

Abstract
Background
We aimed to explore the role of endogenous sulfur dioxide (SO 2) in pulmonary vascular collagen remodeling induced by monocrotaline and its mechanisms.
Methods and Results
A rat model of monocrotaline‐induced pulmonary vascular collagen remodeling was developed and administered with l‐aspartate‐β‐hydroxamate or SO 2 donor. The morphology of small pulmonary arteries and collagen metabolism were examined. Cultured pulmonary arterial fibroblasts stimulated by transforming growth factor β1 (TGF‐β1) were used to explore the mechanism. The results showed that in monocrotaline‐treated rats, mean pulmonary artery pressure increased markedly, small pulmonary arterial remodeling developed, and collagen deposition in lung tissue and pulmonary arteries increased significantly in association with elevated SO 2 content, aspartate aminotransferase (AAT) activity, and expression of AAT1 compared with control rats. Interestingly, l‐aspartate‐β‐hydroxamate, an inhibitor of SO 2 generation, further aggravated pulmonary vascular collagen remodeling in monocrotaline‐treated rats, and inhibition of SO 2 in pulmonary artery smooth muscle cells activated collagen accumulation in pulmonary arterial fibroblasts. SO 2 donor, however, alleviated pulmonary vascular collagen remodeling with inhibited collagen synthesis, augmented collagen degradation, and decreased TGF‐β1 expression of pulmonary arteries. Mechanistically, overexpression of AAT1, a key enzyme of SO 2 production, prevented the activation of the TGF‐β/type I TGF‐β receptor/Smad2/3 signaling pathway and abnormal collagen synthesis in pulmonary arterial fibroblasts. In contrast, knockdown of AAT1 exacerbated Smad2/3 phosphorylation and deposition of collagen types I and III in TGF‐β1–treated pulmonary arterial fibroblasts.
Conclusions
Endogenous SO 2 plays a protective role in pulmonary artery collagen accumulation induced by monocrotaline via inhibition of the TGF‐β/type I TGF‐β receptor/Smad2/3 pathway.
Keywords: collagen deposition, pulmonary artery, Smad2/3 signal, sulfur dioxide, transforming growth factor β
Subject Categories: Pulmonary Hypertension, Vascular Disease
Introduction
Pulmonary hypertension (PH) is a potentially fatal disease characterized by progressive elevation in mean pulmonary arterial pressure, pulmonary vascular remodeling, and right ventricular hypertrophy.1 Among these pathological changes, pulmonary vascular remodeling leads to increased pulmonary vascular resistance, a key factor determining the outcome of PH. Increasing evidence shows that both medial vascular remodeling and adventitial remodeling markedly contribute to pathological change.2 Fibroblasts, the main component of adventitia, respond rapidly following injury stimuli. Such responses include self‐proliferation, secretion of cytokines to exert influences on intima and media (eg, triggering of smooth muscle cell proliferation), modulation of extracellular matrix (ECM), differentiation into myofibroblasts migrating to remodeled media, and recruitment of inflammatory cells.2, 3 Accumulation of ECM within the vascular media and disturbance in the homeostasis of the synthesis and degradation of collagen contribute greatly to the formation of pulmonary vascular structural remodeling. The mechanisms responsible for pulmonary vascular structural remodeling, however, have not yet been clarified.
The discovery of endogenous gaseous signaling molecules opened a new avenue for the study of mechanisms responsible for PH. Gaseous molecules—characterized by sustained generation, rapid dispersion, an extensive role in organisms, and short half‐life—regulate pulmonary vascular homeostasis, and alterations in their endogenous production have been linked to PH progression.4, 5, 6
Sulfur dioxide (SO2), previously recognized as a toxic gas, was recently found to possess a vasorelaxant effect and can be generated endogenously in cardiovascular tissue by metabolism of sulfur‐containing amino acids.7, 8 The exogenous supplement of the SO2 donor could decrease the ratio of media to lumen radius and the proliferative index of smooth muscle cells in the aorta9 and ameliorate the proliferation of medial smooth muscle cells.10 Furthermore, our previous study demonstrated that the changes in endogenous SO2 production were associated with the development of monocrotaline‐induced PH.11 The present study was undertaken to examine the possible role of endogenous SO2 in the regulation of pulmonary vascular collagen deposition in models of monocrotaline‐induced pulmonary vascular remodeling and potential mechanisms.
Methods
Animal Treatments and Groups
All animal care and experimental procedures complied strictly with the Animal Management Rule of the Ministry of Health of the People's Republic of China (Documentation 55, 2001). The protocol was specifically approved by the Animal Research Ethics Committee of Peking University First Hospital (permit number 201215). All surgery was performed under ethyl carbamate anesthesia, and all efforts were made to minimize suffering. Male Wistar rats (n=32, body weight 160±10 g) used in the experiment were obtained from the Experimental Animal Center, Peking University Health Science Center (Beijing, China). The experimental animals were randomly divided into the following 4 groups (n=8 for each group): control, monocrotaline, monocrotaline plus l‐aspartate‐β‐hydroxamate (HDX; an inhibitor of SO2 synthase), and monocrotaline plus SO2. Rats in the monocrotaline, monocrotaline plus HDX, and monocrotaline plus SO2 groups were injected subcutaneously with monocrotaline (Sigma‐Aldrich) at 60 mg/kg on day 1.12 Rats in the monocrotaline plus HDX group were given HDX (Sigma‐Aldrich) orally at 25 mg/kg on days 0, 7, and 14.11 For rats in the monocrotaline plus SO2 group, sodium sulfite (Na2SO3) and sodium bisulfite (NaHSO3; Sigma‐Aldrich), were used as the SO2 donor (molar ratio of Na2SO3 and NaHSO3 was adjusted to 3:1, pH 7.4) and were given daily by intraperitoneal injection at 85 mg/kg for 3 weeks. Rats in the control group received only the same volume of solvent vehicle. Food and water were freely available at all times, and the rats were housed with a 12/12‐hour light/dark cycle. After 3 weeks, rats were injected with 12% ethyl carbamate anesthesia (10 mL/kg intraperitoneally), and lung tissues were harvested for the following analysis.
Lung Tissue Preparation
Three parts of the lung were removed. One part of the left lung was fixed in 4% (wt/vol) paraformaldehyde and routinely processed in 6‐μm–thick paraffin sections for each tissue sample. The other part of the left lung was embedded in Tissue‐Tek O.C.T. Compound (Sakura Finetek) immediately, frozen at −80°C, and processed into 6‐μm–thick paraffin sections for each tissue sample. One part of the right lung was kept in liquid nitrogen for quick freezing and then stored at −80°C, which was used to prepare tissue homogenates.
Collagen Sirius Red Staining
The paraffin sections of lung were initially dewaxed and hydrated and then stained for 1 minute in a Sirius red solution. Subsequently, sections underwent rapid washing in running tap water without counterstaining. The sections were dehydrated, made transparent, and mounted. The samples were observed layer by layer under a polarizing filter microscope (Leica Q550CW). Sirius red is specific for collagen and colors collagen fibrils different colors (yellow, red/orange, or green) according to their direction under polarizing filter microscopy.13
Collagen Masson's Trichrome Staining
Paraffin‐embedded lungs were dewaxed and hydrated and then stained using the Masson's trichrome stain kit (Loogene Biotechnology). The sections were dehydrated, made transparent, and mounted. Results were observed under light microscopy (×200; Olympus). Green is specific for collagen. Muscle fibers were dyed red, and nuclei were dyed dark blue.
Expression of Pulmonary Artery Collagen Types I and III and Transforming Growth Factor β1 by Immunofluorescence and Confocal Microscopy
Frozen sections were placed at room temperature for 30 minutes and then blocked and permeabilized in 5% bovine serum albumin with 0.3% Triton X‐100 (Sigma‐Aldrich) for 30 minutes at 37°C. Slides were incubated with primary antibody for collagen type I analysis (rabbit anti–collagen type I; Abcam), collagen type III analysis (rabbit anti–collagen type III; Sigma‐Aldrich), and transforming growth factor β1 (TGF‐β1) analysis (rabbit anti‐TGF‐β1; Santa Cruz Biotechnology) and mouse anti‐vimentin (Boster Bioengineering) overnight at 4°C and with secondary antibody (Santa Cruz Biotechnology) at room temperature for 1.5 hours. Fluorescent images were captured by the FluoView laser scanning confocal microscope (Olympus).
Lung Tissue Hydroxyproline Content
The content of hydroxyproline in lung tissue was determined, as reported previously.14 Lung tissues homogenized in 2 mol/L NaOH were hydrolyzed in a water bath at 100°C for 20 minutes. The pH value was adjusted to 6.0 to 6.8, and the tissue diluents were clarified by centrifugation at 884 g for 10 minutes. The supernatant was used to assay hydroxyproline using an experimental kit (Jiancheng). Absorbance of each sample was read at 550 nm using a spectrophotometer, and the hydroxyproline content was calculated according to the protocol outlined in the manufacturer's kit. Results were expressed in micrograms per gram of protein (hydroxyproline of tissue homogenate).
Analysis of Lung Tissue Collagen Types I and III by Enzyme‐Linked Immunosorbent Assay
Double‐antibody sandwich enzyme‐linked immunosorbent assays (ELISAs) were used according to the manufacturer's instructions (Rapidbio). Lung tissues were homogenized in buffer and ultracentrifuged for liquid supernatants. Samples (100 μL), collagen type I standards (2000, 1000, 500, 250, 125, 62.5, 31.25, and 0 pg/mL), and collagen type III standards (144, 72, 36, 18, 9, 4.5, 2.25, and 0 ng/mL) were added to the well and incubated for 1 hour at 37°C. Samples were removed, and plates were washed with a washing buffer. Anti–rat collagen types I and III (50 μL) were added to each well and incubated for 60 minutes at 37°C. After 5 additional washing steps, 100 μL of horseradish peroxidase was added to the well and left at 37°C for 60 minutes. The plates were washed 5 times, and 100 μL of tetramethylbenzidine substrate was added to each well and incubated in the dark for 5 to 10 minutes at room temperature. Next, 100 μL of stop solution was added to stop the reaction. Optical density at 492 nm was measured using an ELISA reader (Bio‐Rad Laboratories), and the antigen level (in ng/mL) was calculated from the standard curve.
Procollagen Type I, Procollagen Type III, and TGF‐β1 mRNA Expression by Quantitative Real‐Time Polymerase Chain Reaction
Total RNA in the lung tissue was extracted by the Trizol reagent and reverse transcribed by the oligo(dT) primer and Moloney murine leukemia virus reverse transcriptase (Promega Corp). Next, cDNA was synthesized using TaqMan reverse transcription reagents. Primers and TaqMan probes for the samples were designed with the software Primer Express 3.0 (Applied Biosystems) and synthesized by SBS Company. Quantitative real‐time polymerase chain reaction (PCR) was performed on an ABI PRISM 7300 instrument (Applied Biosystems). The PCR mixture contained 5 μL of 10 times PCR buffer, 5 μL of cDNA template or standard DNA, 4 μL of 2.5 mmol/L each dNTP, 5 U of Taq DNA polymerase, 1 μL of 6‐carboxy‐X‐rhodamine (Invitrogen), 15 pmol of each forward and reverse primer, and 10 pmol TaqMan probe at a total volume of 50 μL. Samples and standard DNA were run in duplicate. The PCR condition was set to predenaturing at 95°C for 5 minutes and then 95°C for 15 seconds and 60°C for 1 minute for 40 cycles. The TaqMan probe was labeled with FAM at the 5′ end and TAMRA at the 3′ end. β‐actin was used to standardize gene expression.
