

Abstract
Background
The prevalence of primary carnitine deficiency (PCD) in the Faroe Islands is the highest reported in the world (1:300). Serious symptoms related to PCD, e.g. sudden death, have previously only been associated to the c.95A > G/c.95A > G genotype in the Faroe Islands. We report and characterize novel mutations associated with PCD in the Faroese population and report and compare free carnitine levels and OCTN2 transport activities measured in fibroblasts from PCD patients with different genotypes.
Methods
Genetic analyses were used to identify novel mutations, and carnitine uptake analyses in cultured skin fibroblasts from selected patients were used to examine residual OCTN2 transporter activities of the various genotypes.
Results
Four different mutations, including the unpublished c.131C > T (p.A44V), the novel splice mutation c.825-52G > A and a novel risk-haplotype (RH) were identified in the Faroese population. The two most prevalent genotypes were c.95A > G/RH (1:600) and c.95A > G/c.95A > G (1:1300). Patients homozygous for the c.95A > G mutation had both the significantly (p < 0.01) lowest mean free carnitine level at 2.03 (SD 0.66) μmol/L and lowest residual OCTN2 transporter activity (4% of normal). There was a significant positive correlation between free carnitine levels and residual OCTN2 transporter activities in PCD patients (R2 = 0.430, p < 0.01).
Conclusion
There was a significant positive correlation between carnitine levels and OCTN2 transporter activities. The c.95A > G/c.95A > G genotype had the significantly lowest mean free carnitine level and residual OCTN2 transporter activity.
Keywords: Primary carnitine deficiency, OCTN2, SLC22A5, The Faroe Islands
1. Introduction
A nationwide screening programme was initiated in the Faroe Islands in 2009, to uncover undiagnosed children and adults suffering from primary carnitine deficiency (PCD, OMIM #212140), following several episodes of sudden death among young Faroese individuals diagnosed post-mortem with PCD [1], [2]. Analysis of data from the screening programme revealed that the prevalence of PCD in the Faroe Islands, a small island community in the North Atlantic Ocean with a population of approximately 50,000 inhabitants, was by far the highest reported in the world (1:300) [2], [3], [4].
PCD is an autosomal recessive disorder of fatty acid β-oxidation caused by a lack of functional organic cation transporter 2 (OCTN2) transporters, which transport carnitine from the extracellular to the intracellular space and also prevent excretion of carnitine in urine [3], [5]. Carnitine is involved in the transfer of long chain fatty acids across the inner mitochondrial membrane for β-oxidation and also participates in several other important cellular processes [6], [7], [8]. Symptoms of PCD range from none to severe cardiac symptoms with cardiomyopathy and arrhythmia, although the most frequent symptom is fatigue [1], [9].
OCTN2 is encoded by the SLC22A5 gene, which is ubiquitously expressed. More than 150 mutations have been reported [5], [10].
Serious symptoms related to PCD, e.g. severe cardiac arrhythmia, reported previously in Faroese patients, only occurred in patients homozygous for the well known c.95A > G (p.N32S) mutation [1], [9]. Although the severity and susceptibility to serious symptoms may differ between the genotypes, all patients with PCD are treated with L-carnitine in the Faroe Islands.
We here report novel SLC22A5 mutations in the Faroe Islands, as well as mean free carnitine levels and OCTN2 transport activities associated with the most common genotypes. We show that severe symptoms, levels of free carnitine and OCTN2 transport activities all correlate with genotype.
2. Materials and methods
2.1. Subjects
The nationwide screening programme included 26,462 individuals or approximately 55% of the total population in the Faroe Islands from August 2009 to January 2011 [2]. Free carnitine was measured in whole blood by tandem mass spectrometry as described previously [2]. Patients verified by genetic analysis to be homozygous or compound heterozygous for mutations causing PCD were included. Heterozygotes and subjects without PCD, who were included in the analyses of carnitine levels and fibroblast studies, were all genetically verified before inclusion. Subjects designated without PCD were though only analysed for the prevalent c.95A > G mutation.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
2.2. Genetic analysis
Molecular genetic analysis was performed in all individuals with carnitine levels below 7 μmol/L [2]. Patients without obvious mutations were haplotyped using the marker D5S1984 located 110 kb upstream of SLC22A5 and a highly polymorphic TG repeat located 50 kb 3′ to SLC22A5 at chr5_131.779.669-131.779.956. Primers and conditions are available upon request.
