

Abstract
Swine dysentery (SD) is a mucohemorrhagic colitis of swine classically caused by infection with the intestinal spirochete Brachyspira hyodysenteriae. Since around 2007, cases of SD have occurred in North America associated with a different strongly beta-hemolytic spirochete that has been molecularly and phenotypically characterized and provisionally named “Brachyspira hampsonii.” Despite increasing international interest, B. hampsonii is currently not recognized as a valid species. To support its recognition, we sequenced the genomes of strains NSH-16T, NSH-24, and P280/1, representing B. hampsonii genetic groups I, II, and III, respectively, and compared them with genomes of other valid Brachyspira species. The draft genome of strain NSH-16T has a DNA G+C content of 27.4% and an approximate size of 3.2 Mb. Genomic indices, including digital DNA-DNA hybridization (dDDH), average nucleotide identity (ANI), and average amino acid identity (AAI), clearly differentiated B. hampsonii from other recognized Brachyspira species. Although discriminated genotypically, the three genetic groups are phenotypically similar. By electron microscopy, cells of different strains of B. hampsonii measure 5 to 10 μm by 0.28 to 0.34 μm, with one or two flat curves, and have 10 to 14 periplasmic flagella inserted at each cell end. Using a comprehensive evaluation of genotypic (gene comparisons and multilocus sequence typing and analysis), genomic (dDDH, ANI, and AAI) and phenotypic (hemolysis, biochemical profiles, protein spectra, antibiogram, and pathogenicity) properties, we classify Brachyspira hampsonii sp. nov. as a unique species with genetically diverse yet phenotypically similar genomovars (I, II, and III). We designate the type strain NSH-16 (= ATCC BAA-2463 = NCTC 13792).
INTRODUCTION
The genus Brachyspira includes Gram-negative aerotolerant anaerobic spirochetes that colonize the intestine of and/or cause disease in a wide range of host species (1). Over several decades, multiple taxonomic changes were applied to members of this genus (originally Treponema, which was then transferred to Serpula, to Serpulina, and finally to Brachyspira) (2–5). Currently, the genus Brachyspira consists of eight valid species, B. hyodysenteriae, B. pilosicoli, B. intermedia, B. innocens, B. murdochii, B. aalborgi, B. alvinipulli, and most recently, B. suanatina (1, 6). This genus also consists of several provisional species (1), of which the most clinically significant is the recently discovered “Brachyspira hampsonii” (7). Within the Brachyspira genus, all currently identified strongly beta-hemolytic species (B. hyodysenteriae, B. suanatina, and the novel B. hampsonii) are known to cause severe mucohemorrhagic diarrhea in pigs, while weakly beta-hemolytic Brachyspira species are either commensals (B. innocens) or are capable of causing diarrhea and/or colitis (B. pilosicoli, B. murdochii, B. intermedia, B. aalborgi, and B. alvinipulli) in pigs, chickens and/or humans (1). B. hyodysenteriae, the most virulent and clinically significant Brachyspira species, has historically also been the most researched or investigated species. It causes swine dysentery (SD), a disease characterized by mucohemorrhagic diarrhea that is most commonly observed in grower-finisher pigs (1). In addition to the adverse impact on the health and welfare of pigs, its negative effect on productivity (such as decreased weight gain and poor feed conversion) leads to significant economic losses to livestock-raising communities and countries (1). The recently validated B. suanatina also causes SD in pigs; however, its isolation has been limited to a few northern European countries (8). The isolation of different bacterial species from clinically and pathologically indistinguishable dysentery cases of pigs highlights the evolving and expanding etiology of SD. Thus, the definition of SD should include all strongly beta-hemolytic Brachyspira species that cause mucohemorrhagic colitis and dysentery in pigs (9). The genetically diverse B. pilosicoli is the primary etiological agent of colonic spirochetosis, a disease characterized by diarrhea and/or colitis in a wide range of host species, including pigs (porcine intestinal spirochetosis [PIS]) (10), chickens (avian intestinal spirochetosis [AIS]) (11), and human beings (human intestinal spirochetosis [HIS]) (12). AIS also can be caused by other Brachyspira species, including B. intermedia (13) and B. alvinipulli (14), while HIS is also caused by B. aalborgi (15). Although long considered to be a commensal, the association of B. murdochii with mild diarrhea and/or colitis in pigs has been reported (16, 17). These Brachyspira-associated disease conditions negatively impact the health and welfare of the affected host species and reduce the productivity of livestock (1).
Clinical SD was rarely reported in North America after the early 1990s, despite continuing to have negative impacts on the health and productivity of pigs in other countries across the world. Outbreaks of bloody diarrhea in commercial swine herds in 2007 signaled the reemergence of this disease in North America (18). Interestingly, the detection of reemergent B. hyodysenteriae in the United States was accompanied by the unexpected discovery of a novel species B. hampsonii from cases of classic mucohemorrhagic diarrhea which were clinically indistinguishable from those caused by B. hyodysenteriae. Preliminary characterization led to the identification and provisional designation of B. hampsonii and its two diverse genetic groups (previously called clades), group I and group II (7). Since the initial identification of B. hampsonii in North American pigs, it has also been detected in pigs in Belgium and Germany (19, 20) and in migratory water birds in Europe and North America (21, 22).
