

Abstract
Amidst the proteomes of human tissues lie subsets of proteins that are closely involved in conserved pathophysiological processes. Much of biomedical research concerns interrogating disease signature proteins and defining their roles in disease mechanisms. With advances in proteomics technologies, it is now feasible to develop targeted proteomics assays that can accurately quantify protein abundance as well as their post-translational modifications; however, with rapidly accumulating number of studies implicating proteins in diseases, current resources are insufficient to target every protein without judiciously prioritizing the proteins with high significance and impact for assay development. We describe here a data science method to prioritize and expedite assay development on highimpact proteins across research fields by leveraging the biomedical literature record to rank and normalize proteins that are popularly and preferentially published by biomedical researchers. We demonstrate this method by finding priority proteins across six major physiological systems (cardiovascular, cerebral, hepatic, renal, pulmonary, and intestinal). The described method is data-driven and builds upon the collective knowledge of previous publications referenced on PubMed to lend objectivity to target selection. The method and resulting popular protein lists may also be useful for exploring biological processes associated with various physiological systems and research topics, in addition to benefiting ongoing efforts to facilitate the broad translation of proteomics technologies.
Keywords: data science, bibliometrics, semantics, proteomics translation, common proteins, human tissue convergence, targeted proteomics
Graphical abstract
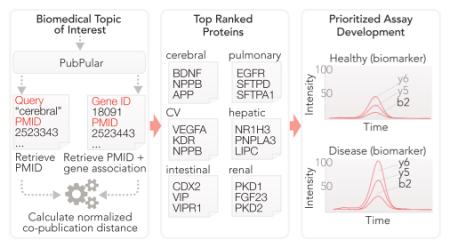
INTRODUCTION
The human proteome comprises interweaved dynamic networks whose differential regulation provides important insights into cellular physiology and disease mechanisms. Targeted proteomics approaches such as multiple-reaction monitoring mass spectrometry (MRM-MS) can now reliably analyze proteins with sensitivity and specificity that rival immunobiological approaches without being restricted by the availability of antibody pairs.1–3 MRM assays can be viewed as reagents, similar to ELISA or Western blots, that once developed can be used broadly by the scientific community. Nevertheless, the adoption of targeted proteomics remains unsatisfactory, and verified assays are available only for few potential disease proteins and biomarker candidates (see PeptideAtlas/PASSEL for list).4 For instance, it has been estimated that the human plasma proteome contains about 4000 canonical proteins based on the latest Human Plasma PeptideAtlas and Guidelines requiring two uniquely mapped peptides of at least nine amino acids in length with excellent spectra. The majority of these proteins currently cannot be readily targeted with MRM-MS experiments to support biomarker discovery. The Plasma Proteome Database catalogues only 279 proteins (~7% of the proteome) with developed MRM-MS assays.5,6 A bottleneck in proteomics translation is thought to be the time- and labor-intensive nature of method development, a task typically undertaken by relatively few specialized laboratories. The success of technology dissemination therefore hinges upon effectively focusing limited resources on select proteins that have high likelihood of having biomedical values or are thus aligned with the interests of biomedical research fields.
Identifying the quintessential proteins can facilitate judicious investments of resources and broader applications of proteomics to advance biomedical research. Such efforts may also benefit gene annotation, database curation, and other fields where resources are limited. Nevertheless, which proteins may constitute good candidate targets is often unclear, and thus far few systematic methods have been demonstrated to objectively identify them. This challenge has spurred recent works from the Biology/Disease Human Proteome Project (B/D-HPP) in the Human Proteome Organization (HUPO). The core mission of the B/D-HPP is to promote broad application of proteomics to researchers looking to understand the molecular mechanisms of human disease.7 Comprising the B/D-HPP are individual initiatives including the Cardiovascular Initiative,8 the Eye Proteome Project,9 and the Diabetes Proteome Project,2 each of which is tasked to promote proteomics adoption in a specific discipline.7 The B/D-HPP initiatives have developed several approaches to vectorize quantitative assay development, among which is to catalog significantly altered proteins from seminal proteomics data sets to identify “priority proteins” that merit follow-up validations by virtue of their disease implications.10 The B/D-HPP maintains one such collection of these priority protein lists on the PeptideAtlas Web site (https://db. systemsbiology.net/sbeams/cgi/PeptideAtlas/proteinList), recommending that these proteins be considered for precedence in method development. Other strategies have also been proposed, such as to aggregate multiple data sets to infer network hub proteins, which because of their high connectivity to other proteins may likelier represent bona fide disease proteins.11,12
Although these approaches can reveal interesting protein candidates, they may be limited by the coverage of the proteomics data sets selected and the models on which the experiments were performed. We hereby suggest a complementary data science-based methodology to identify lists of “popular proteins” by taking in the totality of publications referenced on PubMed. Our method is applicable to diverse fields of inquiry (e.g., cardiovascular, hepatic, cancer, etc.) regardless of their experimental approach. A distinguishing feature is that no subjective classification or inference on functional significance is performed at the step of target prioritization. Instead, the collective intelligence of the scientific community is crowdsourced to identify which proteins emerge as biologically significant by virtue of their selective research interest as a whole. The approach is based on the hypothesis that individual biomedical researchers will collectively make rational decisions to preferentially pursue studies on proteins or pathways that are deemed to be biologically significant, based on either their expert opinions or other sources of data. The overall research popularity of a protein therefore provides essential information on its significance in a particular biological system or concept.
