

Abstract
Congenital leptin deficiency is a rare recessively inherited condition due to homozygous mutations in the LEP gene. To date, only nine mutations have been identified in the LEP gene (p.L72S, p.N103K, p.R105W, p.H118L, p.S141C, c.104_106delTCA, c.135del3bp, c.398delG and c.481_482delCT). In this study we present a novel homozygous nonsense mutation (W121X) in LEP in a twelve year old obese male and his severely obese sister. As this disorder is treatable with recombinant leptin, it is intriguing to report a novel homozygous nonsense mutation in LEP in two obese children of consanguineous parents. These patients showed features in accordance with leptin deficiency.
Keywords: Obesity, LEP, Novel, Mutation, Egypt
1. Introduction
Leptin is a circulating adipocyte-derived hormone which functions centrally to regulate body weight [1]. In 1997, the first human mutation in LEP (the gene that encodes leptin) was reported in two severely obese cousins, from a consanguineous UK family of Pakistani origin [2]. This homozygous single base deletion at codon 133 in the gene coding for leptin caused a frameshift mutation resulting in a truncated protein and undetectable serum leptin levels. Since then several other severely obese patients with missense mutations (p.L72S, p.N103K, p.R105W, p.H118L, p.S141C) and deletion mutations (c.104_106delTCA, c.135del 3 bp, c.398delG and c.481_482delCT) [3], [4], [5], [6], [7], [8], [9], [10], [11] in LEP have been reported. All of these mutations were found in patients from Pakistan, Turkey, Turkmenistan, Egypt, Europe and China where consanguineous marriages are more prevalent. In this study, we report a novel homozygous nonsense mutation associated with undetectable serum leptin levels in an Egyptian family with severe obesity.
2. Research design and methods
2.1. Case history
A twelve-year-old male patient with severe early onset obesity was referred to the Obesity & Endocrinology Clinic at the National Research Centre, Cairo, Egypt. He weighed 85 kg, his height was 140 cm and his BMI was 43 kg/m2. The parents were first cousins. Pedigree analysis revealed 3 similarly affected female sibs two of whom died in early childhood from recurrent infections (Fig. 1). The patient was born with normal birth weight (3.5 kg). Hyperphagia was noticed since birth. Rapid weight gain and hyperphagia continued and his BMI reached 49.7 kg/m2 by the age of 13. His sibling was a 3 month old girl, her weight was 9 kg, her length was 64 cm and her BMI was 22.5 kg/m2. By the age of 2 years her BMI was 42.5 kg/m2. She also had hyperphagia since birth. The height of the mother was 155 cm, weight was 63 kg and BMI was 26 kg/m2. The father's height was 158 cm, weight was 70 kg and the BMI was 28 kg/m2. Clinical examination of both children showed no clinical features suggestive of a pleiotropic genetic syndrome, such as Alstrom's or Prader–Willi syndrome. They both had undetectable serum leptin levels with hyperinsulinaemia (Table 1). MRI of the brain and field of vision were also normal.
Fig. 1.

Pedigree of family with congenital leptin deficiency.
Table 1.
Biological data of the two patients.
| Parameter | Male | Female | Reference range |
|---|---|---|---|
| TSH (l U/ml) | 5 | 3 | 1–39 |
| Free T3 (pg/ml) | 1.5 | 2.5 | 2–5 |
| Free T4 (pg/ml) | 1 | 1.5 | 0.7–1.8 |
| Serum cortisol 9 am (l g/dl) | 10 | 12 | 6.2–19.4 |
| Serum cortisol 9 pm (l g/dl) | 3 | 6.2 | 2.3–11.9 |
| Cortisol after suppression (l g/dl) | 1 | 0.9 | 0.8 < 1.8 |
| ACTH (pg/ml) | 12 | 12.8 | Up to 50 |
| Prolactin (ng/ml) | 10 | 12 | 13 |
| Serum leptin (ng/ml) | Undetectable | Undetectable | Control value 30.91 |
| Insulin (l U/ml) | 40 | 30.8 | 2.6–24.9 |
| Total cholesterol (mg/dl) | 140 | 150 | 110–230 |
TSH: thyroid stimulating hormone, ACTH: adrenocorticotropic stimulating hormone.
2.2. Molecular analysis
This study was approved by the National Research Centre Ethics Committees and written informed consent was obtained from the child's parents. Peripheral blood samples were taken from two affected sibs and from their parents. Polymerase chain reaction (PCR) amplification of genomic DNA was performed using primers spanning the coding exons and flanking introns of the LEP gene (RefSeq NM_000230.2) as described previously [12]. PCR reactions were performed in a final volume of 25 μl containing 100 ng of genomic DNA, 0.2 m M dNTP mix, 0.2 μ M of each primer, 1.5 mM MgCl2 and 2 units of Taq polymerase. The amplicons were purified using the QIAquick PCR purification kit (Applied Biosystems, Warrington, UK) and sequenced using an Applied Biosystems 310 DNA Analyzer.
