

Abstract
Current migratory movements require new strategies for rapidly tracking the transmission of high-risk imported Mycobacterium tuberculosis strains. Whole-genome sequencing (WGS) enables us to identify single-nucleotide polymorphisms (SNPs) and therefore design PCRs to track specific relevant strains. However, fast implementation of these strategies in the hospital setting is difficult because professionals working in diagnostics, molecular epidemiology, and genomics are generally at separate institutions. In this study, we describe the urgent implementation of a system that integrates genomics and molecular tools in a genuine high-risk epidemiological alert involving 2 independent importations of extensively drug resistant (XDR) and pre-XDR Beijing M. tuberculosis strains from Russia into Spain. Both cases involved commercial sex workers with long-standing tuberculosis (TB). The system was based on strain-specific PCRs tailored from WGS data that were transferred to the local node that was managing the epidemiological alert. The optimized tests were available for prospective implementation in the local node 33 working days after receiving the primary cultures of the XDR strains and were applied to all 42 new incident cases. An interpretable result was obtained in each case (directly from sputum for 27 stain-positive cases) and corresponded to the amplification profiles for strains other than the targeted pre-XDR and XDR strains, which made it possible to prospectively rule out transmission of these high-risk strains at diagnosis.
INTRODUCTION
Current massive migratory movements in Europe and elsewhere will likely affect the international distribution of Mycobacterium tuberculosis strains. Multidrug-resistant, extensively resistant, and highly transmissible M. tuberculosis strains, which are restricted mostly to high-prevalence settings, can now cross frontiers more easily and be imported into countries where they are not yet a major problem. In this new scenario, strategies for rapid tracking of the importation and subsequent transmission of these high-risk strains in the host population must be implemented.
Surveillance of the importation/distribution of M. tuberculosis strains is supported mainly by universal genotyping of the isolates and systematic comparison of genotypic profiles in databases comprising whole populations (1, 2). This approach allows us to identify when a strain is imported into a population for the first time and to track subsequent transmission clusters caused by it. An alternative approach involves strain-specific PCRs designed to track specific outbreak strains in specific circumstances (3–5). The development of a strain-specific PCR is possible only when the strain harbors a specific genetic feature (e.g., large deletions [regions of difference] or insertions [mostly IS6110 insertions]) that enables it to be targeted. Consequently, robust strain-specific genetic data could not be obtained until recently, as knowledge of genetic composition was not always sufficient, and the specificity of the selected features was sometimes uncertain.
Whole-genome sequencing (WGS) enables us to identify singular features (strain-specific single-nucleotide polymorphisms [SNPs]) for any M. tuberculosis strain of interest, thus facilitating the design of strain-specific PCRs. We used allele-specific-oligonucleotide (ASO) PCR designed from whole-genome sequencing data to demonstrate the efficiency and flexibility of this new strategy for tracking the transmission of prevalent strains in an area of Spain with a high proportion of TB cases in immigrants (6). Similar approaches have been applied for precise redefinition of a long-term TB outbreak in Switzerland (7).
However, the design of strain-specific PCRs that target specific SNPs to prioritize the prospective surveillance of specific strains requires a complex preanalytical design step. In addition, experiences implementing WGS in clinical laboratories have been limited (8–10) and focused on issues other than tuberculosis, thus leaving most local peripheral laboratories dependent on centers with the capacity for genomic analyses. The resulting delays hamper effective local implementation of surveillance strategies when a genuine epidemiological alert to the presence of a high-risk M. tuberculosis strain is activated.
The aim of our study was to evaluate the feasibility of rapid in situ implementation of a simple surveillance system based on strain-specific PCRs to respond to a serious epidemiological alert, namely, the importation of XDR strains. The strategy was applied in a real-world scenario, that is, diagnostics, molecular epidemiology, and genomic analyses were managed at 3 separate institutions, each in a different city.
(The results of this study were partially presented at the 2016 European Society of Mycobacteriology Meeting, 3 to 6 July 2016, Catania, Italy.)
MATERIALS AND METHODS
Microbiological procedures.
Clinical specimens were processed according to standard methods (11). Susceptibility testing for isoniazid, rifampin, streptomycin, ethambutol, and second-line anti-TB drugs was performed using the mycobacterial growth indicator SIRE system (Becton Dickinson, Sparks, MD, USA) according to standard methods (12).
MIRU-VNTR analysis.
