

Abstract
Breathing is a vital homeostatic behavior and must be precisely regulated throughout life. Clinical conditions commonly associated with inflammation, undermine respiratory function may involve plasticity in respiratory control circuits to compensate and maintain adequate ventilation. Alternatively, other clinical conditions may evoke maladaptive plasticity. Yet, we have only recently begun to understand the effects of inflammation on respiratory plasticity. Here we review some of common models used to investigate the effects of inflammation and discuss the impact of inflammation on nociception, chemosensory plasticity, medullary respiratory centers, motor plasticity in motor neurons and respiratory frequency, and adaptation to high altitude. We provide new data suggesting glial cells contribute to CNS inflammatory gene expression after 24 hours of sustained hypoxia and inflammation induced by 8 hours of intermittent hypoxia inhibits long-term facilitation of respiratory frequency. We also discuss how inflammation can have opposite effects on the capacity for plasticity, whereby it is necessary for increases in the hypoxic ventilatory response with sustained hypoxia but inhibits phrenic long term facilitation after intermittent hypoxia. This review highlights gaps in our knowledge about the effects of inflammation on respiratory control (development, age, and sex differences). In summary, data to date suggest plasticity can be either adaptive or maladaptive and understanding how inflammation alters the respiratory system is crucial for development of better therapeutic interventions to promote breathing and for utilization of plasticity as a clinical treatment.
Keywords: respiratory plasticity, inflammation, neuroplasticity, hypoxia, acclimatization, sensory plasticity, motor plasticity
Introduction
Undermining the respiratory control system poses a critical threat to homeostasis. Many studies have examined how important molecules (like opiates) or diseases (like sleep apnea) cause dysfunction in the respiratory system, but only recently have we begun to recognize the interplay between the immune system and the respiratory system. A common attribute of almost all diseases and disorders, respiratory or other, is inflammation. Since we no longer recognize the central nervous system (CNS) as immune privileged, additional avenues whereby inflammation may undermine breathing are beginning to be understood. Increasing evidence supports a dynamic role for inflammation either by promoting or inhibiting different forms of neuroplasticity. Since respiratory plasticity (sensory or motor) has been proposed to be involved in disease development or treatment, many investigations into the role of inflammation in the respiratory system have focused on these areas. As will be discussed in this review, additional research into the interactions between inflammation and various forms of respiratory plasticity is still needed, in addition to increased focus on how inflammation might alter other aspects of respiratory control (rhythm generation, development, acclimatization). This review will highlight relevant models of inflammation and discuss the complex mechanisms by which inflammation can undermine or promote plasticity. The importance of inflammatory signals in the mechanism for neural plasticity in pain and nociception has been recognized for years (Di Filippo et al., 2008; Liu et al., 2011; Stemkowski and Smith, 2012; Watkins and Maier, 2002; Woolf and Salter, 2000). More recently, interest is growing into the effects of such signals for neural plasticity in other physiological control systems, such as the control of breathing and the heart (Powell and Kou, 2011). Despite the increased interest, many gaps in our knowledge of the effects of inflammation on the respiratory system remain and will be discussed. Understanding how inflammation alters the respiratory system is vital for development of better therapeutic interventions to promote breathing and utilization of plasticity as a clinical treatment.
Models of inflammation
Models of systemic inflammation are routinely used in research, in part, due to the increased appreciation for an underlying role for and the effects of inflammation in many pathologies. Here, we will briefly review some commonly used models of inflammation of particular relevance to research in respiratory control. Each model provides some similar, yet distinct, research advantages, but one common feature remains – all models activate some aspect of the innate immune system. Exogenous models like cytotoxins induce direct cell damage, causing release of cytokines and danger-associated molecular patterns (DAMPs) and inducing endogenous inflammation. Other exogenous models, like lipopolysaccharide (LPS), complete Freund's adjuvant (CFA), and Polyinosinic:polycytidylic acid (poly(I:C)), use components of pathogenic bacteria or viruses to activate receptors and stimulate endogenous immune responses. Relevant physiological perturbations, like hypoxia, can also induce inflammatory responses as discussed below.
There is now substantial evidence that systemic inflammation induces CNS inflammation. CNS inflammation is primarily associated with activation of microglia, the resident CNS immune cells, but other cell types (such as astrocytes) likely also play important roles. Microglia, endothelial cells, neurons, and astrocytes all express functional levels of toll-like receptors (TLRs) which are activated by components of bacteria and viruses and lead to activation of NF-ĸB, stimulating release of cytokines, including IL-1β and TNFα, to cause inflammation. Furthermore, IL-1 and TNF receptors are found on microglia, neurons, astrocytes, and neurovascular endothelial cells (Lampron et al., 2013; Probert, 2015), suggesting inflammation can be sensed and further propagated by multiple cell-types in the CNS. For example, peripherally applied IL-1β induces prostaglandin synthesis in vascular endothelial cells of the blood brain barrier (BBB), which impairs respiration and is important for neonatal respiratory control during systemic infection (Hofstetter and Herlenius, 2005; Siljehav et al., 2015). Overall, the results of systemic inflammation are widespread in the CNS and multiple cell-types play important roles in mediating CNS inflammatory effects. Since systemic inflammation elicits many changes in the CNS, it is probable such CNS changes alter neural control of breathing.
Lipopolysaccharide and CNS inflammation
LPS, a component of gram-negative bacterial cell walls, activates the innate immune response via TLR4. TLR4 activation induces pro-inflammatory gene expression through activation of MAPKs JNK, ERK, p38, and the transcription factor NF-ĸB. Many endogenous ligands also activate TLR4 including proteins released from dead or dying cells, heat shock proteins, and modified low-density lipoproteins (Erridge, 2010; Lehnardt et al., 2008; Ohashi et al., 2000). Since LPS activates endogenous inflammatory pathways and much is known about its signaling cascade, it serves as a relevant ligand to induce systemic inflammation.
The magnitude of LPS effects are dependent on the organism from which LPS originates, and the species, sex, and age of the organism in which it is used (Nemzek et al., 2008). E. coli, salmonella, and other gram negative bacteria contain LPS, but the structure of the lipid component of LPS can differ between organisms, with some LPS molecules having more potent effects, likely due to changes in how they interact with TLRs (Jin et al., 2013; Li et al., 2013; Rietschel et al., 1994). High doses (>10 mg/kg in rats, >7 mg/kg in mice) of LPS cause sepsis and high mortality and even more moderate doses (5-6 mg/kg) of LPS can cause progressive neurodegeneration (Cardoso et al., 2015; Qin et al., 2007), long-term behavioral changes, and decreased neural markers of hippocampal plasticity (Anderson et al., 2015). Importantly, lower doses (typically 0.05-1 mg/kg) induce febrile responses in rodents similar to human responses to LPS (Nemzek et al., 2008; Redl et al., 1993). Furthermore, low doses of LPS increase cytokine expression similarly in humans and rodents, further supporting the relevance of LPS as a model to induce inflammation (Copeland et al., 2005). While some controversy exists about the use of LPS to mimic endotoxemia (Perlman et al., 2013; Seok et al., 2013), use of low-dose LPS as a TLR4 agonist is a reasonable model of low-level inflammation.
While the CNS was historically described as “immune-privileged”, there is now substantial evidence for systemic inflammatory signals altering CNS activity. Some pathogens, immune cells, and pro-inflammatory molecules can either cross the BBB under certain conditions or induce inflammatory signaling within the CNS via cross-talk between endothelial cells, neurons, and glia (for further details refer to Lampron et al., 2013). Circumventricular organs also play a role in CNS responses to inflammation by allowing either LPS or cytokines to interact directly with neurons and glia. For example, the area postrema contains cells that are activated by LPS, TNFα, and IL-1β (Wuchert et al., 2008; Wuchert et al., 2009). Since the capacity for LPS penetration across the BBB is low (Banks and Robinson, 2010), recent research has highlighted multiple alternative transmission mechanisms. Even low-dose LPS (intraperitoneal or intratracheal) is sufficient to induce inflammatory gene and protein expression in the CNS without significant levels of LPS crossing the BBB (Balan et al., 2011; Banks and Robinson, 2010; Laye et al., 1995). Vagal afferents moderate communication of low-level systemic inflammation to the CNS, since vagotomy eliminates the febrile response associated with sickness and inflammation after very low doses of LPS (1ug/kg), but not higher doses. At higher doses, there are likely alternative routes for transmission and induction of CNS inflammation (Romanovsky et al., 1997). This is evident by subdiaphragmatic vagotomy inhibiting CNS gene expression of IL-1β after i.p. LPS (Laye et al., 1995), and cervical vagotomy inhibiting medullary IL-1β after intratracheal LPS (Balan et al., 2011). The mechanisms of this alternative transmission are still active areas of investigation.
Other models of induced inflammation
Viral infections are also common models of inflammation and induce similar inflammatory pathways to LPS-induced inflammation. RNA viruses induce inflammation through activation of TLR3, TLR7, and TLR8. The most common model of viral infections is the viral mimetic, poly(I:C), a synthetic dual-stranded RNA molecule activating TLR3. TLR3 activation elicits similar inflammatory profiles to other TLRs, including activation of NF-kB and MAPKs and increased proinflammatory gene expression. The inflammatory response induced by poly(I:C) is similar to LPS with respect to effects on systemic and CNS inflammatory cytokine expression, febrile response, and HPA axis activation (Arsenault et al., 2013; Cunningham et al., 2007; Gandhi et al., 2007). Furthermore, though poly(I:C) can compromise the BBB (Wang et al., 2004), other mechanisms of peripheral to central inflammatory transmission are likely similar between poly(I:C) and LPS. In summary, there are similar CNS inflammatory consequences of inflammation induced by LPS and poly(I:C), though LPS is a more widely used model. However, based on correlations between maternal viral influenza infection and the risk of offspring developing schizophrenia, poly(I:C) is increasingly being used to study perinatal CNS inflammation (Brown et al., 2004).
Generalized tissue damage and cell death induces systemic and CNS inflammation. Cell damage leads to release of DAMPs, which activate TLR4 to induce systemic inflammation. Bleomycin, a cytotoxin and anti-cancer chemotherapy drug, causes lipid peroxidation and DNA damage and has been used to model the effects of acute lung injury, induce cell damage, and cause inflammation (Tolle and Standiford, 2013). Intrapulmonary administration of bleomycin increased levels of inflammatory cytokines in lung tissue, area postrema and nucleus tractus solitarius (Jacono et al., 2011). Thus, generalized cell damage is another inflammatory model altering areas relevant to the neural control of breathing.