Several methods have been used to characterize the phenotype of B. hampsonii, including growth characterization, identification and qualification of hemolysis on blood agar, biochemical tests (hippurate hydrolysis, production of indole, α-galactosidase, α-glucosidase, and β-glucosidase activities), protein spectrum profiling, antibiogram testing, and characterization of its pathogenic nature. Growth on solid medium (tryptic soy agar containing 5% defibrinated sheep blood) is observed as tiny transparent colonies with underlying strong beta-hemolysis (23) that is most distinct in areas of cuts made in the agar (known as the “ring phenomenon”), while growth in liquid medium (brain heart infusion broth supplemented with 10% fetal bovine serum) is observed as light turbidity in the broth (24). Cultural properties alone are insufficient to differentiate B. hampsonii from other strongly beta-hemolytic Brachyspira species (B. hyodysenteriae and B. suanatina), thus emphasizing the need for other phenotypic or genotypic tests. B. hampsonii isolates were found to be negative for indole production and for hippurate, α-galactosidase, and α-glucosidase activity, with the indole spot test being the most useful test for differentiating B. hampsonii from other strongly beta-hemolytic Brachyspira species (B. hyodysenteriae and B. suanatina are usually indole positive) (7, 25). Although two biochemical profiles for B. hampsonii have been described, neither was absolutely effective in differentiating genetic groups I and II (7). Main spectrum profiles (MSPs) generated with matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) technology were consistently able to identify B. hampsonii and differentiate B. hampsonii from other Brachyspira species (26). Although this method was able to often differentiate between the genetic groups of B. hampsonii, this differential identification was not consistently reliable (26). A study characterizing antibiograms of North American B. hampsonii isolates reported its high susceptibility to several commonly used antimicrobials, including tiamulin, valnemulin, lincomycin, tylosin, doxycycline, and carbadox (27). Although B. hampsonii often demonstrated a more susceptible antibiogram than other Brachyspira species, no clear differences in antibiogram profiles were observed between its genetic groups (27). Finally, several trials have reproduced mucohemorrhagic diarrhea in pigs by oral inoculation of B. hampsonii genetic groups I and II under experimental conditions and have thus confirmed the pathogenic nature of both groups (28–30). The resulting disease was indistinguishable from SD caused by B. hyodysenteriae on the basis of clinical signs and gross pathology. Examination of tissues obtained from the experimentally infected pigs had microscopic lesions consistent with those seen in the mucohemorrhagic colitis induced by B. hyodysenteriae (28–30). Currently, no differences in clinical signs or gross and microscopic pathology have been reported in the SD caused by either genetic group I or II of B. hampsonii.
Several methods have been used to characterize the genotype of B. hampsonii, including gene comparisons and identification of genotypes (7, 31). For the purpose of species delineation in the Brachyspira genus, the NADH oxidase (nox) gene has historically been considered to be more useful than the 16S rRNA gene (32). Both of these genes have been used to identify B. hampsonii and differentiate it from other Brachyspira species, as well as to differentiate between the diverse genetic groups of B. hampsonii (7). The nox gene is also often used as a target for diagnostic tests, such as quantitative PCR (qPCR) and Sanger sequencing (7, 29), to specifically detect B. hampsonii. Genotyping of B. hampsonii from diverse epidemiological origins using the multilocus sequence typing (MLST) approach (31) identified a total of 20 genotypes that clustered into four genetic groups (I, II, III, and IV). It included the commonly reported genetic groups I and II that are frequently isolated from affected North American pigs (7) and occasionally isolated from pigs in Europe (19, 20), and from migratory birds of both North American and European origin (21, 22). It also included the less frequently reported genetic group III, which has been isolated occasionally from pigs and migratory water birds of European origin (20, 21, 33), as well as the rare genetic group IV, which has been detected only in migratory water birds in Europe (21). Overall, B. hampsonii was observed to demonstrate high diversity and a heterogeneous population structure (31). In addition, a Brachyspira genus-wide multilocus sequence analysis (MLSA) approach was used to confirm that B. hampsonii could be differentiated from other Brachyspira species. This study reported clustering of B. hampsonii genetic groups in spite of its diverse nature (31).
Despite the significance of this novel pathogenic species and the information that is currently available, B. hampsonii still remains a proposed species. Therefore, the objective of this study was to support its position as a valid species by providing additional information on its whole-genome sequences, genomic relatedness to other Brachyspira species, and ultrastructural morphology.
MATERIALS AND METHODS
B. hampsonii isolates.
Brachyspira hampsonii strains NSH-16 (ATCC BAA-2463), NSH-24 (ATCC BAA-2464), and P280/1 were selected for study, as they represent the type strains for genetic groups I, II, and III, respectively. Most importantly, strain NSH-16 is also the designated type strain for B. hampsonii (ATCC BAA-2463 = NCTC 13792). Plates of tryptic soy agar (TSA) (BD, Franklin Lakes, NJ, USA) containing 5% defibrinated sheep blood (I-Tek Medical Technologies, MN, USA) were inoculated with pure cultures and incubated under anaerobic conditions at 37°C for 4 days. Growth was observed as zones of strong beta-hemolysis, with observation of the characteristic ring phenomenon. The purity of the isolates was confirmed by phase-contrast microscopy of wet mounts.
Phase-contrast and electron microscopy.
Cells of strains NSH-16T, NSH-24, and P280/1 were grown to mid-log phase on TSA plates and prepared for phase-contrast and electron microscopy as described previously (10). Actively dividing cells were gently harvested from the plates with 1 ml of 0.01 M sodium phosphate buffer at pH 7.0 and centrifuged at 2,000 × g for 3 min. The pellet was resuspended with 1 ml of phosphate buffer and centrifuged at 2,000 × g to wash the cells. Washing was performed three times before resuspending the cells with 0.5 ml of phosphate buffer. For phase-contrast microscopy, the washed cells were adhered to coverslips using 0.1% polyethyleneimine and examined with a Nikon Eclipse 90i microscope under a 100× phase-contrast objective with a Ph3 condenser ring. For electron microscopy, a 0.02-ml sample of the washed cells was negatively stained with an equal volume of 2% phosphotungstic acid (pH 7) before being mounted on a carbon-reinforced 200-mesh copper grid coated with 2% Parlodion. The grids were examined with a Phillips model 410 transmission electron microscope. The cell dimensions and the ultrastructural characteristics of the spirochete were determined from electron micrographs of at least 10 individual cells.
Genome sequencing, assembly, and annotation.
Genomic DNA from each isolate was extracted using the DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA), as per the manufacturer's instructions. For strains NSH-16T and NSH-24, quality control, library preparation, and whole-genome sequencing of the extracted genomic DNA were carried out at the University of Minnesota Genomics Center, Minneapolis, MN. Briefly, the samples were evaluated for quality control and DNA concentrations using the Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Thermo Fisher Scientific, Waltham, MA, USA), and the DNA library was prepared using the Nextera XT DNA library preparation kit (Illumina, CA, USA), as per the manufacturer's instructions. Sequencing was carried out using the MiSeq reagent kit version 3 (Illumina) with a paired-end 2 × 300-bp construct on the MiSeq system (Illumina). This yielded 4,566,767 and 3,603,642 passing filter reads for strains NSH-16T and NSH-24, which corresponded to average genome coverages of approximately 860× and 700×, respectively. Using the default parameters of the De Novo Assembly tool of CLC Genomics Grid Workbench 8.0.2, the reads were quality checked, trimmed based on quality, and assembled de novo to generate contigs. Filters were applied to select and extract a subset of contigs with consensus length of ≥1 kb and coverage of ≥50× in order to generate a draft genome. Whole-genome sequencing of strain P280/1 was performed in Australia by GeneWorks Pty. Ltd. (Thebarton, SA, Australia) under the Illumina Certified Service Provider (CSPro) program. Sequencing was carried out using the TruSeq DNA PCR-free library preparation kit (Illumina), with a paired-end 2 × 75-bp construct on the Genome Analyzer IIx (Illumina), which yielded 14,097,542 reads corresponding to an average genome coverage of approximately 661×. De novo assembly of reads was performed with the SeqMan NGen 3.0 assembly software (DNAStar, Madison, WI, USA) using default parameters to generate a draft genome. All three strains were annotated using the Rapid Annotations using Subsystems Technology (RAST version 2.0) (34) with parameters allowing for frameshift error corrections. The genomes were also annotated using the NCBI Prokaryotic Genome Annotation Pipeline (35).