Mycobacterial interspersed repetitive-unit–variable-number tandem-repeat (MIRU-VNTR) analysis was performed directly from the bacteria in the specimens according to a procedure described elsewhere (13). The MIRU-VNTR loci order was MIRU 02, MIRU 20, MIRU 23, MIRU 24, MIRU 27, MIRU 39, MIRU 04, MIRU 26, MIRU 40, MIRU 10, MIRU 16, MIRU 31, VNTR 42, VNTR 43, ETRA, VNTR 47, VNTR 52, VNTR 53, QUB-11b, 1995, QUB-26, VNTR 46, VNTR 48, and VNTR 49. MIRU-VNTR types were compared using Bionumerics 4.6 (Applied Maths, Sint-Martens-Latem, Belgium).
Whole-genome sequencing.
Instead of performing WGS according to the standard procedure, i.e., from purified DNA obtained from M. tuberculosis subcultured in Lowenstein-Jensen medium, we purified DNA directly from the primary mycobacterial growth indicator tube (MGIT) liquid culture. We included a pretreatment step to minimize interference from human DNA (14). Briefly, the samples were sonicated, inactivated, centrifuged, and, after removing the supernatant, samples were resuspended in 1 ml of saline wash. They were then centrifuged again, and the supernatant was removed. The pellets were resuspended in water, and the DNA was precipitated with 3 M sodium acetate and 96% ethyl alcohol (EtOH). Finally, the DNA was resuspended with Tris-EDTA (TE) and used to prepare libraries.
DNA libraries were generated following the Nextera XT Illumina protocol (Nextera XT Library Prep kit [FC-131-1024]). We used 0.2 ng/μl purified gDNA to initiate the protocol. The multiplexing step was performed using a Nextera XT index kit (FC-131-1096); the index PCR amplification program on the thermal cycler was modified to 15 cycles. Library quality and size distribution were checked by running 1 μl on a 2200 TapeStation bioanalyzer (Agilent Technologies, USA). The indexed libraries were quantified using Qubit fluorometric quantitation using a Qubit 2.0 fluorometer (Life Technologies, USA) and normalized by a manual procedure based on the average fragment size observed. The libraries were sequenced using a 2 by 300 pb paired-end run (MiSeq reagent kit v3 [MS-102-3001]) on a MiSeq sequencer according to manufacturerinstructions (Illumina) and batched (in a pool of 16) per flow cell, with an average per base coverage of 146× (range, 106× to 208×).
SNP calling was performed as indicated elsewhere (15). In summary, after mapping to a reference strain, we extracted all variable positions in the strain of interest. To avoid false-positive calls, a series of quality filter criteria were applied to the data associated with the SNP (coverage, >20×; mapping quality, 20).
Strain-specific SNPs were identified after comparing the SNPs extracted from the WGS data with those from an in-house database of 219 strains representing the geographic and phylogenetic diversity of the M. tuberculosis complex (16). Finally, only those SNPs that passed the quality filters were kept for the analyses and classified as synonymous, nonsynonymous, or intergenic. We finally selected synonymous SNPs for the ASO-PCR analysis to ensure their stability as genetic markers (17).
Strain-specific ASO-PCR design.
We designed an ASO PCR for each of the 2 strains studied (BJR1 and BJR2). We targeted simultaneously several SNPs that were specific to the strains to rule out false negatives, as recommended elsewhere (7). We designed a multiplex ASO PCR to target 4 strain-specific SNPs in BJR1 and 3 strain-specific SNPs in BJR2. One of the primers in each pair was a selective primer that targeted the alleles in the surveyed strain, whereas the remaining primers targeted the alleles from the nonsurveyed strains. In each PCR, our design generated different patterns depending on whether the strain tested was the surveyed strain (BJR1 or BJR2) or another strain.
The assay conditions were the same for these 2 ASO PCRs with 2 exceptions, namely, the primer concentration (0.14 μM for BJR1 and 0.2 μM for BJR2) and the MgCl2 concentration (1.6 mM for BJR1 and 1.8 mM for BJR2). Both PCRs included 1% DMSO, 200 μM dNTPs, and 0.4 μl AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA). The PCR conditions were 95°C for 10 min followed by 30 cycles of 95°C for 1 min, 64°C for 1 min, and 72°C for 1 min, and then 72°C for 10 min.
Allocation of lineages and clonal complexes.
