

Abstract
We examined alpha-galactosidase A (GLA) gene mutations in 74 Japanese families with Fabry disease (FD) to determine the frequency of de novo mutations. In 5 of 74 families (6.8%), the probands had no positive family histories and were diagnosed as de novo because their parents had no mutations in GLA gene. The parents of Fabry patients do not necessarily have mutations in GLA gene which is an important consideration in genetic counseling for FD.
Keywords: Fabry disease, Alpha-galactosidase A, De novo mutation, Novel mutation, W340S, Genetic counseling
1. Introduction
Fabry disease (OMIN 301500, FD) is an X-linked lysosomal storage disorder resulting from a deficiency of alpha-galactosidase A (EC 3.2.1.22; GLA) activity [1]. The estimated incidence of the disease is 1 per 1250–117,000 live male birth [2], [3], [4], [5]. The deficiency of GLA activity leads to the accumulation of the principal substrate globotriaosylceramide (GL3) in various tissues including vascular endothelium, renal glomeruli and tubules, dorsal root ganglia, cardiac myocytes and valves, cornea and skin.
Classically affected male patients with FD have a markedly shortened lifespan with death occurring in the fourth or fifth decade of life. Although the clinical severity of female patients is heterogeneous, most of them present with life-threatening complications in their fifth or sixth decade of life [6], [7], [8], [9], [10]. Some reports have indicated that enzyme replacement therapy (ERT) is also efficacious for female patients with FD [11], [12].
Based on Mendelian inheritance, while mothers of male patients with X-linked disorders are expected to be obligate heterozygotes, sometimes the mothers of the male patients with FD may not be heterozygotes. It is therefore important to diagnose them accurately because they may not require ERT. To determine the frequency of de novo mutations in Fabry families, we examined the GLA gene mutations in patients and families with FD.
2. Material and methods
2.1. Study patients
Study patients were 126 Japanese patients (61 male patients and 65 female patients) from 74 families with FD. They were referred to us for diagnosis of FD between 1999 and 2012 and diagnosed with FD based on gene analysis.
2.2. Gene analysis
Genomic DNA was extracted from leukocytes using blood and cell culture DNA Midi Kit (Qiagen, Hilden, Germany). Each exon and flanking intron sequence of the GLA gene was amplified by PCR using AmpliTaq gold 360 master mix (Applied Biosystems, Foster city, CA, USA), and directly sequenced using the BigDye Terminator Kit, version 3.1 (Applied Biosystems, Foster city, CA, USA).
2.3. In vitro mutagenesis and expression study in Cos-1 cells
Mutation was introduced to normal GLA cDNA using Quick Change Lighting Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA, USA) following the manufacture's protocol and ligated to mammalian expression vector pcDNA 3.1 (Invitrogen, Carlsbad, CA, USA). This plasmid was transfected to Cos-1 cells using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacture's protocol. Forty eight hours after transfection, GLA activities were assayed using a fluorogenic substrate, 4-methylumbelliferyl-α-d-galactopyranoside, as described previously [13].
This study was performed under the approval of the ethical committee of The Jikei University School of Medicine. Written informed consent was obtained from all study subjects or legal guardian.
3. Results
In 74 study families, 50 different disease causing mutations were identified in the exons and intron/exon boundaries. Five patients (from 5 families) had no positive family histories (Fig. 1A). The probands of families 1, 2, 3 and 4 were classically affected male patients and that of family 5 was a female heterozygote patient (Fig. 1B). The mothers of the male probands and the parents of the female proband had no characteristic symptoms of FD. All of the probands in families 1, 2, 3, 4 and 5 had disease-causing mutations. Two mutations (IVS5 − 2 and IVS3 + 1) of the 5 probands were splicing defect and previously reported as disease causing mutations [14], [15]. The 2 missense mutations (c.3G>A and c.605G>A) were previously reported as disease causing mutations [16], [17]. The remaining one missense mutation (c.1019G>C, p.W340S) was a novel mutation. We confirmed the deficiency of GLA activity (2.3% of wild type) by expression study using Cos-1 cells transfected with c.1019G>C mutation and wild type GLA cDNA. These 3 missense mutations (c.3G>A, c.605G>A and c.1019G>C) were confirmed not to be polymorphism based on SNP analysis using NCBI dbSNP and Human Genetic Variation Browser [18], [19], and the amino acid substitutions of these 3 missense mutations changed the GLA structure in the result of PolyPhen-2 test [20]. They were diagnosed as de novo because their mutations in the GLA gene were not detected in their parents and siblings. In the other 69 families, de novo mutations were excluded by family history of affected parents and/or siblings. In this study, the frequency of de novo mutations was 5/74 (6.8%). In this study, the affected family members with positive family histories were not confirmed to have the same disease causing mutations as study patients.