Five patients homozygous for a risk-haplotype (RH) had their entire SLC22A5 locus (approx. 25 kb) sequenced by both Sanger sequencing (one patient) and next generation sequencing (five patients).
2.3. Functional analysis
Carnitine uptake in cultured skin fibroblasts to determine residual OCTN2 transporter activity was studied essentially as described previously [11] in 21 PCD patients with four different genotypes, six carrier subjects heterozygous for the c.95A > G mutation and five subjects without SLC22A5 aberrations (wildtype/wildtype) (Table 2). Fibroblasts were grown in a medium containing Ham F-10 supplemented with 15% fetal calf serum (FCS). The cells were subcultured into 6-well multidishes. Before performing the carnitine uptake studies, the medium was withdrawn and the cell layer was washed three times with phosphate-buffered saline (PBS). PBS (1 mL) containing 3.5 μmol/L L-[methyl-14C]carnitine was added to the cell-layer in each well and incubation at 37 °C was performed for 4 h. All assays were performed in duplicate. A carnitine concentration of 10 mmol/L was used to correct for non-specific uptake. Following incubation discontinuation by removal of the medium the cells were quickly washed six times with ice-cold PBS. The cells were dissolved in 1 mL of 0.2 mol/L NaOH and incubated overnight at 4 °C; 0.75 mL was added to 10 mL Instagel and, after a few hours at room temperature for decay of chemiluminescence, the 14C content was determined in a β-scintillation counter. The amount of protein was determined on two 100 μL aliquots. Carnitine uptake was expressed as pmol/min per mg protein.
Table 2.
Fibroblast analysis in different genotypes to determine residual OCTN2 activity (pmol/min per mg protein). *Genotype not included in statistical analyses due to low number of patients. RH: risk-haplotype. Wildtype: no c.95A > G mutation found. Note, fibroblast analysis was not performed in patients with c.95A > G/c.825-52G > A.
| Genotype | n | Mean | SD | SEM | Minimum | Maximum | 95% CI for mean |
% of normal (wildtype) | |
|---|---|---|---|---|---|---|---|---|---|
| Lower bound | Upper bound | ||||||||
| c.95A > G/c.95A > G | 6 | 0.02 | 0.02 | 0.01 | 0.0 | 0.06 | 0.01 | 0.04 | 4 |
| c.95A > G/RH | 7 | 0.11 | 0.03 | 0.01 | 0.07 | 0.16 | 0.08 | 0.14 | 18 |
| RH/RH | 6 | 0.18 | 0.05 | 0.02 | 0.08 | 0.22 | 0.12 | 0.23 | 29 |
| c.95A > G/wildtype | 6 | 0.29 | 0.14 | 0.06 | 0.20 | 0.55 | 0.15 | 0.43 | 46 |
| wildtype/wildtype | 5 | 0.62 | 0.10 | 0.04 | 0.50 | 0.74 | 0.50 | 0.75 | 100 |
| c.825-52G > A/c.825-52G > A* | 2 | 0.1 | 0.07 | 0.05 | 0.05 | 0.15 | 0.00 | 0.74 | - |
2.4. Statistical analysis
Data analysis was performed using IBM® SPSS® Statistics Version 19 (SPSS Inc., Chicago, IL, USA). Student's t-test was used to test for a significant difference in mean free carnitine and mean residual OCTN2 transporter activity between selected genotypes. Level of significance was set at p < 0.05 (two tailed test).
