men, matching the epidemiology of kuru in the 1960s (46). It was suggested that small variations in consumption practices throughout the region might be associated with differences in kuru prevalence.
The consumption of deceased relatives who had died of kuru was thus proposed as the mode of transmission (47, 48). It is now thought that kuru first arose after a single individual with CJD was consumed, and the latter assumption is supported by the molecular data of virtual identity of the sporadic CJD and kuru (49). The infectious agent was then recycled through the consumption of those who had died of the disease, amplifying the infectious agent and resulting in the epidemic.
Anthropological reports about kuru and cannibalism were presented in 1962 and 1963 (50) and discussed with scientists visiting the anthropological field site but were often met with skepticism, although some visitors later had a change of mind. Gajdusek repeatedly stated that “everyone in the area knew that kuru was infectious and due to cannibalism, from missionaries to bush pilots” but did not pursue this observation as a topic of research, turning instead to laboratory experiments to find a “classical” or a “slow” virus.
After the successful transmission by intracerebral inoculation of brain tissue homogenate in 1966 (1) and a publication proposing the cannibal connection (47), he presented his view more firmly that “even today, we have no evidence that eating bodies caused the spread.” (51). He suggested an alternate non-oral route of transmission from brain tissue rubbed on the body of mourners during mortuary ceremonies and, in particular, the contamination of skin cuts and sores during body preparation and consumption. However, the Fore did not talk about rubbing their bodies with brain tissue when describing earlier events and have recently denied the practice (52). Although the handling of infectious body parts at mortuary feasts could possibly be a means of self-inoculation, Gajdusek had begun to overstate the case.
The notion that cannibalism was not a socially approved custom in Papua New Guinea, or elsewhere, was proposed in 1979 in a publication suggesting that the practice was an invention of the anthropologist, missionary, and adventurer’s imagination (53), a position that is no longer accepted. Some anthropologists may have once thought that the topic was too delicate to discuss, given the image of the cannibal as an icon of primitivism, but well-documented accounts of the practice in Papua New Guinea have recently been published (54, 55).
The Fore still believe that kuru is caused by malicious sorcerers, a theory of disease causation that also takes account of clinical symptoms, age, gender, and epidemic history. The death of adults demands identification of the person responsible. This mode of analysis includes more social information than we include in our medical assessments but tells the same story. The Fore narratives about the spread of kuru from north to south at the turn of the 20th century, although told in a sorcery idiom, provided an important key and a correction to medical assumptions about the nature of the epidemic. A recent investigation of Fore mortuary practices has shown that, by consuming deceased relatives, the Fore were ensuring that the souls of the departed reached the land of the dead, to be reborn as ancestors (52). First associated with population genetics, anthropological research on cannibalism now finds a home with molecular genetics. Kuru is currently said to have exerted a strong selection pressure on the human PrP gene in the context of the Fore practice of endocannibalism (56).
Clinical Manifestations
Kuru is a uniformly fatal and remarkably stereotyped cerebellar ataxic syndrome accompanied by tremor and choreiform and athetoid movements (27, 39, 57–61) (Fig. 1). The progressive dementia that is a cardinal sign of sporadic CJD went practically unnoticed in earlier investigations and, if it occurred at all, was only a very late clinical manifestation. Emotional changes, including inappropriate euphoria and compulsive laughter (hence, the “laughing death” or “laughing disease”) or apprehension and depression, were evident. Kuru was divided into 3 clinical stages: ambulant, sedentary, and terminal. The duration of kuru, as measured from the onset of prodromal signs and symptoms until death, was about 12 months (range, 3–23 months) (54, 62).
FIGURE 1.
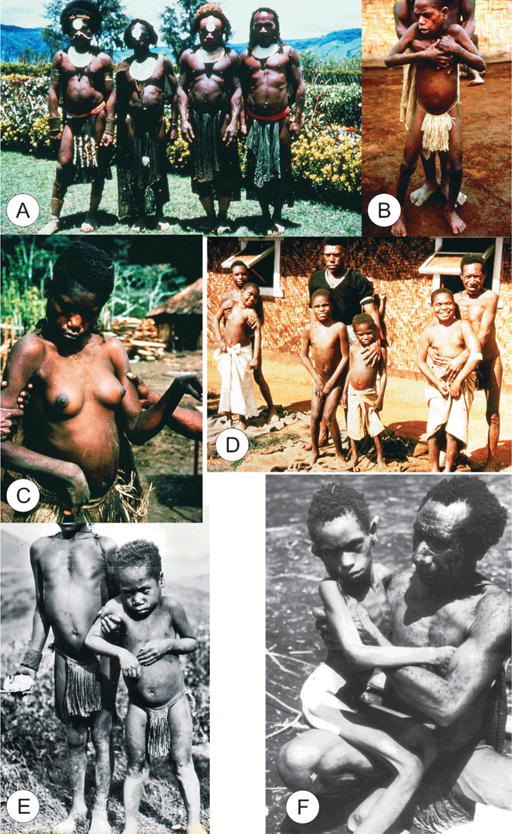
General views of kuru. (A) A group of Fore men, dcg-57-ng-118. (B) A boy with kuru supported by a father. (C) A woman with kuru, dcg-ng-57-346A. (D) A group of boys with kuru. (E) A boy with kuru being supported by an adolescent, dcg-57-png-1148. (F) An advanced stage of kuru. All slides were taken by the late D. Carleton Gajdusek and given to the first author.
There was a vaguely defined prodromal period characterized by the presence of headache and limb pains, frequently in the joints, where knees and ankles came first, followed by elbows and wrists. Occasionally, interphalangeal joints were first affected, with abdominal pains and loss of weight. This period lasted approximately a few months.
The prodromal period was followed by the ambulant stage, the end of which was defined by the patient being unable to walk without support. In early stages, there were subtle signs of unsteadiness of gait that were usually not detected by the observer but only self-diagnosed. During a period of a month or so, this progressed to severe astasia and ataxia, followed by incoordination of the muscles in the trunk and lower limbs. Because patients were well aware that kuru indicated incipient death in approximately a year, they became withdrawn but not demented. A fine shivering tremor, starting in the trunk and amplified by cold and indeed associated with a “goose flesh,” was often followed by titubation and other types of abnormal movements. Attempts to maintain balance were supported by clawing of the toes and curling of the feet. Plantar reflex was always flexor, whereas clonus, more in the ankles than the patella, was a hallmark of the clinical picture. Clonuses could be observed only temporarily and then subsided.