EXPERIMENTAL PROCEDURES
To estimate protein popularity within various fields, we interrogated tissue-specific publications from the >24 million literature records on PubMed using specific search terms (without restriction to the publication date) on the PubMed Web site between May and July of 2015. For instance, publications related to the cardiovascular system were queried with the search terms (“heart” or “cardiac” or “cardiovascular”), which was automatically resolved by PubMed to include any identified MeSH terms and synonyms such that the query reads (“heart”[MeSH Terms] OR “heart”[All Fields]) OR (“heart”[-MeSH Terms] OR “heart”[All Fields] OR “cardiac”[All Fields]) OR (“blood vessels”[MeSH Terms] OR (“blood”[All Fields] AND “vessels”[All Fields]) OR “blood vessels”[All Fields] OR “vascular”[All Fields]) OR (“cardiovascular system”[MeSH Terms] OR (“cardiovascular”[All Fields] AND “system”[All Fields]) OR “cardiovascular system”[All Fields] OR “cardiovascular”[All Fields])). Cerebral system-related publications were similarly queried, using the search term (“brain” or “cerebral”), pulmonary system with (“lung” or “lungs” or “pulmonary”), hepatic system with (“liver” or “hepatic”), renal system with (“kidney” or “kidneys” or “renal”), intestinal system with (“gut” or “intestine” or “intestinal”); the queries were similarly resolved by the PubMed query system to automatically include MeSH term definitions.
We use the Gene2PubMed file to match specific PubMed queries to GeneIDs as previously described13 and count the number of publication records (PMIDs) matching to a unique, species-specific GeneID within a queried topic (e.g., all returned publications from “brain or cerebral”)14 (Figure 1A). The Gene2PubMed file was retrieved on the NCBI FTP server at the time of analysis (June 17, 2015 release), which contained 8 022 914 gene-publication references and was manually curated by the National Library of Medicine (NLM) based on PubMed records as well as information from user submission. To determine the relevance between protein and topic, we define the normalized copublication distance (NCD) of a protein P with a particular queried topic T as
Figure 1.

Topic-specific publication counts in six major organ systems. (A) Computational workflow to automatically derive the number of publications referenced to proteins in our custom PubMed queries. System-relevant publications are retrieved from PubMed with specific search terms (List 1). A cross-reference between PMIDs and GeneIDs is retrieved from the NCBI FTP Web site (List 2). A custom software tool suite matches List 2 to List 1. The software counts the unique occurrences of each protein in each year of a user-specified species and converts GeneID to UniProt/SwissProt accessions. Lastly, the software computes the normalized copublication distance (NCD) between a protein with the queried topic. (B) Summary statistics of mouse (orange) and human (blue) proteins referenced to publications in each system. Left: the total number referenced publications. Right: the total number of distinct proteins with at least five publications for each system. (C) The number of topic-specific publications per protein resembles a logarithm–logarithm relationship with regard to protein rank. The number of referenced article decreases sharply after the top 50 proteins in the queried tissues, with the next 50 proteins accounting for approximately one-third of publications as the first 50.
where T is the set of publications that are linked to any protein within a particular taxonomy and that are retrieved from a queried topic; P is the set of publications linked to a particular protein in all studies; F is the set of all publications linked to any proteins in any topics, containing all PMIDs in the Gene2PubMed file within a particular taxonomy where T ⊆ F and P ⊆ F; and T ∩ P is the set of publications linked to a particular protein within a queried topic. From each query, the software outputs the NCBI GeneIDs that have corresponding UniProt accessions (thus excluding noncoding genes), along with their associated publication frequency and NCD. This method is extended to define the pairwise NCD between two proteins within a topic query such that
For postanalyses, we retrieve Gene Ontology terms associated with the analyzed proteins via the European Bioinformatics Institute QuickGO API. Additional annotations were acquired from Ensembl using the R/Bioconductor package biomaRt. Significant enrichment of annotations in a protein list over the background was calculated with the hypergeometric test, with adjustment of false discovery rate using the Benjamini–Hochberg method. Proteins analyzed are species-specific, identified using their species-specific Entrez GeneIDs or UniProt accessions. Where proteins are compared between species, orthology between mouse and human is defined according to NCBI Homologene.17
A web version of the software tool to retrieve publication count and calculate corresponding NCDs is available at https://heart.shinyapps.io/PubPular/ for free academic and nonprofit use. Documentations are available at the same location.