The Beijing lineage was determined by detecting the specific SNP marker for this lineage (Rv2952, G526A) using PCR and DNA sequencing (17). The short sequence reads were submitted for in silico spoligotyping using the online Total Genotyping Solution for Mycobacterium tuberculosis (TGS-TB) tool (see https://gph.niid.go.jp/tgs-tb).
The clonal complex B0/W148 was determined using in silico analysis for the presence of an IS6110 insertion in the Rv2664-Rv2665 intergenic region, which is a specific marker for this complex (position 2982598 in H37Rv NC_000962.3; positions 2755667–2757021 in strain W-148 NZ_CP012090.1) (4). MiSeq reads were mapped to the complete genomes of reference strain H37Rv (NC_000962.3) and strain W-148 (NZ_CP012090.1) by using the Geneious 9.0 package (Biomatters, Ltd., Auckland, New Zealand).
MiSeq reads of strain BJR2 were mapped to the complete genome of reference strain H37Rv (NC_000962.3), and SNPs were compared with those in the proprietary database at St. Petersburg Pasteur Institute.
Accession number(s).
The sequence data have been deposited in the EBI Sequence Read Archive under accession no. PRJEB15326.
RESULTS AND DISCUSSION
In September 2015, a case of XDR TB and a case of pre-XDR TB (both female patients, one HIV positive and the other HIV negative) were diagnosed 1 week apart in the diagnostic laboratory in Almería, southeast Spain (Table 1). The patients were commercial sex workers who had immigrated to Spain from Russia and had been living in Almería for the last 3 and 2 years, respectively.
TABLE 1.
General features of the cases and strains studied
| Case no. | Strain | Origin | Specimens containing MTB | Resistance profilea | MIRU-VNTR type | Beijing group |
|---|---|---|---|---|---|---|
| 1 | BJR1 | Russia | Sputum, feces, urine, blood, and peritoneal fluid | INH, RIF, STR, ETH, PZA, AMK, CAP, KAN, MOX | 225133273335444432657423 | B0/W148 |
| 2 | BJR2 | Russia | Sputum and feces | INH, RIF, STR, ETH, AK, CAP, KA, MOX, OFL | 225133253335444432656423 | 94-32 |
AMK, amikacin; ETH, ethambutol; INH, isoniazid; KAN, kanamycin; MOX, moxalactam; OFL, ofloxacin; PZA, pyrazinamide; RIF, rifampin; STR, streptomycin.
In the diagnostic laboratory in Almería (diagnostics node), MIRU-VNTR typing was performed directly from stain-positive respiratory specimens. A complete MIRU-VNTR pattern was obtained from BJR1 (225133273335444432657423), whereas no amplification was observed in 9 loci in BJR2. Even with this incomplete MIRU-VNTR analysis, we suspected a transmission event, as the alleles for 15 loci were identical and both strains shared an unusual track (4444) in the loci Mtub04, ETR-C, ETR-A, and Mtub30. The purified DNA and the primary liquid cultures (MGIT) were sent to our laboratory in Madrid (molecular biology node). We completed the MIRU-VNTR type for the isolate with amplification failures, and, contrary to our initial assumption, the second case was infected by a different strain (225133253335444432656423). The 2 MIRU-VNTR types clustered with the Beijing isolates in our database. The presence of the specific SNP marker of the Beijing lineage confirmed that the 2 isolates belonged to this lineage. The 9-signal Beijing spoligotype profile was also predicted using in silico typing based on short sequence reads. A comparison of the MIRU-VNTR patterns with those of strains circulating in Russia (St. Petersburg Pasteur Institute database) and with strains from the MIRU-VNTRplus.org database indicated that they corresponded to the 100-32 type (Beijing B0/W148) clonal cluster, previously defined as a successful Russian clone (18), and to the 94-32 cluster (1065-32 type), respectively, both of which are major Beijing groups found throughout the former Soviet Union (18–23).
The identification of independent importation of a pre-XDR Beijing strain and an XDR Beijing strain (BJR1 and BJR2, respectively) activated the epidemiological alarm, which indicated the possibility of an extremely high-risk transmission based on the following findings: history of commercial sex work, stay of 3 and 2 years in Almería before diagnosis, extremely high bacterial loads (>100/field), and long-standing TB (one case with M. tuberculosis isolated from sputum and feces and the other with M. tuberculosis isolated from sputum, peritoneal, blood, urine, and feces), which were likely due to a long diagnostic delay. These factors supported a highly probable risk of transmission, which activated the epidemiology alert and initiated the implementation of a system to track the potential transmission of these 2 strains in situ at the local diagnostic laboratory in Almería.