Linear regression analysis was used to check for correlation between free carnitine and OCTN2 transport activity in four different PCD genotypes as well as between mean free carnitine levels and mean OCTN2 transporter activities in PCD genotypes, c.95A > G heterozygotes and subjects without PCD.
3. Results
3.1. PCD mutations in the Faroe Islands
Screening for PCD among 26,462 individuals identified 89, 46 male and 43 female, with a mean age of 34 (range 2–79) years [2]. All patients were verified by genetic analysis to be either homozygous or compound heterozygous for four different PCD related mutations and a risk-haplotype. The two published missense mutations c.95A > G (p.N32S) [12] and c.136C > T (p.P46S) [13], the unpublished c.131C > T (p.A44V) and the novel splice-mutation c.825-52G > A were identified. The splice-mutation created a novel 3′ acceptor site leading to inclusion of 50 additional nucleotides in the mRNA and hence a shift in reading frame. cDNA analyses verified the assumed effect, but also showed that the alternative splicing was not complete, allowing for some normal splicing to occur (data not shown).
In a subset of patients with very low free carnitine (< 5 μmol/L) only a single heterozygous SLC22A5 mutation could be identified. Haplotype and segregation analysis using two closely linked microsatellites showed that these patients shared the same haplotype on the mutation negative allele, which we named the RH.
We subsequently identified several individuals with reduced levels of free carnitine without a SLC22A5 mutation who were homozygous for the RH (n = 8). Five of these patients had the entire SLC22A5 locus sequenced (approx. 25 kb), but we were unable to detect a novel or pathogenic variant.
The two genotypes with the highest number of patients were c.95A > G/RH (n = 46; prevalence ≈ 1:600) and c.95A > G/c.95A > G (n = 20; prevalence ≈ 1:1300). The number of patients with each genotype discovered in the Faroe Islands is presented in Table 1.
Table 1.
Mean free carnitine in different PCD genotypes, carriers and people without PCD related mutations. *Genotype not included in statistical analyses due to low number of patients. **Two patients were excluded in the analysis because they had taken L-carnitine supplementation when tested. Three patients with other genotypes were not included in the analysis. RH: risk-haplotype. Wildtype: no c.95A > G mutation found.
| Genotype |
n (total) |
n (included in analysis) |
Mean fC0 |
SD |
Minimum |
Maximum |
95% CI for mean |
|
|---|---|---|---|---|---|---|---|---|
| μmol/L | Lower bound | Upper bound | ||||||
| c.95A > G/c.95A > G | 20 | 20 | 2.03 | 0.66 | 1.25 | 3.34 | 1.72 | 2.34 |
| c.95A > G/RH | 46 | 44** | 3.52 | 0.86 | 1.67 | 5.63 | 3.26 | 3.78 |
| c.95A > G/c.825-52G > A | 10 | 10 | 4.22 | 1.91 | 2.09 | 8.16 | 2.85 | 5.59 |
| RH/RH | 8 | 8 | 5.42 | 0.79 | 4.09 | 6.31 | 4.76 | 6.08 |
| c.95A > G/wildtype | 94 | 94 | 13.02 | 4.12 | 5.60 | 25.34 | 12.18 | 13.87 |
| wildtype/wildtype | 302 | 302 | 21.48 | 6.19 | 7.36 | 44.98 | 20.77 | 22.18 |
| c.825-52G > A/c.825-52G > A* | 2 | 0 | 4.8 | – | 4.00 | 5.60 | – | – |
| c.136C > T/c.825-52G > A* | 1 | 0 | 1.8 | – | – | – | – | – |
| c.95A > G/c.136C > T* | 1 | 0 | 3.2 | – | – | – | – | – |
| c.131C > T/RH* | 1 | 0 | 4.0 | – | – | – | – | – |
3.2. Carnitine level and carnitine uptake
Fig. 1 shows a significant positive correlation between free carnitine levels and residual OCTN2 transporter activities in cultured skin fibroblasts from patients with four different PCD genotypes (R2 = 0.430, p < 0.01). There was also a significant positive correlation between mean free carnitine levels and mean OCTN2 transport activities in PCD genotypes, c.95A > G heterozygotes and subjects without PCD (R2 = 0.968, p < 0.01, Fig. 2). Mean free carnitine was significantly lowest in patients homozygous for the c.95A > G mutation, mean 2.03 μmol/L, followed by those compound heterozygous for the c.95A > G mutation and RH, mean 3.52 μmol/L, (p < 0.01, Table 1). A significant difference in mean free carnitine was also present between patients with PCD and c.95A > G heterozygotes (p < 0.01) as well as between c.95A > G heterozygotes and subjects without a PCD related mutation (p < 0.01, Table 1).