Sequences of the primers and probes were as follows: for procollagen type I, forward 5′‐CTTGTTGCTGAGGGCAACAG‐3′, reverse 5′‐GCAGGCGAGATGGCTTATTC‐3′, TaqMan probe 5′‐ATTCACCTACACTGTCCTTGTCGATGGCTG‐3′; for procollagen type III, forward 5′‐GAAAAAACCCTGCTCGGAATT‐3′, reverse 5′‐ATCCATCTTGCAGCCTTGGT‐3′, TaqMan probe 5′‐AGAGACCTGAAATTCTGCCACCCTGAACTC‐3′; for TGF‐β1, forward 5′‐CACCGGAGAGCCCTGGATA‐3′, reverse 5′‐TCCAACCCAGGTCCTTCCTA‐3′, TaqMan probe 5′‐TGCTTCAGCTCCACAGAGAAGAACTGCTG‐3′; and for β‐actin, forward 5′‐ACCCGCGAGTACAACCTTCTT‐3′, reverse 5′‐TATCGTCATCCATGGCGAACT‐3′, TaqMan probe 5′‐CCTCCGTCGCCGGTCCACAC‐3′.
Pulmonary Artery TGF‐β1 Expression by Immunohistochemical Analysis
Paraffin sections from lung tissues were prepared. The sections were dewaxed and hydrated and then processed with 3% H2O2 in methanol for 12 minutes to block endogenous peroxidase, followed by antigen repair with pepsin for 30 minutes. Sections were incubated with goat serum for 1 hour to prevent unspecific binding of immunoglobulin. TGF‐β1 antibody (Boster Bioengineering) was added and incubated overnight at 4°C in a moist chamber. In sequence, the biotinylated anti–rabbit immunoglobulin G and horseradish peroxidase streptavidin (Santa Cruz Biotechnology) were added at 37°C for 40 minutes, respectively. DAB (ZSGB‐BIO) was added for developing color, and sections were stained in hematoxylin. At the end, sections were dehydrated, made transparent, mounted, and examined under an optical microscope. The brown granules in pulmonary vascular media under the microscope were defined as the positive signals.
Primary Rat Pulmonary Artery Fibroblast Culture
Primary rat pulmonary artery fibroblasts (PAFs) were prepared from male Wistar rats weighing 180 to 220 g. The pulmonary artery was rapidly excised from the rat intraperitoneally, anesthetized with 12% ethyl carbamate (10 mL/kg), and dissected longitudinally to strip away intima and tunica media. Next, the adventitia was snipped into 2‐mm2 pieces before being placed in Petri dishes with DMEM (Invitrogen), supplemented with 20% fetal bovine serum (FBS; Invitrogen), 2 mmol/L l‐glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin under 37°C, 5% CO2, and saturated humidity for cell outgrowth. After 5 to 7 days, confluent cells were subcultured by trypsinization, and the complete medium was changed into one containing 10% FBS. Rat PAFs were determined by positive staining for vimentin and by negative staining for smooth muscle actin and von Willebrand factor. All experiments were carried out between cellular passages 4 and 6.
Primary Rat Pulmonary Artery Smooth Muscle Cell Culture
Pulmonary artery smooth muscle cells (PASMCs) were purchased from Wuhan PriCells Biomedical Technology Co., Ltd (Wuhan, China) and were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen), 2 mmol/L l‐glutamine (Lifeline Cell Technology), 100 U/mL penicillin, 100 μg/mL streptomycin, and primary vascular smooth muscle cell growth supplement (Wuhan PriCells Biomedical Technology Co., Ltd). PASMCs were cultured in an incubator at 37°C with 5% CO2 and saturated humidity, and 4 to 6 generations were used for our experiments.
Lentivirus Harboring Aspartate Aminotransferase 1 (AAT1) or AAT1 Short Hairpin RNA Infection of PAFs and PASMCs
PAFs and PASMCs were seeded in culture flasks of 25 cm2 for virus transfection. When cells were grown to 40% to 50% confluence, lentivirus containing the cDNA encoding AAT1 (Vigene Bioscience, Jinan, China), the short hairpin RNA to AAT1 (Cyagen Bioscience, Guangzhou, China), or vehicle (Vigene Bioscience) were added separately to infect PAFs for 24 hours at a multiplicity of infection of 20. The cells were selected with puromycin (4 μg/mL) to acquire the stably transfected AAT1 or AAT1 short hairpin RNA.
Coculture of PASMCs and PAFs
Transwell (Corning) were used for coculture of PAFs and PASMCs. PAFs were seeded in lower compartments of the transwell and cultured in DMEM supplemented with 10% FBS, 2 mmol/L l‐glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Infected PASMCs were seeded in upper compartments, which were separated from lower compartments by a microporous membrane. PASMCs were cultured in DMEM containing 10% FBS, 2 mmol/L l‐glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and primary vascular smooth muscle cell growth supplement, and all cells were cultured in an incubator at 37°C with 5% CO2 and saturated humidity.
Determination of SO2 Content
SO2 concentrations of PAF supernatant were detected using high‐performance liquid chromatography (Agilent 1200 series; Agilent Technologies).8 Briefly, 100 μL of PAF supernatant was mixed with 70 μL of 0.212 mol/L sodium borohydride in 0.05 mol/L Tris‐HCl (pH 8.5) and incubated at room temperature for 30 minutes. The sample was then mixed with 5 μL of 70 mmol/L monobromobimane in acetonitrile, incubated at 42°C for 10 minutes, and then mixed with 40 μL of 1.5 mol/L perchloric acid. Protein precipitate in the mixture was removed by centrifugation at 12 400 g for 10 minutes at room temperature. The supernatant was then neutralized after being gently mixed with 10 μL of 2.0 mol/L Tris‐HCl. Next, it was centrifuged at 12 400 g for 10 minutes at 26°C. The supernatant (100 μL) was taken into a foil‐wrapped tube, and 10 μL of the sample was added into the high‐performance liquid chromatography column. A buffer of methanol, acetic acid, and water at a ratio of 5.00:0.25:94.75 (by volume, pH 3.4) was used to equilibrate the column. Sulfite‐bimane was examined by excitation at 392 nm and emission at 479 nm.
AAT Activity Analysis
PAFs were homogenized in PBS with an ice‐water bath and centrifuged at 451 g for 10 minutes at 4°C to get the supernatant. Protein content was determined according to Bradford's method,15 and AAT activity was tested by using the AAT activity microplate test kit (Nanjing JianCheng Bioengineering Institute), according to the manufacturer's instructions.
Immunofluorescence and Confocal Microscopy Analysis of Collagen Types I and III in PAFs
PAFs were seeded on coverslips in 6‐well plates to 60% to 70% confluence before they were made quiescent in serum‐free medium for 24 hours and then incubated in DMEM containing 0.5% FBS. The cells were stimulated with or without TGF‐β1 (10 ng/mL) for 72 hours. After treatment, the cells were fixed in 4% formaldehyde for 15 minutes at room temperature and blocked and permeabilized in 5% bovine serum albumin with 0.3% Triton X‐100 (Sigma‐Aldrich) for 30 minutes at 37°C. Coverslips were incubated with rabbit anti–collagen type I (Abcam) or rabbit anti–collagen type III (Sigma‐Aldrich) antibody at 4°C overnight. The coverslips were then washed 3 times with PBS and incubated with FITC‐conjugated goat anti–rabbit immunoglobulin G antibody (Santa Cruz Biotechnology) at room temperature for 2 hours. Fluorescent images were captured by the FluoView laser scanning confocal microscope (Olympus).
Western Blotting Analysis
Western blotting analysis was performed, as described previously.15, 16 Lung samples were homogenized in tissue lysate, followed by centrifugation at 12193 g for 20 minutes at 4°C. The protein content was determined according to Bradford's method,17 with bovine serum albumin used as a standard.
PAFs and PASMCs were seeded in dishes of 60 mm for Western blotting analysis. When cells had grown to 70% to 80% confluence, they were starved in serum‐free medium for 24 hours and then stimulated with or without Na2SO3/NaHSO3 (0–200 μmol/L) or TGF‐β1 (10 ng/mL) for the indicated time. After treatment, the cells were harvested and lysed, as described previously.18
All lung tissue and PAF protein samples were mixed respectively with 2 times loading buffer containing 5% β‐mercaptoethanol and boiled for 5 minutes. Equal amounts of protein were resolved by 10% to 15% polyacrylamide gel under SDS denaturing conditions and transferred onto a nitrocellulose membrane. Nonspecific binding was blocked by incubation in a blocking buffer of 5% milk in Tris‐buffered saline with 1% Tween. The membranes were probed with specific antibodies: mouse anti–matrix metalloproteinase 13 (anti–MMP‐13; Millipore), mouse anti–tissue inhibitor of MMP‐1 (anti–TIMP‐1; Millipore), rabbit anti–signal transducer and activator of transcription 3 (anti‐STAT3; Santa Cruz Biotechnology), rabbit anti–phospho‐STAT3 (Santa Cruz Biotechnology), rabbit anti–TGF‐β1 (Santa Cruz Biotechnology), goat anti‐AAT1 (Sigma‐Aldrich), rabbit anti‐AAT2 (Sigma‐Aldrich), rabbit anti–TGF‐β1 receptor I (Santa Cruz Biotechnology), rabbit anti–collagen type I (Abcam), rabbit anti–collagen type III (Abcam), rabbit anti–phospho‐Smad2 (Cell Signal Technology), rabbit anti–phospho‐Smad3 (Cell Signal Technology), rabbit anti‐Smad2/3 (Cell Signal Technology), rabbit anti–poly (ADP‐ribose) polymerase (anti‐PARP; Cell Signal Technology), mouse anti–proliferating cell nuclear antigen (anti–PCNA; Anbo Biotechnology), rabbit anti–bone morphogenetic protein receptor type 2 (anti–BMPR‐2; Abcam), rabbit anti–nitric oxide synthase 2 (anti‐NOS2; Santa Cruz Biotechnology), mouse anti–β‐actin (Santa Cruz Biotechnology), and mouse anti‐GAPDH (Kang Cheng), followed by detection with respective specific secondary antibodies (Santa Cruz Biotechnology). Immunoreactions were visualized using an enhanced chemiluminescence detection kit (Thermo Scientific).
Detection of Nitric Oxide Concentration in Lung Tissues
Lung tissues were weighted and homogenized in 10 times volume of 0.01 mol/L PBS. After centrifuging at 2000 g/min for 15 minutes, the supernatant was prepared for detection. The nitric oxide (NO) assay kit (Beyotime) was used to detect the NO concentration of lung tissues.