Hypoxia-induced inflammation
The mechanisms by which hypoxia activates inflammatory signals in the CNS are related to O2-sensitive prolyl hydroxylases (PHD). Hypoxia activates NF-ĸB to promote transcription of proinflammatory cytokines when IκB kinase (IKK-β) is disinhibited by PHD (Cummins et al., 2006; Rius et all., 2008). This PHD mechanism of O2-sensitivity is similar to that for hypoxia inducible factor-1α (HIF-1α), which is a master switch for gene expression in hypoxia (Prabhakar and Semenza, 2015). Perhaps it is not surprising that common molecular signals have evolved for two of the most evolutionarily ancient stress responses to tissue injury and hypoxia (Zinkernagel et al., 2007) and that they act synergistically. NF-κB is a critical transcriptional activator of HIF-1α and basal NF-κB activity is required for HIF-1α protein accumulation during hypoxia in the brain of hypoxic animals (Rius et al., 2008). Conversely, HIF-1α promotes the inflammatory response within a hypoxic environment, at least in peripheral immune cells (Cramer et al., 2003; Lipman et al., 2012; Peyssonnaux et al., 2005; Walmsley et al., 2005; Zinkernagel et al., 2007).
To summarize, multiple models are used to study inflammation and many share common mechanisms. LPS is the most commonly used model to study inflammation and acts as a TLR4 agonist at physiologically relevant levels, where it induces both systemic and CNS inflammation. Other models inducing inflammation such as poly(I:C), bleomycin, or CFA, though less commonly utilized, can induce inflammation via TLR activation and display similar increases in CNS inflammatory gene expression. CNS inflammation induced in all of these models is most likely a result of vagal afferent signaling and LPS/cytokine interactions with the BBB at low doses. In addition, hypoxia can stimulate inflammation in the CNS by directly activating similar mechanisms in neurons or glia, or via secondary effects of hypoxic activation of the sympathetic nervous system (see High Altitude below).
CNS inflammation and neuroplasticity
Inflammatory signals can cause or inhibit plasticity at every level of neural control systems, including afferent and efferent arms of a reflex, integration in the CNS, and during different stages of development.
Neuro-immune reprogramming and plasticity
Recent research revealed that perinatal inflammation has potent effects on future immune function. Like adults, neonates challenged with LPS develop a short-term tolerance to a subsequent LPS challenge (Biswas and Lopez-Collazo, 2009). However, there is a critical developmental window between P7 and P28 in which a low dose of LPS or poly(I:C) reprograms the immune system with effects lasting into adulthood (Ellis et al., 2005; Spencer et al., 2011; Spencer et al., 2006). After neonatal inflammation, adults exposed to subsequent inflammatory stimuli have reduced cytokine release, reduced febrile response, and elevated corticosterone levels as a result of long-lasting changes in neural regulation of anti-inflammatory pathways and altered HPA-axis regulation (Mouihate et al., 2010; Shanks et al., 2000). This developing field of research suggests a complex form of plasticity exists within the immune system, whereby activation of the immune system during a critical period induces long-lasting changes in immune function.
Similarly, perinatal inflammation has been implicated in impaired cognitive function and behavior into adulthood. For a detailed review of the many effects of early life information, we direct the readers to recent reviews (Bilbo and Klein, 2012; Bilbo and Schwarz, 2012; Yirmiya and Goshen, 2010). In brief, early-life infection causes lasting impairment of hippocampal plasticity and increases vulnerability for neuropsychiatric disorders (Bilbo et al., 2005; Hornig et al., 1999; Rantakallio et al., 1997). The magnitude and timing of perinatal inflammation, however, can have differential effects on adult behavior, memory, and learning (Bilbo and Schwarz, 2012). For example, high-doses of LPS cause significant long-term structural changes in the brain, including neuron loss and hypomyelination (Cardoso et al., 2015). In contrast, low-dose, neonatal LPS alters future immune responses and causes memory impairment in adults (Bilbo et al., 2005). Low-dose, neonatal LPS exposure also increases adult anxiety-like behaviors, suggesting even minor immune challenges during development can have lasting neural consequences (Walker et al., 2004). Similarly, the perinatal timing of inflammation causes differential effects on behavioral outcomes (Schwarz and Bilbo, 2011). Overall, perinatal inflammation appears to cause long-term detriments in a variety of neural functions, yet we know very little about the effects of perinatal inflammation on any aspect of respiratory control.
Nociception
The effects of inflammation on nociception have been extensively studied (reviewed in Ren and Dubner, 2015; Stemkowski and Smith, 2012; Woolf and Salter, 2000) and provide a framework to reference respiration related studies. Both systemic and local inflammation can induce peripheral and central neural sensitization leading to hyperalgesia and allodynia (Luo et al., 2014). Briefly, peripheral injury and inflammation sensitizes primary nociceptive neurons via inflammatory mediators including bradykinin and prostaglandins (reviewed in Pethő and Reeh, 2012). Peripheral sensitization and increased spontaneous activity of nociceptive neurons cause activity-dependent changes in the spinal dorsal horn second order neurons (Woolf and Salter, 2000), an effect mediated by multiple cell types including microglia, astrocytes, and neurons (Ren and Dubner, 2015). Microglia in the dorsal horn respond rapidly to increased primary afferent activity by increasing in number and changing morphology (McMahon and Malcangio, 2009). They are activated by purinergic signaling and can synthesize and release pro-inflammatory cytokines and mediate sensitization through release of BDNF (Fabbretti, 2013; Old et al., 2015).
More recently, fractalkine, a chemokine primarily released by neurons in the CNS, has been shown to play an important role in the pathogenesis of chronic pain. Fractalkine activates the CX3CR1 receptor, which is exclusively expressed on microglia in the CNS and induces release of inflammatory mediators (Clark and Malcangio, 2014). Intrathecal administration of fractalkine is sufficient to induce mechanical sensitization and, after peripheral nerve injury, a CX3R1 antagonist reduces mechanical pain sensitization, exemplifying the critical role of microglial activation in central sensitization (Milligan et al., 2004). Astrocytes contribute to central sensitization as well, most likely through regulating glutamate transport and releasing cytokines (Gao and Ji, 2010; Old et al., 2015). Together, these studies exemplify an interesting form of inflammation-induced neuroplasticity, which likely bear mechanistic similarities to the role of inflammation in some forms of respiratory sensory plasticity.
Inducing peripheral inflammation with LPS can also cause nociceptive sensitization. In adults and neonates, i.p. doses of LPS cause acute sickness behavior and hyperalgesia (Watkins et al., 1994; Wiertelak et al., 1994) and neonates treated with intrathecal LPS also develop acute hyperalgesia (Moss et al., 2007; Zouikr et al., 2014), while adult male mice treated with intrathecal LPS develop a tactile allodynia lasting at least 7 days (Stokes et al., 2013). While immune responses to LPS are similar in neonates and adults, the long-term effects of LPS in the different age groups are substantially diverse. Adults develop a short-term tolerance to subsequent inflammatory stimuli (Biswas and Lopez-Collazo, 2009). In contrast, adults treated with LPS as neonates display nociceptive sensitivity (Boissé et al., 2005). However, in other models of nociceptive sensitization, neonates exhibit a short-term protection from inflammatory nociceptive plasticity. Spared nerve injury (SNI), a model of neuropathic pain which induces spinal inflammation and hyperalgesia, has no effect on nociceptive sensitivity in mice and rats until 4-5 weeks of age due to central production of anti-inflammatory IL-10 (McKelvey et al., 2015). These studies exemplify the importance of age and cause of inflammation (endogenous – like SNI, or exogenous – like LPS) in determining the long-term effects of inflammation on the nervous system (Schwaller and Fitzgerald, 2014).
Chemosensory Plasticity
Chronic intermittent hypoxia (CIH) induces sensory plasticity in the carotid sinus nerve. Ten days of CIH increases acute hypoxic ventilatory responses and sensitizes the carotid sinus nerve response to hypoxia. CIH also primes the carotid sinus nerve for augmented plasticity whereby acute intermittent hypoxia (IH; 10 × 15 sec 12% O2, interspersed by 5 min of hyperoxia) causes greater sensory long-term facilitation (LTF) in the carotid sinus nerve compared to sensory LTF without CIH preconditioning (Peng et al., 2003). Additionally, CIH also leads to persistently elevated baseline carotid sinus nerve activity and augmented hypoxic ventilatory responses for 21 days, which may be an important step in the pathogenesis of hypertension (Del Rio et al., 2014). Sensory LTF appears independent of hypoxic severity and is reversible by a return to normoxia for 10 days (Peng et al., 2003).
Though the detailed molecular mechanisms of sensory LTF are still incomplete, significant progress has been made in determining the involvement of important molecular players, including inflammatory molecules. Intermittent application of both 5-HT and angiotensin II are sufficient to produce sensory LTF (Peng et al., 2003; Peng et al., 2011; Peng et al., 2006) and 5-HT is released from carotid bodies during acute IH after CIH. Similarly, CIH up-regulates components of the renin-angiotensin system including angiotensin type 1 receptors, which activate NOX2 and are required for sensory LTF (Lam et al., 2014; Peng et al., 2011). Reactive oxygen species (ROS) also have been suggested to contribute to sensory LTF in response to acute IH. ROS are increased by CIH through multiple mechanisms including transcriptional upregulation of NOX2, decreased SOD2 activity (Prabhakar et al., 2015) and induction of inflammation and immune cell infiltration in the carotid body (Lam et al., 2012). Furthermore, antioxidants given during CIH abolish hypoxic sensitization and sensory LTF (Del Rio et al., 2010; Peng et al., 2003; Peng and Prabhakar, 2004; Prabhakar et al., 2015).
While antioxidants and ROS inhibitors abolish many of the effects of CIH (Iturriaga et al., 2015; Peng et al., 2003), anti-inflammatory drugs have no effect on chemosensitivity and sensory LTF (Del Rio et al., 2012; Iturriaga et al., 2015). Thus, though inflammatory cytokines are present in carotid bodies and may modulate carotid sinus nerve activity after CIH, inflammation per se does not significantly contribute to carotid body hypoxic sensitization (Del Rio et al., 2012). However, anti-inflammatory treatment during CIH mitigates CIH-induced hypertension and the elevated baseline carotid sinus nerve activity normally observed after CIH (Del Rio et al., 2012), suggesting a role for inflammatory molecules in modulating carotid body activity. In fact, direct application of IL-1β or LPS to carotid bodies increases carotid sinus nerve frequency (Fernández et al., 2008; Shu et al., 2007), but decreases carotid sinus nerve responses to excitatory stimuli (nicotine and hyperoxia) (Fernández et al., 2008). Thus, although LPS and CIH may both induce carotid body inflammation and modulate afferent activity, the hypoxic sensitization of carotid sinus nerve responses does not appear to depend on inflammation.
