

Abstract
Patients with ARCL-IIA harbor mutations in ATP6V0A2 that codes for an organelle proton pump. The ARCL-IIA syndrome characteristically presents a combined glycosylation defect affecting N-linked and O-linked glycosylations, differentiating it from other cutis laxa syndromes and classifying it as a Congenital Disorder of Glycosylation (ATP6V0A2-CDG). We studied two Mexican Mestizo patients with a clinical phenotype corresponding to an ARCL-IIA syndrome. Both patients presented abnormal transferrin (N-linked) glycosylation but Patient 1 had a normal ApoCIII (O-linked) glycosylation profile. Mutational screening of ATP6V0A2 using cDNA and genomic DNA revealed in Patient 1 a previously reported homozygous nonsense mutation c.187C>T (p.R63X) associated with a novel clinical finding of a VSD. In Patient 2 we found a homozygous c.2293C>T (p.Q765X) mutation that had been previously reported but found that it also altered RNA processing generating a novel transcript not previously identified (r.2176_2293del; p.F726Sfs*10). This is the first report to describe Mestizo patients with molecular diagnosis of ARCL-IIA/ATP6V0A2-CDG and to establish that their mutations are the first to be found in patients from different regions of the world and with different genetic backgrounds.
Keywords: Laxa, Glycosylation, ATP6V0A2, CDG, ARCL, Hispanic
1. Introduction
Inherited cutis laxa syndromes commonly present with wrinkled, pendulous, redundant and inelastic skin. These syndromes include X-linked cutis laxa (XRCL; MIM: 304150), autosomal dominant cutis laxa (ADCL; MIM: 123700), autosomal recessive cutis laxa (ARCL) type IA (MIM: 219100), type IB (MIM: 614437), type IIA (MIM: 219200), type IIB (MIM: 612940), type III (MIM: 219150), Urban–Rifkin–Davis syndrome (URDS; MIM: 613177), macrocephaly alopecia cutis laxa syndrome (MACS; MIM: 613075) and arterial tortuosity syndrome (ATS; MIM: 208050). Mutations in specific genes have been associated with most of these syndromes. Here we report the first two cases of Mexican Mestizo children with a clinical spectrum of ARCL-IIA linked to mutations in ATP6V0A2, [1]. Mutations in ATP6V0A2 are the most frequent known cause of ARCL, followed by mutations in the PYCR1, GORAB and ALDH18A1 genes [2].
ATP6V0A2 encodes for the a2 subunit of the vacuolar H+-ATPase (V-ATPase) [2], [3], a proton pump involved in the maintenance of the pH gradient along the secretory pathway and the regulation of protein transport [4]. Individuals with mutations in ATP6V0A2 have abnormal protein N- and O-linked glycosylations. The presence of glycosylation defects classifies this disease as a Congenital Disorder of Glycosylation or CDG, the ATP6V0A2-CDG [1], [5]. CDGs are a growing group of inherited diseases that cause abnormal glycosylation and in recent years many new subtypes of CDG are linked to genes that indirectly affect the glycosylation machinery, as is the case of mutations in this H+-ATPase or of the Oligomeric Golgi Complex (COG) [6].
Abnormal protein glycosylation in patients with ATP6V0A2-CDG is due to vacuolar H+-ATPase deficiency leading to an increase in Golgi pH that affects glycosyltransferase activity and organelle trafficking causing Golgi fragmentation and possible mislocalization of these enzymes [4], [7], [8]. The pathophysiology of ATP6V0A2-CDG is explained not only by altered sialylation of surface receptors and extracellular matrix proteins, but also by secretory defects involving tropoelastin secretion [4] and upregulated TGF-β downstream signaling [2].
2. Material and methods
2.1. Isoelectric focusing (IEF) of transferrin
Serum (100 μL) was iron saturated at room temperature for 1 h with 5 μL of 0.5 M NaHCO3 and 5 μL of 20 mM FeCl3. One microliter of 10-fold-diluted serum was spotted on polyacrylamide gels (T = 5%, C = 3%) containing 5.7% ampholytes (pH 5–7). After electrophoresis, the gel was covered with 100 μL of anti-transferrin for 30 min at 4 °C. The gel was washed overnight with physiological saline, fixed, stained with Coomassie Brilliant Blue R-250, destained, dried, and photographed [9].