Phosphorylation of Type I TGF‐β Receptor Evaluated by Immunoprecipitation
PAFs were seeded in 100‐mm dishes for immunoprecipitation. When the cells had grown to 70% to 80% confluence, they were starved in serum‐free medium for 24 hours. The cells were pretreated with or without 100 μmol/L Na2SO3/NaHSO3 for 60 minutes and then stimulated with or without TGF‐β1 (10 ng/mL). After 60 minutes, the cells were harvested and lysed, spun at 13 000 g for 10 minutes, and preincubated with protein A/G agarose beads (Thermo Fisher Scientific) for 1 hour at 4°C. Equal amounts of cell lysates subsequently incubated with the antibody against type I TGF‐β receptor (TβRI; Santa Cruz Biotechnology, CA) overnight at 4°C. The antibody–antigen complex was immunoprecipited with protein A/G agarose beads for 4 hours at 4°C. The precipitated proteins were resolved by 10% SDS‐PAGE and then immunoblotted with antibodies against phosphoserine (Abcam).19
Detection of Calcium Levels
The levels of calcium in PAFs and PASMCs were detected with GFP‐Certified FluoForte reagent (Enzo Life Sciences). Cells were washed with PBS for 3 times and incubated in medium supplemented with GFP‐Certified FluoForte reagent at 37°C in the dark for 60 minutes. Results were analyzed using a laser scanning confocal microscope (Olympus).
Detection of Mitochondrial Membrane Potential
Mitochondrial membrane potential in PAFs and PASMCs was detected by using the Mitochondrial Membrane Potential Assay Kit with JC‐1 (Beyotime). Cells seeded in slides were incubated with mixture of JC‐1 working solution and cell medium at 37°C, avoiding light for 20 minutes. After washing 3 times with JC‐1 buffer solution, cell medium was added, and fluorescent images were captured using a laser scanning confocal microscope (Olympus). With high mitochondrial membrane potential, JC‐1 aggregates in the mitochondrial matrix, forming polymer (J‐aggregates), and red fluorescence can be produced. With lower mitochondrial membrane potential, JC‐1 presents in mitochondrial matrix as a monomer with green fluorescence.
Detection of Mitochondrial Permeability Transition Pore Opening Level
The opening level of mitochondrial permeability transition pore (MPTP) in PAFs and PASMCs was measured using the MPTP assay kit (Genmed). First, cell medium was discarded, and preheated cleaning solution was added. Next, cells were incubated with working solution at 37°C and protected from light for 30 minutes. After washing 3 times with 0.01 mol/L PBS, fluorescent images were captured and analyzed using a laser scanning confocal microscope. As a fluorescent probe, calcein AM can be captured easily by mitochondria and presents with green fluorescence. When MPTP opens, calcein is released into cytoplasm, with fluorescent quenching.
Cell Apoptosis Detected by ELISA Assay
The Cell Death Detection ELISAPLUS Kit (Roche) was used to detect cell apoptosis in PAFs and PASMCs. Cells were harvested in cell lysate, and 20 μL of homogenate was added in microplate well. Cell lysate with the same volume was added to microplate well as a background control group. A mixture of 5% anti–DNA‐POD (reconstituted) and 5% anti–histone‐biotin (reconstituted) was added and shaken for 2 hours at room temperature for incubation. After washing by incubation buffer, 100 μL of ABTS substrate solution was added and incubated for 20 minutes. Next, 100 μL of ABTS stop solution was used to terminate the reaction. In the blank control group, only 100 μL of ABTS substrate solution and 100 μL of ABTS stop solution were added. A microtiter plate reader (Bio‐Rad Laboratories) was used to determine the value of each group at 405 nm, with the reference wavelength at 490 nm.20
Detection of Cell Apoptosis by TdT‐Mediated dUTP Nick End Labeling Assay
Cell apoptosis in PAFs and PASMCs was detected by using the In Situ Cell Death Detection Kit, Fluorescein (Roche). After washing with 0.01 mol/L PBS, cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes. Cells were incubated in a terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling, or TUNEL, reaction mixture containing enzyme solution and label solution at 37°C, avoiding light for 60 minutes. After washing 3 times with PBS, the results were observed and analyzed under a laser scanning confocal microscope (Olympus).
Statistics
All data are expressed as mean±SE. We analyzed data with SPSS 17.0 software (IBM Corp). When the data were normally distributed, we used 1‐way ANOVA followed by post hoc analysis (Newman‐Keuls test) for multiple comparison. Otherwise, the Kruskal–Walls rank sum test would be used for comparison.
Results
Effects of Monocrotaline on AAT1 and AAT2 Protein Expression and AAT Activity in Lung Tissue
The study started by establishing a rat model of monocrotaline‐induced pulmonary vascular collagen remodeling. AAT1 protein expression in the lung tissues of the monocrotaline group was markedly upregulated (P<0.05) (Figure 1A); however, AAT2 protein content did not differ between the monocrotaline group and the control group (P>0.05) (Figure 1A).
Figure 1.
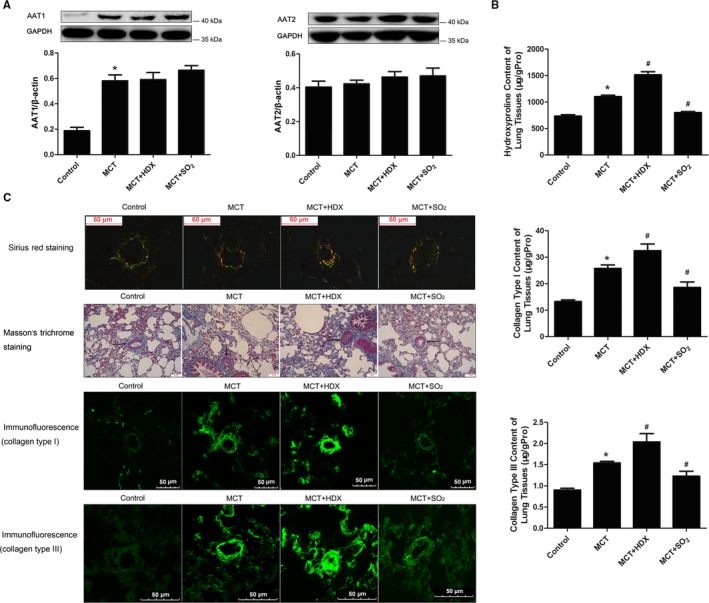
Effects of SO 2 on collagen expression in lung tissue and pulmonary arteries of rats. A, Changes of AAT1 and AAT2 protein expression in lung tissue of rats. B, Collagen content in lung tissues of rats. C, Collagen analysis in pulmonary arteries of rats. Sirius red staining analysis of collagen types I and III in rat pulmonary arteries was observed under a polarizing microscope. For Masson's trichrome staining, green is specific for collagen, muscle fibers were dyed red, and nuclei were dyed dark blue. Results are expressed as mean±SE, n=8. *P<0.05 compared with the control group; # P<0.05 compared with the MCT group. AAT1 indicates aspartate aminotransferase 1; AAT2, aspartate aminotransferase 2; HDX, l‐aspartate‐β‐hydroxamate; MCT, monocrotaline; SO2, sulfur dioxide; μg/gpro, the content of hydroxyproline, collagen type I or collagen type III (μg) in 1 g protein.
SO2 Decreased Total Collagen Content and Collagen Types I and III Content in Lung Tissue
To reveal the mechanism of pulmonary vascular remodeling, ELISA was used to examine hydroxyproline, collagen type I, and collagen type III contents in the lung tissue of rats. Hydroxyproline content in the lung tissue of rats was much higher in the monocrotaline group than in the control group (P<0.05) (Figure 1B). With treatment of SO2 donor for 3 weeks, the hydroxyproline content decreased significantly (P<0.05) (Figure 1B); however, after administration of HDX, the hydroxyproline content of lung tissue was further increased (P<0.05) (Figure 1B).
Evaluation of lung tissue by ELISA demonstrated that collagen types I and III contents were both higher in the monocrotaline group than in controls (P<0.05 for both) (Figure 1B). Their contents rose significantly in the monocrotaline plus HDX group (P<0.05 for both) (Figure 1B) but decreased in the monocrotaline plus SO2 group (P<0.05 for both) (Figure 1B) compared with those of the monocrotaline group.
SO2 Lowered Collagen Types I and III Protein Expression in Pulmonary Arteries
Detection of collagen types I and III protein expression in lung tissue demonstrated that the main changes in collagen types I and III occurred in the pulmonary arteries. Collagen types I and III are typical ECM proteins in remodeled pulmonary arteries visualized using a polarizing microscope after Sirius red staining. The results indicated that spatial accumulation of collagen types I and III in rat pulmonary arteries was more obvious in the monocrotaline group than in the control group. Compared with levels in the monocrotaline group, collagen accumulation increased in the monocrotaline plus HDX group but decreased in the monocrotaline plus SO2 group (Figure 1C). A similar trend was observed using Masson's trichrome staining (Figure 1C).
Immunofluorescent images showed that pulmonary arterial collagen types I and III expression was elevated in monocrotaline‐induced rats. Furthermore, this expression was strengthened in the monocrotaline plus HDX group but weakened in the monocrotaline plus SO2 group compared with the monocrotaline group (Figure 1C).
SO2 Inhibited Collagen Types I and III mRNA Expression in Lung Tissue and Pulmonary Arteries
Because synthesis of collagen types I and III depends on procollagen mRNA expression, we examined the procollagen types I and III mRNA levels in lung tissue using real‐time PCR. The results demonstrated that procollagen types I and III mRNA expression in lung tissue increased in the monocrotaline group (P<0.05 for both) (Figure 2A and 2B). Compared with the monocrotaline group, procollagen types I and III mRNA mounted up further in the monocrotaline plus HDX group (P<0.05 for both) (Figure 2A and 2B) but was markedly reduced in the monocrotaline plus SO2 group (P<0.05 for both) (Figure 2A and 2B).
Figure 2.
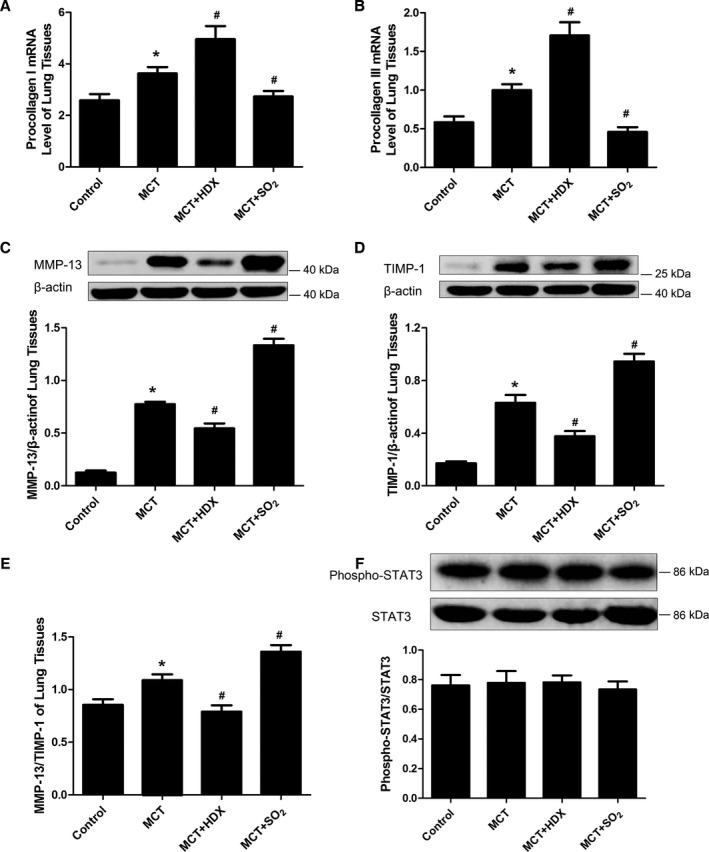
Synthesis and degradation of collagen in lung tissues of rats. A and B, Collagen type I (A) and type III (B) mRNA levels in lung tissues by real‐time polymerase chain reaction analysis. C and D, Protein expression of MMP‐13 (C) and TIMP‐1 (D) in lung tissues, detected by Western blotting. E, The ratio of MMP‐13/TIMP‐1 in lung tissues in rats of different groups. F, Activity of STAT3 in lung tissues of rats. Results are expressed as mean±SE, n=8. *P<0.05 compared with the control group; # P<0.05 compared with the MCT group. HDX indicates l‐aspartate‐β‐hydroxamate; MCT, monocrotaline; MMP‐13, matrix metalloproteinase 13; SO2, sulfur dioxide; STAT3, signal transducer and activator of transcription 3; TIMP‐1, tissue inhibitors of MMP‐1.