Sensory LTF of the carotid sinus nerve is an important form of respiratory plasticity related to inflammation and likely relevant to common pathologies like OSA and hypertension (Del Rio et al., 2014, Dempsey et al., 2010). When inflammation is induced by CIH, treatment with NSAID eliminates elevated carotid baseline activity and hypertension, but not hypersensitivity to hypoxia. However, details of the relationship between inflammation and sensory LTF have not been fully investigated so it is not known where NSAIDs exert an inhibitory effect. Regardless, inflammatory cytokines appear to modulate carotid sinus nerve activity and may alter neural activity in the CNS and contribute to CIH-induced hypertension (Iturriaga et al., 2015).
Chronic sustained hypoxia (CSH) also increases carotid body chemoreceptor O2-sensitivity although this is not necessarily the same mechanism as sensory LTF with CIH. Liu and colleagues showed increased carotid body O2-sensitivity with CSH (8-10 days) in rats was accompanied by an increased presence of immune cells (ED1+) in the carotid body (which are rare in control conditions), and increased cytokine (IL-1β, IL-6, TNFα) and chemokine (MCP-1) mRNA (Liu et al., 2009). Treating animals with dexamethasone or ibuprofen during CSH blunted or blocked these increases in carotid body O2-sensitivity, immune cell invasion and cytokine increases. Cytokines showed differential expression time courses, which were not dependent just on immune cell migration because both type 1 and type 2 carotid body cells (neuronal and glial-like, respectively) produced cytokines (Liu et al., 2009). The significance of inflammatory signaling in different types of cells remains to be investigated, as does the potential consequence of different sources of immune cells migrating into the carotid body. For example, alveolar macrophages are the ‘first responders’ to environmental hypoxia in rats, and induce systemic inflammation by releasing chemokines (MCP-1) in the microvasculature after hypoxic activation in the lungs (Chao et al., 2011a; Chao et al., 2011b). Potential differences between such hypoxic alveolar macrophages and those from, for instance, an ischemic circulatory bed have not been studied. The possibility that such differences also contribute to the opposite effects of inflammation induced by LPS versus inspired hypoxia on chemoreflexes (inhibiting vs. facilitating, respectively), remains to be tested. In a number of animal species, the hypoxic or hypercapnic ventilatory responses are actually attenuated by systemic inflammation (Fernández et al., 2008; Ladino et al., 2007; McDeigan et al., 2003).
While molecular signaling changes contribute to carotid body plasticity, hypoxia and inflammation can also induce morphological changes in the carotid body. For example, CSH induces hyperplasia and hypertrophy of glomus cells, increased expression of tryrosine hydroxylase, and neo-vascularization (Wang et al., 2002; McGregor et al., 1984), but it is not clear if this contributes to functional changes. The effects of anti-inflammatories on carotid body hypertrophy per se during CSH remain to be studied. CIH has no clear effects on glomus cell morphology (Prabhakar et al., 2015), but does induce macrophage infiltration, though no significant structural changes after CIH have been demonstrated in carotid bodies (Lam et al., 2012). Similarly, carotid body inflammation induced by intravenous or direct application of LPS causes immune cell infiltration and disorganizes glomus cell clusters, which may contribute to functional alterations (Fernández et al., 2008; Gauda et al., 2013). Neonatal LPS treatment also induces immune cell infiltration in the carotid body and a small reduction in carotid sinus nerve activity in response to hypoxia (Master et al., 2015). Further investigation is needed to determine whether or not inflammation leads to significant morphological changes in the carotid body.
Plasticity in medullary respiratory centers
Recent experiments indicate that inflammatory signals in respiratory centers of the central nervous system (CNS) contribute to plasticity during CSH. For example, ventilatory acclimatization to hypoxia involves a time-dependent increase in the hypoxic ventilatory response (HVR) during CSH (Pamenter and Powell, 2016) that is blocked by systemic application of the NSAID ibuprofen in rats (Popa et al., 2011). Ibuprofen during CSH also blocks increases in IL-1β and IL-6 expression in the nucleus tractus solitarii (NTS) of rats (Popa et al., 2011). The NTS is an important site for VAH the site of the primary synapse from carotid body chemoreceptors and exhibiting plasticity in glutamatergic neurotransmission that is necessary for VAH (reviewed by Pamenter and Powell, 2016). Together, these data suggest that inflammatory signals in the NTS are important for plasticity in VAH. Further, Popa and co-workers (2011) provide additional arguments that the results cannot be explained just by the known effect of ibuprofen to block increased carotid body O2-sensitivty with CSH (Liu et al., 2009). CSH also increases the ‘CNS gain of the HVR’, which is an increase in ventilatory motor output (measured as integrated phrenic nerve activity or ventilation) for a given level of carotid body stimulation (Dwinell and Powell, 1999; Wilkinson et al., 2010) and this also must be decreased by ibuprofen during CSH to explain the results in rats (Popa et al., 2011). The importance of inflammatory signals for plasticity in VAH is also supported by significant decreases in the HVR of healthy human volunteers given ibuprofen during altitude acclimatization (Basaran et al., 2016), although the site of action cannot be determined in these studies. (see High Altitude below).
Similar to the discussion of the carotid body, the type of cells involved in generating inflammatory signals may be important for plasticity in CNS respiratory centers. Glial cells, specifically microglia and astrocytes, are present in respiratory centers of the brainstem surrounding neural synapses and neighboring blood vessels, and glial cells influence neuronal transmission and central respiratory control (Erlichman et al., 2010; Funk et al., 2015; Huxtable et al., 2010). For example, astrocytes residing in central chemoreceptor areas of the brainstem are chemosensitive and respond to physiological decreases in pH (Kasymov et al., 2013; Gourine et al., 2010). Recently, Tadmouri et al. (2014) used immunohistochemistry to show that microglia and astrocytes in the NTS were activated by 24 hours of CSH, with astrocyte activation peaking at 6 hours, followed by microglia at 24 hours of CSH. They also showed that systemic administration of minocycline, which inhibits microglia among other effects (Moller et al., 2016), blocked a time-dependent increase in ventilation observed following 24 hours of CSH, as well as immunohistochemical signals for microglia activation (Tadmouri et al., 2014). MacFarlane et al. (2015) also found that CSH (5 days) increased expression of microglia in the NTS and dorsal motor nucleus of the vagus, and this change was blocked by minocycline. Here, we present new data to show that minocycline blocks increases in inflammatory signals in the NTS in adult male rats with CSH. These experiments were carried out in accordance with the NIH guidelines for care and use of laboratory animals and approved by the UC San Diego Institutional Animal Care and Use Committee, using the same methods and primers described previously (Popa et al., 2011). Briefly, we used RT-PCR to measure changes in mRNA for cytokines in biopsies of the NTS after 24 hours of CSH with and without minocycline treatment (45 mg/kg i.p.). The results in Figure 1 show that CSH increased mRNA for IL-6 and TNFα in the NTS and these increases were blocked by systemic minocycline treatment. This data supports glial activation as a step leading to increased cytokines and inflammatory signals for ventilatory acclimatization to CSH. However, further experiments are necessary to determine the exact effects of minocycline (cf. Moller et al., 2016), the relative roles of microglia versus astrocytes and if the sequence of their activation is important as suggested by Tadmouri and co-workers (2014).
Figure 1.
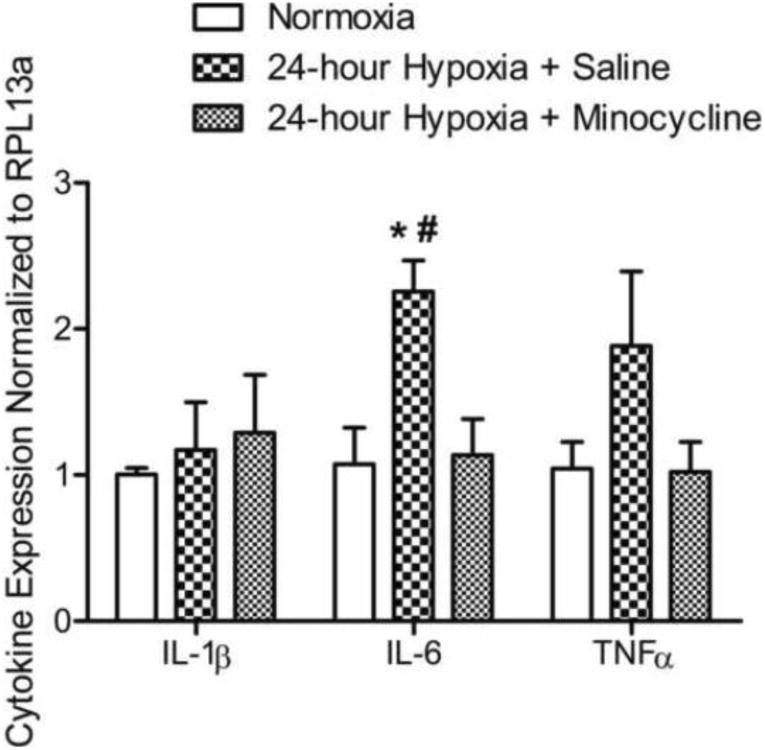
Systemic minocycline administration inhibits cytokine gene expression in the NTS following 24 hours of CSH. Three groups of rats (n=5/group) were exposed to one of the following conditions: 1) Normoxic (controls); 2) 24-hour CSH (10% O2) + i.p. saline; or, 3) 24-hour CSH (10% O2) + i.p. minocycline (45mg/kg). RT-PCR was used to assess cytokine mRNA in the NTS region of the rat brainstem. Exposure to 24-hours of CSH increased both IL-6 and TNF-α mRNA compared to normoxic controls. Systemic minocycline administration blocked the IL-6 and TNF-α mRNA increase. All data presented as mean ± SEM. Two-way ANOVA, Bonferroni post-test; p < 0.05; * indicates difference between normoxia and 24-hour Hypoxia + Saline; # indicates difference between 24-hour Hypoxia + Saline and 24-hour Hypoxia + Minocycline.