Fig. 1.
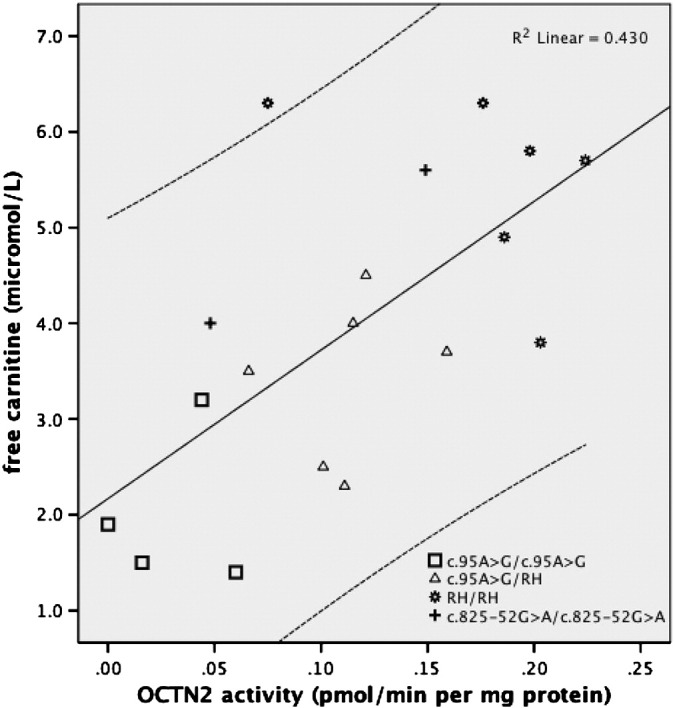
OCTN2 activities plotted against free carnitine levels in four different PCD genotypes.
Fig. 2.
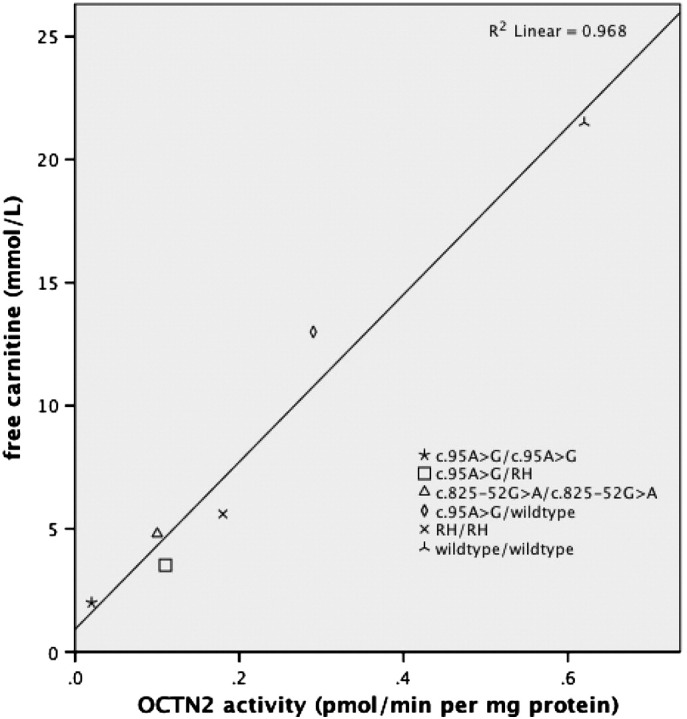
Mean fibroblast carnitine uptake activity plotted against mean free carnitine levels in different genotypes, c.95A > G heterozygotes and subjects without PCD (wildtype/wildtype).