The first step was to analyze the 2 strains using WGS, which required high-quality purified DNA from the M. tuberculosis isolates while excluding DNA from other sources. Therefore, the primary cultures were sent to Madrid (molecular biology node). The day the samples were received in Madrid was considered day 0 (5 October 2015) in the implementation timeline (Fig. 1). As epithelial human cells and accompanying respiratory bacteria can also be found in respiratory specimens, subcultures from the primary culture are generally necessary for elimination of interfering DNA for WGS analysis. As our objective was to ensure rapid implementation of our system, the delay expected from the M. tuberculosis subculture was not acceptable. Instead, we applied WGS directly from the genomic DNA purified from the primary liquid (MGIT) culture. We processed 2 independent cultures for each strain (BJR1 and BJR2) to provide an extra opportunity to complete the WGS analysis in case the readings obtained were not of sufficiently high quality in terms of depth and/or coverage.
FIG 1.

Timeline and distribution of tasks for the different nodes involved in the study.
Once the quantity of human material was reduced, purified enriched M. tuberculosis DNA was sent to our collaborative genomics center in Valencia (genomics node), and after reception and quality assessment (Fig. 1), libraries were prepared following the protocol modifications described elsewhere for analysis of DNA from primary cultures (14).
Once the quality of the WGS readings was shown to not differ from the quality obtained using the standard process, which involved subculture of the primary isolate, we first confirmed that both isolates belonged to the 2 major Beijing groups in Russia, as suggested initially by the MIRU-VNTR analysis. BJR1 was confirmed to belong to the B0/W148 cluster by in silico detection of the specific Rv2664-Rv2665:IS6110 insertion, which has been proposed as a specific marker for this clonal cluster. On the whole, analysis of the short reads mapped to the available complete genome of strain W-148 (representative of B0/W148-cluster) revealed that all IS6110 insertions in this strain were also present in strain BJR1. We also checked and confirmed that strain BJR1 harbored the mutation signatures that are characteristic of B0/W148 group strains circulating in Russia, and that strain BJR2 belonged to the 94-32-cluster based on in silico detection of 23 specific SNPs (I. Mokrousov, E. Chernyaeva, and A. Vyazovaya, unpublished data).
Second, we started the process of identifying specific SNPs for each of the strains (Fig. 1). The SNPs found after comparison with the most recent common ancestor of the M. tuberculosis complex (16, 24) were compared with those from a global database, including 219 strains that are representative of the composition of strains circulating worldwide (16) as the first filter to rule out non-strain-specific SNPs (Fig. 1). Thirteen and 80 SNPs were obtained for the BJR1 and BJR2 strains, respectively.
To further refine our analysis for the BJR2 strain, which had a higher number of putative strain-specific SNPs than did the BJR1 strain (likely due to underrepresentation of 94-32 clonal complex strains in our database), we applied 2 new filters. First, we selected the 18 SNPs mapping essential genes to ensure their stability as markers (17). Then, we compared these 18 SNPs for BJR2 with the sequences available in the St. Petersburg Pasteur Institute for Russian isolates belonging to the 94-32 Beijing group (I. Mokrousov, E. Chernyaeva, and A. Vyazovaya, unpublished data) and those available in the Genome-Wide Mycobacterium tuberculosis Variation (GMTV) database (see http://mtb.dobzhanskycenter.org/). After applying both filters, the number of SNPs considered strain-specific fell from 80 to 8.
To ensure specificity, we decided to simultaneously target 4 and 3 SNPs for the BJR1 and BJR2 strains, respectively (Fig. 2). In this selection, we requested that the SNPs be synonymous to ensure their usefulness as stable genetic markers. The ASO PCR was designed at the molecular biology node and used a multiplex format to interrogate the 4 BJR1 strain-specific SNPs and a different multiplex format to interrogate the 3 BJR2-specific SNPs (Fig. 2). In our design, 2 pairs of primers for each ASO PCR targeted the alleles expected for the surveyed strain (SNPs 1 and 3 for BJR1 and SNPs 5 and 7 for BJR2), whereas the remaining primers targeted the alternative allele found in strains other than those surveyed (non-BJR1 and non-BJR2) (Fig. 2). This design was to ensure the presence of amplification products, regardless of whether the case was infected by BJR1, BJR2, or another strain, to enable detection of potential PCR failures. Finally, primers were designed to produce different sizes for each of the amplicons (Fig. 2). A blind evaluation of the 3 multiplex ASO PCRs enabled us to confirm the patterns expected for the BJR1 and BJR2 isolates and for a selection of unrelated controls (Fig. 3).