Effects of SO2 on Lung Tissue MMP‐13 and TIMP‐1 Protein Expression
We examined MMP‐13 and TIMP‐1 protein content and STAT3 activity in lung tissue by Western blotting. Quantification of band intensities demonstrated that MMP‐13 and TIMP‐1 protein contents in lung tissue of the monocrotaline group were significantly greater than those in the control group (P<0.05 for both) (Figure 2C and 2D) and were confirmed by the ratio of MMP‐13/TIMP‐1 (P<0.05) (Figure 2E). In the monocrotaline plus HDX group, MMP‐13 and TIMP‐1 contents were significantly decreased compared with the monocrotaline group (P<0.05 for both) (Figure 2C and 2D), with the MMP‐13/TIMP‐1 ratio producing similar results (P<0.05) (Figure 2E). Meanwhile, expression of MMP‐13 and TIMP‐1 was higher in the monocrotaline plus SO2 group than in the monocrotaline group (P<0.05 for both) (Figure 2C and 2D), as corroborated by the MMP‐13/TIMP‐1 ratio (P<0.05) (Figure 2E). There was no change in STAT3 activity in different groups (P>0.05) (Figure 2F).
SO2 Repressed TGF‐β1 Expression of Lung Tissue and Pulmonary Arteries
TGF‐β1 protein content in lung tissues was examined by Western blotting. TGF‐β1 content was significantly higher in the monocrotaline group than in the control group (P<0.05) (Figure 3A) and was further increased in the monocrotaline plus HDX group than in the monocrotaline group (P<0.05) (Figure 3A). In the monocrotaline plus SO2 group, however, TGF‐β1 content was significantly lower compared with the monocrotaline group (P<0.05) (Figure 3A). The real‐time PCR analysis demonstrated that TGF‐β1 mRNA expression in lung tissue of the monocrotaline group was greater than that of control group (P<0.05) (Figure 3B). In the monocrotaline plus HDX group, TGF‐β1 mRNA expression was significantly increased compared with the monocrotaline group (P<0.05) (Figure 3B); however, it was downregulated further in the monocrotaline plus SO2 group compared with the monocrotaline group (P<0.05) (Figure 3B).
Figure 3.
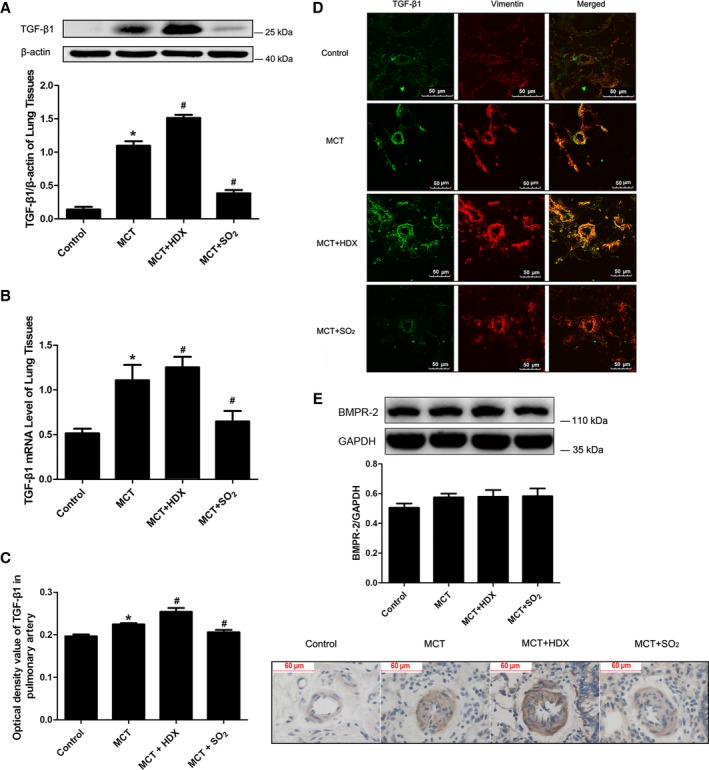
Effects of SO 2 on TGF‐β1 protein expression and mRNA in MCT‐treated rats. A, TGF‐β1 protein expression in lung tissues analyzed by Western blotting. B, TGF‐β1 mRNA expression of pulmonary arteries in different groups analyzed by real‐time polymerase chain reaction. C, TGF‐β1 protein expression in lung tissues analyzed by immunohistochemistry. D, The colocalization of TGF‐β1 and vimentin in small pulmonary arteries by immunofluorescence analysis (×600). E, BMPR‐2 protein expression in lung tissues of rats. Results are expressed as mean±SE, n=8. *P<0.05 compared with the control group; # P<0.05 compared with the MCT group. BMPR‐2 indicates bone morphogenetic protein receptor type 2; HDX, l‐aspartate‐β‐hydroxamate; MCT, monocrotaline; SO2, sulfur dioxide; TGF‐β1, transforming growth factor β1.
By immunohistochemical staining, we observed that TGF‐β1 expression in pulmonary arteries was enhanced in the monocrotaline group, and expression signals were weaker in the monocrotaline plus SO2 group than in the monocrotaline group (Figure 3C). Furthermore, the colocalization of the increased TGF‐β1 expression with vimentin was observed in small pulmonary arteries using immunofluorescence and confocal microscopy (Figure 3D). The results suggested that the main change in TGF‐β1 protein expression took place in PAFs.
Protein expression of BMPR‐2 in lung tissues was detected by Western blotting; however, it did not differ significantly between groups (Figure 3E).
SO2 Inhibited Accumulation of Collagen Types I and III in TGF‐β1–Treated PAFs
TGF‐β1 treatment significantly upregulated protein expression of collagen types I and III (P<0.05 for both) (Figure 4A), but these effects were blocked by SO2 treatment (P<0.05 for both) (Figure 4A). There was no apparent difference in the collagen protein contents of PAFs between the control and SO2 groups (P>0.05 for both) (Figure 4A).
Figure 4.
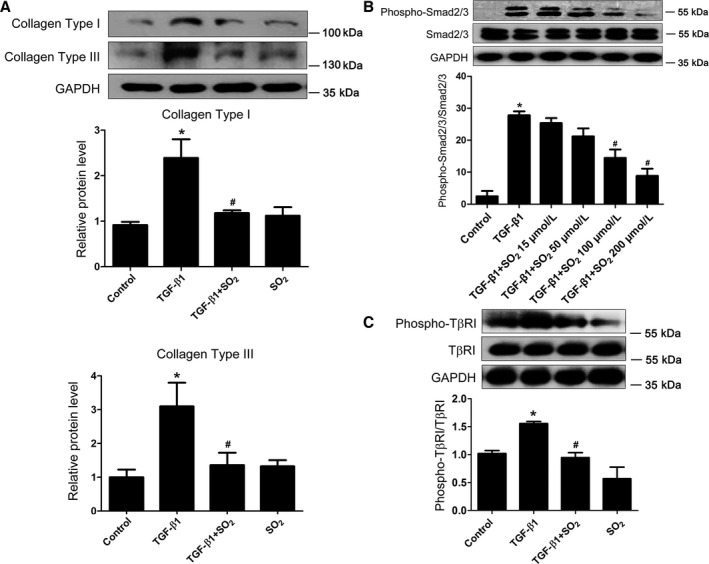
Inhibitory effect of SO 2 on collagen types I and III accumulation and TGF‐β/TβRI/Smad2/3 signal cascades in TGF‐β1–treated PAFs. A, Protein expression of collagen types I and III in TGF‐β1–stimulated PAFs with or without SO 2 pretreatment. B, The phosphorylation of Smad2/3 induced by TGF‐β1 in PAFs with or without SO 2 pretreatment. C, The serine phosphorylation of TβRI induced by TGF‐β1 in PAFs with or without SO 2 pretreatment. Results are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the control group; # P<0.05 compared with the TGF‐β1 group. PAF indicates pulmonary arterial fibroblast; SO2, sulfur dioxide; TGF‐β1, transforming growth factor β1; TβRI, type I transforming growth factor β receptor.
SO2 Repressed Activation of the TGF‐β–Stimulated TGF‐β/TβRI/Smad2/3 Signaling Pathway
Western blotting results showed that TGF‐β1 could significantly stimulate the phosphorylation of Smad2/3 (P<0.05) (Figure 4B); however, the phosphorylation of Smad2/3 induced by TGF‐β1 was significantly inhibited when PAFs were pretreated with SO2 at a concentration of 100 μmol/L for 30 minutes and then treated with TGF‐β1 for 1 hour (P<0.05) (Figure 4B). Considering that TGF‐β1 promoted Smad2/3 phosphorylation via serine phosphorylation and activation of TβRI, we examined the phosphorylation of TβRI in PAFs by immunoprecipitating cell lysates with anti‐TβRI antibodies followed by immunoblotting with anti–phosphoserine antibodies. TGF‐β1 could significantly increase the phosphorylation of TβRI (P<0.05) (Figure 4C), whereas SO2 treatment markedly attenuated serine phosphorylation of TβRI (P<0.05) (Figure 4C). SO2, however, did not influence the total expression of TβRI in PAFs with or without TGF‐β1 stimulation (P>0.05) (Figure 4C).
Increasing Endogenous SO2 Generation by AAT1 Overexpression Repressed Smad2/3 Phosphorylation and Collagen Types I and III Accumulation Induced by TGF‐β1 in PAFs
To further verify the inhibitory effect of endogenous SO2 on collagen remodeling, PAFs were infected with lentivirus, which contains the cDNA encoding AAT1. After 72 hours, AAT activity, AAT1 protein expression, and SO2 concentration were detected. Compared with the vehicle‐lentivirus group and the control group, AAT1 protein expression was markedly enhanced in the group with AAT1 overexpression (P<0.05) (Figure 5A). Likewise, AAT activity in PAFs and SO2 concentration in the supernatant of PAFs were significantly increased in the group with AAT1 overexpression (P<0.05 for both) (Figure 5A).
Figure 5.