2.2. Mass spectrometry
On-column immunoaffinity-ESI mass spectrometry for transferrin and Apolipoprotein CIII (ApoCIII) was performed using an API-5000 triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA).
2.3. Genetic analysis
Genomic DNA from patients and their parents was obtained from peripheral blood using GenElute Blood Genomic DNA Kit (Sigma-Aldrich, St Louis, MO, USA). Total mRNA from the patients was obtained from skin fibroblasts using TRIzol reagent (Life Technologies, Rockville, MD, USA) and complementary DNA (cDNA) was synthesized using M-MLV Reverse Transcriptase (Life Technologies, Rockville, MD, USA). Mutational analysis of ATP6V0A2 was performed by directly sequencing the cDNA based polymerase chain reaction (PCR) products obtained using forward 5′TGCAGTCTGGAGCCCCATAGTG3′ and reverse 5′GTCATAAGGCAGTAATGAAGATGG3′ primers that amplify the twenty exons that encode the gene. Translation of the identified mutations was obtained using pDRAW32 (AcaClone Software). To gain insight into the cause of exon 18 skipping, forward 5′CGTCTGGGGTTTCTGTTCC3′ and reverse 5′CCACAGTGCCCCCTGAGT3′ primers for genomic DNA were designed to amplify exon 18 including the intron/exon junctions. The amplified products were resolved in a 2–2.5% agarose gel by electrophoresis. The desired DNA bands were cut out from the agarose gel under ultraviolet light and purified from the agarose using the GeneJet gel extraction kit (Thermo Scientific, Rockford, IL, USA). Sequencing of PCR products was performed by an ABI Prism 3130xl autoanalyzer (Applied Biosystems, Foster City, CA) and results were visualized using SnapGene Viewer 2.2.2 (GSL Biotech LLC, Chicago, IL, USA). For primer design and mutation nomenclature the NCBI reference sequences for genomic DNA were NC_000012.12 and NM_012463 for coding DNA.
2.4. Patient 1
The patient is a 3 year old child, son of consanguineous parents (siblings) with no known previous pregnancies, miscarriages or relatives affected by cutis laxa. At birth, he was small for his gestational age (36 weeks) with a weight and length of 2190 g and 45 cm (< 5th percentile), respectively; hypotonia, over-folded skin, enlarged fontanelles and distinctive facial features were also present. A heart murmur revealed a ventricular septal defect (VSD) and pulmonary artery banding was performed at 3 months with surgical repair at 18 months. Neurodevelopmental delay, failure to thrive, asthma, recurrent pneumonias, gastroesophageal reflux, inguinal hernia, cryptorchidism, hyperuricemia and microcytic hypochromic anemia were also present. Recurrent urinary tract infections leading to pyelonephritis were reported. Liver enzymes were normal. His karyotype was 46, XY and skin biopsy demonstrated an abnormal, broken, shorted and fuzzy elastic fiber structure. A trans-fontanellar ultrasound showed increased bilateral peri- and paraventricular echogenicities, increased size of choroid plexus, probable Dandy Walker variant as well as hypoplasia of the cerebellar vermis and corpus callosum.
Physical exploration at 8 months of age revealed persistent low weight, height and cephalic circumference (< 5th percentile). Facial manifestations included microcephaly with a wide anterior fontanelle (> 6 cm), droopy skin on the cheeks and prominent nasolabial folds, blue sclerae, mild hypertelorism, epicanthal folds, small nose with anteverted nares, long philtrum, a thin upper lip and retrognathia (Fig. 1A–B). Also observed were markedly loose and wrinkled skin in the abdomen, gluteal zone, arms and legs (Fig. 1C–D) and hyperextensible joints with a single transverse palmar crease and 5th finger clinodactyly (Fig. 1E–F). A follow-up at 3 years revealed an improvement in the skin folds of the neck, abdomen and gluteal area (Fig. 1G–I).