Genome comparisons for species delineation.
Publicly available genomes of various Brachyspira species were obtained from the NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/). These included B. hyodysenteriae (strain B-78T), B. suanatina strain AN4859/03T, B. pilosicoli strain P43/6/78T, B. intermedia strain PWS/AT, B. murdochii strain 56-150T, B. innocens strain B256T, B. alvinipulli strain 911207/C1T, and B. hampsonii (strains 30599 and 30446). The publicly available genome of B. aalborgi strain 513AT was obtained from the MetaHIT Consortium website (http://www.sanger.ac.uk/resources/downloads/bacteria/metahit/). The genome-to-genome distance (GGD) values of B. hampsonii strains NSH-16T, NSH-24, and P280/1 and other Brachyspira species and strains were calculated using the Genome-to-Genome Distance Calculator (GGDC 2.1) Web service (http://ggdc.dsmz.de/distcalc2.php) (36). Similarly, the average nucleotide identity (ANI) values and the average amino acid identity (AAI) values of B. hampsonii strains NSH-16T, NSH-24, and P280/1 and other Brachyspira species and strains were calculated using the EzGenome ANI Web service (http://www.ezbiocloud.net/ezgenome/ani) (37) based on the algorithm of Goris et al. (38), and using the Web-based AAI tool (http://enve-omics.ce.gatech.edu/aai/index) (39) based on two-way AAI calculations, respectively. SpecI, a Web-based species identification tool (http://vm-lux.embl.de/~mende/specI/) (40), was used to extract 40 universal single-copy-marker genes of B. hampsonii strains NSH-16T, NSH-24, and P280/1 and evaluate the average genetic distance of these strains from publicly available complete genomes of valid bacterial species.
Accession number(s).
The whole-genome shotgun projects for B. hampsonii strains NSH-16T, B. hampsonii NSH-24, and B. hampsonii P280/1 have been deposited at DDBJ/ENA/GenBank under the accession numbers LZOF00000000, LZOG00000000, and MDCO00000000, respectively. The versions described in this paper are LZOF01000000, LZOG01000000, and MDCO01000000, respectively.
RESULTS
As determined by phase-contrast and electron microscopy, the shape of the spirochete cells was consistent with that of other Brachyspira species. The cells had slightly tapered ends and one or two flat serpentine curves (Fig. 1). The cells of P280/1 were longer than those of NSH-16T and NSH-24 but were otherwise similar, with 10 to 14 periplasmic flagella inserted subterminally at each end of the cell, with a total of 20 to 28 flagella per cell (Fig. 2). The cells of P280/1 were 10.49 ± 0.41 μm long, whereas those of NSH-16T and NSH-24 were 5.43 ± 0.34 and 5.06 ± 0.37 μm long, respectively (Table 1). The mean cell widths for the strains varied from 0.28 to 0.34 μm.
FIG 1.
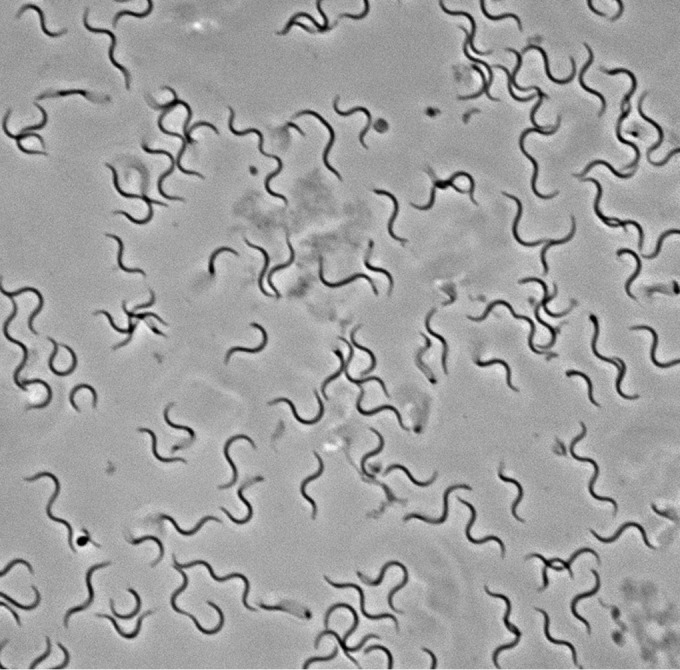
Phase-contrast micrograph of B. hampsonii strain NSH-24 cells viewed at ×100 showing one to two flat serpentine coils and slightly tapered ends.
FIG 2.
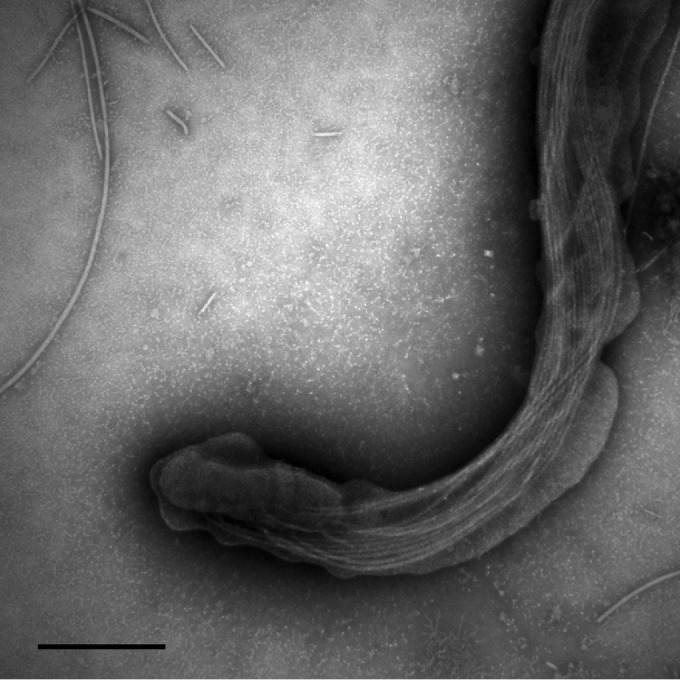
Electron micrograph of negatively stained B. hampsonii strain NSH-16T showing 12 periplasmic flagella at one end of the cell. The cell was viewed at ×60,000 magnification, and the scale bar represents 500 nm.