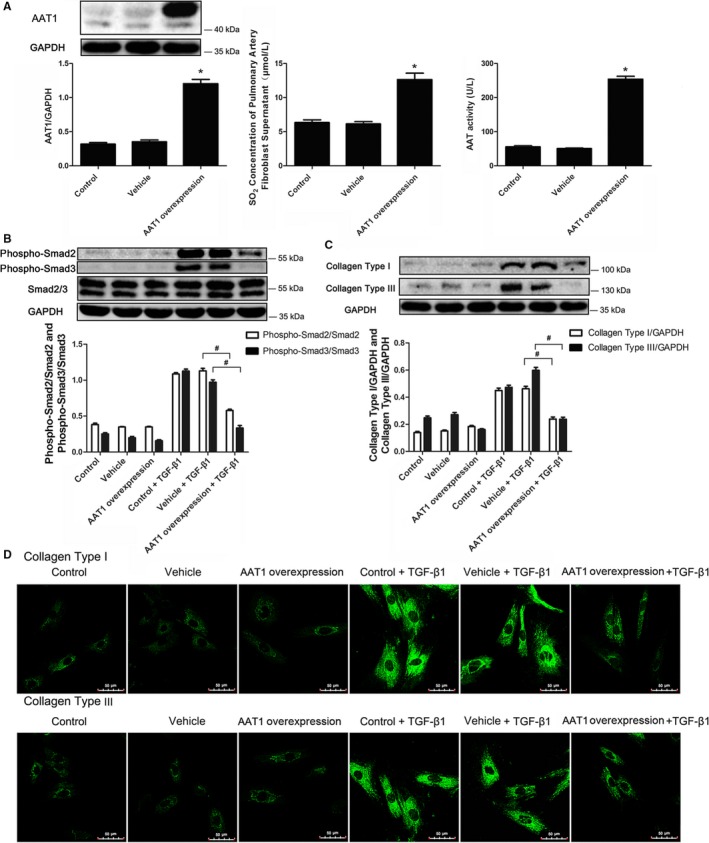
Endogenous SO 2 inhibited the Smad2/3 signaling pathway and collagen deposition. A, AAT1 protein expression, SO 2 content, and AAT activity in PAFs overexpressing AAT1. B, The phosphorylation of Smad2/3 induced by TGF‐β1 in PAFs overexpressing AAT1. C and D, Western blotting (C) and immunofluorescence analysis (D) of collagen types I and III induced by TGF‐β1 in PAFs with AAT1 overexpression. Results are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the vehicle group; # P<0.05 compared with the vehicle plus TGF‐β1 group. AAT1 indicates aspartate aminotransferase 1; PAF, pulmonary arterial fibroblast; SO2, sulfur dioxide; TGF‐β1, transforming growth factor β1.
Western blotting analysis showed that AAT1 overexpression markedly prevented phosphorylation of Smad2 and Smad3 stimulated by TGF‐β1 (P<0.05 for both) (Figure 5B). Subsequently, both Western blotting and immunofluorescence revealed that the TGF‐β1–induced collagen types I and III content was inhibited in PAFs overexpressing AAT1 (P<0.05 for both) (Figure 5C and 5D).
Decreasing Endogenous SO2 Generation by AAT1 Knockdown Aggravated Smad2/3 Phosphorylation and Collagen Types I and III Deposition Induced by TGF‐β1 in PAFs
PAFs were infected with lentivirus, which contains the cDNA encoding short hairpin RNA to AAT1. Compared with the vehicle‐lentivirus and control groups, AAT1 protein expression was reduced in the AAT1 knockdown group (P<0.05) (Figure 6A). Likewise, AAT activity in PAFs and SO2 concentration in the supernatant of PAFs were also decreased in the AAT1 knockdown group (P<0.05 for both) (Figure 6A).
Figure 6.
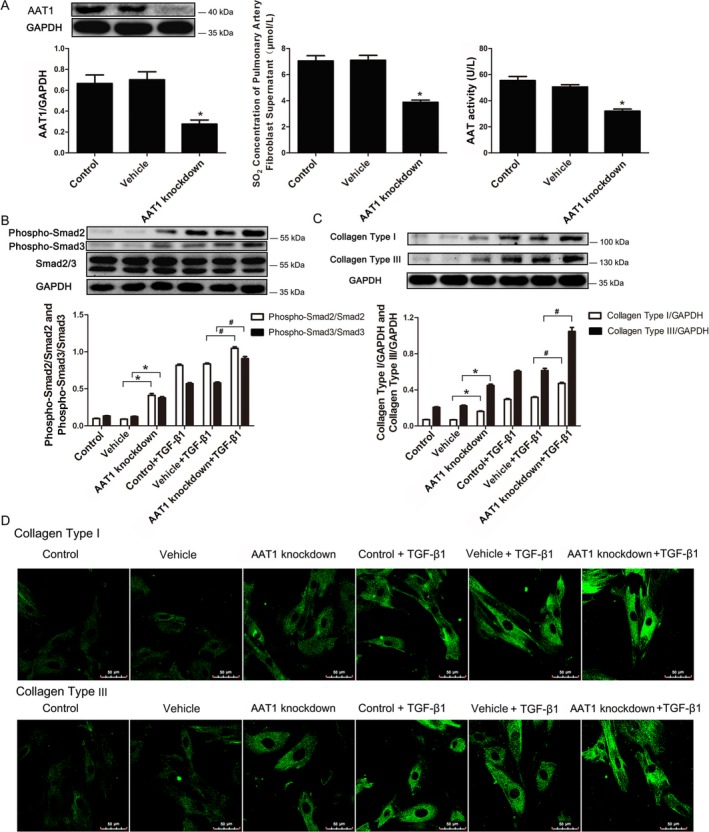
Decreased endogenous SO 2 content further exacerbated activation of the Smad2/3 signaling pathway and collagen accumulation. A, AAT1 protein expression, SO 2 content, and AAT activity in PAFs by AAT1 knockdown. B, Phosphorylation of Smad2/3 induced by TGF‐β1 in PAFs with AAT1 knockdown. C and D, Western blotting (C) and immunofluorescence analysis (D) of collagen types I and III protein expression in PAFs with knockdown of AAT1. Results are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the vehicle group; # P<0.05 compared with the vehicle plus TGF‐β1 group. AAT1 indicates aspartate aminotransferase 1; PAF, pulmonary arterial fibroblast; SO2, sulfur dioxide; TGF‐β1, transforming growth factor β1.
Compared with the vehicle group, TGF‐β1 stimulated phosphorylation of Smad2/3 (P<0.05 for both) (Figure 6B), which was further exacerbated by AAT1 knockdown (P<0.05 for both) (Figure 6B). With stimulation of TGF‐β1, the content of collagen types I and III was increased, and was further stimulated by AAT1 knockdown. (Figure 6C and 6D).
Reduction of Endogenous SO2 in PASMCs Increased Collagen Accumulation in PAFs
After the coculture of PASMCs and PAFs for 72 hours, cells were harvested, and collagen I and III protein expression in PAFs was detected by Western blotting. We found that with inhibition of endogenous SO2 generation in PASMCs, accumulation of collagen types I and III in PAFs was activated significantly (P<0.05 for both) (Figure 7). After we increased endogenous SO2 in PASMCs, collagen accumulation did not change in PAFs (P>0.05) (Figure 7).
Figure 7.
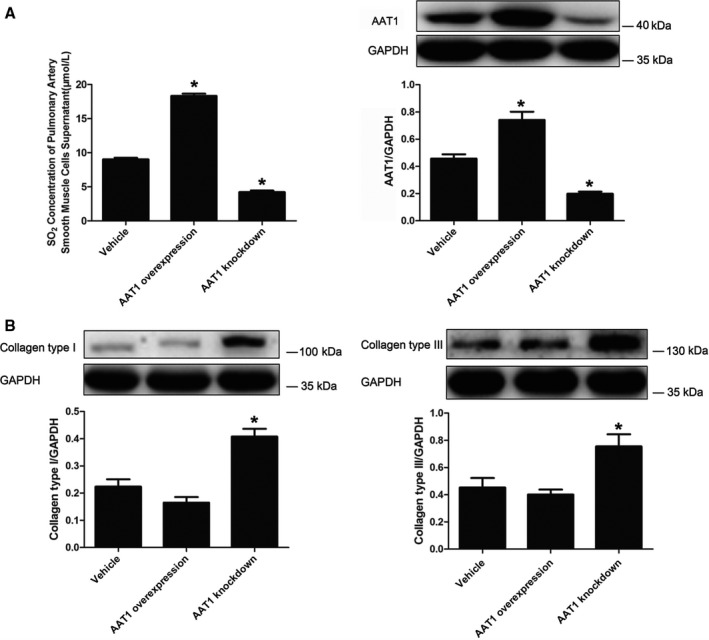
Reduction of endogenous SO 2 in PASMCs increased collagen accumulation in PAFs. A, Changes in the SO 2/AAT1 system in PASMCs. B, Collagen accumulation in PAFs. Results are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the vehicle group. AAT1 indicates aspartate aminotransferase 1; PAF, pulmonary arterial fibroblast; PASMC, pulmonary artery smooth muscle cell; SO2, sulfur dioxide.
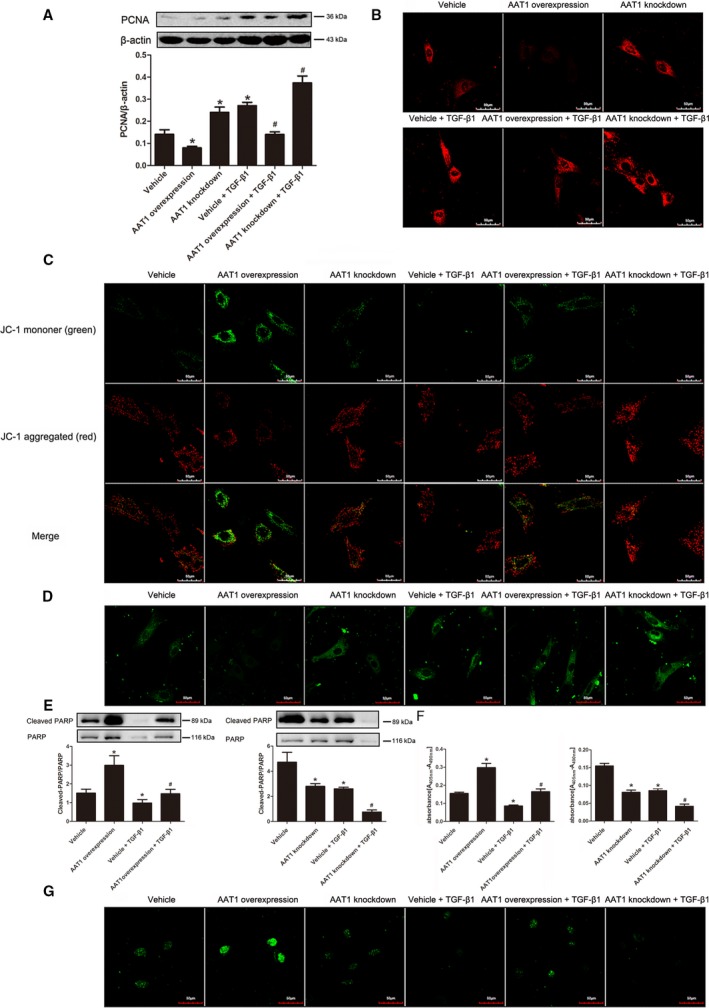
SO2 Inhibited Cell Proliferation in PAFs
PCNA is closely related to cell DNA synthesis and plays an important role in the initiation of cell proliferation. With the increase in SO2 content by overexpressing AAT1, protein expression of PCNA was decreased significantly (P<0.05) (Figure 8A). TGF‐β1 increased the expression of PCNA in PAFs, but this was prevented by SO2 (P<0.05) (Figure 8A). After we reduced SO2 content by knockdown of AAT1, expression of PCNA was increased (P<0.05) (Figure 8A).