Fig. 1.

Patient 1. A and B. Microcephaly, mild hypertelorism, droopy skin on the cheeks of the face and neck, epicanthal folds, small nose with anteverted nares, long philtrum, thin upper lip and retrognathia and blue sclera. C. Loose and wrinkled skin in the abdomen, arms and legs. D. Increased gluteal folds E. Hyperextensible joints. F. and 5th finger clinodactyly G–I. Phenotype at 3 years old.
The phenotype correlation [10] suggested that this patient had an ARCL-IIA cutis laxa syndrome, particularly because of the presence of motor nervous system abnormalities, cardiovascular abnormalities and patent anterior fontanel which distinguishes it from ARCL-IIB and the absence of athetoid movements and corneal opacification present in the closely resembling ARCL-III.
To further confirm if this patient had the characteristic hypoglycosylation seen in ATP6V0A2-CDG, serum transferrin glycosylation (N-glycoprotein) was analyzed by IEF and revealed an abnormal type II profile (Fig. 2) which indicates truncated glycans of transferrin and points to a Golgi compartment disturbance [11]. Subsequently, transferrin and Apolipoprotein CIII (ApoCIII, O-glycoprotein) were profiled by mass spectrometry, confirming the hypoglycosylation of serum transferrin but revealing normal glycosylation of serum ApoCIII (Fig. 3). Although ATP6V0A2-CDG patients commonly have both abnormal transferrin and ApoCIII hypoglycosylation [1], [7], the clinical phenotype and transferrin hypoglycosylation hinted to mutations in ATP6V0A2. The mutational analysis of ATP6V0A2 using patient cDNA revealed a single ATP6V0A2 transcript with a nonsense mutation in exon 2 that codes for a premature stop codon (c.187C>T; p.R63X). Sanger sequencing on patient genomic DNA confirmed that the c.187C>T was homozygous and analysis of familial DNA confirmed that it was inherited in a recessive fashion (Fig. 4A). Screening of parental gDNA confirmed the recessive transmission. This mutation was previously reported, also in a homozygous state, in only two Turkish patients with ARCL-IIA [1], [12].
Fig. 2.
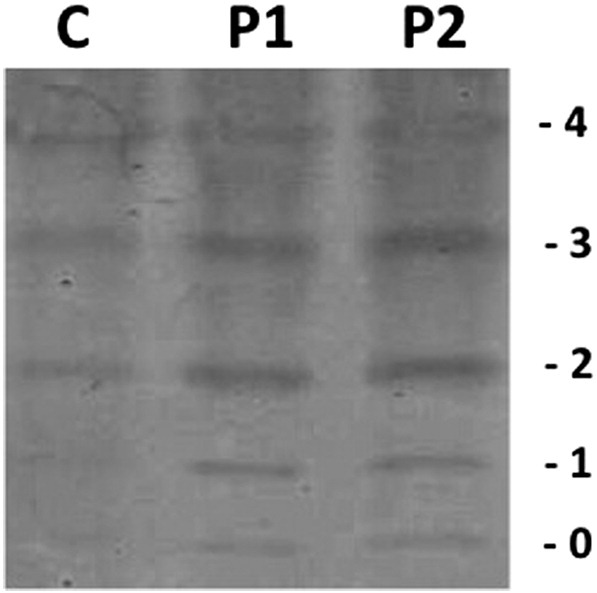
IEF of serum transferrin showing an abnormal type II profile for both patients. C, control sample; P1, Patient 1; P2, Patient 2. Numbers in the edge correspond to the number of sialic acids in the transferrin isoforms.
Fig. 3.

Mass spectrometry of serum transferrin and ApoCIII. A. Transferrin profiles of Patients 1 and 2 showing an increase of trisialylated transferrin compared to a normal control. B. ApoCIII profile of Patient 1 showing a normal glycosylation profile and Patient 2 showing an abnormal profile compared to a normal control. C. Transferrin and ApoCIII glycoform ratios of profiles shown in A and B with the reference range values obtained from a series of independent controls.
Fig. 4.