TABLE 1.
Comparison of mean cell dimensions of B. hampsonii strains
| Dimension (in μm) by strain | Mean | SD |
|---|---|---|
| NSH-16T | ||
| Length | 5.43 | 0.43 |
| Width | 0.34 | 0.01 |
| NSH-24 | ||
| Length | 5.06 | 0.37 |
| Width | 0.28 | 0.03 |
| P280/1 | ||
| Length | 10.49 | 0.41 |
| Width | 0.33 | 0.01 |
The final assembly of the B. hampsonii strain NSH-16T genome resulted in 77 contigs comprising approximately 3.16 Mb, with a G+C content of 27.4%. Eleven large contigs >100 kb in size and another 43 contigs 10 to 100 kb in size comprised 97.4% of the assembled B. hampsonii strain NSH-16T genome. The final assembly of B. hampsonii strain NSH-24 resulted in 178 contigs comprising approximately 2.97 Mb, with a G+C content of 27.5%. One large contig of >100 kb in size and another 93 contigs 10 to 100 kb in size comprised 88% of the assembled B. hampsonii strain NSH-24 genome. Assembly of the B. hampsonii strain P280/1 genome resulted in 16 contigs of 3,186,631 bp, with a G+C content of 27.5%. The general genomic features of B. hampsonii strains NSH-16T, NSH-24, and P280/1 are shown in Table 2.
TABLE 2.
Genome assembly statistics and annotation features of B. hampsonii strains
| Genome feature | NSH-16T | NSH-24 | P280/1 |
|---|---|---|---|
| Genome status | Draft | Draft | Draft |
| Total assembly size (bp) | 3,161,271 | 2,969,002 | 3,186,631 |
| No. of contigs | 77 | 178 | 16 |
| N50 (bp) | 88,495 | 29,547 | 690,165 |
| L50 | 13 | 29 | 2 |
| G+C content (%) | 27.4 | 27.5 | 27.5 |
| No. of subsystems | 309 | 307 | 309 |
| No. of coding sequences | 2,822 | 2,576 | 2,945 |
| No. of predicted RNAs | 36 | 35 | 39 |
The GGD, ANI, and AAI values comparing B. hampsonii strains NSH-16T, NSH-24, and P280/1 with other Brachyspira species and strains are shown in Tables 3, 4, and 5, respectively. Comparisons of B. hampsonii GGD values with other Brachyspira species, between B. hampsonii genetic groups, and within B. hampsonii genetic groups were approximately 20 to 35%, 50 to 57%, and 99%, respectively. A similar trend was observed when using the ANI method, where interspecies, intergenetic group, and intragenetic group comparisons yielded nucleotide identities of approximately 75 to 88%, 93 to 94.5%, and 100%, respectively. The AAI method yielded mostly similar results for interspecies, intergenetic group, and intragenetic group comparisons, with amino acid identities of approximately 72 to 90%, 94 to 95%, and 100%, respectively. SpecI was unable to categorize B. hampsonii as any previously recognized valid bacterial species (Table 6). Interestingly, it identified B. hampsonii genetic group I as a closest match to B. hyodysenteriae, and genetic groups II and III as closest matches to B. intermedia.
TABLE 3.
Genome-to-genome distance comparisons of B. hampsonii and other valid Brachyspira species
| Reference genome | Genome-to-genome distance (% [model CI {%}])a |
||
|---|---|---|---|
| B. hampsonii NSH-16T | B. hampsonii NSH-24 | B. hampsonii P280/1 | |
| B. hampsonii NSH-16T | 100 (100–100) | 50.5 (47.9–53.2) | 53.2 (50.5–55.9) |
| B. hampsonii 30599 | 98.8 (98.2–99.2) | 51.3 (48.7–54) | 53.9 (51.2–56.5) |
| B. hampsonii NSH-24 | 50.5 (47.9–53.2) | 100 (100–100) | 57.2 (54.4–59.9) |
| B. hampsonii 30446 | 50.2 (47.6–52.9) | 99.6 (99.3–99.8) | 56.9 (54.1–59.7) |
| B. hampsonii P280/1 | 53.2 (50.5–55.9) | 57.2 (54.4–59.9) | 100 (100–100) |
| B. hyodysenteriae B-78T | 34.6 (32.2–37.2) | 34 (31.5–36.5) | 34.2 (31.8–36.7) |
| B. suanatina AN4859/03T | 34.7 (32.2–37.2) | 34.1 (31.7–36.6) | 34.3 (31.9–36.8) |
| B. intermedia PWS/AT | 35 (37.2–42.3) | 34.4 (31.9–36.9) | 34.6 (32.2–37.1) |
| B. murdochii 56-150T | 30.2 (27.8–32.7) | 29.6 (27.2–32.1) | 30.2 (27.8–32.7) |
| B. innocens B256T | 29.7 (27.3–32.2) | 29.5 (27.1–32) | 29.8 (27.4–32.3) |
| B. alvinipulli 911207/C1T | 25.8 (23.5–28.3) | 25.6 (23.3–28.1) | 25.6 (23.2–28) |
| B. pilosicoli P43/6/78T | 24.9 (22.6–27.4) | 24.7 (22.3–27.1) | 24.9 (22.6–27.4) |
| B. aalborgi 513AT | 20.9 (18.6–23.3) | 20.2 (18–22.6) | 21.1 (18.8–23.5) |
GGD values have been calculated using formula 2, which is recommended for the GGDC method calculation, as it is independent of the length of genomes and thus robust against the use of draft genomes (http://ggdc.dsmz.de/formhelp.php). CI, confidence interval.
TABLE 4.