RESULTS
Our overarching goal is to systematically establish lists of highly published proteins pertaining to individual biomedical disciplines, for which expedited quantitative assay and other focused resource developments would create immediate beneficial impact to large numbers of researchers. We recently demonstrated that the PubMed literature record and the Gene2PubMed file may be used to estimate the popularity of a protein in the heart13 and in the eye.9 Here we greatly expanded on this approach to the research focus in six major physiological systems—cerebral, cardiovascular, intestinal, hepatic, renal, and pulmonary (Figure 1A). We queried >24 million PubMed records for articles related to each system going back to 1966.
On average, each query returns ~0.6–1.6 million articles, out of which ~10 000 to ~40 000 unique articles are referenced to at least one protein (Figure 1B). The six queried systems are on average associated with a total of ~10 000 proteins (min: 4836; max: 16570), with an average of ~1900 (min: 768; max: 3774) proteins being referenced to five or more publications. The protein counts are in the decreasing order of cerebral > cardiovascular > hepatic > renal > pulmonary > intestinal (Figure 1B). We observe that the top 50 proteins (ranked according to publication number) in each system disproportionally account for ~17% of all referenced publications in each system. Publication counts decrease sharply thereafter, with the next 50 proteins (rank 51–100) accounting for approximately only one-third as many publications as the top 50 proteins (~5% of total publications) (Figure 1C). The total number of studies linked to mouse proteins is comparable to that of human, with mouse proteins having slightly fewer (~28%) publications.
We observe that a small number of proteins are broadly studied across multiple fields, as reflected by the high multiplicity of their occurrences in the top-50 list in the six systems (Figure 2A). Notable examples include cellular tumor antigen p53 (TP53), vascular endothelial growth factor A (VEGFA), interleukin 6 (IL6), and tumor necrosis factor (TNF). We further observe that functional classifications of the most published proteins by Gene Ontology suggest that the primary research interests in renal, intestinal, and pulmonary research all revolve around carcinogenesis pathways or immune responses. For instance, the top-ranked protein TP53 from the “liver or hepatic” query is a major tumor suppressor functioning in DNA repair and apoptosis heavily investigated in heptocarcinoma research but also highly published in other systems. Promiscuity of these proteins obscures other proteins that may be more relevant to a particular topic despite their lower overall publication counts.
Figure 2.

Identifying topic-relevant significant proteins using normalized copublication distance (NCD). (A) The multiplicity of occurrence of the top 50 proteins in each of the six examined systems is shown. Proteins with a multiplicity of six (e.g., TP53) are found in the top 50 most published proteins in all six examined systems and are colored in dark brown. Proteins with a multiplicity of one (e.g., BDNF in the cerebral system) are in the top 50 in only one of the six organ systems queried. (B) NCD normalizes the number of referenced publications in a particular topic for a particular protein by the total number of referenced publications of that protein to any topic. In contrast with ranking proteins by total publication count, normalized copublication distance down-ranks proteins that are of general interest with large numbers of publications in multiple fields (e.g., certain proteins in tumorigenesis pathways) and promotes query-specific proteins, such that top-ranked proteins by NCD (right) are mostly organ-specific. (C) The distribution of NCD values for proteins in a query (black line) follows a normal Gaussian distribution (red line), with a mean of 1.0 and standard deviation (sd) of 0.1. (D) The graphs show the number of publications for each protein referenced in a queried tissue (ordinates), plotted against the NCD between the protein and the tissue (abscissae). Line and shade: locally weighted scatterplot smoothing regression and 95% confidence interval thereof, respectively. Proteins with significant NCD (Z ≤ −1.96) are colored in blue. The top protein in each query is labeled in red text.
To determine the specificity of a protein to a topic, we normalized the system-specific publication counts of a protein by its total publication counts. The resulting metric, termed here normalized copublication distance and hitherto abbreviated as NCD where applicable, is a measurement of the semantic similarity (also known as semantic distance) of the topic versus the protein, similar to other metrics such as normalized compression distance and normalized Google similarity distance,15 the latter of which is intended to compute the relatedness in meanings between two terms as they appear in a corpus of textual knowledge and whose formalism is based on information theory and Kolmogorov complexity. The normalization eliminates the multiplicity of protein occurrences in the queried system (Figure 2B) and down-ranks proteins with high publication count in the systems such as TP53, TNF, and apolipoprotein E (APOE) in the cardiovascular system, but some proteins remain highly ranked (e.g., VEGFA), likely because it remains preferentially studied therein. The distributions of NCD in each queried organ system approximate normality (Figure 2C). We estimate the significance of copublication by Z scores. The NCD is nonlinearly related to publication counts (Figure 2D) and furthermore is not significantly correlated with the abundance or ease of detection of a protein in most queried topics (Supporting Figure S1). For certain applications, NCD may be harnessed in conjunction with publication counts to present a complementary view of the subject and may be adjusted by the citation counts of articles (Supporting Figure S2). For example, identifying highly published proteins may be useful for assay development that intends to target the broadest audience possible in multiple fields, whereas NCD may be used to pinpoint topic-specific proteins.