Sanger sequencing chromatograms showing mutations. A. Patient 1 showing the homozygous mutation in genomic DNA (c.187C>T) and his parents showing it in a heterozygous fashion. B. Patient 2 showing exon 18 skipping in cDNA. C. Patient 2 showing a homozygous mutation in genomic DNA (c.2293C>T) and his parents showing it in a heterozygous fashion. The control sequence is from a healthy individual.
2.5. Patient 2
The patient is a 10 month old male, son of 4th degree consanguineous relatives with a previous pregnancy that ended in miscarriage with no known relatives affected by cutis laxa. At birth, he was small for his gestational age (39 weeks) with a weight and length of 2350 g and 45 cm, respectively (< 5th percentile). Hypotonia and neurodevelopmental delay were present. Furrowing of the skin, enlarged fontanelles and distinctive facial features were also present. No heart murmurs, hyperuricemia, abnormal liver enzymes or anemia was detected. His karyotype was 46, XY and skin biopsy also demonstrated an abnormal, broken, shorted and fuzzy elastic fiber structure.
Physical exploration revealed a persistently low weight, height and cephalic circumference (< 5th percentile), a wide anterior fontanelle (> 6 cm), droopy skin on the cheeks of the face, small nose with anteverted nares, long philtrum, marked nasolabial folds and large everted ears with helix hypoplasia (Fig. 5A). Also observed were markedly loose and wrinkled skin in the neck, abdomen, gluteal zone, arms and legs (Fig. 5B–C) and hyperextensible joints with a single transverse palmar crease. No umbilical hernias were found. The left leg was shortened by acetabular dysplasia. A brain MRI showed widened Virchow–Robin spaces in the white matter with no other apparent malformations (Fig. 5D). This phenotype also suggested an ARCL-IIA cutis laxa syndrome leading to transferrin IEF analysis.
Fig. 5.

Patient 2. A. Facial features are droopy skin on the cheeks, small nose with anteverted nares, long philtrum and marked nasolabial folds. B and C. Loose and wrinkled skin in the abdomen. D. Axial brain MRI showing widened Virchow–Robin spaces in the white matter.
Serum transferrin IEF revealed an abnormal type II profile (Fig. 2). Transferrin and Apolipoprotein CIII were profiled by mass spectrometry and confirmed a combined hypoglycosylation defect (Fig. 3).
PCR amplification and sequencing of ATP6V0A2 using cDNA revealed the presence of two transcripts, a full length transcript with a previously reported nonsense mutation in exon 18 that causes a premature stop codon (c.2293C>T; p.Q765X) [1], [7] and a transcript lacking exon 18 (r.2176_2293del) (see Fig. 4B). The translation effect of exon 18 skipping is a frame shift and the appearance of a premature stop codon (p.F726Sfs*10). Analysis of genomic DNA revealed no intronic mutations in the 5′ or 3′ boundary of exon 18 nor its deletion but did show that the patient was homozygous for the c.2293C>T mutation (see Fig. 4C). Screening of parental genomic DNA confirmed the recessive transmission.
3. Discussion
The molecular basis of cutis laxa syndromes associated with protein hypoglycosylation can be caused by at least two gene defects, mutations in the ATP6V0A2, as was confirmed in both patients reported here, or mutations in the COG7 that encodes for a subunit of the Oligomeric Golgi Complex. COG7-CDG is characteristically associated with hyperthermia, severe liver involvement and coagulation defects and lethality not present in either of the cases we report [12], [13].
No reports in the literature exist to our knowledge regarding patients of Mestizo or Mexican ethnicity with ATP6V0A2-CDG. There is only a report of a Hispanic patient that interestingly harbors the same mutation as Patient 2 but no information was given regarding ethnicity or nationality [1]. Few cases of CDG have been reported in Latin America most probably because of subdiagnosis and/or lack of specialized laboratories able to perform transferrin IEF.