Similarly, more experiments are necessary to determine which inflammatory signals increase O2-sensitivity in carotid bodies and ventilation. Hypoxia increases NF-κB as described above, and this can promote the transcription of IL-1β and other pro-inflammatory cytokines (Stanimirovic et al., 2001). IL-1β can directly affect CNS neurons (Aleksandrova et al., 2015) including those in the NTS (Jacono et al., 2011) and IL-1β is the major inducer of cyclooxygenase-2 (COX-2) in the CNS (Samad et al., 2001). COX-2 induction leads to increased production and release of prostaglandin E2 (PGE2) that can affect breathing via EP3 receptors in the NTS (Herlenius, 2011). PGE2 effects are of interest for plasticity in chemoreflexes because they are important for increased excitability in sensory nerve endings with hyperalgesia (Watkins and Maier, 2002). In fact, the first studies of inflammatory signals in carotid body sensitization from chronic hypoxia in Fidone's laboratory (Liu et al., 2009) were based on the hypothesis that the mechanisms of hyperalgesia increased carotid body O2-sensitivity with chronic hypoxia. Further, the rate-limiting enzyme for PGE2 production (mPGES-1) is O2-sensitive and induced by hypoxia (Herlenius, 2011). Hence, this pathway could be blocked by the COX-1 and COX-2 inhibitor ibuprofen that also blocks the increase in carotid body and ventilatory O2 sensitivity with CSH. Therapeutic levels of ibuprofen can block NF-κB induced transcription of cytokines independently of inhibiting COX in isolated cell experiments also (Scheuren et al., 1998; Stuhlmeier et al., 1999), although this effect is not observed in every case (Yamamoto et al., 1999).
Respiratory Motor Plasticity
As discussed above, peripheral injury and inflammation can induce plasticity in the spinal dorsal horn and sensory neurons, but CNS inflammation in the spinal ventral horn inhibits motor plasticity. One commonly studied form of respiratory motor plasticity, phrenic long-term facilitation (pLTF, induced by acute IH (AIH), 3 × 5 min episodes of hypoxia; (Devinney et al., 2013; Gonzalez-Rothi et al., 2015) is exquisitely sensitive to even low levels of systemic inflammation, since a low systemic dose of LPS induces CNS inflammation and abolishes pLTF for at least 24 hours (Huxtable et al., 2013). Increased inflammatory gene expression was observed immediately following LPS, but returned to baseline levels by 24 hours, despite the continued absence of pLTF. The effects of LPS were reversed by systemic application of an NSAID (ketoprofen), further supporting the hypothesis that LPS-induced inflammation abolishes pLTF.
We have also shown another, arguably more physiological, stimulus induces systemic inflammation and abolishes pLTF. Eight hours of intermittent hypoxia (2 min episodes, 10.5% O2, followed by 16 hours of normoxia (IH-1) eliminated pLTF but was restored after systemic NSAID administration (Huxtable et al., 2015). Furthermore, IH-1 was shown to upregulate gene expression of IL-1β in microglia from the cervical spinal cord immediately after 8 hours of IH and for at least 16 hours post-IH. In CNS homogenates from the cervical spinal cord, only iNOS gene expression was elevated after IH-1 (despite a transient increase in IL-6 immediately after IH), indicating differential cellular contributions to the inflammatory response. To gain a greater understanding of the molecular pathway associated with pLTF inhibition, we targeted p38 MAPK, a common downstream signaling molecule activated by inflammation and which promotes further inflammation (Kumar et al., 2003). Like ketoprofen, spinal inhibition of p38 MAPK restored pLTF after IH-1, but suggested spinal inflammation is key to undermining pLTF (Huxtable et al., 2015). Since gene expression data suggests involvement of both microglia and other cell types, we used immunohistochemistry to begin to visualize changes in different cell types. Immunohistochemistry revealed activated (phosphorylated) p38 MAPK was predominantly localized to back-labeled phrenic motor neurons and nearby microglia in the ventral horn of the cervical spinal after IH-1-induced CNS inflammation. Thus, IH-1-induced inflammation activates p38 MAPK in phrenic motor neurons or nearby microglia to inhibit pLTF. While IH-1 elicits spinal inflammation and abolishes pLTF (Huxtable et al., 2015), seven days of IH (5 min hypoxia, 5 min normoxia, 12 hours) enhances pLTF and ventilatory LTF (Ling et al., 2001; McGuire et al., 2003). These differences may be due to the magnitude of inflammatory signaling or increased expression of trophic factors known to stimulate respiratory plasticity. For example, while one or three nights of IH induces inflammatory gene expression in spinal microglia (Huxtable et al., 2015; Smith et al., 2013), inflammatory gene expression in the spinal cord is not elevated after 14 consecutive nights of IH or after 4 weeks of thrice weekly IH (Peters et al., 2015; Smith et al., 2013).
Integrative examples
Inflammation may be relevant to respiratory control in a number of physiological and pathological conditions and here we consider two examples.
Respiratory Frequency Plasticity with Intermittent Hypoxia
While inflammation clearly has a profound effect on pLTF, it is less clear how inflammation effects frequency plasticity, defined as plasticity in phrenic burst frequency, which potentially translates to respiratory or breathing frequency and likely occurs in the central pattern generator in the medulla. (see Baker-Herman et al. 2008 for a review on frequency plasticity). Frequency plasticity is also evident after AIH, but is more variable than pLTF (Baker-Herman and Mitchell, 2008). Systemic inflammation, induced by a high or low-dose of systemic LPS, had no effect on frequency plasticity 3 hours post-LPS (Huxtable et al., 2013; Vinit et al., 2011). However, 24 hours after a low-dose of LPS, frequency plasticity was abolished and was not restored by the NSAID ketoprofen (Huxtable et al., 2013). The lack of restoration of frequency plasticity may be due to an independent or nonspecific effect of ketoprofen, which also reduced frequency plasticity in control animals (Huxtable et al., 2013). Importantly, frequency plasticity was only present in one of four saline-treated control groups reported in the studies cited above, suggesting the general variability of frequency plasticity following AIH likely masks the effects of inflammation on frequency plasticity (Baker-Herman & Mitchell, 2008).
To further explore the effects of inflammation on frequency plasticity, we analyzed frequency responses from our recent investigations about the effects IH-1 (Huxtable et al., 2015). Briefly, rats were exposed to IH-1 or normoxia and studied the next day. IH-1 decreased the frequency response during the short-term hypoxic ventilatory response (normoxia: 15 ± 1 bursts/min from baseline, n = 7; IH-1: 11 ± 1 bursts/min, n = 6; t-test, p < .05, data not shown). In a separate group, ketoprofen treatment after IH-1 restored the hypoxic frequency response (normoxia + ketoprofen: 16 ± 2 bursts/min from baseline, n = 7; IH-1 + ketoprofen: 19 ± 3 bursts/min, n = 6; t-test, p = .38, data not shown). Frequency plasticity was absent in normoxia and IH-1 treated animals; however, IH-1 caused a significant decrease in frequency relative to normoxia treated rats (normoxia: 3 ± 1 bursts/min from baseline, n = 7; IH-1: -3 ± 3 bursts/min, n = 6; time controls: -3 ± 2 bursts/min, n = 5; two-way repeated measures ANOVA, p = .02, data not shown). Furthermore, ketoprofen had no effect on normoxia and IH-1 groups (normoxia: 1 ± 2 bursts/min from baseline, n = 7; IH-1: 4 ± 2 bursts/min, n = 6; time controls: -6 ± 2 bursts/min, n = 6; two-way repeated measures ANOVA, p = .24, data not shown).
In a separate group, rats were treated with IH-1 or normoxia, with and without intrathecal application of a p38 MAPK inhibitor 15 minutes prior to AIH. The data presented in figure 2 have not previously been published, and were collected using IACUC approved methods as outlined in our recent publication (Huxtable et al., 2015). Figure 2A shows the short-term hypoxic frequency responses to acute hypoxia and figure 2B shows frequency changes 60 minutes after AIH. Neither IH-1 nor p38 inhibition significantly altered the short-term hypoxic frequency response. IH-1, however, blunted frequency plasticity, which was restored by intrathecal p38 MAPK inhibition (normoxia/vehicle: 9 ± 2 bursts/min from baseline, n = 9; normoxia/p38: 10 ± 3 bursts/min, n = 7; IH-1/vehicle: 3 ± 4 bursts/min, n = 6; IH-1/p38: 11 ± 3 bursts/min, n = 7; time control/vehicle: 4 ± 3 bursts/min, n = 8; time control/p38: 3 ± 3 bursts per min, n = 5, two-way repeated measures ANOVA, p < .05). While frequency plasticity likely originates in rhythm generating neurons in the medulla, these data suggest that IH-1 activated p38 MAPK can alter expression of frequency plasticity. Interestingly, spinal application of serotonin also induces frequency plasticity, supporting a possible spinal mechanism to frequency plasticity (Baker-Herman & Mitchell, 2008). Taken together, the effects of systemic inflammation on respiratory frequency plasticity are variable and require further investigation. Other isolated preparations containing respiratory rhythm generating areas (i.e. rhythmic slices and brainstem spinal cord preparations) would be advantageous for studying the effects of inflammation on frequency plasticity (Blitz and Ramirez, 2002; Bocchiaro and Feldman, 2004)
Figure 2.
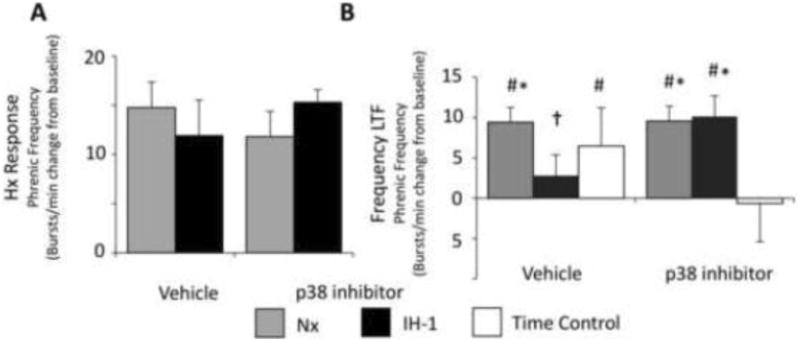
Frequency plasticity is blunted by IH-1 and is restored after spinal treatment with a p38 MAPK inhibitor (SB202190, 1 mM, intrathecal). A) Phrenic nerve frequency responses during the short-term hypoxic response were not altered by IH-1 or intrathecal p38 inhibition. B) Frequency plasticity was evident in normoxia controls (n = 9) and was not altered by p38 inhibition (n = 7). Frequency plasticity was eliminated by IH-1 (n = 6), but was restored after p38 MAPK inhibition (n = 7). Time controls with intrathecal vehicle application (n = 7) did show increased phrenic frequency after 90 minutes relative to baseline, which was absent after the p38 inhibitor (n=5). (Two-way RM ANOVA, Fisher LSD post-test; # indicates difference from baseline, * indicates difference from p38 time controls, † indicates difference from normoxia vehicle controls.)