In the early stages of the disease, usually only the patient was aware of the insidious onset of gait incoordination. On examination, ataxia was only evident when the patient stood on one leg. The Romberg sign was almost always negative (2 of 34 kuru cases in Alpers’ series [63]), but with disease progression, ataxia became marked and the Romberg sign became positive. Indeed, the patients could not stand with feet close together. The gait of kuru was characteristically cerebellar (i.e. wide based) and staggering. Intention tremor was detected in more than 50% of cases in Alpers’ series in the first stage of kuru but was constantly present through the second stage. Dysarthria appeared early. Resting tremor was a cardinal sign of kuru; the major component of it was ataxic, and it was enhanced by muscular activity. When the patient became motionless, it subsided.
Horizontal convergent strabismus was a typical sign, particularly in younger patients; nystagmus was common but the papillary responses were preserved. Facial hemispasm and supranuclear facial palsies of different kinds were also common.
The second sedentary (sitting) stage began when the patient was unable to walk without constant support and ended when the affected person was unable to sit without it. Postural instability, severe ataxia, tremor, and dysarthria progressed endlessly through this stage. Deep reflexes were increased, but the plantar reflex was still flexor and the Babinski sign has never been seen.
In the third stage, the patient was bedridden, incontinent, and covered with urine and feces, with dysphasia and primitive reflexes, and eventually succumbed in a state of severe emaciation, generalized muscle wasting, and fasciculation, spontaneous or evoked by tapping. Some symptoms of dementia were eventually observed. A strong grasp reflex occurred, as well as fixed dystonic postures, athetosis, and chorea.
Neuropathology
Surprisingly, systematic examinations of kuru brains have been performed on only a few dozen cases (Figs. 2–4). Information on cases with sufficient detail is presented in Table. Cases described collectively are discussed herein. The first systematic examination of kuru neuropathology (12 cases) was published by Klatzo et al (62) in 1959 (Table). Neuronal alterations observed in many nuclei throughout the neuraxis, the anterior motor neurons of the spinal cord, brainstem, cerebellum, and cerebral cortex were totally nonspecific in nature (64); nonetheless, they were sufficient for Klatzo to draw a seminal parallel between kuru and CJD in a letter dated September 13, 1957 (65). Klatzo obtained kuru brains that Gajdusek collected and sent to Joseph Smadel, then scientific director of the National Institutes of Health.
FIGURE 2.
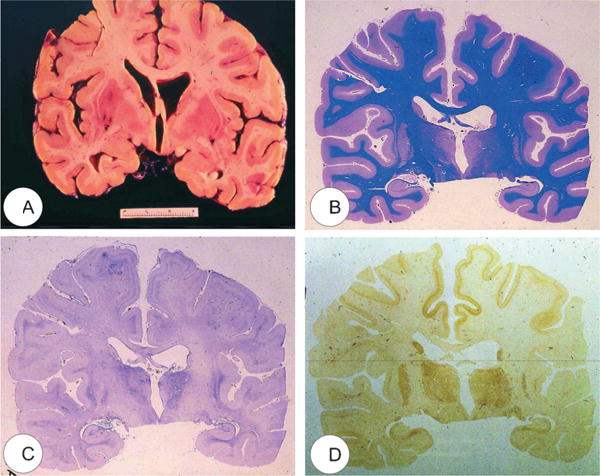
Macroscopic view of the kuru brain (“K”) (64). (A) Coronal section. (B) Luxol fast blue stain. (C) Kanzler stain to detect hyperplastic astrocytes. (D) Immunohistochemistry for the transmissible spongiform encephalopathy–specific prion protein PrPTSE.
FIGURE 4.
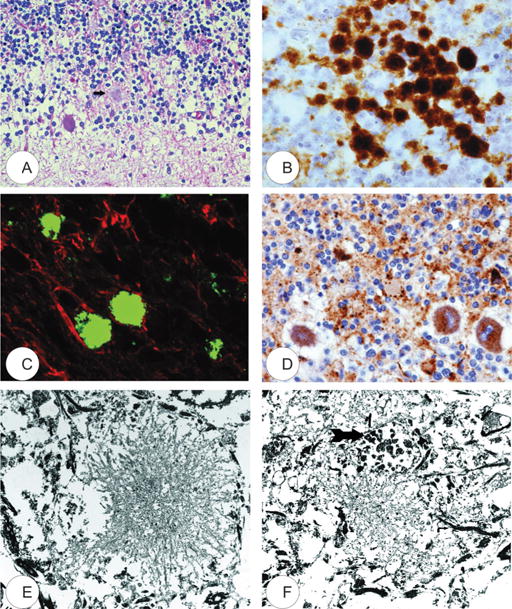
Kuru plaques. (A) Hematoxylin and eosin stain (arrow). (B) Transmissible spongiform encephalopathy–specific prion protein (PrPTSE) immunohistochemistry. (C) Confocal laser microscopy; anti-PrPTSE antibodies are labeled green; anti–glial fibrillary acidic protein is labeled red. (D) p62 accumulation within Purkinje cells and neurites. (E, F) Transmission electron microscopy. A dystrophic neurite (arrow) is present in (F). All figures are from case “K” (64).
TABLE.