Average nucleotide identity of B. hampsonii and all valid Brachyspira species
| Reference genome | ANI (%) |
||
|---|---|---|---|
| B. hampsonii NSH-16T | B. hampsonii NSH-24 | B. hampsonii P280/1 | |
| B. hampsonii NSH-16T | 100 | 93.39 | 93.70 |
| B. hampsonii 30599 | 99.83 | 93.33 | 93.72 |
| B. hampsonii NSH-24 | 92.82 | 100 | 94.50 |
| B. hampsonii 30446 | 92.82 | 99.9 | 94.44 |
| B. hampsonii P280/1 | 93.52 | 94.52 | 100 |
| B. hyodysenteriae B-78T | 88 | 87.63 | 87.83 |
| B. suanatina AN4859/03T | 88.06 | 87.7 | 87.96 |
| B. intermedia PWS/AT | 88.19 | 87.8 | 88.01 |
| B. murdochii 56-150T | 84.71 | 84.3 | 84.74 |
| B. innocens B256T | 84.35 | 84.5 | 84.59 |
| B. alvinipulli 911207/C1T | 82.07 | 81.75 | 81.86 |
| B. pilosicoli P43/6/78T | 78.26 | 78.25 | 78.16 |
| B. aalborgi 513AT | 74.88 | 74.91 | 74.78 |
TABLE 5.
Average amino acid identity of B. hampsonii and all valid Brachyspira species
| Reference genome | AAI (%) |
||
|---|---|---|---|
| B. hampsonii NSH-16T | B. hampsonii NSH-24 | B. hampsonii P280/1 | |
| B. hampsonii NSH-16T | 100 | 94.07 | 94.09 |
| B. hampsonii 30599 | 99.72 | 94.03 | 94.16 |
| B. hampsonii NSH-24 | 94.09 | 100 | 95.04 |
| B. hampsonii 30446 | 94.07 | 99.95 | 95.12 |
| B. hampsonii P280/1 | 94.08 | 95.04 | 100 |
| B. hyodysenteriae B-78T | 89.59 | 89.11 | 89.06 |
| B. suanatina AN4859/03T | 89.27 | 88.92 | 88.88 |
| B. intermedia PWS/AT | 89.58 | 88.95 | 88.94 |
| B. murdochii 56-150T | 84.87 | 84.90 | 85.42 |
| B. innocens B256T | 84.41 | 84.78 | 84.55 |
| B. alvinipulli 911207/C1T | 80.98 | 80.61 | 80.50 |
| B. pilosicoli P43/6/78T | 75.04 | 75.05 | 74.96 |
| B. aalborgi 513AT | 71.57 | 71.62 | 71.55 |
TABLE 6.
Comparison of B. hampsonii to known valid bacterial genomes using SpecI
| Query genome (genomovar) | Result | Closest match |
|
|---|---|---|---|
| NCBI taxonomy name | Avg % identity | ||
| B. hampsonii NSH-16T (I) | Was not assigned a species | Brachyspira hyodysenteriae WA1 | 93.31 |
| B. hampsonii NSH-24 (II) | Was not assigned a species | Brachyspira intermedia PWS/A | 92.92 |
| B. hampsonii P280/1 (III) | Was not assigned a species | Brachyspira intermedia PWS/A | 93.34 |
DISCUSSION
Since the initiation of bacterial taxonomy in the late 19th century, the accepted taxonomic practices for delineation of novel species have evolved with the advent of new technologies and scientific methods. Initially, bacteria were classified on phenotypic characteristics, such as growth requirements, morphology, pathogenicity, physiology, and biochemical activity. Gradually, chemotaxonomy, numerical taxonomy, conventional DDH, DNA G+C content, and eventually 16S rRNA gene sequencing provided further methods of species differentiation. A detailed review of the history of bacterial taxonomy has been provided by Schleifer (41). Most recently, whole-genome sequencing has facilitated several additional approaches to species delineation, including comparison of genome indexes, gene content, and multiple gene aligned sequence data sets (42). The utility of DNA G+C content comparison is limited, as members of several bacterial genera show a high conservation of G+C content, and thus this method serves mostly as an exclusionary determinant (41). Of the abovementioned genotypic methods, conventional DDH and 16S rRNA gene sequencing have been widely used for differentiating bacterial species over the last several years (42). Although 16S rRNA gene sequencing is an effective way to differentiate bacterial species because of its genetically and functionally highly stable nature, this method is not useful for some bacterial species that have multiple rRNA operons in a single genome or show a high degree of conservation within a genus (41). Further, conventional DDH is known to be laborious, error-prone with low reproducibility, expensive, and not equally applicable to all bacterial genera (41). Thus, methods evaluating whole-genome sequence similarity, such as digital DDH, were proposed, as they overcome many of the drawbacks while maintaining a good correlation with conventional DDH and 16S rRNA sequencing for species delineation (38, 43). Given the plethora of methods available, current prokaryotic taxonomy is often based on polyphasic combinations of phenotypic, genotypic, genomic, and/or chemotaxonomic characteristics (41).
This study confirms that the ultrastructure of B. hampsonii cells is similar to that of other Brachyspira species, such as B. hyodysenteriae. The genomes of B. hampsonii strains NSH-16T, NSH-24, and P280/1 show similar G+C content, which falls within the general range of G+C content currently identified for members of the Brachyspira genus (∼27% to 28%). This is not surprising, as diverse Brachyspira species show limited variation in their average chromosomal G+C content (6, 44–47). The approximated genome size also falls within the range of most members of the Brachyspira genus (range, ∼2.7 Mb to ∼3.4 Mb) (6, 44–47). Applying the recommended <70% threshold value for DDH (48) to the GGD results, and the <95 to 96% threshold (49, 50) to the ANI and AAI results, these B. hampsonii strains did not fall under the classification of any previously recognized Brachyspira species. Further, the 96.5% threshold for similarity to universal marker genes (40) was also unable to assign these strains to any known bacterial or archaeal species. These genomic indices add to the already-existing information supporting the position of B. hampsonii as a novel species. Surprisingly, based upon several universal marker genes, the closest matches identified for the various genetic groups differed (i.e., B. hyodysenteriae for genetic group I and B. intermedia for genetic groups II and III). A similar observation was made by the use of whole-genome sequence data, wherein B. hampsonii showed the closest identity to B. hyodysenteriae, B. intermedia, and B. suanatina, followed by B. murdochii and B. innocens. This was in contrast to previous studies (7, 21, 31) which identified B. hampsonii to be most genetically related to B. murdochii and/or B. innocens. Since those studies (7, 21, 31) evaluated only a few conserved genes, it is likely that the genetic relatedness of the overall genome was underrepresented. The use of whole-genome data in this study provides the opportunity to make more detailed and extensive comparisons between B. hampsonii and other species. Future studies comparing the core genomes of various Brachyspira species will help identify with which species B. hampsonii shares common ancestors.