High Altitude
Hypoxia exposure during ascent to high altitude fuels systemic responses that are necessary to maintain homeostasis in low a low-oxygen environment. With initial exposure to high altitude, cortisol is released from the adrenal cortex, mediated by the HPA axis (Humpeler et al., 1980; Sawhney et al., 1991). Cortisol, which is immunosuppressive, gradually returns to sea level values with acclimatization to hypoxia, as an individual's arterial oxygen content improves with acclimatization. In humans, IL-6 is elevated following both acute and chronic altitude exposure (Klausen et al., 1997; Mazzeo, 2005; Mazzeo et al., 2001). While classically thought of as a pro-inflammatory cytokine, IL-6 may have some anti-inflammatory properties as well (Scheller et al., 2011) including promotion of angiogenesis (Motro et al., 1990), and VEGF induction (Cohen et al., 1996). IL-6 may also influence the increase in red blood cell number through modulation of erythropoietin production (Faquin et al., 1992).
Several studies have shown increases in inflammatory cytokines in humans at high altitude (Klausen et al., 1997; Mazzeo et al., 2001) and these may contribute to respiratory plasticity. A recent double-blind, placebo controlled, cross-over trial found that the increase in isocapnic HVR observed with placebo in healthy volunteers over 48 hours at high altitude (3,800 m) was significantly less with ibuprofen treatment (Basaran et al., 2016). This is predictable from the animal studies showing decreased carotid body and ventilatory O2-sensitivity with ibuprofen during CSH (Liu et al., 2009; Popa et al., 2011). However, despite these effects of ibuprofen on the isocapnic HVR, other mechanisms (e.g. CO2 changes) apparently compensated to prevent decreases in total and alveolar ventilation or arterial oxygenation while breathing ambient air at this altitude. This agrees with studies on the effects of ibuprofen as a treatment for headache with acute mountain sickness (AMS). Gertsch and co-workers (Gertsch et al., 2010; Lipman et al., 2012) found no significant difference in SaO2 with ibuprofen versus placebo. Hence, although NSAIDs may blunt plasticity in the isocapnic HVR during altitude acclimatization, this is not a contraindication for their use to treat AMS. Randomized controlled trials find significantly fewer sojourners have AMS when taking ibuprofen versus placebo (Lipman et al., 2012). However, the consequences of anti-inflammatory drugs on cardiopulmonary chemoreflexes in other pathological conditions, such as chronic hypoxemia with or without CO2 retention in chronic lung disease, remain to be investigated. Also, the effects of anti-inflammatory drugs administered after plasticity in chemoreflexes has been established versus the effects studied to date by administering them during chronic hypoxia, remain to be investigated.
Conclusion and Clinical Relevance
Here, we have reviewed some of the common models used to investigate the effects on inflammation and begun discussions about the impact inflammation has on nociception, chemosensory plasticity, medullary respiratory centers, motor plasticity, frequency plasticity, and adaptation to high altitude (summarized in Fig. 3). Additional research is necessary to understand the conditions in and mechanisms by which plasticity is adaptive or maladaptive in the respiratory system. As discussed, inflammation can have opposite effects on the capacity for plasticity, demonstrating the location of plasticity is an important determinant. It promotes plasticity in the spinal dorsal horn, but inhibits plasticity in the spinal ventral horn. The dose and duration of inflammation stimuli is also important in determining the effect on plasticity. For example, the dose of hypoxia, and thus subsequent inflammation, can alter plasticity expression. One night of intermittent hypoxia (IH-1) abolishes pLTF (Huxtable et al., 2015), but seven days of intermittent hypoxia enhances pLTF (Ling et al., 2001). Such differing results may be explained by changes in inflammatory and neurotrophic signaling during repetitive hypoxic episodes (Huxtable et al., 2015; Peters et al., 2015; Smith et al., 2013). Additionally, treatment with anti-inflammatories blunts ventilatory acclimatization to CSH in humans (Basaran et al., 2016), but during CIH does not inhibit hypoxic sensitization (Del Rio et al., 2012; Iturriaga et al., 2015), suggesting the role of inflammation in CSH and CIH are distinct. These results highlight the complexity of interactions between inflammatory stimuli and the respiratory system, but also the importance of further investigations to tease out the mechanistic intricacies.
Figure 3.
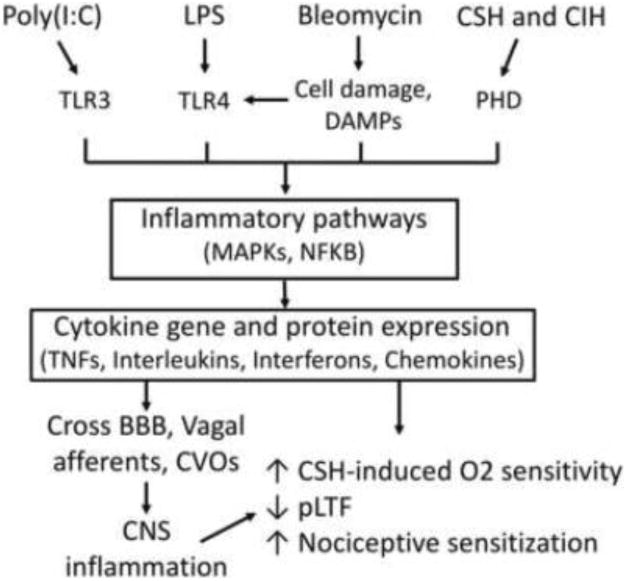
Models of inflammation and effects on plasticity. Multiple models of inflammation converge on pro-inflammatory pathways that alter the expression of plasticity in respiratory and non-respiratory systems.
There is no question inflammation (acute to chronic, minor to severe) occurs in almost every clinical disease and disorder, from the respiratory system and beyond. Yet before reducing maladaptive plasticity (like nociception) or promoting adaptive (like respiratory motor) plasticity can be exploited as a therapeutic intervention, we must first understand how it is caused or undermined, respectively.
Other areas of further investigation into the effect of inflammation during development, aging, and between sexes are also necessary before better therapeutic interventions can be developed. Neonatal infections cause a significant proportion of death in the first week of life (Chan et al., 2015) and account for approximately one third of all neonatal deaths (Black et al., 2010), while those neonates that survive neonatal infection are now recognized to have increased vulnerability to short- and long-term neurodevelopmental disability (Dammann et al., 2002; Ferreira et al., 2014; Stoll et al., 2002). At birth, the neonate must transition from the sterile environment of the mother to an environment filled with pathogens, microbes, and toxins, and must also begin robust, rhythmic breathing. Respiratory problems, such as apneas, represent a significant problem for neonatologists and characterizing this problem represents a significant clinical need (Martin et al., 2012). In fact, recurrent apneas are prevalent in 50% of premature infants (Poets et al., 1994; Poets and Southall, 1994), where infection is particularly common - up to 65% have at least one infection during hospitalization (Stoll et al., 2002; Stoll et al., 2004). Despite this common clinical problem, we know virtually nothing about how early life infection alters functioning and plasticity of the respiratory system. Evidence is beginning to emerge that infants with high incidence of apneas are predisposed for obstructive sleep apnea (OSA) (McNamara and Sullivan, 2000), yet research lags significantly behind in understanding this link. Determining how neonatal inflammation impairs the respiratory system will ultimately lead to better treatments to promote breathing in all age groups and perhaps elucidate links between neonatal conditions and adult respiratory insufficiency.
There are also clear effects of sex and age on respiratory plasticity. pLTF is diminished with age in male rats (Zabka et al., 2001) and testosterone restores hypoglossal LTF (Nelson et al., 2011). In female rats, pLTF is enhanced with age (Zabka and Behan, 2001) and both male and female elderly rats display increased pLTF after CIH (McGuire et al., 2003; Zabka et al., 2003). Systemic inflammation is suggested to increase with age and is related to detrimental aging processes and incidence of chronic diseases (Chung et al., 2009). Furthermore, sex hormones (specifically estrogen which has anti-inflammatory effects) are suggested to be a cause of sexual dimorphism in neurological diseases and may account for some sex-differences in respiratory plasticity (Czlonkowska et al., 2005). Interestingly, in patients with inflammatory respiratory diseases, males are more susceptible to acute inflammation and women are more susceptible to chronic inflammation (Casimir et al., 2013), further suggesting sexual dimorphic characteristics of the effects of inflammation, and possibly plasticity. The potential for sex-differences in the effects of inflammation on respiratory plasticity have scarcely been investigated. To develop effective treatments for inflammatory diseases compromising breathing we need to further recognize the importance of sex and age differences in inflammatory responses.
Much remains to be learned about the role inflammation plays in altering various aspects of respiratory control. In this review, we have highlighted some of the recent studies and pointed out gaps in our knowledge. This research direction is ever changing and developing. The next few years will likely show exciting developments in these areas and many beyond.
Highlights.
Reviews the impact of inflammation on different forms of respiratory plasticity
Inflammation contributes to chronic hypoxia induced oxygen sensitivity
Inflammation abolishes phrenic long-term facilitation
Highlights gaps in knowledge and relevance to clinical conditions and treatments
Acknowledgments
We thank Drs. G.S. Mitchell and J.J. Watters for their support of and contributions to the data presented in this review. Authors JAS and FLP would like to thank Dr. Laura E. Crotty-Alexander, Dr. Mark Fuster, and Scott C Johns for the use of their PCR machine.