3. Results
Plasma leptin levels were undetectable in these children. Direct sequencing of the LEP gene in the two affected sibs disclosed a novel homozygous nonsense mutation p.Trp121X (TGG to TAG) in exon 3 of the LEP gene (Fig. 2). Sequencing analysis of parental DNA revealed that the mutation was present in heterozygous form in both parents. The mutation was not detected in 100 alleles of 50 healthy Egyptian children and is likely to yield a truncated protein (due to nonsense-mediated mRNA decay), that is not secreted.
Fig. 2.
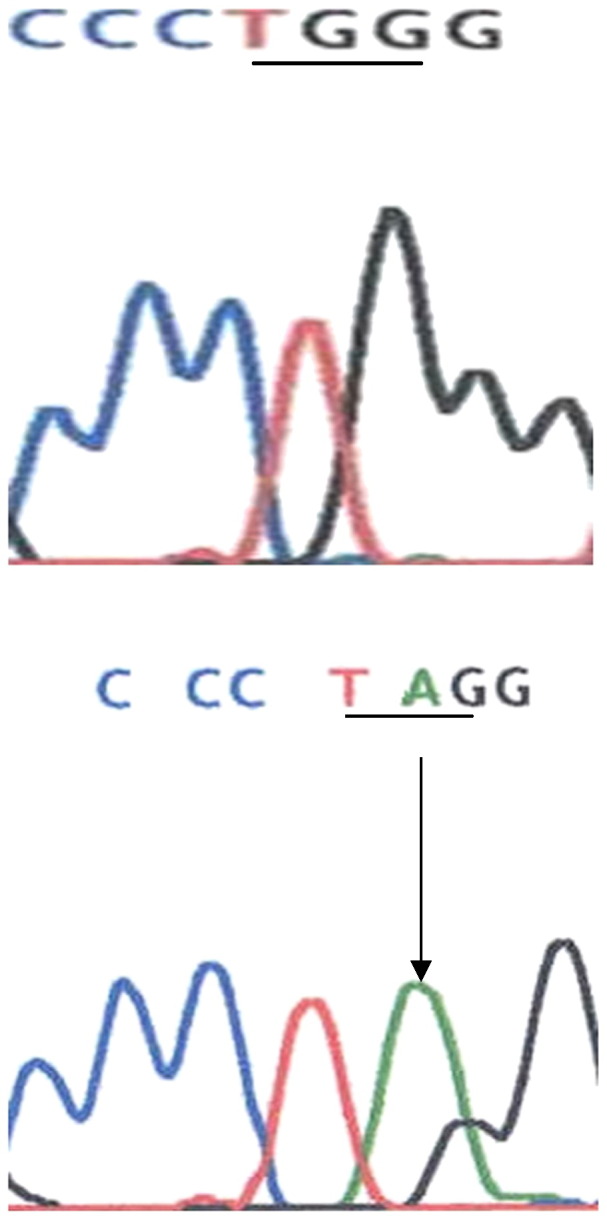
Direct sequencing of PCR-amplified products of exon 3 of the LEP gene revealed a nonsense mutation p.Trp121X (TGG to TAG).
4. Discussion
Congenital leptin deficiency in humans is a rare autosomal recessive monogenic obesity syndrome that results from mutations in the leptin gene [2]. In this study, we report a novel nonsense W121X (c.223G.A) mutation in two siblings from a consanguineous Egyptian family. The patient was born of normal birth weight to Egyptian parents who were first cousins. The clinical features are consistent with previous reports as the patient was intensely hyperphagic with food seeking behaviour and aggressive behaviour when food was denied. The patient had a history of recurrent respiratory tract infections. Two younger siblings had died in this family suffered from recurrent respiratory infections and both were severely obese. Children with leptin deficiency have previously been shown to have profound abnormalities of T cell number and function [3], consistent with high rates of childhood infection and a high reported rate of childhood mortality from infection in obese Turkish subjects [13].
The patient was hyperinsulinaemic consistent with the severity of obesity (40 IU/ml) but was normoglycaemic (4.5 mmol/l). Furthermore, plasma thyroid stimulating hormone (TSH) and free thyroxine were within the normal range (Table 1). Elevated TSH levels have been reported in four leptin deficient children and in one instance led to the initiation of thyroxine therapy which was withdrawn after normalization of TSH following leptin administration [3], [11].