The carnitine uptake studies in cultured skin fibroblasts showed that the mean residual OCTN2 transporter activity was lowest in c.95A > G homozygotes with a mean of 0.02 pmol/min per mg protein or 4% of normal followed by the c.95A > G/RH patients, mean 0.11 pmol/min per mg protein or 18% of normal (p < 0.01, Fig. 2, Table 2). c.95A > G heterozygotes had a significantly lower residual transporter activity, mean 0.29 or 46% of normal, compared to individuals without a PCD related mutation, mean 0.62, (p < 0.01, Fig. 2, Table 2).
4. Discussion
We can for the first time report a significant positive correlation between residual OCTN2 transporter activities in fibroblasts and free carnitine levels in PCD patients with different genotypes (Fig. 1). There was thus a direct correlation between the functional capacity of the OCTN2 transporter and the level of carnitine in PCD patients. There was also a significant positive correlation between mean OCTN2 transporter activities and mean free carnitine levels in PCD patients, c.95A > G heterozygotes and subjects without PCD (Fig. 2).
All but three Faroese PCD patients were either homozygous or compound heterozygous for the c.95A > G mutation, the novel c.825-52G > A mutation or the novel RH (Table 1). Patients homozygous for c.95A > G had a very low residual OCTN2 transport activity of only 4% compared to normal (Table 2). Residual OCTN2 transport activity measured in cultured fibroblasts from c.95A > G homozygous patients has previously been reported twice in the literature—in an Asian girl with 21% residual activity and in a Faroese boy with 3% compared to normal [12], [14]. This discrepancy between the Asian girl and the Faroese patients could be due to a difference in promoter polymorphisms influencing transcriptional regulation between ethnic groups [15]. Our data show that the c.95A > G homozygous genotype must be considered severe. Furthermore, previous reports from the Faroe Islands of severe symptoms related to PCD, e.g. cardiac arrest, have all occurred in c.95A > G homozygous patients, indicating a correlation between very low OCTN2 transporter activity/low carnitine levels and severe symptoms in PCD patients, although there have been previous conflicting reports in the literature about such a correlation [1], [9], [12], [16], [17], [18], [19].
The genotype c.95A > G/RH was the most prevalent PCD related genotype in the Faroe Islands (Table 1). Eight patients with low carnitine levels were found to be homozygous for the RH. Although the entire 25 kb SLC22A5 locus was sequenced no novel or obvious pathogenic aberration could be associated with the RH. Nor did extensive cDNA analysis reveal any obvious missplicing (data not shown). As the RH undoubtedly is associated with decreased carnitine levels and OCTN2 transporter activity, we assume that the underlying genetic aberration is located in an enhancer element closely linked to SLC22A5.