Top-ranked proteins by NCD in each system are shown in Figure 3 and in further detail in Supporting Table S1. We suggest that these proteins represent targets of exceptional interest to their respective disciplines of biomedical research. In the cardiovascular system, the top protein is VEGFA in both human and mouse, which also has the highest numbers of publications. In the cerebral system, the top-ranked protein in both human and mouse is brain-derived neurotrophic factor (BDNF), a neurotrophin associated with neuron survival, memory formation, and learning. In the hepatic system, the top-ranked mouse protein is leptin (LEP), a hormone involved in the regulation of hunger, energy metabolism, and obesity. In the renal system, the top protein in both human and mouse is polycystin-1 (PKD1), a developmental glycoprotein associated with polycystic kidney diseases. In the pulmonary system, the top human protein is epidermal growth factor receptor (EGFR), a cell surface receptor tyrosine kinase that transduces to multiple pathways including Akt and STAT, which is activated in multiple types of lung cancers; the top ranked mouse protein is pulmonary surfactant-associated protein C (SFTPC), which forms the surfactant that lines the lung tissue. In the intestinal system, the top-ranked human protein is homeobox CDX-2, a homeobox protein that directs the formation of small and large intestines in human and is a marker in colon cancer; the top-ranked mouse protein is adenomatous polyposis coli protein (APC), a regulator of Wnt signaling mutated in familial adenomatous polyposis and other sporadic colorectal cancers. When comparing the rankings of some human proteins to their mouse orthologs, we observe a distinction in their rankings in mouse and human queries. We observe that only moderate correlation exists between the NCD of the top-50 human proteins and their mouse orthologs (Spearman’s correlation coeficient ρ = 0.27 to 0.60), which we attribute to disparate emphases between interests of basic research in animal models and clinical applicability in human. Network analysis of the cardiovascular protein list corroborates the mechanistic emphasis of the mouse proteins that appears to occupy regulatory hubs within protein networks.13 In contrast, protein nodes highly studied in human are more peripheral in network connectivity, likely due to their status as downstream targets more closely linked to overt pathophysiological phenotypes.
Figure 3.

High-impact proteins in six organ systems in (A) human and (B) mouse. The gene names and protein names of the top five proteins, as determined by their normalized copublication distance within the queried organ system in the literature, are shown. The identities of the top proteins in each system indicate both organ-system-specific as well as species-specific differences in the focus of biomedical research.
We next interrogated whether the current approach is applicable to other queryable topics such as disease terms. If the described method returns a valid list of essential proteins that are pertinent to understanding a subject of inquiry, a more specific query such as on a particular disorder should return a list of proteins that are identifiably implicated in the pathogenic mechanism of the disease based on prior knowledge. We queried five prevalent multifactorial diseases that are not primarily caused by mutations in single genes: Alzheimer’s disease, Parkinson’s disease, myeloid leukemia, cardiac arrhythmia, and atherosclerosis (Supporting Table S2). Two of the topics were neuro-degenerative diseases in the cerebral system and two were cardiovascular diseases, hence we could validate whether distinct sets of disease-associated proteins may be acquired. Indeed, searching for cardiac arrhythmia returned five cardiac ion channels in the top five proteins, NaV1.5 (SCN5A), hERG (KCNH2), KV7.1 (KCNQ1), delayed rectifier potassium channel subunit IsK (KCNE1), and ryanodine receptor (RYR2), whereas the atherosclerosis search term returned serum paraoxonase (PON1), cholesteryl ester transfer protein (CETP), C-reactive protein (CRP), oxidized low-density lipoprotein receptor (OLR1), and ATP-binding cassette subfamily A1 (ABCA1). In the brain, the five most relevant proteins in Alzheimer’s disease were amyloid beta A4 protein (APP), APOE, presenilin-1 (PSEN1), microtubule-associated protein tau (MAPT), and beta-secretase 1 (BACE1), whereas the top five proteins in Parkinson’s disease were alpha-synuclein (SNCA/PARK1), leucin-rich repeat serine/threonine-protein kinase 2 (LRRK2/PARK8), E3-ubiquitin ligase Parkin (PARK2), mitochondrial serine/threonine protein kinase (PINK1), and protein DJ-1 (PARK7). Manual reverse queries of these proteins returned publications on each of their corresponding disease, suggesting the method is able to correctly identify specific proteins of significance from the literature. The result also suggests a flexible way to acquire protein functional annotations based on disease-specific or other topical keywords and may be particularly useful in higher-level physiology keywords (e.g., arrhythmia) that are not typically encompassed in Gene Ontology and other databases.