Fig. 1.
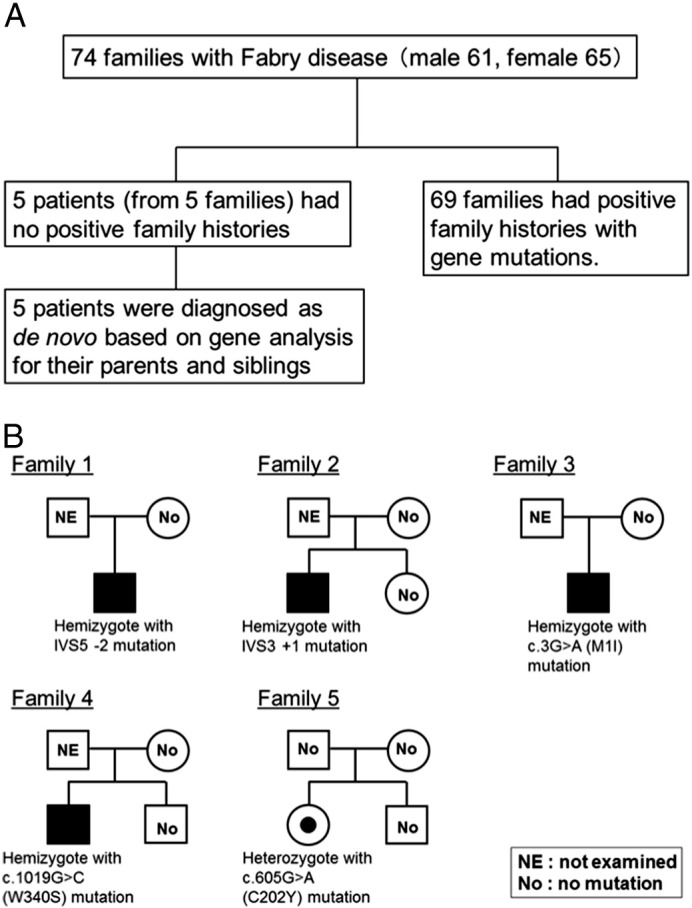
A. Study patients. B. The family trees of de novo cases. The probands of families 1, 2, 3 and 4 were classically affected male patients and that of family 5 was a female heterozygote patient. The parents of probands in these 5 families had no characteristic symptoms of FD. All of the probands had disease-causing mutations and these mutations were not detected from their parents and siblings.
4. Discussion
More than 550 different disease-causing mutations on the GLA gene have been reported [21]. Although several papers reported the de novo mutation of Fabry patients [22], [23], [24], [25], the frequency of de novo mutations of FD has been unclear. Rodriguez-Mari et al. studied 22 families with Fabry disease and detected a de novo mutation [24] and the frequency of de novo mutation in Spain was 4.5% (1 of 22 families). We did not know the frequency of de novo mutation in Japanese patient with FD. In this study, 5 of 74 (6.8%) families with FD had de novo mutations on the GLA gene. The frequency of de novo mutation in Japanese Fabry families is very similar to that in Spanish Fabry families. There might be no ethnic difference in frequency of de novo mutation in GLA gene. To reach this conclusion, we should know the frequency of de novo mutation in other ethnic groups.