It was noteworthy to find the c.187C>T (p.R63X) mutation in Patient 1 since it had previously been reported in two Turkish patients, indicating that the mutation is not restricted to a particular ethnic population as no known Eurasian ancestry was reported by his family [1], [12]. To our knowledge this is the second mutation in ATP6V0A2 to be reported in different regions of the world with different genetic backgrounds [2]. The first is the c.2293C>T (p.Q765X) mutation found in Patient 2 and previously reported in three individuals: a homozygous Hispanic individual [1], and two other individuals, one homozygous (Bahrain, Patient 14 as described in Table 1) and another heterozygous (Turkey, Patient 17 as described in Table 1) [7] (see Table 1).
Table 1.
Comparative clinical and mutational characteristics of patients reported here with other patients harboring the same mutations.
| Patient 1 | Patient 3a | Patient 2 | Patient 14b | Patient 17b | |
|---|---|---|---|---|---|
| cDNA mutation | c.187C>T | c.187C>T | c.2293C>T | c.2293C>T | c.2293C>T; c.1827_1828_insG |
| Protein mutation | p.R63X | p.R63X | p.Q765X; p.F726Sfs*10 | p.Q765X | p.Q765X; p.W609fsX625 |
| Cutis laxa | + | + | + | + | + |
| Large fontanelles | + | + | + | + | + |
| Microcephaly | + | + | + | + | + |
| Eye anomalies | + | + | − | + | + |
| Hypotonia | + | + | + | + | + |
| Neurodevelopmental delay | + | + | + | + | + |
| Delayed growth | + | + | + | − | + |
| Joint laxity | + | + | + | + | + |
| Hernias | + | − | − | + | − |
| CNS anomalies | + | + | + | + | − |
| Congenital cardiac anomalies | + | − | − | − | − |
| Congenital urogenital anomalies | + | + | − | − | − |
| Coagulation anomalies | − | − | − | + | − |
| Liver function anomalies | − | − | − | − | − |
| Congenital joint anomalies | − | + | + | − | + |
| Recurrent urinary tract infections | + | + | − | − | − |
| Seizures | − | + | − | − | − |
| Abnormal transferrin | + | + | + | n.d | n.d |
| Abnormal ApoCIII | − | + | + | n.d | n.d |
For comparison purposes (+/−) represents the presence or absence of the clinical finding, no grading was used as patients compared to our Patients 1 and 2 were studied by another group. N.D., non determined.
Patients 14 and 17 has been reported previously [7].
Patient 1 had a normal ApoCIII glycosylation profile which is frequently considered an uncommon finding in patients with ATP6V0A2-CDG. Only one of the two Turkish patients with a homozygous p.R63X mutation had been analyzed for ApoCIII and was determined to be abnormally glycosylated [1]. Analysis of 21 patients from previous ATP6V0A2-CDG patient-series reports where transferrin and ApoCIII glycosylation profiles were analyzed indicates that 23% of patients present with a normal ApoCIII profile, whereas abnormal glycosylation of transferrin is present in all patients > 6 months of age [1], [7]. The above data indicate that ApoCIII is far less reliable compared to transferrin to detect hypoglycosylation in patients with ATP6V0A2-CDG.
Of the two Turkish patients with the c.187C>T (p.R63X) mutation found in Patient 1 only one has been clinically described in detail in the literature as Patient 3 [12]. Both individuals shared most of the clinical findings, nonetheless no pelvis hypoplasia or seizures were found in the Mexican Mestizo patient whereas the Turkish patient did not show VSD (Table 1). Clinically, Patient 2 with the c.2293C>T (p.Q765X/p.F726Sfs*10) mutation shared most features of previously reported Patients 14 and 17 with the same mutation, nonetheless no coagulation anomalies were found as reported for Patient 14 (Table 1).
In regard to Patient 2, we propose that the previously reported c.2293C>T (p.Q765X) mutation also induces nonsense alternative splicing through disruption of the normal donor splice site (AC/GU to AU/GU) leading to exon 18 skipping (r.2176_2293del) causing a frame shift and a premature stop codon (p.F726Sfs*10) (Fig. 6). Because the c.2293C>T mutation yields two transcripts it should be described as r.[2293c>u, 2176_2293del]. It is possible that the novel transcript lacking exon 18 was not identified in previously reported patients as the screening is usually performed on genomic DNA.