The genus Brachyspira is unique and complicated, as it consists of a variety of species that can infect a wide range of host species with different abilities to cause disease, yet each shows varying degrees of ability to be differentiated by phenotypic and genotypic characteristics. For instance, the low variation in 16S rRNA gene sequence and DNA G+C content (<1%) would be insufficient to differentiate the various species within the Brachyspira genus. On the other hand, genetically and phenotypically diverse species (B. hyodysenteriae, B. suanatina, and B. hampsonii) all infect a single host species (pig), occupy the same ecological niche (the colon), and cause a clinically and pathologically indistinguishable disease (SD). Thus, a comprehensive and conservative approach that evaluates information on a variety of genotypic and phenotypic properties as well as ecological characteristics should be applied in delineating species within the Brachyspira genus. While both genotypic and phenotypic data clearly support B. hampsonii as a novel species, they provide ambiguous interpretations for whether the various genetic groups represent one or multiple novel species. Specifically, although the genomic indices (GGD, ANI, and AAI values) comparing B. hampsonii genetic groups I, II, and III to each other are significantly lower than the threshold of species differentiation, they are also significantly higher than the values obtained in a comparison of either of the genetic groups with other Brachyspira species. This depicts a situation wherein based on genomic information, one could identify the genetic groups of B. hampsonii as three closely related species. Tindall et al. (42) recommends that the <70% threshold for DDH (and by correlation, other genome sequence identity methods) should not be used as a strict boundary for species delineation. A species can include strains with DDH values of <50% if these strains are not clearly distinguishable based on other properties, such as phenotypic characteristics (42). Ursing et al. (51) recommend that such genomic groups be classified as genomovars of a single species, with the possibility for reclassification as different species once clear and stable discriminative phenotypic properties are identified. Although the genotypic properties (i.e., gene sequence comparisons [7], MLST [31], MLSA [31], GGD, ANI, and AAI) reliably discriminate several genetic groups of B. hampsonii, currently, analysis of the available phenotypic properties (i.e., beta-hemolysis on blood agar [7], biochemical profiles [7], MALDI protein spectra [26], antibiograms [27], and pathogenicity [28–30]) is unable to clearly and consistently differentiate them. Thus, based on a comprehensive genotypic, phenotypic, and genomic evaluation, we propose that Brachyspira hampsonii sp. nov. should be considered a single novel species with multiple genomovars. To that effect, the various B. hampsonii genetic groups (31) (previously called clades [7]) should henceforth be referred to as genomovars, such that genetic groups I, II, and III be replaced by the terms genomovars I, II, and III, respectively.
Description of Brachyspira hampsonii sp. nov.
Brachyspira hampsonii (hamp.so'ni.i N.L. masc. gen. n. hampsonii of Hampson), in recognition of David J. Hampson for his extensive work on the Brachyspira genus, as first proposed by Chander et al. (7).
Brachyspira hampsonii sp. nov. is a Gram-negative oxygen-tolerant anaerobe and strongly beta-hemolytic spirochete. B. hampsonii cells measure 5 to 10 μm by 0.25 to 0.38 μm, have slightly tapered ends, and have one to two flat serpentine coils. Each spirochete cell has 10 to 14 periplasmic flagella inserted at each end of the cell. Growth occurs after inoculated agar (stationary) or broth (rotating at ∼80 rpm) has been incubated at 37°C for 4 days under anaerobic (80% N2, 10% CO2, 10% H2) conditions. Growth on tryptic soy agar containing 5% defibrinated sheep blood is observed as tiny transparent colonies with underlying strong beta-hemolysis that is most distinct in areas of cuts made in the agar (known as the ring phenomenon). Growth in brain heart infusion broth containing 10% fetal bovine serum is observed as light turbidity. Strains are indole negative, hippurate negative, α-galactosidase negative, α-glucosidase negative, and either positive or negative for β-glucosidase. Strains of this species colonize pigs in which they induce swine dysentery characterized by mucohemorrhagic diarrhea. They also are recorded as naturally colonizing species of waterfowl, including feral ducks and geese. They are highly susceptible to the antimicrobials tiamulin, valnemulin, and carbadox. Strains of this species can be genetically differentiated from other Brachyspira species by the use of 16S rRNA and nox gene sequencing, MLST, and whole-genome sequencing, as well as species-specific PCRs based on the nox gene. The draft genome of B. hampsonii sp. nov. strain NSH-16T has a DNA G+C content of 27.4% and an approximate genome size of 3.2 Mb. Multiple genotypic (MLST, 16S rRNA, and nox gene sequence comparisons), genomic (GGD, ANI, and AAI), and phenotypic measures (hemolysis, biochemical profiles, MALDI, and antibiograms) support the taxonomic classification of Brachyspira hampsonii sp. nov. They also support the detection of several genetically diverse yet phenotypically similar groups that have now been designated genomovars (I, II, and III). The type strain for Brachyspira hampsonii sp. nov. is NSH-16. The type strains for B. hampsonii sp. nov. genomovar I, B. hampsonii sp. nov. genomovar II, and B. hampsonii sp. nov. genomovar III are designated NSH-16, NSH-24, and P280/1, respectively. B. hampsonii sp. nov. strain NSH-16T (= ATCC BAA-2463 = NCTC 13792) and B. hampsonii sp. nov. strain NSH-24 (= ATCC BAA-2464 = NCTC 13793) have been deposited with two recognized culture collections in two different countries (ATCC, USA, and NCTC, United Kingdom).
ACKNOWLEDGMENTS
We thank the Minnesota Supercomputing Institute for providing computing resources and Ying Zhang for her guidance with genome assembly. We thank Anibal Armien (College of Veterinary Medicine, University of Minnesota) and Robert Cook of the Faculty of Medicine, Dentistry and Health Sciences at the University of Western Australia for their assistance with the electron microscopy.
Funding Statement
The funders had no role in study design, data collection, data analysis, interpretation of findings, preparation of the manuscript, or the decision to publish.