Validation of our results requires interrogating whether the acquired list of popular proteins accurately portrays the important proteins in each system. This is challenged by the paucity of gold standards: We note from anecdotal experience that the significance of a protein to a particular field typically cannot be objectively quantified a priori even by individuals with expert knowledge in the field and that the reason for the current work is to introduce an objective metric to what is hitherto a subjective endeavor. Nevertheless, we demonstrate that the top-protein lists possess several expected properties. We compared our list of popular disease proteins to Gene Prospector,16 which uses a custom literature database including genome-wide association studies (GWAS) and meta-analyses to identify genetic loci implicated in complex diseases. We compared our results for Alzheimer’s disease and cardiac arrhythmia and found significant correlation in disease gene implications (ρ: 0.22, P: 6.4 × 10−9 for Alzheimer’s disease; ρ: 0.43, P: 6.4 × 10−11 for cardiacarrhythmia; ρ: correlation coeficient, P: significance of correlation), suggesting the method here identifies proteins that are functionally implicated in disease by orthogonal means. We further matched the results against a gold standard positive of manually curated gene list in the PDGene database,17,21 a Parkinson’s disease gene database that ranks proteins by meta-analysis significance of multiple genetic association studies on Europeans. Although PDGene does not capture proteins implicated in familial or experimental models of Parkinson’s diseases (e.g., PINK1/Parkin/PARK7), our method here nevertheless identifies 10 out of 21 of the top proteins in PDGene, plus others that are omitted in the database (correlation between our result (NCD) and the PDGene results (log P): ρ: 0.68, P: 0.035). Taken together, these analyses demonstrate the top NCD list likely captures proteins of bona fide biological significance within specific topics.
Furthermore, we demonstrate that the top proteins are highly and significantly enriched with the expected Gene Ontology annotations, which corroborates their likely bona fide importance to the queried topics. We include only proteins with at least 10 publications in the same topic as background for comparison to avoid biases in annotation completeness (mean GO terms per protein = 13.6 vs 18.4). Functional annotations suggest that the primary cardiovascular research interests intersect with biological processes including cardiac muscle contraction (GO:0060048, 14.0× enriched, PB–H < 3.0 × 10−11), angiogenesis (GO:0001525, 9.2× enriched, PB–H < 9.2 × 10−9), and heart development (GO: 0007507, 4.7× enriched, PB–H < 2.1 × 10−6). Functional annotations of the top cerebral proteins intersected with biological processes including ionotropic glutamate receptor signaling pathway (GO:0035235; 24.0× enriched, PB–H < 9.6 × 10−9), synaptic transmission (GO:0007268, 4.8× enriched, PB–H < 1.1 × 10−6), and learning (GO:0007612, 9.7× enriched, PB–H < 2.1 × 10−5). Top hepatic proteins intersected with biological processes including small-molecule metabolic process (GO:0044281; 6.4× enriched, PB–H < 1.1 × 10−21), bile acid and bile salt transport (GO:0015271, 10.6× enriched, PB–H < 2.0 × 10−5), and xenobiotic metabolic process (GO:0006805, 4.3× enriched, PB–H < 3.3 × 10−5). Top renal proteins intersected with biological processes including transmembrane transport (GO:0055085, 6.2× enriched, PB–H < 1.1 × 10−16), excretion (GO:0007588, 7.7× enriched, PB–H < 3.3 × 10−6), and kidney development (GO:0001822, 3.7× enriched, PB–H < 4.9 × 10−4). Top intestinal proteins intersected with biological processes including transmembrane transport (GO:0055085, 5.1× enriched, PB–H 1.4 × 10−4), digestion (GO:0007586, 4.4× enriched, PB–H < 5.2 × 10−2), and G-protein-coupled receptor signaling pathway (GO:0007186, 2.9× enriched, PB–H < 6.3 × 10−2). Top pulmonary proteins intersected with biological processes including respiratory gaseous exchange (GO:0007585, 62.0× enriched, PB–H < 1.5 × 10−5), and alveolar lamellar body (GO:0004984, inf. enriched, PB–H < 2.8 × 10−2). The observed tissues specificity was preserved when top proteins from each queried topic were compared with those of all six systems, suggesting the literature-derived list corroborated with annotation databases in identifying proteins involved in important pathways.
Next, we extended the method to consider the pairwise NCD (NCD*P1,P2,T) between two proteins within a particular topic (e.g., angiotensinogen (AGT) vs angiotensin converting enzyme (ACE) in the cardiovascular system). Two proteins are considered to have a finite pairwise NCD* from each other if they are referenced to at least one identical PubMed publication within a particular query. Proteins that are closely related (e.g., belonging to the same supramolecular complex) should therefore be expected to have a low NCD*. Hierarchical clustering of the protein–protein NCD* matrix in the cerebral and cardiovascular systems corroborated that shared publication readily recapitulates known functional clusters and biological pathways (Figure 4A).