FIG 2.

SNPs and alleles targeted in each of the 2 multiplex ASO PCRs designed to track the BJR1 and BJR2 strains. The expected patterns for the BJR1, BJR2, and any other strains expected in each of the 2 ASO PCRs are shown in the gels on the right. The samples were all run on the same gel using the same molecular weight (MW) markers but were separated into two graphics for clarity.
FIG 3.
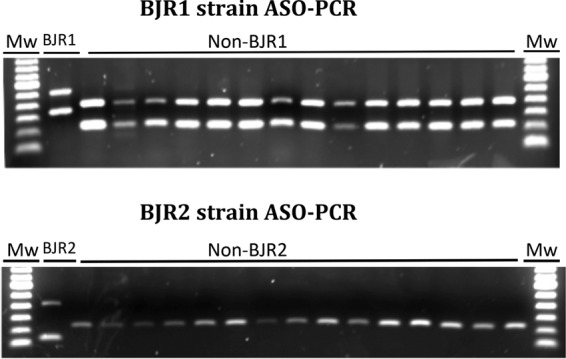
Amplification patterns obtained for the BJR1 and BJR2 multiplex ASO PCRs when applied to the control BJR1 and BJR2 strains and to randomly selected non-BJR1 and non-BJR2 strains. Mw, molecular weight marker.
Once the multiplex ASO PCRs were considered to be optimized, they were transferred to the diagnostics node in Almeria. In our search for maximum efficiency in the surveillance scheme, which sought to identify the strains surveyed at diagnosis, we evaluated the performance of the multiplex ASO PCRs when applied to DNA purified directly from decontaminated smear-positive specimens (using Qiagen kits) and found the same interpretable patterns (data not shown).
The final step was to start the prospective survey of the 2 imported Beijing strains on all newly diagnosed cases. Between November 2015 and June 2016, multiplex ASO PCR was applied to 27 respiratory specimens from incident smear-positive patients and to 15 isolates from non-smear-positive patients. From 22 of 27 smear-positive cases, we obtained an interpretable result within the first 72 h after observation of acid-fast bacilli. All but 1 of the 5 specimens without a result corresponded to specimens with scarce acid-fast bacilli, which would have been considered stain-negative specimens under standard circumstances because a modification of the auramine staining method with significantly improved sensitivity (25) is routinely applied in the Almería laboratory (diagnostics node). Moreover, in all 15 incident smear-negative cases and in the 5 cases with no initial results, the multiplex ASO PCRs were applied on DNA extracted from the cultured isolates. Interpretable results were obtained in all 42 isolates and corresponded to the amplification profiles for non-BJR1/non-BJR2 strains, which made it possible to quickly rule out secondary cases resulting from exposure to the imported high-risk XDR cases.
Our strategy has an added value when applied to track XDR strains. Expensive drug susceptibility testing that is performed in diagnostic laboratories can be avoided. Given that the acquisition of drug resistance in TB is essentially stable (i.e., there is no reversion to susceptibility), genetic markers for TB clones are surrogates for not only such clones but also of the drug resistance determinants they harbor. In practice, this means that performing the standard drug susceptibility testing can be unnecessary if multidrug-resistant/XDR clones are diagnosed by specific low-cost PCR tests.
The final optimized tests were available for prospective implementation at the local node 33 working days after receiving the primary culture. This period is probably longer than would be necessary to implement an equivalent system if the clinical diagnostics laboratory, the molecular epidemiology laboratory, and the genomics analysis unit were in a single institution. However, the scenario we described, where 3 separate collaborative nodes are responsible for each part of the process, is closer to real-world practice. In this context, we believe that the response time is reasonable and this shows that even in nonideal networks of activity, such as ours, a local, simple, low-cost, and efficient tool can be implemented to ensure specific in situ surveillance of high-risk strains at the diagnosis of each new case.
Tailoring PCRs that target SNPs from relevant strains is a novel, low-cost, rapid, and transferable strategy for in situ monitoring of high-risk TB transmission. Our strategy ensures that epidemiological surveillance is implemented at the diagnosis of TB. This approach represents a major transformation in the way the transmission of this infection is analyzed.