REFERENCES
- 1.Hampson DJ. 2012. Brachyspiral colitis, p 680–696. In Zimmerman, J, Karriker, L, Ramirez, A, Schwartz, K, Stevenson, G (ed), Diseases of swine, 10th ed Wiley-Blackwell, Chichster, United Kingdom. [Google Scholar]
- 2.Harris DL, Glock RD, Christensen CR, Kinyon JM. 1972. Inoculation of pigs with Treponema hyodysenteriae (new species) and reproduction of the disease. Vet Med Small Anim Clin 67:61–64. [PubMed] [Google Scholar]
- 3.Stanton TB, Jensen NS, Casey TA, Tordoff LA, Dewhirst FE, Paster BJ. 1991. Reclassification of Treponema hyodysenteriae and Treponema innocens in a new genus, Serpula gen. nov., as Serpula hyodysenteriae comb. nov. and Serpula innocens comb. nov. Int J Syst Bacteriol 41:50–58. doi: 10.1099/00207713-41-1-50. [DOI] [PubMed] [Google Scholar]
- 4.Stanton TB. 1992. Proposal to change the genus designation Serpula to Serpulina gen. nov. containing the species Serpulina hyodysenteriae comb. nov. and Serpulina innocens comb. nov. Int J Syst Bacteriol 42:189–190. doi: 10.1099/00207713-42-1-189. [DOI] [PubMed] [Google Scholar]
- 5.Ochiai S, Adachi Y, Mori K. 1997. Unification of the genera Serpulina and Brachyspira, and proposals of Brachyspira hyodysenteriae comb. nov., Brachyspira innocens comb. nov. and Brachyspira pilosicoli comb. nov. Microbiol Immunol 41:445–452. [DOI] [PubMed] [Google Scholar]
- 6.Mushtaq M, Zubair S, Råsbäck T, Bongcam-Rudloff E, Jansson DS. 2015. Brachyspira suanatina sp. nov., an enteropathogenic intestinal spirochaete isolated from pigs and mallards: genomic and phenotypic characteristics. BMC Microbiol 15:208. doi: 10.1186/s12866-015-0537-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chander Y, Primus A, Oliveira S, Gebhart CJ. 2012. Phenotypic and molecular characterization of a novel strongly hemolytic Brachyspira species, provisionally designated “Brachyspira hampsonii. ” J Vet Diagn Invest 24:903–910. [DOI] [PubMed] [Google Scholar]
- 8.Råsbäck T, Jansson DS, Johansson KE, Fellstrom C. 2007. A novel enteropathogenic, strongly haemolytic spirochaete isolated from pig and mallard, provisionally designated “Brachyspira suanatina” sp. nov. Environ Microbiol 9:983–991. doi: 10.1111/j.1462-2920.2006.01220.x. [DOI] [PubMed] [Google Scholar]
- 9.Burrough ER. Swine dysentery: etiopathogenesis and diagnosis of a reemerging disease. Vet Pathol, in press. doi: 10.1177/0300985816653795. [DOI] [Google Scholar]
- 10.Trott DJ, Stanton TB, Jensen NS, Duhamel GE, Johnson JL, Hampson DJ. 1996. Serpulina pilosicoli sp. nov., the agent of porcine intestinal spirochetosis. Int J Syst Bacteriol 46:206–215. doi: 10.1099/00207713-46-1-206. [DOI] [PubMed] [Google Scholar]
- 11.McLaren AJ, Trott DJ, Swayne DE, Oxberry SL, Hampson DJ. 1997. Genetic and phenotypic characterization of intestinal spirochetes colonizing chickens and allocation of known pathogenic isolates to three distinct genetic groups. J Clin Microbiol 35:412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trivett-Moore NL, Gilbert GL, Law CLH, Trott DJ, Hampson DJ. 1998. Isolation of Serpulina pilosicoli from rectal biopsy specimens showing evidence of intestinal spirochetosis. J Clin Microbiol 36:261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampson DJ, McLaren AJ. 1999. Experimental infection of laying hens with Serpulina intermedia causes reduced egg production and increased faecal water content. Avian Pathol 28:113–117. doi: 10.1080/03079459994821. [DOI] [PubMed] [Google Scholar]
- 14.Stanton TB, Postic D, Jensen NS. 1998. Serpulina alvinipulli sp. nov., a new Serpulina species that is enteropathogenic for chickens. Int J Syst Bacteriol 48:669–676. [DOI] [PubMed] [Google Scholar]
- 15.Mikosza ASJ, La T, Brooke CJ, Lindboe CF, Ward PB, Heine RG, Guccion JG, De Boer WB, Hampson DJ. 1999. PCR amplification from fixed tissue indicates frequent involvement of Brachyspira aalborgi in human intestinal spirochetosis. J Clin Microbiol 37:2093–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komarek V, Maderner A, Spergser J, Weissenbock H. 2009. Infections with weakly haemolytic Brachyspira species in pigs with miscellaneous chronic diseases. Vet Microbiol 134:311–317. doi: 10.1016/j.vetmic.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Jensen TK, Christensen AS, Boye M. 2010. Brachyspira murdochii colitis in pigs. Vet Pathol 47:334–338. doi: 10.1177/0300985809359054. [DOI] [PubMed] [Google Scholar]
- 18.Mirajkar NS, Gebhart CJ. 2014. Understanding the molecular epidemiology and global relationships of Brachyspira hyodysenteriae from swine herds in the United States: a multi-locus sequence typing approach. PLoS One 9:e107176. doi: 10.1371/journal.pone.0107176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahu M, de Jong E, De Pauw N, Vande Maele L, Vandenbroucke V, Vandersmissen T, Miry C, Pasmans F, Haesebrouck F, Martel A, Boyen F. 2014. First isolation of “Brachyspira hampsonii” from pigs in Europe. Vet Rec 174:47.2-47. [DOI] [PubMed] [Google Scholar]
- 20.Rohde J, Habighorst-Blome K, Seehusen F. 2014. “Brachyspira hampsonii” clade I isolated from Belgian pigs imported to Germany. Vet Microbiol 168:432–435. doi: 10.1016/j.vetmic.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Lobo FJ, Hidalgo A, Garcia M, Arguello H, Naharro G, Carvajal A, Rubio P. 2013. First identification of “Brachyspira hampsonii” in wild European waterfowl. PLoS One 8:e82626. doi: 10.1371/journal.pone.0082626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubin JE, Harms NJ, Fernando C, Soos C, Detmer SE, Harding JCS, Hill JE. 2013. Isolation and characterization of Brachyspira spp. including “Brachyspira hampsonii” from lesser snow geese (Chen caerulescens caerulescens) in the Canadian Arctic. Microb Ecol 66:813–822. doi: 10.1007/s00248-013-0273-5. [DOI] [PubMed] [Google Scholar]
- 23.ATCC. Product sheet: Brachyspira hampsonii (ATCC BAA-2464). American Type Culture Collection, Manassas, VA. [Google Scholar]
- 24.ATCC. Product sheet: Brachyspira hampsonii (ATCC® BAA-2463™). American Type Culture Collection, Manassas, VA. [Google Scholar]