Fig. 6.
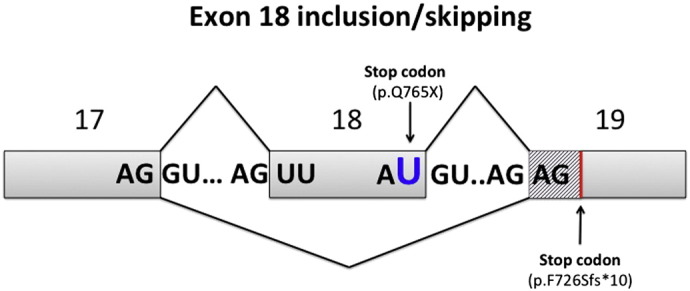
Skipping or inclusion of exon 18. The c.2293C>T (r.2293c>u) mutation in exon 18 induces a premature stop codon (p.Q765X) but is also able to generate a nonsense alternative splicing mutation possibly through disruption of the normal donor splice site in exon 18 (AC/GU to AU/GU) leading to its skipping (r.2176_2293del) causing a frame shift (stripe pattern) and a premature stop codon (p.F726Sfs*10).
4. Summary
Screening for ATP6V0A2 mutations is indispensable in patients presenting with an ARCL-IIA cutis laxa syndrome once hypoglycosylation of serum transferrin has been established. ApoCIII is a less reliable marker of hypoglycosylation compared to transferrin in patients with ATP6V0A2-CDG. In Patient 1 novel findings include VSD and a normal ApoCIII profile not previously reported in patients with the c.187C>T mutation (p.R63X). Also, we establish that the c.187C>T (p.R63X) and c.2293C>T (p.Q765X) ATP6V0A2 mutations are the first to be found in patients from different regions of the world and with different genetic backgrounds.
Acknowledgments
IMD was supported by the Latin American Society for Glycobiology fund for CDG diagnosis and the CDG International Fund. HHF is supported by The Rocket Fund, and the National Institutes of Health (R01DK55615). DBB was supported by the Mexican National Council for Science and Technology (CONACYT) scholarship fund 298079. We thank Genos Medica Laboratories (México City, Mexico) for the establishment of fibroblasts from skin biopsies.
References
- 1.Kornak U. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008;40(1):32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 2.Fischer B. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum. Genet. 2012;131(11):1761–1773. doi: 10.1007/s00439-012-1197-8. [DOI] [PubMed] [Google Scholar]
- 3.Mohamed M. Metabolic cutis laxa syndromes. J. Inherit. Metab. Dis. 2011;34(4):907–916. doi: 10.1007/s10545-011-9305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guillard M. Vacuolar H+-ATPase meets glycosylation in patients with cutis laxa. Biochim. Biophys. Acta. 2009;1792(9):903–914. doi: 10.1016/j.bbadis.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Jaeken J. CDG nomenclature: time for a change! Biochim. Biophys. Acta. 2009;1792(9):825–826. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosnoblet C. Glycosylation disorders of membrane trafficking. Glycoconj. J. 2013;30(1):23–31. doi: 10.1007/s10719-012-9389-y. [DOI] [PubMed] [Google Scholar]
- 7.Hucthagowder V. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 2009;18(12):2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivinoja A. Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J. Cell. Physiol. 2009;220(1):144–154. doi: 10.1002/jcp.21744. [DOI] [PubMed] [Google Scholar]
- 9.Niehues R. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Invest. 1998;101(7):1414–1420. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berk D.R. Cutis laxa: a review. J. Am. Acad. Dermatol. 2012;66(5):842. doi: 10.1016/j.jaad.2011.01.004. (e1–17) [DOI] [PubMed] [Google Scholar]
- 11.Jaeken J. Congenital disorders of glycosylation. Ann. N. Y. Acad. Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- 12.Morava E. Defective protein glycosylation in patients with cutis laxa syndrome. Eur. J. Hum. Genet. 2005;13(4):414–421. doi: 10.1038/sj.ejhg.5201361. [DOI] [PubMed] [Google Scholar]
- 13.Wu X. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10(5):518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]