Neuropathology of Kuru Cases Examined
| Case No. (Reference) |
Spinal Cord | Medulla | Thalamus, Hypothalonus | Striatum | Cerebellum | Cerebral Cortex |
|---|---|---|---|---|---|---|
| Case 18 | Anterior horn cell chromatolysis, microglial rosettes, astrocytosis in anterior and lateral horns | Interior and accessory olives: vacuoles, chromatolysis, spinal nucleus of the 5th nerve, lateral cuneate n.; med and lat. ventr. nuclei markedly affected; binucleated neurons. Reticular formation: chromatolysis, intensive astrocytosis of pons | Pronounced changes, astrocytosis | Vacuoles of neurons astrocytosis numerous plaques | Purkinje cell ballooning, torpedos, neuronophagia by microglial cells, gliosis more dense in vermis, kuru plaques | Only slight changes astrocytosis, kuru plaques; mostly in the insular cortex |
| “Y” 5, ♂ (66) Case 6 | Neurons generally well preserved; diffuse astrocytosis | Astrocytosis more prominent in the gray matter | Severe neuronal damage; intensive astrocytosis | Large neurons vacuolated | Purkinje cells, well preserved, some axonal swelling, no plaques | Neurons more affected in deeper layers; intense astrocytosis specially in parietal cortex and Ammon horn |
| “I” (66) 6 ♀ Case 7 | Minimal spongiform change | Inf olives shrunken, lateral arcuate and vestibular n., pale devoid of Nissl substance | Neurons hyperchromatic, shrunken, severe astrocytosis | No abnormalities, severe astrocytosis | Minimal changes, torpedos frequent, no plaques, astrocytosis | |
| “M” 6 ♀ (66) Case 16 | Appeared relatively spared | Astrocytosis | Neurons vacuolated Astrocytosis |
Some loss of Purkinje cells, torpedos, astrocytosis, numerous plaques | Widespread changes, foci of neuronal disappearance, pyramidal cells distorted, astrocytosis, microglial proliferation | |
| “W” 7 ♂ (66) Case 8 | Anterior horn cells frequently chromatolytic and vacuolated, diffuse astrocytosis | Nonspecific neural changes, diffuse astrocytosis | Neurons devoid of Nissl substance, intense gliosis, | Neurons vacuolated, intense gliosis | Mostly pale, Purkinje cells, intense astrocytosis, numerous plaques | Cortical and subcortical gliosis, rodlike microglial cells |
| “A” 7 ♂ (66) Case 3 | Neurons well preserved | Neurons well preserved | Neurons well preserved, astrocytosis | Neurons well preserved, astrocytosis | Purkinje cells devoid of the Nissl substance, no plaques, astrocytosis | Astrocytosis in deeper cortical layers, neurons well preserved |
| “N” 9 ♀ (66) Case 24 | Anterior horn cells vacuolated | Severe changes in olives. Nuclei: vestibular, spinal V, ambiquus raphe, lateral reticular, arcuate, more rarely affected; nXII dorsal, n. X facial abducens, no abnormalities | Severe changes extremely vacuolated, severe astrocytosis, many microglia | Large cells vacuolated | Purkinje cells well preserved, numerous torpedos, numerous plaques, intensive astrocytosis | Occasional neuronal degeneration, rodlike microgliosis |
| “A” 11 ♀ (66) Case 11 | Neurons well preserved but astrocytosis pronounced | Neurons well preserved, dense astrocytosis around olives, lateral arcuate n. | Well preserved slight increase of astrocytosis | Well preserved slight increase astrocytosis | Purkinje cells chromatolytic and vacuolated, numerous torpedos, intensive astrocytosis, microglial cells proliferation, no plaques | Chromatolytic neurons rodlike, microglial cells |
| “A” ♀ 13 (66) “E” | Spongiform change, small number of plaques | Spongiform change; astrocytosis | Very numerous plaques | Small number of plaques, no evident spongiform change | ||
| 14 ♀ (75) Case 10 | Anterior motor neuron vacuolation | Olivary neurons pale. Neurons of cuneate and vestibular n. chromatolytic and shrunken | Neurons well preserved, severe astrocytosis | Vacuolated neurons, severe astrocytosis | Purkinje cells—devoid of the Nissl substance, torpedos, severe astrocytosis and microglial proliferation; no plaques | Occasional neurons degenerated |
| ‘I’ 17, ♀ (66) “KA” | Astrocytosis in ventral horns | Astrocytosis | Focal astrocytosis | Torpedos. No plaques, degeneration of Purkinje cells | Spongiform change, plaques | |
| 23 ♀ (75) “M” 28 ♀ (75) Case 17 | Normal, except a few binucleated neurons, astrocytosis in white matter | Vacuolated neurons in various nuclei, gliosis in the white matter | Diffusely involved, astrocytosis | Diffusely involved, large cells vacuolated | Torpedos, plaques Purkinje cells vacuolated, numerous plaques |
Neurons showed widespread changes, astrocytosis, severe microgliosis |
| “I” 30, ♀ (66) KAKULAS 1 (71) | Few changes | Relatively few lesions | Severe changes | Loss of neurons neuronophagia and satellitosis, astrocytosis, microgliosis, plaques | Astrocytosis, microgliosis, torpedos, occasional plaques in molecular layer, typical plaques in the granular layer | Some swollen neurons, axonal swellings, astrocytosis and microgliosis, numerous plaques |
| ♀ 30 “N” | Relatively few changes | Neuronal loss, satellitosis, neuronophagia and astrocytosis | Neuronal loss, satellitosis, neuronophagia and astrocytosis | Moderate loss of Purkinje cells, numerous axonal swellings, torpedos, numerous plaques in the granular cell layer but occasional in the molecular layer | Occasional swollen neurons; rare axonal swelling; small number of plaques | |
| ♀ 30 (71) 42 ♂ (72) | Severe spongiform change, moderate astrocytosis | Frontal cortex: Moderate spongiform change, mild gliosis, no plaques; precentral gyrus, occasional plaques; occipital cortex, more plaques | ||||
| Case 2 | Anterior motor neurons vacuolated, astrocytosis | Slight changes, diffuse astrocytosis | Well preserved | Well preserved | Purkinje cells—reduction of the Nissl substance, numerous torpedos, numerous plaques | Intact |
| “Y” 45, ♀, US (66) Case 4 | Nonspecific change, astrogliosis | Nuclei of vestibular intercalatus, raphe, spinal V, lateral cuneate, pontobulbar arcuate—chromatolytic and vacuolated, microglial proliferation | Anterior and ventrolateral nuclei affected, astrocytosis, microglial proliferation, numerous plaques | Large cells severely vacuolated; satellitosis, astrocytosis, microgliosis, proliferation | Purkinje cells affected, intensive astrocytosis, microglial proliferation, numerous plaques | Occasional cells affected, numerous plaques |
| “Y” 50, ♀, US (66) KAKULAS 2 (71) | Few changes | Few changes | Severe changes | Loss of large cells, astrocytosis, microgliosis | Purkinje cell loses, torpedos, plaques | Pigmentary degeneration, satellitosis, astrocytosis, many kuru plaques |
| “K” ♀, 50 “K” | Remarkable little spongiform change | Moderate spongiform change, neuronal loss more severe than in striatum; severe astrocytosis; no plaques | Spongiform degeneration more intense and severe, widespread neuronal lost; very severe gliosis; small plaques | Spongiform change; Purkinje cell degeneration, torpedos; Plaques occasionally seen in the granular cell layer; dense astrocytosis | Frontal: spongiform change mild to moderate, occasional plaques, brisk astrocytosis | |