Supported by: NIH HL111598, Francis Family Foundation (AGH), University of Oregon (ADH, AGH), NIH RO1 HL081823 (FLP), NIH T32 5T32HL098062 (JAS)
Abbreviations
- CNS
central nervous system
- DAMPs
danger-associated molecular patterns
- LPS
lipopolysaccharide
- CFA
complete Freund's adjuvant
- Poly(I:C)
Polyinosinic:polycytidylic acid
- TLR
Toll-like receptors
- BBB
blood brain barrier
- PHD
prolyl hydroxylases
- IKK-β
IκB kinase
- SNI
spared nerve injury
- CIH
chronic intermittent hypoxia
- LTF
long-term facilitation
- CSH
chronic sustained hypoxia
- IL
interleukin
- TNF
tumor necrosis factor
- COX
cyclyooxengenase
- PGE
prostaglandin
- pLTF
phrenic long-term facilitation
- AIH
acute intermittent hypoxia
- IH-1
8 hours of intermittent hypoxia followed by 16 hours normoxia
- HVR
hypoxic ventilatory response
- AMS
acute mountain sickness
- MAPK
mitogen-activated protein kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- Aleksandrova NP, Danilova GA, Aleksandrov VG. Cyclooxygenase pathway in modulation of the ventilatory response to hypercapnia by interleukin-1beta in rats. Respir Physiol Neurobiol. 2015;209:85–90. doi: 10.1016/j.resp.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Anderson ST, Commins S, Moynagh PN, Coogan AN. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain, Behavior, and Immunity. 2015;43:98–109. doi: 10.1016/j.bbi.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Arsenault D, St-Amour I, Cisbani G, Rousseau LS, Cicchetti F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain, behavior, and immunity. 2013;38:77–90. doi: 10.1016/j.bbi.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respiratory physiology & neurobiology. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan KV, Kc P, Hoxha Z, Mayer CA, Wilson CG, Martin RJ. Vagal afferents modulate cytokine-mediated respiratory control at the neonatal medulla oblongata. Respiratory physiology & neurobiology. 2011;178:458–464. doi: 10.1016/j.resp.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain. 2010 doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basaran KE, Villongco M, Ho B, Ellis E, Zarndt R, Antonova J, Hopkins SR, Powell FL. Ibuprofen Blunts Ventilatory Acclimatization to Sustained Hypoxia in Humans. PLoS One. 2016;11:e0146087. doi: 10.1371/journal.pone.0146087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. COX-2 as a multifunctional neuronal modulator. Nat Med. 2001;7:414–415. doi: 10.1038/86477. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Klein SL. Special Issue: The neuroendocrine–immune axis in health and disease. Hormones and Behavior. 2012;62:187–190. doi: 10.1016/j.yhbeh.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Frontiers in neuroendocrinology. 2012;33:267–286. doi: 10.1016/j.yfrne.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends in immunology. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, Jha P, Campbell H, Walker CF, Cibulskis R, Eisele T, Liu L, Mathers C Child Health Epidemiology Reference Group of, W.H.O., Unicef. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet. 2010;375:1969–1987. doi: 10.1016/S0140-6736(10)60549-1. [DOI] [PubMed] [Google Scholar]
- Blitz DM, Ramirez JM. Long-term modulation of respiratory network activity following anoxia in vitro. Journal of neurophysiology. 2002;87:2964–2971. doi: 10.1152/jn.2002.87.6.2964. [DOI] [PubMed] [Google Scholar]
- Bocchiaro CM, Feldman JL. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc Natl Acad Sci U S A. 2004;101:4292–4295. doi: 10.1073/pnas.0305712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissé L, Spencer SJ, Mouihate A, Vergnolle N, Pittman QJ. Neonatal immune challenge alters nociception in the adult rat. Pain. 2005;119:133–141. doi: 10.1016/j.pain.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Archives of general psychiatry. 2004;61:774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, McGavern DB, Brites D. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. Journal of neuroinflammation. 2015;12:82. doi: 10.1186/s12974-015-0299-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimir GJ, Lefèvre N, Corazza F, Duchateau J. Sex and inflammation in respiratory diseases: a clinical viewpoint. Biology of Sex Differences. 2013;4:16. doi: 10.1186/2042-6410-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GJ, Lee AC, Baqui AH, Tan J, Black RE. Prevalence of early-onset neonatal infection among newborns of mothers with bacterial infection or colonization: a systematic review and meta-analysis. BMC Infectious Diseases. 2015;15:1–16. doi: 10.1186/s12879-015-0813-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Donham P, van Rooijen N, Wood JG, Gonzalez NC. Monocyte chemoattractant protein-1 released from alveolar macrophages mediates the systemic inflammation of acute alveolar hypoxia. Am J Respir Cell Mol Biol. 2011a;45:53–61. doi: 10.1165/rcmb.2010-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Wood JG, Gonzalez NC. Alveolar macrophages initiate the systemic microvascular inflammatory response to alveolar hypoxia. Respir Physiol Neurobiol. 2011b;178:439–448. doi: 10.1016/j.resp.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Frontiers in Cellular Neuroscience. 2014;8 doi: 10.3389/fncel.2014.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996;271:736–741. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D the to Investigators, I. Acute inflammatory response to endotoxin in mice and humans. Clinical and diagnostic laboratory immunology. 2005;12:60–67. doi: 10.1128/CDLI.12.1.60-67.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain, behavior, and immunity. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Czlonkowska A, Ciesielska A, Gromadzka G, Kurkowska-Jastrzebska I. Estrogen and cytokines production - the possible cause of gender differences in neurological diseases. Current pharmaceutical design. 2005;11:1017–1030. doi: 10.2174/1381612053381693. [DOI] [PubMed] [Google Scholar]
- Dammann O, Kuban KC, Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Mental retardation and developmental disabilities research reviews. 2002;8:46–50. doi: 10.1002/mrdd.10005. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. The European respiratory journal. 2010;36:143–150. doi: 10.1183/09031936.00158109. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Iturriaga R. Carotid body potentiation during chronic intermittent hypoxia: implication for hypertension. Frontiers in physiology. 2014;5:434. doi: 10.3389/fphys.2014.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Parga MJJ, Madrid C, Iturriaga R. Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. The European respiratory journal. 2012;39:1492–1500. doi: 10.1183/09031936.00141511. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann N Y Acad Sci. 2013;1279:143–153. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo M, Sarchielli P, Picconi B, Calabresi P. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends in pharmacological sciences. 2008;29:402–412. doi: 10.1016/j.tips.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Dwinell MR, Powell FL. Chronic hypoxia enhances the phrenic nerve response to arterial chemoreceptor stimulation in anesthetized rats. J Appl Physiol (1985) 1999;87:817–823. doi: 10.1152/jappl.1999.87.2.817. [DOI] [PubMed] [Google Scholar]
- Ellis S, Mouihate A, Pittman QJ. Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. The FASEB journal. 2005 doi: 10.1096/fj.04-3569fje. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Leiter JC, Gourine AV. ATP, glia and central respiratory control. Respir Physiol Neurobiol. 2010;173:305–311. doi: 10.1016/j.resp.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? Journal of leukocyte biology. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- Fabbretti E. ATP P2X3 receptors and neuronal sensitization. Frontiers in cellular neuroscience. 2013;7:236. doi: 10.3389/fncel.2013.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faquin WC, Schneider TJ, Goldberg MA. Effect of inflammatory cytokines on hypoxia-induced erythropoietin production. Blood. 1992;79:1987–1994. [PubMed] [Google Scholar]
- Fernández R, González S, Rey S, Cortés PP, Maisey KR, Reyes EPP, Larraín C, Zapata P. Lipopolysaccharide-induced carotid body inflammation in cats: functional manifestations, histopathology and involvement of tumour necrosis factor-alpha. Experimental physiology. 2008;93:892–907. doi: 10.1113/expphysiol.2008.041152. [DOI] [PubMed] [Google Scholar]
- Ferreira RC, Mello RR, Silva KS. Neonatal sepsis as a risk factor for neurodevelopmental changes in preterm infants with very low birth weight. Jornal de pediatria. 2014;90:293–299. doi: 10.1016/j.jped.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Fields DP, Mitchell GS. Spinal metaplasticity in respiratory motor control. Frontiers in neural circuits. 2015 doi: 10.3389/fncir.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk GD, Rajani V, Alvares TS, Revill AL. Neuroglia and their roles in central respiratory control; an overview. … and Physiology Part A: …. 2015 doi: 10.1016/j.cbpa.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Hayley S, Gibb J, Merali Z, Anisman H. Influence of poly I:C on sickness behaviors, plasma cytokines, corticosterone and central monoamine activity: moderation by social stressors. Brain, behavior, and immunity. 2007;21:477–489. doi: 10.1016/j.bbi.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauda EB, Shirahata M, Mason A, Pichard LE, Kostuk EW, Chavez-Valdez R. Inflammation in the carotid body during development and its contribution to apnea of prematurity. Respiratory physiology & neurobiology. 2013;185:120–131. doi: 10.1016/j.resp.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Gertsch JH, Lipman GS, Holck PS, Merritt A, Mulcahy A, Fisher RS, Basnyat B, Allison E, Hanzelka K, Hazan A, Meyers Z, Odegaard J, Pook B, Thompson M, Slomovic B, Wahlberg H, Wilshaw V, Weiss EA, Zafren K. Prospective, double-blind, randomized, placebo-controlled comparison of acetazolamide versus ibuprofen for prophylaxis against high altitude headache: the Headache Evaluation at Altitude Trial (HEAT) Wilderness Environ Med. 2010;21:236–243. doi: 10.1016/j.wem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rothi EJ, Lee KZ, Dale EA, Reier PJ, Mitchell GS, Fuller DD. Intermittent Hypoxia and Neurorehabilitation. Journal of Applied Physiology. 2015 doi: 10.1152/japplphysiol.00235.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlenius E. An inflammatory pathway to apnea and autonomic dysregulation. Respir Physiol Neurobiol. 2011;178:449–457. doi: 10.1016/j.resp.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Hofstetter AO, Herlenius E. Interleukin-1beta depresses hypoxic gasping and autoresuscitation in neonatal DBA/1lacJ mice. Respiratory physiology & neurobiology. 