c.95A > G heterozygotes are prevalent in the Faroe Islands and their mean residual OCTN2 transporter activity and mean free carnitine level are approximately half of those in individuals without PCD (Fig. 2). Reduced OCTN2 transporter activities and carnitine levels in heterozygotes have been reported previously with comparable values [20], [21], [22], [23], [24], [25]. A few reports in the literature have indicated an adverse effect, especially cardiac, related to PCD carriers in both humans and in animal models [26], [27], [28]. A large study of a cohort of people with hypertrophic cardiomyopathy did not support such a relationship and our previous findings of very limited structural cardiac involvement in patients homozygous for PCD indicate that a causal relationship between PCD carrier status and cardiac involvement is unlikely [9], [29]. PCD carriers should though avoid carnitine-lowering drugs. Individuals diagnosed as carriers of a PCD related mutation are not prescribed L-carnitine supplementation in the Faroe Islands—still many heterozygotes have tried over-the-counter L-carnitine and some report improved stamina and reduced symptoms of fatigue. Meanwhile some PCD patients diagnosed with especially less severe genotypes have not experienced any symptoms prior to L-carnitine supplementation and no effect of the treatment [9]. This leads to the question if all patients diagnosed either as homozygous or compound heterozygous for PCD mutations should be routinely treated with large doses of L-carnitine supplementation, which might have long-term adverse effects? [30]
The clear indication of a genotype–phenotype association with the genotype c.95A > G/c.95A > G exhibiting the significantly lowest mean residual OCTN2 transporter activity and mean carnitine levels, while also being the only genotype in the Faroe Islands associated with severe symptoms, further highlights the question, if all genotypes should be treated the same way. A study is underway in the Faroe Islands to investigate if previous sudden deaths among younger individuals can be attributed to other genotypes than homozygosity for c.95A > G. If this proves not to be the case, then treatment in the Faroe Islands might primarily be focused towards the most serious genotype, while treatment of the other less severe genotypes could be individualized towards relieving minor symptoms, e.g. fatigue. How in general to define and classify different genotypes with regards to severity though remains a question.
Implementation of neonatal screening programmes around the world to detect disorders of fatty acid oxidation has led to an increase in the diagnosis of both infants and previously undiagnosed symptomatic and asymptomatic mothers with PCD [19]. The need for an individualized treatment strategy thus seems to be growing, which would ideally take into account the genotypic properties and lack or presence of PCD related symptoms in the patients.
5. Conclusion
We have found a significant positive correlation between carnitine levels and OCTN2 transport activities. Homozygosity for c.95A > G causes the significantly lowest carnitine levels and residual OCTN2 transport activity and is solely associated with severe symptoms in the Faroese population indicating a genotype–phenotype correlation. Future treatment strategies for patients suffering from PCD might be focused towards patients with severe genotypes, while L-carnitine supplementation in other patients could be individualized in order to relieve possible minor symptoms.
6. Limitations
As there were only two patients homozygous for the c.825-52G > A splice mutation and three patients with other genotypes they were not considered when analysing statistically a potential difference among the genotypes; however we do not believe this to have any influence on the conclusions drawn. Carnitine uptake analysis was not performed on patients compound heterozygous for the c.95A > G mutation and the c.825-52G > A splice mutation, but we do not expect these patients to have a lower residual OCTN2 activity than patients homozygous for the c.95A > G mutation. Subjects designated without PCD were only examined for the most prevalent and severe c.95A > G mutation—there might potentially be some subjects with other PCD related mutations among the cohort; this would though only have a marginal affect.
Conflict of interest
Jan Rasmussen, Allan M. Lund, Lotte Risom, Flemming Wibrand, Hannes Gislason, Lars Køber, Olav W. Nielsen and Morten Dunø have no conflicts of interest to report.
Proof that informed consent was obtained is available upon request
Acknowledgments
The authors would like to thank the Genetic Biobank in the Faroe Islands for its support and the Faroese National Research Council.