The frequency of de novo mutations has been reported to be high in X-linked disorders such as Duchenne muscular dystrophy and hemophilia A and approximately one-third of mutations of these two diseases are expected to arise de novo[26], [27], [28]. The size and structure of the gene and its position within the genome may contribute to the frequency of the de novo mutations. In Duchenne muscular dystrophy, the high rate of de novo mutations is thought to be related to the unusually large size (2400 kb) of dystrophin gene. The presence of CpG dinucleotides also reportedly increases mutational frequency among single base pair substitutions (CG-to-TG or CG-to-CA transitions). In hemophilia A, the high rate of de novo mutations is thought to be caused by the rich CpG dinucleotide content in the human factor VIII gene. In Fabry patients, the relatively low frequency of de novo mutations of GLA gene might be caused by its smaller gene size (12 kb) and lower contents of CpG dinucleotides.
There are three limitations in this study. First, we did not analyze all parents of the study patients. In some families, the de novo mutations were excluded by existence of FD patients in the same family based on the information of the client physicians. Therefore, we might under estimate the frequency of de novo mutations in FD. Second, we did not confirm that the parents are biological father or mother. Third, we did not address to the germline mosaicism. In these points of view, there are some biases in this study.
5. Conclusion
The parents of Fabry patients do not necessarily have mutations in GLA gene and this is important when considering genetic counseling of FD.
Conflict of interest
T. Ohashi has active research support from Genzyme Corporation, Shire Pharmaceuticals and Dainippon Sumitomo Pharma.
Y. Eto has active research support from Genzyme Corporation.
H. Ida has active research support from Genzyme Corporation and Dainippon Sumitomo Pharma.
These activities have been fully disclosed and are managed under Memorandum of Understanding with the Conflict of Interest Resolution Board of The Jikei University School of Medicine.
Acknowledgments
We thank Dr. Cheng, Genzyme Corporation for reviewing the manuscript. We also thank our colleagues in the Department of Gene Therapy for their excellent technical assistance. This work was supported by a Grant for Research on Measures for Intractable Disease from the Japanese Ministry of Health, Welfare, and Labor (H22-nanchi-ippan-002).
References
- 1.Desnick R.J., Ioannou Y.A., Eng C.M. α-Galactosidase A deficiency: Fabry disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. McGraw-Hill Inc.; New York: 2001. pp. 3733–3774. [Google Scholar]
- 2.Meikle P.J., Hopwood J.J., Clague A.E., Carey W.F. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 3.Spada M., Pagliardini S., Yasuda M., Tukel T., Thiagarajan G., Sakuraba H., Pnonzone A., Desnick R.J. High incidence of later onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006;79:21–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwu W.L., Chien Y.H., Lee N.C., Chiang S.C., Dobrovolny R., Huang A.C., Yeh H.Y., Chao M.C., Lin S.H., Kitagawa T., Desnick R.J., Hsu L.W. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G>A (IVS4 + 919G>A) Hum. Mutat. 2009;30:1397–1405. doi: 10.1002/humu.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue T., Hattori K., Ihara K., Ishii A., Nakamura K., Hirose S. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J. Hum. Genet. 2013;58:548–552. doi: 10.1038/jhg.2013.48. [DOI] [PubMed] [Google Scholar]