- 25.Fellström C, Karlsson M, Pettersson B, Zimmerman U, Gunnarsson A, Aspan A. 1999. Emended descriptions of indole negative and indole positive isolates of Brachyspira (Serpulina) hyodysenteriae. Vet Microbiol 70:225–238. doi: 10.1016/S0378-1135(99)00146-7. [DOI] [PubMed] [Google Scholar]
- 26.Warneke HL, Kinyon JM, Bower LP, Burrough ER, Frana TS. 2014. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry for rapid identification of Brachyspira species isolated from swine, including the newly described “Brachyspira hampsonii.”. J Vet Diagn Invest 26:635–639. [DOI] [PubMed] [Google Scholar]
- 27.Mirajkar NS, Davies PR, Gebhart CJ. 2016. Antimicrobial susceptibility patterns of Brachyspira species isolated from swine herds in the United States. J Clin Microbiol 54:2109–2119. doi: 10.1128/JCM.00834-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa MO, Hill JE, Fernando C, Lemieux HD, Detmer SE, Rubin JE, Harding JCS. 2014. Confirmation that “Brachyspira hampsonii” clade I (Canadian strain 30599) causes mucohemorrhagic diarrhea and colitis in experimentally infected pigs. BMC Vet Res 10:129. doi: 10.1186/1746-6148-10-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubin JE, Costa MO, Hill JE, Kittrell HE, Fernando C, Huang Y, O'Connor B, Harding JCS. 2013. Reproduction of mucohaemorrhagic diarrhea and colitis indistinguishable from swine dysentery following experimental inoculation with “Brachyspira hampsonii” strain 30446. PLoS One 8:e57146. doi: 10.1371/journal.pone.0057146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilberts BL, Arruda PH, Kinyon JM, Madson DM, Frana TS, Burrough ER. 2014. Comparison of lesion severity, distribution, and colonic mucin expression in pigs with acute swine dysentery following oral inoculation with “Brachyspira hampsonii” or Brachyspira hyodysenteriae. Vet Pathol 51:1096–1108. doi: 10.1177/0300985813516646. [DOI] [PubMed] [Google Scholar]
- 31.Mirajkar NS, Bekele AZ, Chander YY, Gebhart CJ. 2015. Molecular epidemiology of novel pathogen “Brachyspira hampsonii” reveals relationships between diverse genetic groups, regions, host species, and other pathogenic and commensal Brachyspira species. J Clin Microbiol 53:2908–2918. doi: 10.1128/JCM.01236-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atyeo RF, Stanton TB, Jensen NS, Suriyaarachichi DS, Hampson DJ. 1999. Differentiation of Serpulina species by NADH oxidase gene (nox) sequence comparisons and nox-based polymerase chain reaction tests. Vet Microbiol 67:47–60. doi: 10.1016/S0378-1135(99)00030-9. [DOI] [PubMed] [Google Scholar]
- 33.Neef NA, Lysons RJ, Trott DJ, Hampson DJ, Jones PW, Morgan JH. 1994. Pathogenicity of porcine intestinal spirochetes in gnotobiotic pigs. Infect Immun 62:2395–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatusova T, Ciufo S, Federhen S, Fedorov B, McVeigh R, O'Neill K, Tolstoy I, Zaslavsky L. 2015. Update on RefSeq microbial genomes resources. Nucleic Acids Res 43:D599–D605. doi: 10.1093/nar/gku1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee I, Kim YO, Park SC, Chun J. 2016. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103. doi: 10.1099/ijsem.0.000760. [DOI] [PubMed] [Google Scholar]
- 38.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J Prepr 4:e1900v1. [Google Scholar]
- 40.Mende DR, Sunagawa S, Zeller G, Bork P. 2013. Accurate and universal delineation of prokaryotic species. Nat Methods 10:881–884. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 41.Schleifer KH. 2009. Classification of Bacteria and Archaea: past, present and future. Syst Appl Microbiol 32:533–542. doi: 10.1016/j.syapm.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Tindall BJ, Rosselló-Móra R, Busse HJ, Ludwig W, Kämpfer P. 2010. Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol 60:249–266. doi: 10.1099/ijs.0.016949-0. [DOI] [PubMed] [Google Scholar]
- 43.Auch AF, von Jan M, Klenk H-P, Göker M. 2010. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Håfström T, Jansson DS, Segerman B. 2011. Complete genome sequence of Brachyspira intermedia reveals unique genomic features in Brachyspira species and phage-mediated horizontal gene transfer. BMC Genomics 12:395. doi: 10.1186/1471-2164-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellgard MI, Wanchanthuek P, La T, Ryan K, Moolhuijzen P, Albertyn Z, Shaban B, Motro Y, Dunn DS, Schibeci D, Hunter A, Barrero R, Phillips ND, Hampson DJ. 2009. Genome sequence of the pathogenic intestinal spirochete Brachyspira hyodysenteriae reveals adaptations to its lifestyle in the porcine large intestine. PLoS One 4:e4641–e4641. doi: 10.1371/journal.pone.0004641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wanchanthuek P, Bellgard MI, La T, Ryan K, Moolhuijzen P, Chapman B, Black M, Schibeci D, Hunter A, Barrero R, Phillips ND, Hampson DJ. 2010. The complete genome sequence of the pathogenic intestinal spirochete Brachyspira pilosicoli and comparison with other Brachyspira genomes. PLoS One 5:e11455. doi: 10.1371/journal.pone.0011455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pati A, Sikorski J, Gronow S, Munk C, Lapidus A, Copeland A, Glavina Del Tio T, Nolan M, Lucas S, Chen F, Tice H, Cheng J-F, Han C, Detter JC, Bruce D, Tapia R, Goodwin L, Pitluck S, Liolios K, Ivanova N, Mavromatis K, Mikhailova N, Chen A, Palaniappan K, Land M, Hauser L, Chang Y-J, Jeffries CD, Spring S, Rohde M, Göker M, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk H-P. 2010. Complete genome sequence of Brachyspira murdochii type strain (56-150T). Stand Genomic Sci 2:260–269. doi: 10.4056/sigs.831993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wayne LG, Brenner DJ, Colwell RR, Grimont Pa D, Kandler O, Krichevsky MI, Moore LH, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Truper HG. 1987. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464. doi: 10.1099/00207713-37-4-463. [DOI] [Google Scholar]
- 49.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konstantinidis KT, Tiedje JM. 2005. Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187:6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ursing JB, Rossellomora RA, Garciavaldes E, Lalucat J. 1995. Taxonomic note: a pragmatic approach to the nomenclature of phenotypically similar genomic groups. Int J Syst Bacteriol 45:604. doi: 10.1099/00207713-45-3-604. [DOI] [Google Scholar]