References
- 1.Rasmussen J., Nielsen O.W., Lund A.M., Kober L., Djurhuus H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J. Inherit. Metab. Dis. 2013;36:35–41. doi: 10.1007/s10545-012-9488-8. [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen O.W. Nielsen, Janzen N., Duno M., Kober L., Steuerwald U., Lund A.M. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J. Inherit. Metab. Dis. 2013;37:215–222. doi: 10.1007/s10545-013-9606-2. [DOI] [PubMed] [Google Scholar]
- 3.Longo N., Amat di San Filippo C., Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C. Semin. Med. Genet. 2006;142C:77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magoulas P.L., El-Hattab A.W. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2012;7:68. doi: 10.1186/1750-1172-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nezu J., Tamai I., Oku A., Ohashi R., Yabuuchi H., Hashimoto N., Nikaido H., Sai Y., Koizumi A., Shoji Y., Takada G., Matsuishi T., Yoshino M., Kato H., Ohura T., Tsujimoto G., Hayakawa J., Shimane M., Tsuji A. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999;21:91–94. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell M.E. Carnitine metabolism in human subjects. I. Normal metabolism. Am. J. Clin. Nutr. 1978;31:293–306. doi: 10.1093/ajcn/31.2.293. [DOI] [PubMed] [Google Scholar]
- 7.Rebouche C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004;1033:30–41. doi: 10.1196/annals.1320.003. [DOI] [PubMed] [Google Scholar]
- 8.Steiber A., Kerner J., Hoppel C.L. Carnitine: a nutritional, biosynthetic, and functional perspective. Mol. Aspects Med. 2004;25:455–473. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen L. Kober, Lund A.M., Nielsen O.W. Primary Carnitine deficiency in the Faroe Islands: health and cardiac status in 76 adult patients diagnosed by screening. J. Inherit. Metab. Dis. 2013;37:223–230. doi: 10.1007/s10545-013-9640-0. [DOI] [PubMed] [Google Scholar]
- 10.ARUP . University of Utah; 2013. SLC22A5 Database. [Google Scholar]
- 11.Christensen E., Vikre-Jorgensen J. Six years' experience with carnitine supplementation in a patient with an inherited defective carnitine transport system. J. Inherit. Metab. Dis. 1995;18:233–236. doi: 10.1007/BF00711776. [DOI] [PubMed] [Google Scholar]
- 12.Lamhonwah A.M., Olpin S.E., Pollitt R.J., Vianey-Saban C., Divry P., Guffon N., Besley G.T., Onizuka R., De Meirleir L.J., Cvitanovic-Sojat L., Baric I., Dionisi-Vici C., Fumic K., Maradin M., Tein I. Novel OCTN2 mutations: no genotype–phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am. J. Med. Genet. 2002;111:271–284. doi: 10.1002/ajmg.10585. [DOI] [PubMed] [Google Scholar]
- 13.Schimmenti L.A., Crombez E.A., Schwahn B.C., Heese B.A., Wood T.C., Schroer R.J., Bentler K., Cederbaum S., Sarafoglou K., McCann M., Rinaldo P., Matern D., di San Filippo C.A., Pasquali M., Berry S.A., Longo N. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol. Genet. Metab. 2007;90:441–445. doi: 10.1016/j.ymgme.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Lund A.M., Joensen F., Hougaard D.M., Jensen L.K., Christensen E., Christensen M., Norgaard-Petersen B., Schwartz M., Skovby F. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J. Inherit. Metab. Dis. 2007;30:341–349. doi: 10.1007/s10545-007-0527-9. [DOI] [PubMed] [Google Scholar]
- 15.Tahara H., Yee S.W., Urban T.J., Hesselson S., Castro R.A., Kawamoto M., Stryke D., Johns S.J., Ferrin T.E., Kwok P.Y., Giacomini K.M. Functional genetic variation in the basal promoter of the organic cation/carnitine transporters OCTN1 (SLC22A4) and OCTN2 (SLC22A5) J. Pharmacol. Exp. Ther. 2009;329:262–271. doi: 10.1124/jpet.108.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christensen E., Holm J., Hansen S.H., Sorensen N., Nezu J., Tsuji A., Skoby F. Sudden infant death following pivampicillin treatment in a patient with carnitine transporter deficiency. J. Inherit. Metab. Dis. 2000;23(Suppl. 1) [Google Scholar]