Figure 4.

Popular protein networks. (A) Pairwise normalized copublication distance matrices of top proteins in the cardiovascular and the cerebral system are shown. Cells in the heat map represent the normalized copublication distance between each protein–protein pair via their copublication history (red: greater number of copublications). Proteins may be clustered into identifiable pathways that are known to play significant roles in the physiology of each system, as shown on the left, suggesting the described method of using literature records to identify essential protein readily recapitulates known biology (PB–H: Benjamini–Hochberg adjusted P value of enrichment). (B) Proximal proteins of ten of the top proteins in the cardiovascular system are visualized in protein–protein interaction network graphs. The color of each node denotes the normalized copublication distance of a protein to cardiovascular research, where darker colors denote a protein is more preferentially found in cardiovascular publications compared with other fields. The size of nodes denotes publication counts in cardiovascular-relevant publications; size increases with increasing publication count. Selected hub genes and highly published cardiovascular proteins are labeled in black; in addition, proteins in the network with fewer than 10 publications are labeled in blue and represent proteins that are associated with popular proteins via protein–protein interactions but are themselves yet to be heavily investigated.
Finally, we note that the method presented here may be used to identify not only proteins that are popular but also proteins that have potential biological significance but currently are not associated with high numbers of publications. As it has been remarked that biomedical research is disproportionately focused on a few proteins for which high-quality reagents are available,18 the present method may be used to reorient research efforts toward currently neglected proteins. We note that such proteins may function in similar biological contexts as the popular proteins or associate with them via protein–protein interactions. To understand the biological contexts of the popular proteins, we analyzed top proteins using their membership within known protein–protein interaction networks from Reactome.19,20,22,23
Major functional clusters surrounding the focal points of research are visualized in networks (Figure 4B) using several popular cardiovascular proteins as examples. A number of connected functional clusters in the cardiac proteome can be discerned, including contractile proteins (proximal to TNNI3 and TNNT2), VEGF signaling (proximal to VEGFA, FLT1, KDR, and HIF1A), nitric oxide/PKA signaling (proximal to NOS3), and ion channels (proximal to KCNQ1, KCNH2, SCN5A). Remarkably, although the proximal proteins are functionally related to the popular proteins, suggesting they are involved in biological processes that are considered of interest to cardiovascular research, many are associated with relatively few dedicated publications. For example, tropomyosin 4 alpha chain (TPM4) is functionally associated with cardiac troponins (TNNT2 and TNNI3) and is highly expressed in the heart but has only five referenced cardiovascular publications on Gene2PubMed at the time of analysis. Therefore, the presented method, in conjunction with network analysis and tissue expression profile data, may also be used to identify under-investigated proteins that may be network neighbors to essential proteins and predict protein targets that may in time become popular in or significant to research within a particular field.
DISCUSSION
Quantitative proteomics assays, such as MRM assays, have the advantage that they can be developed for any number of protein sequences encoded by the genome and are not dependent on antibody availability unless enrichment is required and then only a single antibody is required. Their effective dissemination for clinical and hypothesis-testing applications is becoming a pressing objective that promises a transformative impact on biomedical research, both within individual laboratories and across the research community as a whole. The objective of prioritizing technology development bares resemblance to the candidate disease gene prioritization problem (e.g., within specific loci in genetic linkage studies), which has invited successful bioinformatics methods that analyze sequences features or bibliometrics to prioritize relatively small gene lists. Here we suggest that popular and important proteins in a broad query topic may be similarly identified from PubMed queries and protein-reference coenrichment. The general approach and protein lists presented here may be used to guide rational experimental design in optimizing MRM transitions for any given topic for which there have been literature publications.
Our analysis suggests that biomedical investigations are highly specific to individual fields of inquiry with respect to the proteins studied. The priority of protein assay development in, for instance, cardiovascular research applications differs from that for pulmonary applications. Prioritized development of quantitative assays for tissue-specific proteins should therefore be explored to facilitate broader applications and expedited protein characterizations. Identifying the important protein in each field may also help focus resources and manpower toward improving annotations on the proteins for which a large body of information is available and toward which community interest is gravitated. Moreover, the publication records of proteins may be overlaid on existing interaction networks to identify potential gaps in scientific knowledge. In addition to developing quantitative assays for popular proteins, we note that many proteins currently not intensely studied are nevertheless likely to have important biological functions. In the long run, therefore, proteomics assays ought also to be tailored for under-studied proteins and pathways, which will have the potential to greatly propel the pace of research in particular areas. Hence the current method may be reoriented to support rational method development for currently under-studied protein, which, for example, may be functionally associated (e.g., interaction neighbors) with the top proteins but are themselves associated with only few published articles. Both the popular proteins and their corresponding networks therefore constitute attractive targets that may be prioritized by the proteomics community for the development and optimization of high-impact quantitative assays.