Figure 8.
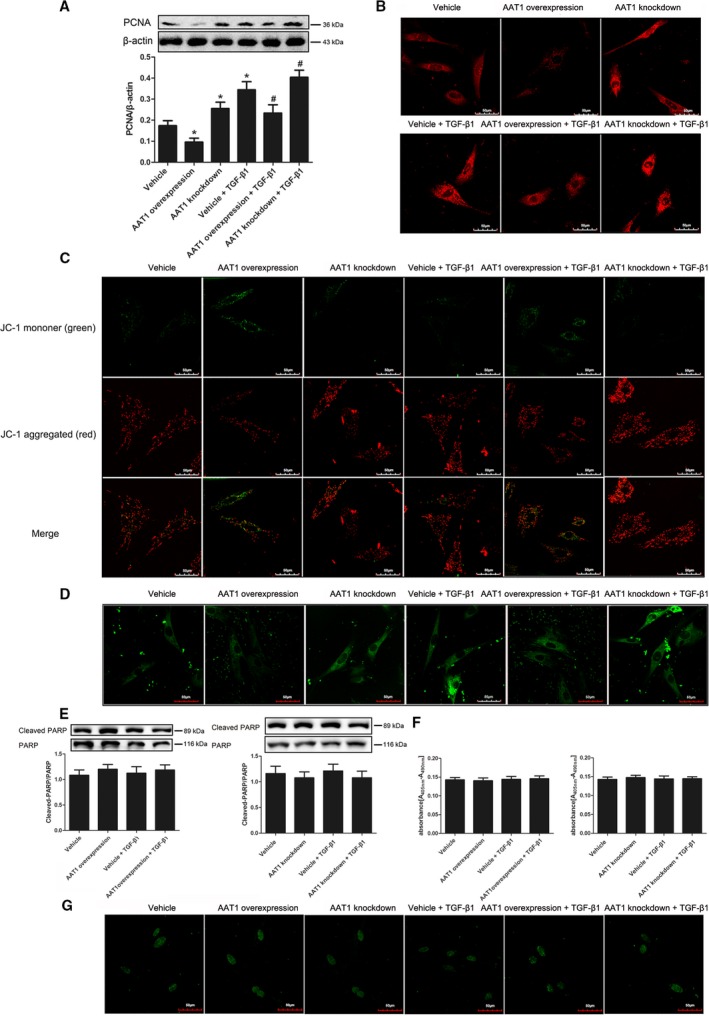
Changes of cell proliferation and apoptosis in PAFs. A, Protein expression changes of PCNA in PAFs detected by Western blotting. B, Calcium level in PAFs (×600). C, Mitochondrial membrane potential of PAFs was detected using the JC‐1 kit. D, The opening level of mitochondrial permeability transition pore in PAFs. E, The activity of PARP in PAFs detected by Western blot. F, Cell apoptosis detected by using an enzyme‐linked immunosorbent assay. G, Cell apoptosis examined by the TUNEL method (×600). Data are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the vehicle group; # P<0.05 compared with the vehicle plus TGF‐β1 group. AAT1 indicates aspartate aminotransferase 1; PAF, pulmonary arterial fibroblast; PARP, poly (ADP‐ribose) polymerase; PCNA, proliferating cell nuclear antigen; TGF‐β1, transforming growth factor β1; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling.
SO2 Decreased the Calcium Level in PAFs
To investigate the effects of SO2 on calcium level, we used GFP‐Certified FluoForte reagent to examine the calcium level in PAFs. Results showed that with the increase of SO2 generation, calcium in PAFs was reduced significantly (Figure 8B). After we inhibited the generation of endogenous SO2, the calcium level in PAFs was increased (Figure 8B).
SO2 Increased Mitochondrial Dysfunction of PAFs
To measure the function of mitochondria in PASMCs, we used the Mitochondrial Membrane Potential Assay Kit with JC‐1 to detect the mitochondrial membrane potential and the MPTP assay kit to detect the opening level of the MPTP channel. Results showed that SO2 decreased mitochondrial membrane potential in PAFs but increased the opening level of MPTP (Figure 8C and 8D). With knockdown of AAT1 to decrease SO2 generation, mitochondrial membrane potential in PAFs was increased, but the opening level of MPTP was decreased (Figure 8C and 8D).
SO2 Had No Effects on Cell Apoptosis in PAFs
To investigate the effects of SO2 on cell apoptosis, we used Western blotting, ELISA, and TUNEL assay to detect PARP activity and cell apoptosis. We found that with stimulation of TGF‐β1 and intervention in endogenous SO2 generation, there were no changes in PARP activity and cell apoptosis in PAFs detected by ELISA and TUNEL assay (Figure 8E through 8G).
SO2 Inhibited Cell Proliferation in PASMCs
To detect cell proliferation in PASMCs, protein expression of PCNA was examined by Western blotting. Results showed that TGF‐β1 could stimulate the expression of PCNA in PASMCs, but protein expression of PCNA was decreased significantly with overexpression of AAT1 (P<0.05) (Figure 9A). In contrast, with the inhibition of SO2 generation, the expression of PCNA was increased (P<0.05) (Figure 9A).
Figure 9.
Effects of sulfur dioxide on proliferation and cell apoptosis in PASMCs. A, PCNA protein expression in PASMCs. B, Content of intracellular calcium in PASMCs (×600). C, Mitochondrial membrane potential in PASMCs (×600). D, The opening level of mitochondrial permeability transition pore in PASMCs (×600). E, PARP activity in PASMCs detected by Western blot. F and G, Cell apoptosis detected by using an enzyme‐linked immunosorbent assay (F) and the TUNEL method (G) (×600). Results are expressed as mean±SE for 3 separate experiments (n=3), each in triplicate. *P<0.05 compared with the vehicle group; # P<0.05 compared with the vehicle plus TGF‐β1 group. AAT1 indicates aspartate aminotransferase 1; PCNA, proliferating cell nuclear antigen; PARP, poly (ADP‐ribose) polymerase; PASMC, pulmonary artery smooth muscle cell; TGF‐β1, transforming growth factor β1; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling.
SO2 Decreased the Calcium Level in PASMCs
GFP‐Certified FluoForte reagent was used to detected calcium levels in PASMCs. With increasing content of SO2, calcium levels in PASMCs were decreased significantly with reduction of fluorescence density in PASMCs. Meanwhile, calcium levels were increased with the inhibition of SO2 generation (Figure 9B).
SO2 Increased Mitochondrial Dysfunction in PASMCs
The Mitochondrial Membrane Potential Assay Kit with JC‐1 and MPTP assay kit were used to detect mitochondrial membrane potential and MPTP channel opening level. We found that with increasing content of SO2, mitochondrial membrane potential in PASMCs was decreased but the opening level of MPTP was increased (Figure 9C and 9D). With the inhibition of SO2 generation, however, mitochondrial membrane potential in PASMCs was increased, and the opening level of MPTP was reduced significantly (Figure 9C and 9D).
SO2 Promoted Cell Apoptosis of PASMCs
To investigate the effects of SO2 on cell apoptosis, we used Western blotting, ELISA, and TUNEL assay to detect PARP activity and cell apoptosis. Stimulated by TGF‐β1, PARP activity was decreased (P<0.05) (Figure 9E) along with the reduction of nucleosomes in PASMCs examined by ELISA (P<0.05) (Figure 9F) and the weakened fluorescence in the nuclei of PASMCs (Figure 9G). Furthermore, the effects of TGF‐β1 were aggravated by the decrease of SO2 (Figure 9E–9G). With the increase of SO2, however, the activity of PARP, the amount of nucleosomes in PASMCs, and the fluorescence density in nuclei of PASMCs were increased significantly (Figure 9E–9G).
Effects of Monocrotaline on NO Generation and NOS2 Protein Expression in Lung Tissue
To investigate the changes in NO generation, we detected NO concentration and NOS2 protein expression in lung tissues of rats. Results showed that NO concentration and NOS2 protein expression were decreased in monocrotaline‐treated rats compared with control rats (P<0.05 for both) (Figure 10). In monocrotaline‐treated rats with inhibition of SO2 generation, NO concentration and NOS2 protein expression in lung tissues were clearly increased (P<0.05 for both) (Figure 10); however, they did not change in monocrotaline‐treated rats when SO2 was supplied (P>0.05) (Figure 10).
Figure 10.
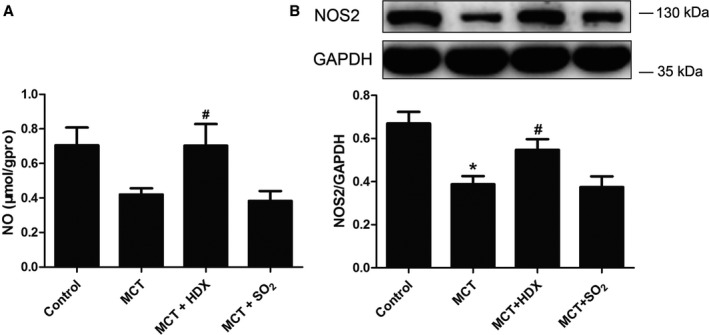
Changes in the NO/NOS2 system in lung tissues of rats. A, NO concentration in lung tissues of rats. B, Protein expression of NOS2 in lung tissues of rats, detected by Western blotting. Results are expressed as mean±SE, n=8. *P<0.05 compared with the control group; # P<0.05 compared with the MCT group. HDX indicates l‐aspartate‐β‐hydroxamate; MCT, monocrotaline; NO, nitric oxide; NOS2, nitric oxide synthase 2; SO2, sulfur dioxide; μmol/gpro, the content of NO (μmol) in 1 g protein.
Discussion
Pulmonary vascular structural remodeling is characterized by structural and functional changes in pulmonary vasculature, such as the proliferation of PASMCs and excess collagen synthesis in the lungs.21, 22, 23, 24 The mechanisms responsible for the development of monocrotaline‐induced pulmonary vascular remodeling remain unclear. Our previous studies and many other investigations indicated that endogenous gaseous messenger molecules NO, CO, and H2S played important pathophysiological roles in PH.4, 5, 6, 25 Nevertheless, the effects of gaseous messenger molecules do not completely explain the mechanisms of pulmonary vascular remodeling.25, 26 SO2 is a well‐known common air pollutant, and its toxic effects have been studied extensively over the past few decades. Less recognized, however, is the fact that SO2 is also a biological gas that is endogenously generated by the metabolism of sulfur‐containing amino acids via a series of reactions catalyzed by AAT and other enzymes. SO2 exerts important effects on the cardiovascular system, such as vasorelaxant and negative inotropic effects.8, 27, 28 Our previous studies found that SO2 could alleviate abnormal collagen accumulation in small pulmonary arteries of hypoxic pulmonary hypertensive rats and in a mechanical stretch‐stimulated cell model.29, 30 In our previous research,11 the pulmonary vascular structural remodeling induced by monocrotaline was confirmed by an increase in the relative medial thickness and area of pulmonary arteries. With inhibition of endogenous SO2 production by HDX, pulmonary arterial remodeling became worse in the monocrotaline plus HDX group than in the monocrotaline group, indicating that inhibition of endogenous SO2 could aggravate pulmonary vascular remodeling in monocrotaline‐treated rats. We found in this study that the endogenous SO2/AAT pathway was upregulated in this pathological change of monocrotaline‐induced PH, with an increase in AAT1 protein expression in the lung tissues. The results indicated that upregulation of the endogenous SO2/AAT pathway might be involved in the mechanisms for pulmonary vascular structural remodeling and play a protective role in the development of monocrotaline‐induced pulmonary vascular remodeling.