ACKNOWLEDGMENTS
We thank Thomas O'Boyle for proofreading the manuscript.
This study was funded by the Ministry of Economy and Competitiveness ISCIII-FIS (grants PI15/01554 and PI13/01207) and cofunded by ERDF (FEDER) Funds from the European Commission, “A way of making Europe.” L.P. was supported by M. Servet CP15/00075. I.C. was supported by MINECO (SAF2013-43521-R). I.M. acknowledges support from Russian Science Foundation (project 14-14-00292). I.C. was supported by MINECO research grant SAF2013-43521-R and European Research Council (ERC) grant 638553-TB-ACCELERATE.
We declare no conflicts of interest.
REFERENCES
- 1.Allix-Beguec C, Fauville-Dufaux M, Supply P. 2008. Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 46:1398–1406. doi: 10.1128/JCM.02089-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambregts-van Weezenbeek CS, Sebek MM, van Gerven PJ, de Vries G, Verver S, Kalisvaart NA, van Soolingen D. 2003. Tuberculosis contact investigation and DNA fingerprint surveillance in the Netherlands: 6 years' experience with nation-wide cluster feedback and cluster monitoring. Int J Tuberc Lung Dis 7:S463–470. [PubMed] [Google Scholar]
- 3.Plikaytis BB, Marden JL, Crawford JT, Woodley CL, Butler WR, Shinnick TM. 1994. Multiplex PCR assay specific for the multidrug-resistant strain W of Mycobacterium tuberculosis. J Clin Microbiol 32:1542–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mokrousov I, Narvskaya O, Vyazovaya A, Otten T, Jiao WW, Gomes LL, Suffys PN, Shen AD, Vishnevsky B. 2012. Russian “successful” clone B0/W148 of Mycobacterium tuberculosis Beijing genotype: a multiplex PCR assay for rapid detection and global screening. J Clin Microbiol 50:3757–3759. doi: 10.1128/JCM.02001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millan-Lou MI, Alonso H, Gavin P, Hernandez-Febles M, Campos-Herrero MI, Copado R, Canas F, Kremer K, Caminero JA, Martin C, Samper S. 2012. Rapid test for identification of a highly transmissible Mycobacterium tuberculosis Beijing strain of sub-Saharan origin. J Clin Microbiol 50:516–518. doi: 10.1128/JCM.06314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Lago L, Martinez Lirola M, Herranz M, Comas I, Bouza E, Garcia-de-Viedma D. 2015. Fast and low-cost decentralized surveillance of transmission of tuberculosis based on strain-specific PCRs tailored from whole-genome sequencing data: a pilot study. Clin Microbiol Infect 21:249.e1–249.e9. doi: 10.1016/j.cmi.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Stucki D, Ballif M, Bodmer T, Coscolla M, Maurer AM, Droz S, Butz C, Borrell S, Langle C, Feldmann J, Furrer H, Mordasini C, Helbling P, Rieder HL, Egger M, Gagneux S, Fenner L. 2015. Tracking a tuberculosis outbreak over 21 years: strain-specific single-nucleotide polymorphism typing combined with targeted whole-genome sequencing. J Infect Dis 211:1306–1316. doi: 10.1093/infdis/jiu601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long SW, Williams D, Valson C, Cantu CC, Cernoch P, Musser JM, Olsen RJ. 2013. A genomic day in the life of a clinical microbiology laboratory. J Clin Microbiol 51:1272–1277. doi: 10.1128/JCM.03237-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, Shendure J, Salipante SJ. 2015. A year of infection in the intensive care unit: prospective whole-genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet 11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mellmann A, Bletz S, Boking T, Kipp F, Becker K, Schultes A, Prior K, Harmsen D. 24 August 2016. Real-time genome sequencing of resistant bacteria provides precision infection control in an institutional setting. J Clin Microbiol doi: 10.1128/JCM.00790-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfyffer GE. 2011. Mycobacterium: general characteristics, laboratory detection and staining procedures, p 472–502. In Versalovic JCK, Funke G, Jorgensen JH, Landry ML, Warnock DW (ed), Manual of clinical microbiology, 10th ed, ASM Press, Washington, DC. [Google Scholar]
- 12.Clinical and Laboratory Standards Institute. 2011. Susceptibility testing of Mycobacteria, Nocardiae, and other aerobic Actinomycetes. Approved Standard 2nd ed Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed] [Google Scholar]