2005;146:135–146. doi: 10.1016/j.resp.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Hornig M, Weissenbock H, Horscroft N, Lipkin WI. An infection-based model of neurodevelopmental damage. Proc Natl Acad Sci U S A. 1999;96:12102–12107. doi: 10.1073/pnas.96.21.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpeler E, Skrabal F, Bartsch G. Influence of exposure to moderate altitude on the plasma concentraton of cortisol, aldosterone, renin, testosterone, and gonadotropins. Eur J Appl Physiol Occup Physiol. 1980;45:167–176. doi: 10.1007/BF00421324. [DOI] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Peterson TJ, Watters JJ, Mitchell GS. Intermittent Hypoxia-Induced Spinal Inflammation Impairs Respiratory Motor Plasticity by a Spinal p38 MAP Kinase-Dependent Mechanism. J Neurosci. 2015;35:6871–6880. doi: 10.1523/JNEUROSCI.4539-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Vinit S, Watters JJ, Mitchell GS. Systemic LPS induces spinal inflammatory gene expression and impairs phrenic long-term facilitation following acute intermittent hypoxia. Journal of applied physiology (Bethesda, Md: 1985) 2013;114:879–887. doi: 10.1152/japplphysiol.01347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Zwicker JD, Alvares TS, Ruangkittisakul A, Fang X, Hahn LB, Posse de Chaves E, Baker GB, Ballanyi K, Funk GD. Glia contribute to the purinergic modulation of inspiratory rhythm-generating networks. J Neurosci. 2010;30:3947–3958. doi: 10.1523/JNEUROSCI.6027-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturriaga R, Moya EA, Del Rio R. Inflammation and oxidative stress during intermittent hypoxia: the impact on chemoreception. Experimental physiology. 2015;100:149–155. doi: 10.1113/expphysiol.2014.079525. [DOI] [PubMed] [Google Scholar]
- Jacono FJ, Mayer CA, Hsieh YHH, Wilson CG, Dick TE. Lung and brainstem cytokine levels are associated with breathing pattern changes in a rodent model of acute lung injury. Respiratory physiology & neurobiology. 2011;178:429–438. doi: 10.1016/j.resp.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Nation RL, Li J, Nicolazzo JA. Species-Dependent Blood-brain Barrier Disruption of Lipopolysaccharide: Amelioration by Colistin in vitro and in vivo. Antimicrobial Agents and Chemotherapy. 2013 doi: 10.1128/AAC.00765-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasymov V, Larina O, Castaldo C, Marina N, Patrushev M, Kasparov S, Gourine AV. Differential Sensitivity of Brainstem versus Cortical Astrocytes to Changes in pH Reveals Functional Regional Specialization of Astroglia. The Journal of Neuroscience. 2013;33:435–441. doi: 10.1523/JNEUROSCI.2813-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausen T, Olsen NV, Poulsen TD, Richalet JP, Pedersen BK. Hypoxemia increases serum interleukin-6 in humans. Eur J Appl Physiol Occup Physiol. 1997;76:480–482. doi: 10.1007/s004210050278. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Ladino J, Bancalari E, Suguihara C. Ventilatory Response to Hypoxia during Endotoxemia in Young Rats: Role of Nitric Oxide. Pediatric Research. 2007;62:134–138. doi: 10.1203/PDR.0b013e318098721a. [DOI] [PubMed] [Google Scholar]
- Lam SYY, Liu Y, Ng KMM, Lau CFF, Liong EC, Tipoe GL, Fung MLL. Chronic intermittent hypoxia induces local inflammation of the rat carotid body via functional upregulation of proinflammatory cytokine pathways. Histochemistry and cell biology. 2012;137:303–317. doi: 10.1007/s00418-011-0900-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SYY, Liu Y, Ng KMM, Liong EC, Tipoe GL, Leung PS, Fung MLL. Upregulation of a local renin-angiotensin system in the rat carotid body during chronic intermittent hypoxia. Experimental physiology. 2014;99:220–231. doi: 10.1113/expphysiol.2013.074591. [DOI] [PubMed] [Google Scholar]
- Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 2013;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Laye S, Bluthe RM, Kent S, Combe C, Medina C, Parnet P, Kelley K, Dantzer R. Subdiaphragmatic vagotomy blocks induction of IL-1 beta mRNA in mice brain in response to peripheral LPS. Am J Physiol. 1995;268:R1327–1331. doi: 10.1152/ajpregu.1995.268.5.R1327. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Schott E, Trimbuch T, Laubisch D, Krueger C, Wulczyn G, Nitsch R, Weber JR. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee D, Madrenas J. Evolving Bacterial Envelopes and Plasticity of TLR2-Dependent Responses: Basic Research and Translational Opportunities. Frontiers in Immunology. 2013;4 doi: 10.3389/fimmu.2013.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001b;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipman GS, Kanaan NC, Holck PS, Constance BB, Gertsch JH Group, P. Ibuprofen prevents altitude illness: a randomized controlled trial for prevention of altitude illness with nonsteroidal anti-inflammatories. Ann Emerg Med. 2012;59:484–490. doi: 10.1016/j.annemergmed.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Liu X, He L, Dinger B, Gonzalez C, Stensaas L, Fidone S. A chronic pain: inflammation-dependent chemoreceptor adaptation in rat carotid body. Respir Physiol Neurobiol. 2011;178:362–369. doi: 10.1016/j.resp.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, He L, Stensaas L, Dinger B, Fidone S. Adaptation to chronic hypoxia involves immune cell invasion and increased expression of inflammatory cytokines in rat carotid body. American journal of physiology Lung cellular and molecular physiology. 2009;296:66. doi: 10.1152/ajplung.90383.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Kuner T, Kuner R. Synaptic plasticity in pathological pain. Trends in neurosciences. 2014;37:343–355. doi: 10.1016/j.tins.2014.04.002. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mayer CA, Litvin DG. Microglia modulate brainstem serotonergic expression following neonatal sustained hypoxia exposure: implications for Sudden Infant Death Syndrome. J Physiol Dec. 2015;12 doi: 10.1113/JP271845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RJ, Di Fiore JM, Macfarlane PM, Wilson CG. Physiologic basis for intermittent hypoxic episodes in preterm infants. Advances in experimental medicine and biology. 2012;758:351–358. doi: 10.1007/978-94-007-4584-1_47. [DOI] [PubMed] [Google Scholar]
- Master ZR, Kesavan K, Mason A, Shirahata M, Gauda EB. Effect of Lipopolysaccharide Exposure on Structure and Function of the Carotid Body in Newborn Rats. Advances in experimental medicine and biology. 2015;860:115–121. doi: 10.1007/978-3-319-18440-1_13. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS. Altitude, exercise and immune function. Exerc Immunol Rev. 2005;11:6–16. [PubMed] [Google Scholar]
- Mazzeo RS, Donovan D, Fleshner M, Butterfield GE, Zamudio S, Wolfel EE, Moore LG. Interleukin-6 response to exercise and high-altitude exposure: influence of alpha-adrenergic blockade. J Appl Physiol (1985) 2001;91:2143–2149. doi: 10.1152/jappl.2001.91.5.2143. [DOI] [PubMed] [Google Scholar]
- McDeigan GE, Ladino J, Hehre D, Devia C, Bancalari E, Suguihara C. The Effect of Escherichia coli Endotoxin Infusion on the Ventilatory Response to Hypoxia in Unanesthetized Newborn Piglets. Pediatric Research. 2003;53:950–955. doi: 10.1203/01.PDR.0000064581.94126.1C. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. Journal of applied physiology (Bethesda, Md: 1985) 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGregor KH, Gil J, Lahiri S. A morphometric study of the carotid body in chronically hypoxic rats. J Appl Physiol. 1984;57:1430–1438. doi: 10.1152/jappl.1984.57.5.1430. [DOI] [PubMed] [Google Scholar]
- McKelvey R, Berta T, Old E, Ji RRR, Fitzgerald M. Neuropathic pain is constitutively suppressed in early life by anti-inflammatory neuroimmune regulation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:457–466. doi: 10.1523/JNEUROSCI.2315-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- McNamara F, Sullivan CE. Pediatric origins of adult lung diseases. 3: the genesis of adult sleep apnoea in childhood. Thorax. 2000;55:964–969. doi: 10.1136/thorax.55.11.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. The European journal of neuroscience. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Möller T, Bard F, Bhattacharya A, Biber K, Campbell B, Dale E, Eder C, Gan L, Garden GA, Hughes ZA, Pearse DD, Staal RG, Sayed FA, Wes PD, Boddeke HW. Critical data-based re-evaluation of minocycline as a putative specific microglia inhibitor. Glia. 2016 Jun 1; doi: 10.1002/glia.23007. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Moss A, Beggs S, Vega-Avelaira D, Costigan M, Hathway GJ, Salter MW, Fitzgerald M. Spinal microglia and neuropathic pain in young rats. Pain. 2007;128:215–224. doi: 10.1016/j.pain.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Motro B, Itin A, Sachs L, Keshet E. Pattern of interleukin 6 gene expression in vivo suggests a role for this cytokine in angiogenesis. Proc Natl Acad Sci U S A. 1990;87:3092–3096. doi: 10.1073/pnas.87.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouihate A, Galic MA, Ellis SL, Spencer SJ, Tsutsui S, Pittman QJ. Early life activation of toll-like receptor 4 reprograms neural anti-inflammatory pathways. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:7975–7983. doi: 10.1523/JNEUROSCI.6078-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson NR, Bird IM, Behan M. Testosterone restores respiratory long term facilitation in old male rats by an aromatase-dependent mechanism. The Journal of physiology. 2011 doi: 10.1113/jphysiol.2010.198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemzek JA, Hugunin KM, Opp MR. Modeling sepsis in the laboratory: merging sound science with animal well-being. Comparative medicine. 2008;58:120–128. [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Burkart V, Flohé S. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. The Journal of Immunology. 2000 doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Old E, Clark A, Malcangio M. The Role of Glia in the Spinal Cord in Neuropathic and Inflammatory Pain. In: Schaible HG, editor. Pain Control. Springer; Berlin Heidelberg: 2015. pp. 145–170. [DOI] [PubMed] [Google Scholar]