| ♂ 58 (79) “K” | Neuronal loss, satellitosis, neuronophagia and astrocytosis | Loss of both large and small cells and astrocytosis; a large plaque | Loss of Purkinje cells, torpedos, dendritic enlargements, astrogliosis and microgliosis, numerous plaques | Loss of neurons, pigmentary degeneration, satellitosis, plaques common, astrogliosis and microgliosis | ||
| ♀ Middle-aged (71) |
Neurons were shrunken and either hyperchromatic or pale, with dispersion of the Nissl substance or contained intracytoplasmic vacuoles similar to those that had been described in scrapie (66). Swollen neurons were occasionally described (67, 68). In the striatum, some neurons were vacuolated to such a degree that they looked “moth-eaten” (“a group of bubbles” [69]). Neuronophagia was observed. A few binucleated neurons were visible, and torpedo formation was noticed in the Purkinje cell layer, along with “empty baskets” that marked the sites of depopulated Purkinje cells. In the medulla, neurons of the vestibular nuclei and the lateral cuneatus were frequently affected. The spinal nucleus of the trigeminal nerve and nuclei of 6th, 7th, and motor nucleus of the 6th cranial nerves were affected less frequently, whereas nuclei of the 12th cranial nerve, the dorsal nucleus of 10th cranial nerve, and nucleus ambiguous were relatively spared. In the cerebral cortex, the deeper layers were affected more than the superficial layers and neurons in the hippocampal formation were normal. In the cerebellum, the paleocerebellar structures (vermis and flocculonodular lobe) were most severely affected; spinal cord pathology was most pronounced in the corticospinal and spinocerebellar tracts. Fowler et al (67) described severe demyelination in the lateral corticospinal tract and degeneration of dorsal and ventral spinocerebellar tracts. The posterior columns were normal. The anterior horn large neurons of both cervical and lumbosacral expansions were practically intact except for the occasional cell with chromatolysis, vacuolation, and swelling. Astroglial and microglial proliferation was widespread; microglial cells formed rosettes and appeared as rod or amoeboid types or as macrophages (gitter cells). Myelin degradation was observed in 10 of 12 cases. Interestingly, although vacuoles were noted by Klatzo, the significance of spongiform change was not appreciated (62), but “small spongy spaces” were noticed in 7 of 13 cases studied by Beck and Daniel (68). It must be stressed, however, that the spongiform change is poorly visible in thick celloidin blocks stained with Nissl stain, as opposed to paraffin blocks stained with hematoxylin and eosin.
The most striking neuropathologic feature of kuru is the presence of numerous amyloid plaques described as “spherical bodies with a rim of radiating filaments” and found in 6 of 12 cases studied by Klatzo et al (62) and in “about three quarters” of the 13 cases of Beck and Daniel (68); they then became known as “kuru plaques” (67, 69–71). Kuru plaques measured 20 to 60 Hm in diameter, were round or oval, and consisted of a dark-stained core, with delicate radiating periphery surrounded by a pale “halo.” Kuru plaques were most numerous in the granular cell layer of the cerebellum, basal ganglia, thalamus, and cerebral cortex in that order of frequency. Kuru plaques, as all amyloids, are metachromatic and stain with periodic acid-Schiff, Alcian blue, and Congo red, and a proportion are weakly argentophilic when impregnated according to Bielschowsky or von Braunmühl techniques. Klatzo et al (62) reported that plaques were most readily visualized by Holmes’ silver impregnation method. Of historical interest, another unique disease reported by Seitelberger as “a peculiar hereditary disease of the central nervous system in a family from lower Austria” (germ. Eigenartige familiar-hereditare Krankenheit des Zentralnervensystems in einer niederoosterreichen Sippe) (72) was mentioned by Neumann et al (73), who were thus the first authors to suggest a connection between kuru and GSS. Indeed, GSS was transmitted to nonhuman primates in 1981 (74).
In 1996, the appearance of a variant form of CJD characterized by numerous amyloid plaques, including “florid” or “daisy” plaques (a kuru plaque surrounded by a rim of spongiform vacuoles), provoked the question of whether variant CJD is a modern form of kuru (71). We (64) and others (75–77) studied kuru using modern PrP immunohistochemistry. We were privileged to examine a case of a young male kuru patient named “K” from the South Fore region whose brain tissue had transmitted the disease to chimpanzees, and McLean et al (76, 77) examined a series of 11 cases of kuru. In contrast to the classical studies described above, both articles stressed the presence of typical spongiform change present, as in sporadic CJD, in deep cortical layers (III–V) of the cingulate, entorhinal and insular cortices, and in the subiculum. The occipital cortex is variably affected (77). Spongiform change was also observed in the putamen and caudate, and some putaminal neurons contained intraneuronal vacuoles. Spongiform change was prominent in the molecular layer of the cerebellum, in periaqueductal gray matter, basal pontis, central tegmental area, and inferior olivary nucleus. The spinal cord showed only a minimal spongiform change.
Immunohistochemical studies revealed that PrPTSE was present not only as kuru plaques but also in synaptic and perineuronal sites (64) and in the spinal cord within substantia gelatinosa. The latter location was reminiscent of that typical of iatrogenic CJD cases after peripheral inoculation (78). Brandner et al (79) studied one very recent case of kuru and confirmed our findings.
In the frontal cortex, spongiform change was of moderate intensity, and glial fibrillary acidic protein (GFAP)–immunoreactive astrocytes were present. The PrPTSE immunohistochemistry revealed mostly synaptic deposits and plaques of varying size and shape. Larger confluent vacuoles were visible in the caudate nuclei. In the cerebellum, plaques were seen in granular cell layers but were not that numerous. The latter case has been neuropathologically compared with known subtypes of CJD and it seems the most similar to type 2 or 3 129 MV type of CJD of the Collinge et al (80) classification or type 2 CJD of the Parchi et al (22) classification.
In our previous study on amyloid plaques encountered in prion diseases (71), we demonstrated that kuru plaques morphologically and ultrastructurally resemble those that are characteristic of sporadic CJD subtype 3 according to the Parchi and Gambetti classification (22) but were remarkably different from florid plaques in variant CJD and multicentric plaques in GSS.