- 17.Osa E., Simonsen H. Carnitine transporter deficiency in two Faeroese children. Ugeskr. Laeger. 2004;166:4612–4613. [PubMed] [Google Scholar]
- 18.Wang Y., Taroni F., Garavaglia B., Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype–phenotype correlation. Hum. Mutat. 2000;16:401–407. doi: 10.1002/1098-1004(200011)16:5<401::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Rose E.C., di San Filippo C.A., Ndukwe Erlingsson U.C., Ardon O., Pasquali M., Longo N. Genotype–phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012;33:118–123. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komlosi K., Magyari L., Talian G.C., Nemes E., Kaposzta R., Mogyorosy G., Mehes K., Melegh B. Plasma carnitine ester profile in homozygous and heterozygous OCTN2 deficiency. J. Inherit. Metab. Dis. 2009;32(Suppl. 1):S15–S19. doi: 10.1007/s10545-009-0926-1. [DOI] [PubMed] [Google Scholar]
- 21.Stanley C.A., DeLeeuw S., Coates P.M., Vianey-Liaud C., Divry P., Bonnefont J.P., Saudubray J.M., Haymond M., Trefz F.K., Breningstall G.N. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann. Neurol. 1991;30:709–716. doi: 10.1002/ana.410300512. [DOI] [PubMed] [Google Scholar]
- 22.Tein I., De Vivo D.C., Bierman F., Pulver P., De Meirleir L.J., Cvitanovic-Sojat L., Pagon R.A., Bertini E., Dionisi-Vici C., Servidei S. Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr. Res. 1990;28:247–255. doi: 10.1203/00006450-199009000-00020. [DOI] [PubMed] [Google Scholar]
- 23.Eriksson B.O., Lindstedt S., Nordin I. Hereditary defect in carnitine membrane transport is expressed in skin fibroblasts. Eur. J. Pediatr. 1988;147:662–663. doi: 10.1007/BF00442488. [DOI] [PubMed] [Google Scholar]
- 24.Scaglia F., Wang Y., Singh R.H., Dembure P.P., Pasquali M., Fernhoff P.M., Longo N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet. Med. 1998;1:34–39. doi: 10.1097/00125817-199811000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Garavaglia B., Uziel G., Dworzak F., Carrara F., DiDonato S. Primary carnitine deficiency: heterozygote and intrafamilial phenotypic variation. Neurology. 1991;41:1691–1693. doi: 10.1212/wnl.41.10.1691. [DOI] [PubMed] [Google Scholar]
- 26.Xiaofei E., Wada Y., Dakeishi M., Hirasawa F., Murata K., Masuda H., Sugiyama T., Nikaido H., Koizumi A. Age-associated cardiomyopathy in heterozygous carrier mice of a pathological mutation of carnitine transporter gene, OCTN2. J. Gerontol. A Biol. Sci. Med. Sci. 2002:B270–B278. doi: 10.1093/gerona/57.7.b270. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi R., Asai T., Murakami H., Murakami R., Tsuzuki M., Numaguchi Y., Matsui H., Murohara T., Okumura K. Pressure overload-induced cardiomyopathy in heterozygous carrier mice of carnitine transporter gene mutation. Hypertension. 2007;50:497–502. doi: 10.1161/HYPERTENSIONAHA.107.088609. [DOI] [PubMed] [Google Scholar]
- 28.Sarafoglou K., Tridgell A.H., Bentler K., Redlinger-Grosse K., Berry S.A., Schimmenti L.A. Cardiac conduction improvement in two heterozygotes for primary carnitine deficiency on L-carnitine supplementation. Clin. Genet. 2010;78:191–194. doi: 10.1111/j.1399-0004.2009.01368.x. [DOI] [PubMed] [Google Scholar]
- 29.Amat di San Filippo C., Taylor M.R., Mestroni L., Botto L.D., Longo N. Cardiomyopathy and carnitine deficiency. Mol. Genet. Metab. 2008;94:162–166. doi: 10.1016/j.ymgme.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koeth R.A., Wang Z., Levison B.S., Buffa J.A., Org E., Sheehy B.T., Britt E.B., Fu X., Wu Y., Li L., Smith J.D., DiDonato J.A., Chen J., Li H., Wu G.D., Lewis J.D., Warrier M., Brown J.M., Krauss R.M., Tang W.H., Bushman F.D., Lusis A.J., Hazen S.L. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]