Consequently, we designed the following studies to clarify the underlying mechanisms. Pulmonary vascular structural remodeling is a key pathological basis of the development of PH.31, 32 Previous studies reported that SO2 could significantly inhibit vascular smooth muscle cell proliferation and transformation from the contractile to the synthetic phenotype in hypoxic PH.24, 33 Considering that SO2 might be a potential factor in reversing vascular remodeling, we evaluated the metabolism of collagen, the main component of vascular ECM. The concentrations of hydroxyproline (representing total collagen content) and collagen types I and III in lung tissues and pulmonary arteries increased markedly in monocrotaline‐treated rats. After endogenous SO2 was inhibited by HDX, aberrant accumulation of both collagen types I and III was noticeably aggravated, which suggested that inhibition of endogenous SO2 could exacerbate collagen accumulation in pulmonary arteries of monocrotaline‐treated rats. When the endogenous SO2 was supplied with Na2SO3/NaHSO3, the concentrations of collagen types I and III in rat lung tissues and pulmonary arteries were evidently lessened, suggesting that augmented endogenous SO2 content could alleviate accumulation of collagen in monocrotaline‐treated rats. These results indicated that SO2 regulates monocrotaline‐induced pulmonary vascular remodeling, likely by downregulating the content of collagens, especially collagen types I and III.
The stability of vascular collagen depends on the homeostasis of its synthesis and degradation. Procollagen mRNA level is an important element of collagen synthesis. In the present study, we found that procollagen types I and III mRNA levels in lung tissues presented similar alterations in the protein expression of collagen types I and III in those groups. These results implied that SO2 could affect the transcription of procollagen types I and III mRNA to attenuate synthesis of collagen types I and III. Collagen degradation is an active process that depends on the action of proteases, including MMPs. MMP activity is modulated by counterregulatory TIMPs.34 TIMP‐1 is synthesized by most types of connective tissue cells and inhibits collagenases, stromelysin, and gelatinases.35 MMPs associate with TIMPs and form high‐affinity noncovalent 1:1 complexes. Disruption of the balance of TIMPs and MMPs would result in abnormal accumulation of ECM collagen. As a member of the STAT protein family, STAT3 was activated in endothelial cells and played an important role in the proliferative pulmonary vascular lesions in idiopathic arterial PH.36 During chronic liver injury, STAT3 was partially in control of TIMP‐1 production in hepatocytes.37 In this study, the balance of MMP‐13 and TIMP‐1 in lung tissue was disturbed by monocrotaline injections, and the imbalance was aggravated by reducing SO2 content using HDX. In contrast, after SO2 treatment, MMP‐13, TIMP‐1, and the MMP‐13/TIMP‐1 ratio were all increased. Changes in MMP‐13 and TIMP‐1 in lung tissues were related to the changes in STAT3. Such findings demonstrate that SO2 could reduce abnormal ECM collagen accumulation in monocrotaline‐induced pulmonary vascular remodeling, likely through the inhibition of collagen synthesis and the enhancement of its degradation to reconstruct its metabolic balance.
Among the molecules to regulate matrix deposition, TGF‐β1 is probably the best studied. TGF‐β1 belongs to the TGF‐β family of multifunctional molecules that play great roles in a variety of physiological activities including growth, differentiation, immunosuppression, proliferation, inflammation, tissue remodeling, and wound healing.38, 39, 40 Disruption or overexpression of various members of this family would result in disordered vascular and lung development and aberrant expression of other molecules that are important for pulmonary vascular development.41, 42 Previous studies demonstrated that overexpression of TGF‐β1 could stimulate neointimal hyperplasia and fibrosis,43 increase the production of collagen and other ECM components, and inhibit degradation of ECM components.44, 45, 46 More important, overexpression of TGF‐β1 in humans was reportedly linked to the development of hypertension.47 BMPR‐2 was closely related to pulmonary arterial hypertension48; therefore, to examine possible mechanisms by which SO2 regulated pulmonary vascular collagen remodeling, we studied the possible impact of SO2 on TGF‐β1 and BMPR‐2. In our study, we found that SO2 did not have an impact on BMPR‐2 protein expression, but the elevated levels of TGF‐β1 and TGF‐β1 mRNA induced by monocrotaline administration were both reduced by SO2 supplement. The latter finding gives strong support to our view that SO2 could probably affect the transcription of lung tissue TGF‐β1 expression to repress its biological function and to regulate collagen remodeling in the pulmonary arteries of monocrotaline‐induced pulmonary vascular remodeling.
TGF‐β1 is a well‐known cytokine that is believed to trigger pulmonary medial smooth muscle cell growth and to induce fibroblast transformation into myofibroblasts, enhancing production of the ECM and especially collagens.2, 3, 49 The myofibroblasts could migrate into media and bring collagen deposition into pulmonary vascular walls.2, 3 The above results showed that TGF‐β1 was colocalized with vimentin, a marker of fibroblasts, in the monocrotaline‐induced pulmonary vascular remodeling model. Consequently, we next used TGF‐β1 to stimulate PAFs from adventitia to further explore this mechanism. We demonstrated that endogenously derived SO2 could inhibit expression of collagen types I and III in cultured PAFs induced by TGF‐β1, leading to our investigation of the influence of SO2 on the TGF‐β1 signal transduction pathway in PAFs. In human pulmonary arterial hypertension, the TGF‐β1 signaling pathway was activated.50 This pathway was initiated by TGF‐β1 binding of the type II TGF‐β receptor, which led to serine phosphorylation and activation of TβRI. The activated TβRI, in turn, promoted serine phosphorylation of Smad2 and Smad3, their association with Smad4, translocation to the nucleus, and transcription of profibrotic genes.51, 52, 53 For experimental or hypoxic PH, the Smad2/3 signaling pathway is very important for the onset and development of PH, especially for the homeostasis of pulmonary vasculature.52, 54 In our present study, the results showed that increasing the endogenous SO2 level by AAT1 overexpression or by supplementation with an SO2 donor could inhibit serine phosphorylation of TβRI, leading to the repression of Smad2/3 activation and to a decrease in collagen deposition in PAFs with TGF‐β1 stimulation, which was in accordance to the biological function of TGF‐β1 signal.55 In contrast, decreasing endogenous SO2 by AAT1 knockdown significantly aggravated the phosphorylation of Smad2/3 and collagen remodeling in PAFs with TGF‐β1 treatment, suggesting that upregulation of the endogenous SO2/AAT1 pathway might play a protective role in maintaining pulmonary vascular collagen homeostasis. The data presented in the present study indicated that endogenous SO2 inhibited abnormal ECM production of PAFs, which might at least be mediated by inactivation of the TGF‐β/TβRI/Smad2/3 pathway.
Intercellular communication is an important way to regulate cell function. Our coculture studies showed that the inhibition of SO2 in PASMCs activated collagen accumulation in PAFs, suggesting that communications between PASMCs and PAFs could regulate the function of PAFs, such as collagen accumulation.
NO is an important gaseous signaling molecule in the cardiovascular system that is widely involved in the regulation of vascular function. Our study found that the NO/NOS2 pathway was significantly reduced in the lung tissue of rats after monocrotaline intervention. In monocrotaline‐treated rats with inhibition of SO2 generation using HDX, the NO/NOS2 pathway was significantly increased. These results suggested that SO2 could inhibit the NO/NOS2 pathway in the lung tissue of rats.
The proliferation and apoptosis of PASMCs is also an important pathological process of pulmonary vascular structural remodeling.56, 57, 58 In our study, we detected cell proliferation, mitochondrial function, apoptosis, and intracellular calcium levels of PASMCs and PAFs. Results showed that SO2 decreased proliferation and increased mitochondrial dysfunction of PASMCs and PAFs and indicated that SO2 was involved in proliferation and apoptosis of PASMCs.
In conclusion, this study demonstrated that SO2 could significantly alleviate excessive pulmonary artery collagen accumulation induced by monocrotaline. The mechanisms underlying this inhibitory effect on vascular collagen deposition might include a role for endogenous SO2 in regulating expression of TGF‐β1 and inhibiting its activation of TGF‐β/TβRI/Smad2/3 signaling cascades in PAFs. Endogenous SO2 repressed PAFs from secreting collagen and from triggering alteration of ECM composition in media and led to reverse of pulmonary vascular remodeling.
Author Contribution
Conception and design of the work: Yu, Liu, Tang, Jin and Junbao Du. Acquisition of data: Yu, Liu, Tang, Liang, Shuxu Du, Ochs, Stella Chen, Selena Chen, Jin, Junbao Du and Huang. Analysis and interpretation of data: Yu, Liu, Huang, Ochs, Stella Chen, Selena Chen and Liang. Drafting the article or revising it critically for important intellectual content: Yu, Liu, Liang, Huang, Ochs, Stella Chen, Selena Chen, Tang, Jin and Junbao Du. Final approval of the version: Jin and Junbao Du.
Sources of Funding
This work was supported by National Natural Science Foundation of China (31130030 and 81121061) and the Major Basic Research Program of China (2012CB517806 and 2011CB503904).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003910 doi: 10.1161/JAHA.116.003910)
Contributor Information
Yaqian Huang, Email: yaqianhuang@126.com.
Junbao Du, Email: junbaodu1@126.com.
Hongfang Jin, Email: jinhongfang51@126.com.