Moreover, in kuru, in contrast to plaques in variant CJD and GSS, we observed GFAP-positive astrocytic cell processes around plaques and at the peripheral part of plaques. This phenomenon was seen, albeit to a slightly lesser degree, in sporadic CJD. Immunohistochemistry for HLA-DR revealed the presence of positive cells around kuru plaques. In kuru and sporadic CJD, we also observed some distended β-amyloid precursor protein–positive processes, but they were dispersed throughout the cortex and not around the plaques. This finding is consistent with the presence of dystrophic neurites at the periphery of kuru plaques in kuru and sporadic CJD, as seen by electron microscopy. An interesting observation was made by immunohistochemistry for hyperphosphorylated microtubule-associated protein (MAP)-tau using AT8 antibody. In kuru and sporadic CJD, minimal immunoreactivity of MAP-tau was observed at the periphery of the plaques, that is, again, the pattern of MAP-tau immunoreactivity was different from that of variant CJD and GSS, in which the expression of this protein was much more robust. Immunochemistry using antibodies against phosphorylated epitopes of neurofilaments showed loss of axons in the cerebellum that was more severe than in other prion diseases. Confocal laser microscopy confirmed the concentration of GFAP-positive astrocytic processes at the plaque periphery. There was no colocalization of MAP-tau and PrPSc. We observed no colocalization of PrPd and β-amyloid precursor protein either inside the plaques or in the neuropil.
Our immunohistochemistry confirmed the presence of microglial cells, astrocytic processes, and dystrophic neurites at the periphery of kuru plaques and a striking resemblance of the amyloid plaques in kuru with those in the plaque subtype of sporadic CJD.
Electron Microscopy
The amount of ultrastructural data on kuru-affected brains is extremely limited probably because of severe constraints on obtaining well-fixed material. Field et al (81) used formalin-fixed brains of 5 kuru cases to demonstrate degeneration of Purkinje cells, torpedo formation, and proliferation of Bergmann glia. They described a typical fine structure of amyloid plaques as well as numerous Hirano bodies. Peat and Field (82) reported on “cytoplasmic lamellar bodies,” which subsequently were shown to be perfectly normal cell constituents (83). Lampert et al (84) used kuru-infected chimpanzee at the level of the first and the second passages. They mentioned spongiform change and astrocytic gliosis by light microscopy as well as central chromatolysis, lipid-laden macrophages, and occasionally, lymphocytic cuffs. Electron microscopy provides the background of the spongiform change in a form of membrane-bound “empty” spaces. Membranes were single or, as in autophagy vacuoles, duplicated. Lampert, who had already published an article of degenerating neurites (85), also mentioned these structures that are now considered to be related to autophagy (86). Thus, Lampert’s work preceded the current view that autophagy is one of the most important phenomena observed in all prion diseases (87, 88). The hallmark of TSE is the vacuole, which is a membrane-bound intracellular electron-lucent space. The histogenesis of vacuoles is not well understood, and most of the ultrastructural studies have an inability to detect subcellular organelles from which vacuoles originate. We suggested that vacuoles are formed relatively abruptly with no detectable transitional stages (Gibson and Liberski, unpublished data). It is tempting to speculate that vacuolation in TSEs is somehow related to type III programmed cell death characterized by the presence of large membrane-bound intracellular empty spaces without the participation of lysosomes. It is entirely plausible that the autophagic process leading to cell death through the expansion of the autophagic vacuole(s) left behind “spongiform” vacuoles as remnants of neuronal processes. Lampert et al suggested that the primary target is the neuron, whereas astrocytic proliferation is merely a reactive phenomenon. It is of utmost interest that as early as 1969, Lampert et al (having cited an earlier article by Arstila et al [88]) suggested that the spongiform change is somehow related to autophagy (84). Those pioneer data were confirmed by Beck et al (89), who also reported membrane lamination observed before full-blown pathology could be appreciated. From examination of the chimpanzee infected with the “K” kuru brain, the typical autophagic vacuoles were illustrated, but the process was not mentioned.
To elucidate the structure of amyloid plaques, we reversed the material from the “K” case for electron microscopy (64). The fine structure of plaques was surprisingly well preserved, and it corresponds to that described by light microscopy (71). Occasionally, dystrophic neurites were seen in the vicinity.
CONCLUSIONS
Kuru was the first slow virus disease of humans discovered in a remote tribe of Papua New Guinea. Through the insight, endurance, persistence, and genius of D. Carleton Gajdusek and subsequent collaborators and followers, the new type of human infectious pathogen, called “a slow virus,” was discovered. With the passage of time, the slow virus changed clothes to become a “prion.” That discovery opened a new field of neurodegenerations viewed as protein conformational disorders. Also, the discovery of transmissibility of kuru and subsequent transmission of CJD would set the stage for the timely discovery of variant CJD almost 50 years later, without which its cause might have remained a mystery for a very long time.
FIGURE 3.
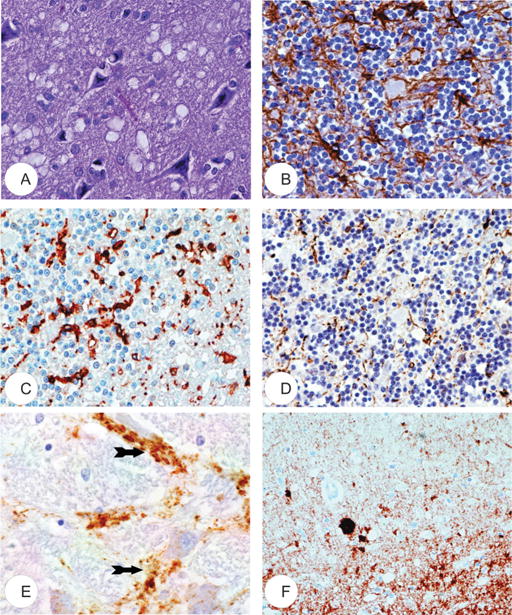
Neuropathology of kuru. (A) Typical spongiform change. Hematoxylin and eosin stain. (B) Immunohistochemistry for glial fibrillary acidic protein demonstrates astrocyte proliferation. (C) Microglial cells are detected by anti-HLA-DR immunohistochemistry. (D) Numerous enlarged neurites detected using anti-neurofilament protein antibody. (E) Perineuronal accumulation (arrows) of transmissible spongiform encephalopathy–specific prion protein PrPTSE. (F) Synaptic accumulation of PrPTSE; an amyloid plaque (arrow) is also visible. (A, E) are from the cerebral cortex; (B–D, F) are from the cerebellum. All are from case “K” (64).
Acknowledgments
Pawel Liberski, Beata Sikorska, and Johannes Hainfellner are supported by the Austrian Agency for International Cooperation in Education and Research (OeAD).