Normal pubertal development does not occur in adults with leptin deficiency, with biochemical evidence of hypogonadotropic hypogonadism [10], [13]. It is noticeable that our older patient had basal gonadotropin concentrations in the prepubertal range and there was no development of secondary sexual characteristics.
In conclusion, we report a second novel mutation of the LEP gene in Egyptian patients. Although congenital leptin deficiency is rare, administration of recombinant human leptin in patients with congenital leptin deficiency represents a rational mechanism based therapy for severe obesity and has the potential to provide substantial clinical benefits for the patients concerned [14], [15].
Acknowledgements
ISF was supported by the Wellcome Trust, the MRC and the UK NIHR Cambridge Biomedical Research Centre.
References
- 1.Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J.M. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 2.Montague C.T., Farooqi I.S., Whitehead J.P., Soos M.A., Rau H., Wareham N.J., Sewter C.O., Digby J.E., Mohammed S.N., Hurst J.A., Cheetham C.H., Earley A.R., Barnett A.H., Prins J.B., O'Rahilly S. Congenital leptin deficiency is associated with severe early onset obesity in humans. Nature. 1997;387:903–907. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 3.Farooqi I.S., Matarese G., Lord G.M., Keogh J.M., Lawrence E., Agwu C., Sanna V., Jebb S.A., Perna F., Fontana S., Lechler R.I., DePaoli A.M., O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank S., Heni M., Moss A., von Schnurbein J., Fritsche A., Häring H.U., Farooqi S., Preissl H., Wabitsch M. Leptin therapy in a congenital leptin-deficient patient leads to acute and long-term changes in homeostatic, reward, and food-related brain areas. J. Clin. Endocrinol. Metab. 2011;96:E1283–E1287. doi: 10.1210/jc.2010-2713. [DOI] [PubMed] [Google Scholar]
- 5.Mazen I., El-Gammal M., Abdel-Hamid M., Amr K. A novel homozygous missense mutation of the leptin gene (N103K) in an obese Egyptian patient. Mol. Genet. Metab. 2009;97:305–308. doi: 10.1016/j.ymgme.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Fatima W., Shahid A., Imran M., Manzoor J., Hasnain S., Rana S., Mahmood S. Leptin deficiency and leptin gene mutations in obese children from Pakistan. Int. J. Pediatr. Obes. 2011;6:419–427. doi: 10.3109/17477166.2011.608431. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y., Hong N., Liu X., Wu B., Tang S., Yang J., Hu C., Jia W. A novel mutation in leptin gene is associated with severe obesity in Chinese individuals. BioMed Res. Int. 2014;2014:912052. doi: 10.1155/2014/912052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeed S., Butt T.A., Anwer M., Arslan M., Froguel P. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol. Genet. Metab. 2012;106:121–126. doi: 10.1016/j.ymgme.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Chekhranova M.K., Karpova S.K., Iatsyshina S.B., Pankov IuA. A new mutation c.422C>G (p.S141C) in homo- and heterozygous forms of the human leptin gene. Bioorg. Khim. 2008;34:854–856. doi: 10.1134/s1068162008060198. [DOI] [PubMed] [Google Scholar]
- 10.Strobel A., Issad T., Camoin L., Ozata M., Strosberg A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998;18:213–215. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 11.Gibson W.T., Farooqi I.S., Moreau M., DePaoli A.M., Lawrence E., O'Rahilly S., Trussell R.A. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab. 2004;89:4821–4826. doi: 10.1210/jc.2004-0376. [DOI] [PubMed] [Google Scholar]
- 12.Shigemoto M., Nishi S., Ogawa Y., Isse N., Matsuoka N., Tanaka T., Azuma N., Masuzaki H., Nishimura H., Yoshimasa Y., Hosoda K., Nakao K. Molecular screening of both the promoter and the protein coding regions in the human ob gene in Japanese obese subjects with non-insulin-dependent diabetes mellitus. Eur. J. Endocrinol. 1997;137:511–513. doi: 10.1530/eje.0.1370511. [DOI] [PubMed] [Google Scholar]
- 13.Ozat M., Ozdemi I.C., Licini J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 1999;84:3686–3695. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 14.Farooqi I.S., Jebb S.A., Langmack G., Lawrence E., Cheetham C.H., Prentice A.M., Hughes I.A., McCamish M.A., O'Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 15.Licinio J., Caglayan S., Ozata M., Yildiz B.O., de Miranda P.B., O'Kirwan F., Whitby R., Liang L., Cohen P., Bhasin S., Krauss R.M., Veldhuis J.D., Wagner A.J., DePaoli A.M., McCann S.M., Wong M.L. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4531–4536. doi: 10.1073/pnas.0308767101. [DOI] [PMC free article] [PubMed] [Google Scholar]