A limitation of the presented approach is the dificulty to distinguish less intensively investigated pathways that may nevertheless be important to disease pathogenesis. Another potential limitation is its dependence on the completeness and accuracy of biocuration effort of Gene2PubMed. The coenrichment of terms or ontologies to deduce functional relationship is a well-established method in data-mining,11,21 and several previous works have examined the use of literature records including Gene Ontology, Gene2PubMed and others to compare gene sets or infer gene set functions.22 The Gene2MeSH service by NCIBI (http://gene2mesh.ncibi.org), for example, allows identification of genes associated with medical subject headings (MeSH) using contingency table based statistics to inform on gene enrichment across MeSH terms. The approach described here differs from these prior efforts in that it is not restricted to particular vocabularies (e.g., MeSH) or concepts (disease terms). As with these other existing efforts, however, the approach described here would be biased against particular topics or time periods for which manual curation (in our case, that of Gene2PubMed) is relatively less complete. We also do not currently distinguish investigations at the protein level from those in their cognate genes or transcripts; however, we note that many publications may have been limited to transcript-level measurements in part because of the general unavailability of protein research reagents,18 which is, in fact, the very challenge the HUPO B/ D-HPP initiatives aim to address. Nevertheless, future works may benefit from additional data sources that distinguish the disease relevance of transcripts, proteins, splice isoforms, and post-translational modifications (e.g., to include only publications that concern a particular modification site). Such distinctions may be achieved via the use of automated text-mining of keywords and abstracts, which have already been demonstrated to complement the coverage of existing Gene2PubMed curation.22–24
In summary, we describe a data science approach to rationally target proteomics assay development in various biomedical disciplines by identifying proteins that show close semantic relationship to a queried topic in the literature record. The method is scalable between queries of a few retrievable publications (e.g., searching for a particular disease in a particular journal) to those of entire organ system. Our analysis identifies highly investigated proteins in six major human and mouse organ systems (cardiovascular, cerebral, hepatic, renal, pulmonary, and intestinal). Although the biomedical significance of priority proteins may only be validated after careful investigations in various models and cohorts, the current study should provide a ready-made list of high-impact proteins for which proteomics assays will be of broad interest.
Supplementary Material
Supporting Table S1: Complete lists of popular proteins in the cerebral, cardiovascular, hepatic, renal, intestinal, and pulmonary system in human and in the mouse, ranked by publication counts and normalized copublication distance. (XLS)
Supporting Table S2: Lists of popular proteins in Alzheimer’s disease, Parkinson’s disease, myeloid leukemia, cardiac arrhythmia, and atherosclerosis. (XLS)
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) Grants: U54-GM114833 (to P.P.); K99-HL127302 (to M.P.Y.L.); and P01-HL107153, P01-HL112730, and R01-HL111362 (to J.E.V.E.).
ABBREVIATIONS
- MRM
multiple-reaction monitoring
- B/D-HPP
the Biology/ Disease Human Proteome Project
- HUPO
Human Proteome Organization
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.6b00095.
Supporting Methods; Supporting Figure S1: Relationship between normalized copublication distance and protein relative abundance; Supporting Figure S2: Relationship between normalized copublication distance and total publication citation counts. (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Grote E, Fu Q, Ji W, Liu X, Van Eyk JE. Using pure protein to build a multiple reaction monitoring mass spectrometry assay for targeted detection and quantitation. Methods Mol. Biol. 2013;1005:199–213. doi: 10.1007/978-1-62703-386-2_16. [DOI] [PubMed] [Google Scholar]
- (2).Li X.-j., Hayward C, Fong P-Y, Dominguez M, Hunsucker SW, Lee LW, McLean M, Law S, Butler H, Schirm M, Gingras O, Lamontagne J, Allard R, Chelsky D, Price ND, Lam S, Massion PP, Pass H, Rom WN, Vachani A, Fang KC, Hood L, Kearney P. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci. Transl. Med. 2013;5:207ra142. doi: 10.1126/scitranslmed.3007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Nimeus-Malmstrom E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 2012;4:142ra94–142ra94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nanjappa V, Thomas JK, Marimuthu A, Muthusamy B, Radhakrishnan A, Sharma R, Ahmad Khan A, Balakrishnan L, Sahasrabuddhe NA, Kumar S, Jhaveri BN, Sheth KV, Kumar Khatana R, Shaw PG, Srikanth SM, Mathur PP, Shankar S, Nagaraja D, Christopher R, Mathivanan S, Raju R, Sirdeshmukh R, Chatterjee A, Simpson RJ, Harsha HC, Pandey A, Prasad TS. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014;42:D959–65. doi: 10.1093/nar/gkt1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Plasma Proteome Database. http://www.plasmaproteomedatabase.org/ (accessed Jan 1, 2015)