References
- 1.Gajdusek DC, Gibbs CJ, Alpers MP. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature. 1966;209:794–96. doi: 10.1038/209794a0. [DOI] [PubMed] [Google Scholar]
- 2.Gibbs CJ, Jr, Gajdusek DC, Asher DM, et al. Creutzfeldt-Jakob disease (spongiform encephalopathy): Transmission to chimpanzee. Science. 1968;161:388–89. doi: 10.1126/science.161.3839.388. [DOI] [PubMed] [Google Scholar]
- 3.Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197:943–60. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB. Prions. In: Jörnvall H, editor. Nobel Lectures, Physiology or Medicine 1996–2000. Singapore: World Scientific Publishing Co; 2003. [Google Scholar]
- 5.Wüthrich K. NMR studies of structure and function of biological macromolecules. In: Frängsmyr T, editor. Les Prix Nobel The Nobel Prizes 2002. Stockholm, Sweden: Nobel Foundation; 2003. [Google Scholar]
- 6.Liberski PP, Brown P. Disease-specific particles without prion protein in prion diseases—Phenomenon or epiphenomenon? Neuropathol Appl Neurobiol. 2007;33:395–97. doi: 10.1111/j.1365-2990.2007.00867.x. [DOI] [PubMed] [Google Scholar]
- 7.Gajdusek CD. Observation on the early history of kuru investigation. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Disease of the Nervous System. Vol. 1. New York, NY: Academic Press; 1979. pp. 7–35. [Google Scholar]
- 8.Gajdusek DC, editor. Correspondence on the Discovery and Original Investigations on Kuru Smadel–Gajdusek Correspondence 1955–1958. Bethesda, MD: US Department of Health, Education, and Welfare; Public Health Service, National Institutes of Health; 1976. 2nd printing. [Google Scholar]
- 9.Gibbs CJ., Jr . Spongiform encephalopathies—Slow, latent, and temperate virus infections—In retrospect. In: Prusiner SB, Collinge J, Powell J, Anderton B, editors. Prion diseases of human and animals. London, UK: Ellis Horwood Limited; 1992. pp. 53–62. [Google Scholar]
- 10.Hadlow WJ. Scrapie and kuru. Lancet. 1959;2:289–90. [Google Scholar]
- 11.Hadlow WJ. The scrapie-kuru connection: Recollection of how it came about. In: Prusiner SB, Collinge J, Powell J, Anderton B, editors. Prion Diseases of Human and Animals. London, UK: Ellis Horwood Ltd.; 1992. pp. 40–46. [Google Scholar]
- 12.Gibbs CJ, Jr, Gajdusek DC. Attempts to demonstrate a transmissible agent in kuru, amyotrophic lateral sclerosis, and other subacute and chronic progressive nervous system degenerations of man. In: Gajdusek DC, Gibbs CJ Jr, Alpers M, editors. Slow, Latent, and Temperate Virus Infections. Washington, DC: US Department of Health, Education, and Welfare; 1965. pp. 39–48. (NINDB Monograph 2). [Google Scholar]
- 13.Gajdusek DC, Gibbs CJ., Jr Transmission of kuru from man to Rhesus monkeys (Macaca mulatta) 81/2 years after inoculation. Nature. 1972;240:351. doi: 10.1038/240351a0. [DOI] [PubMed] [Google Scholar]
- 14.Peterson DA, Wolfe LG, Deinhardt F, et al. Transmission of kuru and Creutzfeldt-Jakob disease to Marmoset monkeys. Intervirology. 1973;2:14–19. doi: 10.1159/000149399. [DOI] [PubMed] [Google Scholar]
- 15.Masters CL, Alpers MP, Gajdusek DC, et al. Experimental kuru in the gibbon and Sooty mangabey and Creutzfeldt-Jakob disease in the pigtailed macaque. J Med Primatol. 1976;5:205–9. doi: 10.1159/000459951. [DOI] [PubMed] [Google Scholar]
- 16.Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, et al. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–29. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 17.Diener TO. Viroids and Viroid Diseases. New York, NY: Wiley & Sons; 1979. [Google Scholar]
- 18.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–44. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 19.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 20.Diringer H, Gelderblom H, Hilmert H, et al. Scrapie infectivity, fibrils and low-molecular- weight protein. Nature. 1983;306:476–78. doi: 10.1038/306476a0. [DOI] [PubMed] [Google Scholar]
- 21.Wadsworth JD, Joiner S, Linehan JM, et al. Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc Natl Acad Sci USA. 2008;105:3885–90. doi: 10.1073/pnas.0800190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–78. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 23.Goldfarb LG, Petersen RB, Tabaton M, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–8. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 24.Cervenakova L, Goldfarb L, Garruto R, et al. Phenotype-genotype studies in kuru: Implications for new variant Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 1998;95:13239–41. doi: 10.1073/pnas.95.22.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer MS, Dryden AJ, Hughes JT, et al. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–42. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 26.Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991;337:1441–42. doi: 10.1016/0140-6736(91)93128-v. [DOI] [PubMed] [Google Scholar]
- 27.Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea. The endemic occurrence of “kuru” in the native population. N Engl J Med. 1957;257:974–78. doi: 10.1056/NEJM195711142572005. [DOI] [PubMed] [Google Scholar]
- 28.Bennett JH, Rhodes FA, Robson HN. Observations on kuru. I. A possible genetic basis Australas. Ann Med. 1958;7:269–75. doi: 10.1111/imj.1958.7.4.269. [DOI] [PubMed] [Google Scholar]
- 29.Bennett JH, Rhodes FA, Robson HN. A possible genetic basis for kuru. Am J Hum Genet. 1959;11:169–87. [PMC free article] [PubMed] [Google Scholar]
- 30.Gajdusek DC, Alpers MP. Genetic studies in relation to kuru. I. Cultural, historical, and demographic background. Am J Hum Gen. 1972;24:1–38. [PMC free article] [PubMed] [Google Scholar]
- 31.Kofler A, Braun A, Jenkins T, et al. Characterization of mutants of the vitamin-D–binding protein/group specific component: GC aborigine (1A1) from Australian aborigines and South African blacks, and 2A9 from south Germany. Vox Sang. 1995;68:50–54. doi: 10.1111/j.1423-0410.1995.tb02545.x. [DOI] [PubMed] [Google Scholar]