- 6.MacDermot K.D., Holmes A., Miners A.H. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001;38:769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deegan P.B., Baehner A.F., Barba Romero M.A., Hughes D.A., Kampmann C., Beck M. Natural history of Fabry disease in females in the Fabry outcome survey. J. Med. Genet. 2006;43:347–352. doi: 10.1136/jmg.2005.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eng C.M., Fletcher J., Wilcox W.R., Waldek S., Scott C.R., Sillence D.O., Breunig F., Charrow J., Germain D.P., Nicholls K., Banikazemi M. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 2007;30:184–192. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi M., Ohashi T., Sakuma M., Ida H., Eto Y. Clinical manifestation and natural history of Japanese heterozygous females with Fabry disease. J. Inherit. Metab. Dis. 2008 doi: 10.1007/s10545-007-0740-6. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 10.Schiffmann R., Warnock D.G., Banikazemi M., Bultas J., Linthorst G.E., Packman S., Sorensen S.A., Wilcox W.R., Desnick R.J. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transplant. 2009;24:2102–2111. doi: 10.1093/ndt/gfp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baehner F., Kampmann C., Whybra C., Miebach E., Wiethoff C.M., Beck M. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J. Inherit. Metab. Dis. 2003;26:617–627. doi: 10.1023/b:boli.0000005658.14563.77. [DOI] [PubMed] [Google Scholar]
- 12.Linthorst G.E., Hollak C.E., Donker-Koopman W.E., Strijland A., Arts J.M. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 13.Desnick R.J., Allen K.Y., Desnick S.J., Raman M.K., Bemlohr R.W., Krivit W. Fabry's disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine and leukocytes. J. Lab. Clin. Med. 1973;81:157–171. [PubMed] [Google Scholar]
- 14.Shabbeer J., Yasuda M., Benson S.D., Desnick R.J. Fabry disease: identification of 50 novel alpha-galactosidase-A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum. Genomics. 2006;2:297–309. doi: 10.1186/1479-7364-2-5-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashton-Prolla P., Tong B., Shabbeer J., Astrin K.H., Eng C.M., Desnick R.J. Fabry disease: twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J. Investig. Med. 2000;48:227–235. [PubMed] [Google Scholar]
- 16.Blanch L.C., Meaney C., Morris C.P. A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the alpha-galactosidase A gene. Hum. Mutat. 1996;8:38–43. doi: 10.1002/(SICI)1098-1004(1996)8:1<38::AID-HUMU5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Eng C.M., Ashley G.A., Burgert T.S., Enriquez A.L., D'Souza M., Desnick R.J. Fabry disease: thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol. Med. 1997;3:174–182. [PMC free article] [PubMed] [Google Scholar]
- 18.http://www.ncbi.nlm.nih.gov/snp/
- 19.http://www.genome.med.kyoto-u.ac.jp/SnpDB/
- 20.http://genetics.bwh.harvard.edu/pph2/index.shtml
- 21.The Human Gene Mutation Database (HGMD) http://www.hgmd.cf.ac.uk/ac/index.php
- 22.Iemolo F., Pizzo F., Albeggiani G., Zizzo C., Colomba P., Scalia S., Bartolotta C., Duro G. De novo mutation in a male patient with Fabry disease: a case report. BMC Res. Notes. 2014;7:11. doi: 10.1186/1756-0500-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brokalaki E.I., Hentschke M., Grabbe S., Jansen T. Fabry disease in female patient due to a de novo point mutation at position 691 of exon 5. Eur. J. Med. Res. 2006;11:306–308. [PubMed] [Google Scholar]
- 24.Rodriguez-Mari A., Coll M.J., Chabas A. Molecular analysis in Fabry disease in Spain: fifteen novel GLA mutations and identification of a homozygous female. Hum. Mutat. 2003;22:258. doi: 10.1002/humu.9172. [DOI] [PubMed] [Google Scholar]
- 25.Germain D.P., Salard D., Fellmann F., Azibi K., Caillaud C., Bernard M.C., Poenaru L. Identification of a de novo mutation (G373D) in the alpha-galactosidase A gene in a patient with Fabry disease. Hum. Mutat. 2001;17:353. doi: 10.1002/humu.41. [DOI] [PubMed] [Google Scholar]
- 26.Beaudet A.L., Scriver C.R., Sly W.S., Valle D. Genetics, biochemistry and molecular bases of variant human phenotypes. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. McGraw-Hill Inc.; New York: 2001. pp. 3–45. [Google Scholar]
- 27.Lee T., Takeshima Y., Kusunoki N., Awano H., Yagi M., Matsuo M., Iijima K. Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J. Hum. Genet. 2014;59:46–50. doi: 10.1038/jhg.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haldane J.B. The rate of spontaneous mutation of a human gene. J. Genet. 1935;31:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]