References
- 1. Levine DJ. Diagnosis and management of pulmonary arterial hypertension: implications for care. Respir Care. 2006;51:368–381. [PubMed] [Google Scholar]
- 2. Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda). 2006;21:134–145. [DOI] [PubMed] [Google Scholar]
- 4. Kan P, Zhang CY, Fan J, Tang CS, Du JB. Inhalation of nebulized nitroglycerin, a nitric oxide donor, for the treatment of pulmonary hypertension induced by high pulmonary blood flow. Heart Vessels. 2006;21:169–179. [DOI] [PubMed] [Google Scholar]
- 5. Gong LM, Du JB, Shi L, Shi Y, Tang CS. Effects of endogenous carbon monoxide on collagen synthesis in pulmonary artery in rats under hypoxia. Life Sci. 2004;74:1225–1241. [DOI] [PubMed] [Google Scholar]
- 6. Zhang CY, Du JB, Bu DF, Yan H, Tang XY, Tang CS. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun. 2003;302:810–816. [DOI] [PubMed] [Google Scholar]
- 7. Balazy M, Abu‐Yousef IA, Harpp DN, Park J. Identification of carbonyl sulfide and sulfur dioxide in porcine coronary artery by gas chromatography/mass spectrometry, possible relevance to EDHF. Biochem Biophys Res Commun. 2003;311:728–734. [DOI] [PubMed] [Google Scholar]
- 8. Du SX, Jin HF, Bu DF, Zhao X, Geng B, Tang CS, Du JB. Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol Sin. 2008;29:923–930. [DOI] [PubMed] [Google Scholar]
- 9. Zhao X, Jin HF, Du SX, Tang CS, Du JB. Effect of sulfur dioxide on blood pressure and pulmonary vascular structure in spontaneous hypertensive rat. Chin Pharm Bull. 2008;24:327–330. [Google Scholar]
- 10. Liu D, Huang Y, Bu D, Liu AD, Holmberg L, Jia Y, Tang C, Du J, Jin H. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death Dis. 2014;5:e1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin HF, Du SX, Zhao X, Wei HL, Wang YF, Liang YF, Tang CS, Du JB. Effects of endogenous sulfur dioxide on monocrotaline‐induced pulmonary hypertension in rats. Acta Pharmacol Sin. 2008;29:1157–1166. [DOI] [PubMed] [Google Scholar]
- 12. Lee YS, Byun J, Kim JA, Lee JS, Kim KL, Suh YL, Kim JM, Jang HS, Lee JY, Shin IS, Suh W, Jeon ES, Kim DK. Monocrotaline‐induced pulmonary hypertension correlates with upregulation of connective tissue growth factor expression in the lung. Exp Mol Med. 2005;37:27–35. [DOI] [PubMed] [Google Scholar]
- 13. Kuttan R, Di Ferrante N. Sirius‐red‐collagen interaction: a method for the measurement of collagen and bacterial collagenase activity. Biochem Int. 1980;1:455–460. [Google Scholar]
- 14. Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem. 1996;29:225–229. [DOI] [PubMed] [Google Scholar]
- 15. Wei H, Zhang R, Jin H, Liu D, Tang X, Tang C, Du J. Hydrogen sulfide attenuates hyperhomocysteinemia‐induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid Redox Signal. 2010;12:1079–1091. [DOI] [PubMed] [Google Scholar]
- 16. Zmijewski JW, Zhao X, Xu Z, Abraham E. Exposure to hydrogen peroxide diminishes NF‐kappaB activation, IkappaB‐alpha degradation, and proteasome activity in neutrophils. Am J Physiol Cell Physiol. 2007;293:C255–C266. [DOI] [PubMed] [Google Scholar]
- 17. Lorne E, Zmijewski JW, Zhao X, Liu G, Tsuruta Y, Park YJ, Dupont H, Abraham E. Role of extracellular superoxide in neutrophil activation: interactions between xanthine oxidase and TLR4 induce proinflammatory cytokine production. Am J Physiol Cell Physiol. 2008;294:C985–C993. [DOI] [PubMed] [Google Scholar]
- 18. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 19. Chen X, Wang H, Liao HJ, Hu W, Gewin L, Mernaugh G, Zhang S, Zhang ZY, Vega‐Montoto L, Vanacore RM, Fässler R, Zent R, Pozzi A. Integrin‐mediated type II TGF‐β receptor tyrosine dephosphorylation controls SMAD‐dependent profibrotic signaling. J Clin Invest. 2014;124:3295–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zong Y, Huang Y, Chen S, Zhu M, Chen Q, Feng S, Sun Y, Zhang Q, Tang C, Du J, Jin H. Downregulation of endogenous hydrogen sulfide pathway is involved in mitochondrion‐related end othelial cell apoptosis induced by high salt. Oxid Med Cell Longev. 2015;2015:754670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Botney MD, Liptay MJ, Kaiser LR, Cooper JD, Parks WC, Mecham RP. Active collagen synthesis by pulmonary arteries in human primary pulmonary hypertension. Am J Pathol. 1993;143:121–129. [PMC free article] [PubMed] [Google Scholar]
- 22. Yi ES, Kim H, Ahn H, Strother J, Morris T, Masliah E, Hansen LA, Park K, Friedman PJ. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension: a morphometric and immunohistochemical study. Am J Respir Crit Care Med. 2000;162:1577–1586. [DOI] [PubMed] [Google Scholar]
- 23. Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol. 1988;255:1484–1491. [DOI] [PubMed] [Google Scholar]
- 24. Werchan PM, Summer WR, Gerdes AM, McDonough KH. Right ventricular performance after monocrotaline‐induced pulmonary hypertension. Am J Physiol. 1989;256:H1328–H1336. [DOI] [PubMed] [Google Scholar]
- 25. Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57:585–630. [DOI] [PubMed] [Google Scholar]
- 26. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. [DOI] [PubMed] [Google Scholar]
- 27. Stipanuk MH. Metabolism of sulfur containing amino acids. Annu Rev Nutr. 1986;6:179–209. [DOI] [PubMed] [Google Scholar]
- 28. Wang XB, Jin HF, Tang CS, Du JB. The biological effect of endogenous sulfur dioxide in the cardiovascular system. Eur J Pharmacol. 2011;670:1–6. [DOI] [PubMed] [Google Scholar]
- 29. Tian Y, Tang XY, Jin HF, Tang CS, Du JB. Effect of sulfur dioxide on pulmonary vascular structure of hypoxic pulmonary hypertensive rats. Zhonghua Er Ke Za Zhi. 2008;46:675–679. [PubMed] [Google Scholar]
- 30. Liu J, Yu W, Liu Y, Chen S, Huang Y, Li X, Liu C, Zhang Y, Li Z, Du J, Tang C, Du J, Jin H. Mechanical stretching stimulates collagen synthesis via down‐regulating SO2/AAT1 pathway. Sci Rep. 2016;6:21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation: an ultrastructural study. Lab Invest. 1978;38:188–200. [PubMed] [Google Scholar]
- 32. Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol. 1979;236:H818–H827. [DOI] [PubMed] [Google Scholar]
- 33. Prabha M, Jin HF, Tian Y, Tang CS, Du JB. Mechanisms responsible for pulmonary hypertension. Chin Med J (Engl). 2008;121:2604–2609. [PubMed] [Google Scholar]
- 34. Willenbrock F, Murphy G. Structure‐function relationships in the tissue inhibitors of metalloproteinases. Am J Respir Crit Care Med. 1994;150:S165–S170. [DOI] [PubMed] [Google Scholar]
- 35. Dollery CM, McEwan JR, Henney AM. Matrix metalloproteinases and cardiovascular disease. Circ Res. 1995;77:863–868. [DOI] [PubMed] [Google Scholar]
- 36. Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A. Hyperproliferative apoptosis‐resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. [DOI] [PubMed] [Google Scholar]
- 37. Wang H, Lafdil F, Wang L, Yin S, Feng D, Gao B. Tissue inhibitor of metalloproteinase 1 (TIMP‐1) deficiency exacerbates carbon tetrachloride‐induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP‐1 production. Cell Biosci. 2011;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grande JP. Role of transforming growth factor‐beta in tissue injury and repair. Proc Soc Exp Biol Med. 1997;214:27–40. [DOI] [PubMed] [Google Scholar]
- 39. Heldin CH, Miyazono K, ten Dijke P. TGF‐beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. [DOI] [PubMed] [Google Scholar]
- 40. de Caestecker M. The transforming growth factor‐beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. [DOI] [PubMed] [Google Scholar]
- 41. Pauling MH, Vu TH. Mechanisms and regulation of lung vascular development. Curr Top Dev Biol. 2004;64:73–99. [DOI] [PubMed] [Google Scholar]
- 42. Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor‐beta superfamily. Endocr Rev. 2002;23:787–823. [DOI] [PubMed] [Google Scholar]
- 43. Nabel EG, Shum L, Pompili VJ, Yang ZY, San H, Shu HB, Liptay S, Gold L, Gordon D, Derynck R. Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc Natl Acad Sci USA. 1993;90:10759–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, Gauldie J. Transfer of tumor necrosis factor‐alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor‐beta1 and myofibroblasts. Am J Pathol. 1998;153:825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, Senior RM, Elias JA. Interleukin‐13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta1. J Exp Med. 2001;194:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruiz‐Ortega M, Rodríguez‐Vita J, Sanchez‐Lopez E, Carvajal G, Egido J. TGF‐beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. [DOI] [PubMed] [Google Scholar]
- 47. Li B, Khanna A, Sharma V, Singh T, Suthanthiran M, August P. TGF beta1 DNA polymorphisms, protein levels and blood pressure. Hypertension. 1999;33:271–275. [DOI] [PubMed] [Google Scholar]
- 48. Wang J, Zhang C, Liu C, Wang W, Zhang N, Hadadi C, Huang J, Zhong N, Lu W. Functional mutations in 5′UTR of the BMPR‐2 gene identified in Chinese families with pulmonary arterial hypertension. Pulm Circ. 2016;6:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang YL, Dai AG, Li QF, Hu RC. Transforming growth factor‐beta1 induces transdifferentiation of fibroblasts into myofibroblasts in hypoxic pulmonary vascular remodeling. Acta Biochim Biophys Sin (Shanghai). 2006;38:29–36. [DOI] [PubMed] [Google Scholar]
- 50. Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth factor‐beta signaling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340–1348. [DOI] [PubMed] [Google Scholar]
- 51. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature. 2003;425:577–584. [DOI] [PubMed] [Google Scholar]
- 52. Zakrzewicz A, Kouri FM, Nejman B, Kwapiszewska G, Hecker M, Sandu R, Dony E, Seeger W, Schermuly RT, Eickelberg O, Morty RE. The transforming growth factor‐beta/Smad2/3 signalling axis is impaired in experimental pulmonary hypertension. Eur Respir J. 2007;29:1094–1104. [DOI] [PubMed] [Google Scholar]
- 53. Wang YW, Liou NH, Cherng JH, Chang SJ, Ma KH, Fu E, Liu JC, Dai NT. siRNA‐targeting transforming growth factor‐β type I receptor reduces wound scarring and extracellular matrix deposition of scar tissue. J Invest Dermatol. 2014;134:2016–2025. [DOI] [PubMed] [Google Scholar]
- 54. Graham BB, Chabon J, Gebreab L, Poole J, Debella E, Davis L, Tanaka T, Sanders L, Dropcho N, Bandeira A, Vandivier RW, Champion HC, Butrous G, Wang XJ, Wynn TA, Tuder RM. Transforming growth factor‐β signaling promotes pulmonary hypertension caused by Schistosoma mansoni . Circulation. 2013;128:1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hough C, Radu M, Doré JJ. Tgf‐beta induced erk phosphorylation of smad linker region regulates smad signaling. PLoS One. 2012;7:e42513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting surviving selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2015;115:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils‐Bonnet S, Tremblay E, Couture C, Michelakis ED, Provencher S, Bonnet S. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129:786–797. [DOI] [PubMed] [Google Scholar]
- 58. Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils‐Bonnet S, Tremblay È, Nadeau V, Lambert C, Paradis R, Provencher S, Bonnet S. Bromodomain‐containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circ Res. 2015;117:525–535. [DOI] [PubMed] [Google Scholar]