- 32.Kitchin D, Bearn AG, Alpers MP, et al. Genetic studies in relation to kuru. III. Distribution of the inherited serum group-specific protein (Gc) phenotypes in New Guineans: An association of kuru and the Gc Ab phenotype. Am J Hum Gen. 1972;24:72–85. [PMC free article] [PubMed] [Google Scholar]
- 33.Wiesenfeld SL, Gajdusek DC. Genetic studies in relation to kuru. VI. Evaluation of increased liability to kuru in Gc, Ab-Ab individuals. Am J Hum Gen. 1975;27:498–504. [PMC free article] [PubMed] [Google Scholar]
- 34.Goldfarb LG, Brown P, Goldgaber D, et al. Patients with Creutzfeldt-Jakob disease and kuru lack the mutation in the PRIP gene found in Gersrmann-Sträussler syndrome, but they show a different double-allele mutation in the same gene. Am J Hum Genet. 1989;45(Suppl):A189. [Google Scholar]
- 35.Goldfarb LG, Brown P, Gajdusek DC. The molecular genetics of human transmissible spongiform encephalopathy. In: Prusiner SB, Collinge J, Powell J, Anderton B, editors. Prion Diseases of Humans and Animals. Chichester, UK: Ellis Horwood; 1992. pp. 139–53. [Google Scholar]
- 36.Goldfarb LG, Cervenakova L, Gajdusek DC. Genetic studies in relation to kuru: An overview. Curr Mol Med. 2004;4:375–84. doi: 10.2174/1566524043360627. [DOI] [PubMed] [Google Scholar]
- 37.Lee H-S, Brown P, Cervenakova L, et al. Increased susceptibility to kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis. 2001;183:192–96. doi: 10.1086/317935. [DOI] [PubMed] [Google Scholar]
- 38.Mead S, Whitfield J, Poulter M, et al. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med. 2009;361:2056–65. doi: 10.1056/NEJMoa0809716. [DOI] [PubMed] [Google Scholar]
- 39.Collinge CJ, Whitfield J, McKintosh E, et al. Kuru in the 21st century—An acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. doi: 10.1016/S0140-6736(06)68930-7. [DOI] [PubMed] [Google Scholar]
- 40.Mead S, Stumpf MP, Whitfield J, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300:640–43. doi: 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- 41.Hardy J, Scholz S, Evans W, et al. Prion genotypes in Central America suggest selection for the V129 allele. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:33–35. doi: 10.1002/ajmg.b.30248. [DOI] [PubMed] [Google Scholar]
- 42.Lindenbaum S. Kuru Sorcery. Disease and Danger in the New Guinea Highlands. Palo Alto, CA: Mayfield Publishing Company; 1979. [Google Scholar]
- 43.Glasse RM. The Spread of Kuru Among the Fore: A Preliminary Report. Bethesda, MD: Department of Public Health, Territory PNG, National Institutes of Health; 1962. p. 18. [Google Scholar]
- 44.Mathews JD. PhD thesis. University of Melbourne; Melbourne, Victoria, Australia: 1971. Kuru: A Puzzle in Culture and Environmental Medicine. [Google Scholar]
- 45.Whitfield JT. PhD dissertation. University College; London, UK: 2010. Mortuary Practices, Genetics and Other Factors Relevant to the Transmission of Kuru in Papua New Guinea. [Google Scholar]
- 46.Glasse RM, Lindenbaum S. Kuru at Wanitabe. In: Hornabrook RW, editor. Essays on Kuru. Berks. E. W. Classey; Faringdon, Oxon, UK: 1976. pp. 38–52. [Google Scholar]
- 47.Mathews JD, Glasse RM, Lindenbaum S. Kuru and cannibalism. Lancet. 1968;2:449–52. doi: 10.1016/s0140-6736(68)90482-0. [DOI] [PubMed] [Google Scholar]
- 48.Alpers MP. Kuru: Implications of its transmissibility for the interpretation of its changing epidemiologic pattern. In: Bailey OT, Smith DE, editors. The Central Nervous System, Some Experimental Models of Neurological Diseases. Baltimore, MD: Lippincott, Williams & Wilkins; 1968. pp. 234–51. [Google Scholar]
- 49.Alpers MP, Rail L. Kuru and Creutzfeldt-Jakob disease: Clinical and etiological aspects. Proceedings of the Australian Association of Neurologists. 1971;8:7–15. [PubMed] [Google Scholar]
- 50.Glasse RM. Cannibalism in the kuru region. Bethesda, MD: US Department of Health, Education, and Welfare; Public Health Service, National Institutes of Health; 1962. p. 14. [Google Scholar]
- 51.Lindenbaum S. Cannibalism, kuru and anthropology. Folia Neuropathol. 2009;47:138–44. [PubMed] [Google Scholar]
- 52.Whitfield JP, Pako WH, Collinge J, et al. Mortuary rites of the South Fore and kuru. Phil Trans R Soc B. 2008;363:3721–24. doi: 10.1098/rstb.2008.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arens W. The Man-Eating Myth. New York, NY: Oxford University Press; 1979. [Google Scholar]
- 54.Knauft BM. Good Company and Violence. Sorcery and Social Action in a Lowland New Guinea Society. Berkeley, CA: University of California Press; 1985. [Google Scholar]
- 55.Gillison G. A New Guinea Highlands Mythology. Chicago, IL: Chicago University Press; 1993. Between Culture and Fantasy. [Google Scholar]
- 56.Mead S, Whitfield J, Poulter M, et al. Genetic susceptibility, evolution and the kuru epidemic. Phil Trans R Soc B. 2008;363:741–46. doi: 10.1098/rstb.2008.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alpers MP. Kuru: Age and Duration Studies. Vol. 12 Adelaide Department of Medicine; 1964. [Google Scholar]
- 58.Beasley A. Fore experiences on the kuru patrols. Oceania. 2009;79:34–52. [Google Scholar]
- 59.Collinge CJ, Whitfield J, McKintosch E, et al. A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Phil Trans R Soc B. 2008;363:3725–39. doi: 10.1098/rstb.2008.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collinge J. Lessons of kuru research: Background to recent studies with some personal reflections. Phil Trans R Soc B. 2008;363:3689–96. doi: 10.1098/rstb.2008.0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prusiner SB, Gajdusek DC, Alpers MP. Kuru with incubation periods exceeding two decades. Ann Neurol. 1982;12:1–9. doi: 10.1002/ana.410120102. [DOI] [PubMed] [Google Scholar]