- 13.Alonso M, Herranz M, Martinez Lirola M, INDAL-TB Group, Gonzalez-Rivera M, Bouza E, Garcia de Viedma D. 2012. Real-time molecular epidemiology of tuberculosis by direct genotyping of smear-positive clinical specimens. J Clin Microbiol 50:1755–1757. doi: 10.1128/JCM.00132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Votintseva AA, Pankhurst LJ, Anson LW, Morgan MR, Gascoyne-Binzi D, Walker TM, Quan TP, Wyllie DH, Del Ojo Elias C, Wilcox M, Walker AS, Peto TE, Crook DW. 2015. Mycobacterial DNA extraction for whole-genome sequencing from early positive liquid (MGIT) cultures. J Clin Microbiol 53:1137–1143. doi: 10.1128/JCM.03073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Lago L, Comas I, Navarro Y, Gonzalez-Candelas F, Herranz M, Bouza E, Garcia de Viedma D. 2014. Whole-genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis 209:98–108. doi: 10.1093/infdis/jit439. [DOI] [PubMed] [Google Scholar]
- 16.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. 2013. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet 45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stucki D, Malla B, Hostettler S, Huna T, Feldmann J, Yeboah-Manu D, Borrell S, Fenner L, Comas I, Coscolla M, Gagneux S. 2012. Two new rapid SNP-typing methods for classifying Mycobacterium tuberculosis complex into the main phylogenetic lineages. PLoS One 7:e41253. doi: 10.1371/journal.pone.0041253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mokrousov I. 2013. Insights into the origin, emergence, and current spread of a successful Russian clone of Mycobacterium tuberculosis. Clin Microbiol Rev 26:342–360. doi: 10.1128/CMR.00087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skiba Y, Mokrousov I, Ismagulova G, Maltseva E, Yurkevich N, Bismilda V, Chingissova L, Abildaev T, Aitkhozhina N. 2015. Molecular snapshot of Mycobacterium tuberculosis population in Kazakhstan: a country-wide study. Tuberculosis 95:538–546. doi: 10.1016/j.tube.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Mokrousov I, Valcheva V, Sovhozova N, Aldashev A, Rastogi N, Isakova J. 2009. Penitentiary population of Mycobacterium tuberculosis in Kyrgyzstan: exceptionally high prevalence of the Beijing genotype and its Russia-specific subtype. Infect Genet Evol 9:1400–1405. doi: 10.1016/j.meegid.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Blum MG, Rusch-Gerdes S, Mokrousov I, Aleksic E, Allix-Beguec C, Antierens A, Augustynowicz-Kopec E, Ballif M, Barletta F, Beck HP, Barry CE 3rd, Bonnet M, Borroni E, Campos-Herrero I, Cirillo D, Cox H, Crowe S, Crudu V, Diel R, Drobniewski F, Fauville-Dufaux M, Gagneux S, Ghebremichael S, Hanekom M, Hoffner S, Jiao WW, Kalon S, Kohl TA, Kontsevaya I, Lillebaek T, Maeda S, Nikolayevskyy V, Rasmussen M, Rastogi N, Samper S, Sanchez-Padilla E, Savic B, Shamputa IC, Shen A, Sng LH, Stakenas P, Toit K, Varaine F, Vukovic D, Wahl C, Warren R, Supply P, Niemann S, Wirth T. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 47:242–249. doi: 10.1038/ng.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo T, Comas I, Luo D, Lu B, Wu J, Wei L, Yang C, Liu Q, Gan M, Sun G, Shen X, Liu F, Gagneux S, Mei J, Lan R, Wan K, Gao Q. 2015. Southern east Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc Natl Acad Sci U S A 112:8136–8141. doi: 10.1073/pnas.1424063112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vyazovaya A, Mokrousov I, Solovieva N, Mushkin A, Manicheva O, Vishnevsky B, Zhuravlev V, Narvskaya O. 2015. Tuberculous spondylitis in Russia and prominent role of multidrug-resistant clone Mycobacterium tuberculosis Beijing B0/W148. Antimicrob Agents Chemother 59:2349–2357. doi: 10.1128/AAC.04221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet 42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cejudo-Garcia MA, Munoz-Davila MJ, Martinez-Lirola MJ. 2013. UV-fixed-thick-blotch preparation improves sensitivity of auramine staining. J Clin Microbiol 51:3400–3402. doi: 10.1128/JCM.00974-13. [DOI] [PMC free article] [PubMed] [Google Scholar]