- (7).Aebersold R, Bader GD, Edwards AM, van Eyk JE, Kussmann M, Qin J, Omenn GS. The biology/disease-driven human proteome project (B/D-HPP): enabling protein research for the life sciences community. J. Proteome Res. 2013;12(1):23–7. doi: 10.1021/pr301151m. [DOI] [PubMed] [Google Scholar]
- (8).Lam MP, Vivanco F, Scholten A, Hermjakob H, Van Eyk J, Ping P. HUPO 2011: The new Cardiovascular Initiative - integrating proteomics and cardiovascular biology in health and disease. Proteomics. 2012;12(6):749–51. doi: 10.1002/pmic.201270015. [DOI] [PubMed] [Google Scholar]
- (9).Semba RD, Enghild JJ, Venkatraman V, Dyrlund TF, Van Eyk JE. The Human Eye Proteome Project: Perspectives on an emerging proteome. Proteomics. 2013;13:2500–2511. doi: 10.1002/pmic.201300075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Aebersold R, Bader GD, Edwards AM, van Eyk J, Kussman M, Qin J, Omenn GS. Proteomics; Highlights of B/D-HPP and HPP Resource Pillar Workshops at 12th Annual HUPO World Congress of Proteomics; Yokohama, Japan. September 14–18, 2013; 2014. pp. 975–88. [DOI] [PubMed] [Google Scholar]
- (11).Bromberg Y. Chapter 15: Disease gene prioritization. PLoS Comput. Biol. 2013;9(4):e1002902. doi: 10.1371/journal.pcbi.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, Carmeliet P, Moreau Y. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24(5):537–44. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- (13).Lam MP, Venkatraman V, Cao Q, Wang D, Dincer TU, Lau E, Su AI, Xing Y, Ge J, Ping P, Van Eyk JE. Prioritizing proteomics assay development for clinical translation. J. Am. Coll. Cardiol. 2015;66(2):202–4. doi: 10.1016/j.jacc.2015.04.072. [DOI] [PubMed] [Google Scholar]
- (14). BD2KPubMed http://www.heartproteome.org/pubmed/ (accessed Apr 10, 2015)
- (15).Cilibrasi RL, Vitanyi PMB. The Google Similarity Distance. IEEE Trans. Knowl. Data Eng. 2007;19(3):370–383. [Google Scholar]
- (16).Yu W, Wulf A, Liu T, Khoury MJ, Gwinn M. Gene Prospector: an evidence gateway for evaluating potential susceptibility genes and interacting risk factors for human diseases. BMC Bioinf. 2008;9:528. doi: 10.1186/1471-2105-9-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, Maraganore DM, Martin ER, Martinez M, Myers RH, Nalls MA, Pankratz N, Payami H, Satake W, Scott WK, Sharma M, Singleton AB, Stefansson K, Toda T, Tung JY, Vance J, Wood NW, Zabetian CP, Me Genetic Epidemiology of Parkinson’s Disease, C. International Parkinson’s Disease Genomics, C. Parkinson’s Disease, G. C. Wellcome Trust Case Control, C. Young P, Tanzi RE, Khoury MJ, Zipp F, Lehrach H, Ioannidis JP, Bertram L, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8(3):e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011;470(7333):163–5. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- (19).Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D’Eustachio P, Stein L. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers. 2012;4:1180–211. doi: 10.3390/cancers4041180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, Matthews L, May B, Milacic M, Rothfels K, Shamovsky V, Webber M, Weiser J, Williams M, Wu G, Stein L, Hermjakob H, D’Eustachio P. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44(D1):D481–7. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Perez-Iratxeta C, Bork P, Andrade MA. Association of genes to genetically inherited diseases using data mining. Nat. Genet. 2002;31(3):316–9. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- (22).Qiao N, Huang Y, Naveed H, Green CD, Han JD. CoCiter: an efficient tool to infer gene function by assessing the significance of literature co-citation. PLoS One. 2013;8(9):e74074. doi: 10.1371/journal.pone.0074074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Minguez P, Al-Shahrour F, Montaner D, Dopazo J. Functional profiling of microarray experiments using text-mining derived bioentities. Bioinformatics. 2007;23(22):3098–9. doi: 10.1093/bioinformatics/btm445. [DOI] [PubMed] [Google Scholar]
- (24).Liu Y, Liang Y, Wishart D. PolySearch2: a significantly improved text-mining system for discovering associations between human diseases, genes, drugs, metabolites, toxins and more. Nucleic Acids Res. 2015;43:W535–W542. doi: 10.1093/nar/gkv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Table S1: Complete lists of popular proteins in the cerebral, cardiovascular, hepatic, renal, intestinal, and pulmonary system in human and in the mouse, ranked by publication counts and normalized copublication distance. (XLS)
Supporting Table S2: Lists of popular proteins in Alzheimer’s disease, Parkinson’s disease, myeloid leukemia, cardiac arrhythmia, and atherosclerosis. (XLS)