- 62.Klatzo I, Gajdusek DC. Pathology of kuru. Lab Invest. 1959;8:799–847. [PubMed] [Google Scholar]
- 63.Alpers MP. Kuru: A Clinical Study. Washington, DC: US Department of Health, Education, and Welfare; 1964. pp. 1–38. [Google Scholar]
- 64.Hainfellner J, Liberski PP, Guiroy DC, et al. Pathology and immunohistochemistry of a kuru brain. Brain Pathol. 1997;7:547–54. doi: 10.1111/j.1750-3639.1997.tb01072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klatzo I. The Life Story of a Survivor. Cologne, Germany: Max Planck Institute for Neuronal Research; 2008. [Google Scholar]
- 66.Innes JRM, Saunders LZ. Comparative Neuropathology. New York, NY: Academic Press; 1962. p. 839. [Google Scholar]
- 67.Fowler M, Robertson EG. Observations on kuru. III. Pathological features in five cases. Australas Ann Med. 1959;8:16–26. doi: 10.1111/imj.1959.8.1.16. [DOI] [PubMed] [Google Scholar]
- 68.Beck E, Daniel PM. Kuru and scrapie compared: Are they examples of system degeneration? In: Gajdusek DC, Gibbs CJ Jr, Alpers MP, editors. Slow, Latent, and Temperate Virus Infections. Washington, DC: US Department of Health, Education, and Welfare; 1965. pp. 85–93. [Google Scholar]
- 69.Kakulas BA, Lecours A-R, Gajdusek DC. Further observations on the pathology of kuru. J Neuropathol Exp Neurol. 1967;26:85–97. doi: 10.1097/00005072-196701000-00007. [DOI] [PubMed] [Google Scholar]
- 70.Scrimgeour EM, Masters CL, Alpers MP, et al. A clinicopathological study of case of kuru. J Neurol Sci. 1983;59:265–75. doi: 10.1016/0022-510x(83)90044-8. [DOI] [PubMed] [Google Scholar]
- 71.Sikorska B, Liberski PP, Sobów T, et al. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: A comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease. Neuropathol Appl Neurobiol. 2009;35:46–59. doi: 10.1111/j.1365-2990.2008.00959.x. [DOI] [PubMed] [Google Scholar]
- 72.Seitelberger F. Eigenartige familiar-hereditare Krankheit des Zetralnervensystems in einer niederosterreichischen Sippe. Wien Klein Wochen. 1962;74:687–91. [Google Scholar]
- 73.Neumann MA, Gajdusek DC, Zigas V. Neuropathologic findings in exotic neurologic disorder among natives of the Highlands of New Guinea. J Neuropathol Exp Neurol. 1964;23:486–507. doi: 10.1097/00005072-196407000-00007. [DOI] [PubMed] [Google Scholar]
- 74.Masters CL, Gajdusek DC, Gibbs CJ., Jr Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Sträussler syndrome. With an analysis of the various forms of amyloid plaque deposition in the virus induced spongiform encephalopathies. Brain. 1981;104:559–88. doi: 10.1093/brain/104.3.559. [DOI] [PubMed] [Google Scholar]
- 75.Lantos B, Bhata K, Doey LJ, et al. Is the neuropathology of new variant Creutzfeldt-Jakob disease and kuru similar? Lancet. 1997;350:187–88. doi: 10.1016/s0140-6736(05)62355-0. [DOI] [PubMed] [Google Scholar]
- 76.McLean CA, Ironside JW, Alpers MP, et al. Comparative neuropathology of kuru with the new variant of Creutzfeldt-Jakob disease: Evidence for strain of agent predominating over genotype of host. Brain Pathol. 1998;8:428–37. doi: 10.1111/j.1750-3639.1998.tb00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McLean CA. The neuropathology of kuru and variant Creutzfeldt-Jakob disease. Phil Trans R Soc. 2008;363:3685–87. doi: 10.1098/rstb.2008.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goodbrand IA, Ironside JW, Nicolson D, et al. Prion protein accumulations in the spinal cords of patients with sporadic and growth hormone-associated Creutzfeldt-Jakob disease. Neurosci Lett. 1995;183:127–30. doi: 10.1016/0304-3940(94)11131-2. [DOI] [PubMed] [Google Scholar]
- 79.Brandner S, Whitfield J, Boone K, et al. Central and peripheral pathology of kuru: Pathological analysis of a recent case and comparison with other forms of human prion diseases. Phil Trans R Soc B. 2008;363:3755–63. doi: 10.1098/rstb.2008.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Collinge J, Sidle KCL, Meads J, et al. Molecular analysis of prion strain variation and the etiology of new variant CJD. Nature. 1996;383:685–70. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 81.Field EJ, Mathews JD, Raine CS. Electron microscopic observations on the cerebellar cortex in kuru. J Neurol Sci. 1969;8:209–24. doi: 10.1016/0022-510x(69)90111-7. [DOI] [PubMed] [Google Scholar]
- 82.Peat A, Field EJ. An unusual structure in kuru brain. Acta Neuropathol (Berl) 1970;15:288–92. doi: 10.1007/BF00686775. [DOI] [PubMed] [Google Scholar]
- 83.Liberski PP, Gibson PH. Cerebellar lamellar bodies in two strains of murine scrapie. J Comp Pathol. 1987;97:491–93. doi: 10.1016/0021-9975(87)90028-4. [DOI] [PubMed] [Google Scholar]
- 84.Lampert PW, Earle KM, Gibbs CJ, Jr, et al. Experimental kuru encephalopathy in chimpanzees and spider monkey. J Neuropathol Exp Neurol. 1969;28:353–70. doi: 10.1097/00005072-196907000-00001. [DOI] [PubMed] [Google Scholar]
- 85.Lampert PW. A comparative electron microscopic study of reactive, degenerating regenerating, and dystrophic axons. J Neuropathol Exp Neurol. 1967;26:345–68. doi: 10.1097/00005072-196707000-00001. [DOI] [PubMed] [Google Scholar]
- 86.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: An immunoelectron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 87.Liberski PP, Sikorska B, Bratosiewicz-Wasik J, et al. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: From apoptosis to autophagy. Int J Biochem Cell Biol. 2004;36:2473–90. doi: 10.1016/j.biocel.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 88.Arstila AU, Trump BF. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am J Pathol. 1968;53:687–733. [PMC free article] [PubMed] [Google Scholar]
- 89.Beck E, Bak IJ, Christ JF, et al. Experimental kuru in the Spider monkey: Histopathological and ultrastructural studies of the brain during early stages of incubation. Brain. 1975;98:595–612. doi: 10.1093/brain/98.4.595. [DOI] [PubMed] [Google Scholar]