- Pamenter ME, Powell FL. Time domains of the hypoxic ventilatory response and their molecular basis. Compr Physiol. 2016;6:1345–1385. doi: 10.1002/cphy.c150026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. Journal of Applied Physiology. 2004 doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Raghuraman G, Khan SA. Angiotensin II evokes sensory long-term facilitation of the carotid body via NADPH oxidase. Journal of Applied …. 2011 doi: 10.1152/japplphysiol.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. The Journal of …. 2006 doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman H, Budinger GR, Ward PA. Humanizing the mouse: in defense of murine models of critical illness. American journal of respiratory and critical care medicine. 2013;187:898–900. doi: 10.1164/rccm.201303-0489ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters ME, Kimyon RS, Mitchell GS, Watters JJ. Repetitive acute intermittent hypoxia does not promote generalized inflammatory gene expression in the rat CNS. Respiratory Physiology & Neurobiology. 2015;218:1–10. doi: 10.1016/j.resp.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethő G, Reeh PW. Sensory and Signaling Mechanisms of Bradykinin, Eicosanoids, Platelet-Activating Factor, and Nitric Oxide in Peripheral Nociceptors. Physiological Reviews. 2012;92:1699–1775. doi: 10.1152/physrev.00048.2010. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poets CF, Samuels MP, Southall DP. Epidemiology and pathophysiology of apnoea of prematurity. Biology of the neonate. 1994;65:211–219. doi: 10.1159/000244055. [DOI] [PubMed] [Google Scholar]
- Poets CF, Southall DP. Recent developments in research into sudden infant death. Thorax. 1994;49:196–197. doi: 10.1136/thx.49.3.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa D, Fu Z, Go A, Powell FL. Ibuprofen blocks time-dependent increases in hypoxic ventilation in rats. Respir Physiol Neurobiol. 2011;178:381–386. doi: 10.1016/j.resp.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK, Nanduri J. Comprehensive Physiology Comprehensive Physiology. 2015;5 doi: 10.1002/cphy.c140039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Semenza GL. Oxygen Sensing and Homeostasis. Physiology (Bethesda) 2015;30:340–348. doi: 10.1152/physiol.00022.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience. 2015;302:2–22. doi: 10.1016/j.neuroscience.2015.06.038. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JSS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantakallio P, Jones P, Moring J, Von Wendt L. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol. 1997;26:837–843. doi: 10.1093/ije/26.4.837. [DOI] [PubMed] [Google Scholar]
- Redl H, Bahrami S, Schlag G, Traber DL. Clinical detection of LPS and animal models of endotoxemia. Immunobiology. 1993;187:330–345. doi: 10.1016/S0171-2985(11)80348-7. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Current opinion in pharmacology. 2015;26:16–25. doi: 10.1016/j.coph.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Padova DF. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA, Simons CT, Székely M, Kulchitsky VA. The vagus nerve in the thermoregulatory response to systemic inflammation. The American journal of physiology. 1997;273:13. doi: 10.1152/ajpregu.1997.273.1.R407. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sawhney RC, Malhotra AS, Singh T. Glucoregulatory hormones in man at high altitude. Eur J Appl Physiol Occup Physiol. 1991;62:286–291. doi: 10.1007/BF00571554. [DOI] [PubMed] [Google Scholar]
- Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- Scheuren N, Bang H, Munster T, Brune K, Pahl A. Modulation of transcription factor NF-kappaB by enantiomers of the nonsteroidal drug ibuprofen. Br J Pharmacol. 1998;123:645–652. doi: 10.1038/sj.bjp.0701652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller F, Fitzgerald M. The consequences of pain in early life: injury-induced plasticity in developing pain pathways. The European journal of neuroscience. 2014;39:344–352. doi: 10.1111/ejn.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. The Immune System and the Developing Brain. Colloquium Series on The Developing Brain. 2011;2:1–128. [Google Scholar]
- Seok J, Warren SH, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb PJ, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Abouhamze A, Balis UGJ, Camp DG, De AK, Harbrecht BG, Hayden DL, Kaushal A, O'Keefe GE, Kotz KT, Qian W, Schoenfeld DA, Shapiro MB, Silver GM, Smith RD, Storey JD, Tibshirani R, Toner M, Wilhelmy J, Wispelwey B, Wong WH. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks N, Windle RJ, Perks PA, Harbuz MS, Jessop DS, Ingram CD, Lightman SL. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5645–5650. doi: 10.1073/pnas.090571897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HFF, Wang BRR, Wang SRR, Yao W, Huang HPP, Zhou Z, Wang X, Fan J, Wang T, Ju G. IL-1beta inhibits IK and increases [Ca2+]i in the carotid body glomus cells and increases carotid sinus nerve firings in the rat. The European journal of neuroscience. 2007;25:3638–3647. doi: 10.1111/j.1460-9568.2007.05586.x. [DOI] [PubMed] [Google Scholar]
- Siljehav V, Hofstetter AM, Leifsdottir K, Herlenius E. Prostaglandin E2 Mediates Cardiorespiratory Disturbances during Infection in Neonates. The Journal of pediatrics. 2015 doi: 10.1016/j.jpeds.2015.08.053. [DOI] [PubMed] [Google Scholar]
- Smith SM, Friedle SA, Watters JJ. Chronic intermittent hypoxia exerts CNS region-specific effects on rat microglial inflammatory and TLR4 gene expression. PloS one. 2013;8 doi: 10.1371/journal.pone.0081584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, Galic MA, Pittman QJ. Neonatal programming of innate immune function. American journal of physiology. Endocrinology and metabolism. 2011;300:8. doi: 10.1152/ajpendo.00516.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, Martin S, Mouihate A, Pittman QJ. Early-life immune challenge: defining a critical window for effects on adult responses to immune challenge. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2006;31:1910–1918. doi: 10.1038/sj.npp.1301004. [DOI] [PubMed] [Google Scholar]
- Stanimirovic D, Zhang W, Howlett C, Lemieux P, Smith C. Inflammatory gene transcription in human astrocytes exposed to hypoxia: roles of the nuclear factor-kappaB and autocrine stimulation. J Neuroimmunol. 2001;119:365–376. doi: 10.1016/s0165-5728(01)00402-7. [DOI] [PubMed] [Google Scholar]
- Stemkowski PL, Smith PA. Sensory neurons, ion channels, inflammation and the onset of neuropathic pain. Can J Neurol Sci. 2012;39:416–435. doi: 10.1017/s0317167100013937. [DOI] [PubMed] [Google Scholar]
- Stokes JA, Corr M, Yaksh TL. Spinal toll-like receptor signaling and nociceptive processing: regulatory balance between TIRAP and TRIF cascades mediated by TNF and IFNbeta. Pain. 2013;154:733–742. doi: 10.1016/j.pain.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, Oh W, Bauer CR, Korones SB, Shankaran S, Laptook AR, Stevenson DK, Papile LA, Poole WK. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N Engl J Med. 2002;347:240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- Stoll BJ, Hansen NI, Adams-Chapman I, Fanaroff AA, Hintz SR, Vohr B, Higgins RD National Institute of Child, H., Human Development Neonatal Research, N. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. Jama. 2004;292:2357–2365. doi: 10.1001/jama.292.19.2357. [DOI] [PubMed] [Google Scholar]
- Stuhlmeier KM, Li H, Kao JJ. Ibuprofen: new explanation for an old phenomenon. Biochem Pharmacol. 1999;57:313–320. doi: 10.1016/s0006-2952(98)00301-3. [DOI] [PubMed] [Google Scholar]
- Tadmouri A, Champagnat J, Morin-Surun MP. Activation of microglia and astrocytes in the nucleus tractus solitarius during ventilatory acclimatization to 10% hypoxia in unanesthetized mice. Journal of neuroscience research. 2014;92:627–633. doi: 10.1002/jnr.23336. [DOI] [PubMed] [Google Scholar]
- Tolle LB, Standiford TJ. Danger-associated molecular patterns (DAMPs) in acute lung injury. The Journal of Pathology. 2013;229:145–156. doi: 10.1002/path.4124. [DOI] [PubMed] [Google Scholar]
- Vinit S, Windelborn JA, Mitchell GS. Lipopolysaccharide attenuates phrenic long-term facilitation following acute intermittent hypoxia. Respiratory physiology & neurobiology. 2011;176:130–135. doi: 10.1016/j.resp.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FR, March J, Hodgson DM. Endotoxin exposure in early life alters the development of anxiety-like behaviour in the Fischer 344 rat. Behavioural brain research. 2004;154:63–69. doi: 10.1016/j.bbr.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZY, Bisgard GE. Chronic hypoxia-induced morphological and neurochemical changes in the carotid body. Microsc Res Tech. 2002;59:168–177. doi: 10.1002/jemt.10191. [DOI] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Tolllike receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature Medicine. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain research. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- Wiertelak EP, Smith KP, Furness L, Mooney-Heiberger K, Mayr T, Maier SF, Watkins LR. Acute and conditioned hyperalgesic responses to illness. Pain. 1994;56:227–234. doi: 10.1016/0304-3959(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Huey K, Dinger B, He L, Fidone S, Powell FL. Chronic hypoxia increases the gain of the hypoxic ventilatory response by a mechanism in the central nervous system. J Appl Physiol (1985) 2010;109:424–430. doi: 10.1152/japplphysiol.01311.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Wuchert F, Ott D, Murgott J, Rafalzik S, Hitzel N, Roth J, Gerstberger R. Rat area postrema microglial cells act as sensors for the toll-like receptor-4 agonist lipopolysaccharide. Journal of neuroimmunology. 2008;204:66–74. doi: 10.1016/j.jneuroim.2008.07.017. [DOI] [PubMed] [Google Scholar]
- Wuchert F, Ott D, Rafalzik S, Roth J, Gerstberger R. Tumor necrosis factor-alpha, interleukin-1beta and nitric oxide induce calcium transients in distinct populations of cells cultured from the rat area postrema. Journal of neuroimmunology. 2009;206:44–51. doi: 10.1016/j.jneuroim.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274:27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, Behavior, and Immunity. 2010;25 doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Zabka AG, Behan M. Selected contribution: Time-dependent hypoxic respiratory responses in female rats are influenced by age and by the estrus cycle. Journal of Applied …. 2001 doi: 10.1152/jappl.2001.91.6.2831. [DOI] [PubMed] [Google Scholar]
- Zabka AG, Behan M, Mitchell GS. Long term facilitation of respiratory motor output decreases with age in male rats. The Journal of Physiology. 2001 doi: 10.1111/j.1469-7793.2001.0509i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabka AG, Mitchell GS, Olson EB. Selected contribution: chronic intermittent hypoxia enhances respiratory long-term facilitation in geriatric female rats. Journal of Applied …. 2003 doi: 10.1152/japplphysiol.00476.2003. [DOI] [PubMed] [Google Scholar]
- Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med (Berl) 2007;85:1339–1346. doi: 10.1007/s00109-007-0282-2. [DOI] [PubMed] [Google Scholar]
- Zouikr I, Tadros MA, Barouei J, Beagley KW, Clifton VL, Callister RJ, Hodgson DM. Altered nociceptive, endocrine, and dorsal horn neuron responses in rats following a neonatal immune challenge. Psychoneuroendocrinology. 2014;41:1–12. doi: 10.1016/j.psyneuen.2013.11.016. [DOI] [PubMed] [Google Scholar]