

Abstract
Background
Experimental uremic cardiomyopathy causes cardiac fibrosis and is causally related to the increased circulating levels of the cardiotonic steroid, marinobufagenin (MBG), which signals through Na/K‐ATPase. Rapamycin is an inhibitor of the serine/threonine kinase mammalian target of rapamycin (mTOR) implicated in the progression of many different forms of renal disease. Given that Na/K‐ATPase signaling is known to stimulate the mTOR system, we speculated that the ameliorative effects of rapamycin might influence this pathway.
Methods and Results
Biosynthesis of MBG by cultured human JEG‐3 cells is initiated by CYP27A1, which is also a target for rapamycin. It was demonstrated that 1 μmol/L of rapamycin inhibited production of MBG in human JEG‐2 cells. Male Sprague‐Dawley rats were subjected to either partial nephrectomy (PNx), infusion of MBG, and/or infusion of rapamycin through osmotic minipumps. PNx animals showed marked increase in plasma MBG levels (1025±60 vs 377±53 pmol/L; P<0.01), systolic blood pressure (169±1 vs 111±1 mm Hg; P<0.01), and cardiac fibrosis compared to controls. Plasma MBG levels were significantly decreased in PNx‐rapamycin animals compared to PNx (373±46 vs 1025±60 pmol/L; P<0.01), and cardiac fibrosis was substantially attenuated by rapamycin treatment.
Conclusions
Rapamycin treatment in combination with MBG infusion significantly attenuated cardiac fibrosis. Our results suggest that rapamycin may have a dual effect on cardiac fibrosis through (1) mTOR inhibition and (2) inhibiting MBG‐mediated profibrotic signaling and provide support for beneficial effect of a novel therapy for uremic cardiomyopathy.
Keywords: cardiac fibrosis, cardiomyopathy, cardiovascular diseases, fibrosis, heart failure
Subject Categories: Basic Science Research, Animal Models of Human Disease, Fibrosis, Cardiomyopathy
Introduction
The high mortality rate in patients with chronic renal failure is ultimately attributed to severe cardiovascular disease.1 This uremic cardiomyopathy is characterized by cardiac hypertrophy, diastolic dysfunction, and cardiac fibrosis along with elevated circulating concentrations of the cardiotonic steroid marinobufagenin (MBG), a ligand of Na/K‐ATPase. MBG belongs to a family of bufadienolides previously described in amphibians.2 We have shown that MBG is elevated in patients with renal failure3 and in rats subjected to partial nephrectomy (PNx), and those with pharmacological administration of MBG developed a similar cardiomyopathy as observed in patients, whereas active immunization against MBG attenuated this in PNx.4, 5 In PNx rats, we also observed increased cardiac and plasma levels of carbonylated proteins as well as evidence for signaling through Na/K‐ATPase, such as activation of Src and mitogen‐activated protein kinase.4, 5 A recent report from our group has shown that treatment with a monoclonal antibody directed against MBG drastically reduced cardiac fibrosis in PNx animals.6
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase composed of 2 signaling complexes, mTORC1 and mTORC2.7 The mTORC1 complex is involved in cellular proliferation and growth, whereas mTORC2 is involved in the regulation of the cytoskeleton.8 The mTOR pathway has been implicated in the progression of many different forms of renal disease, including experimentally induced uremic cardiomyopathy and development of renal fibrosis.9, 10, 11 MBG has been shown to activate the mTOR pathway in cardiac myocytes.12 Treatment with rapamycin (an mTORC1 inhibitor) has been shown to attenuate inflammation, fibrosis, and cardiac hypertrophy in experimental models of renal disease.9, 10 Rapamycin is also a competitive inhibitor of cytochrome P450, family 27, subfamily a, polypeptide 1 (CYP27A1), a key rate‐limiting enzyme of the bile acid pathway.13 A recent report by Fedorova et al provides compelling evidence that the alternative bile acid pathway, initiated by CYP27A1 from cholesterol, is responsible for the production of MBG in mammals, specifically in rat adrenocortical cells and tissue and in human placental chorionic epithelial cells (JEG‐3).14
Based on this background, the primary goals of the present study were to (1) examine the effects of rapamycin on MBG production in vitro on a cell line known to synthesize MBG (ie, JEG‐3 placental cells15) and (2) determine the effects of rapamycin on cardiac fibrosis and MBG production in vivo using the rat PNx model of uremic cardiomyopathy. In addition, given that we have shown that MBG induces increases in cardiac procollagen‐1 and reactive oxygen species (ROS),4, 5 we sought to determine the effect of rapamycin on procollagen‐1 and ROS production in cardiac fibroblasts.
Methods
Animal Studies
All animal experimentation described in this article was conducted in accord with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals under protocols approved by the University of Toledo Institutional Animal Care and Use Committee. Male Sprague‐Dawley rats (250–300 g) were used for these studies. Rats were divided into 6 groups. In the first group, PNx (n=10) was performed as we have previously described.5 In the second group, PNx was performed and rapamycin (Rapa) was administered (0.2 mg/kg per day; n=6).16 The third group received both rapamycin (0.2 mg/kg per day) and MBG (10 μg/kg per day; n=8). The fourth and fifth groups were administered MBG alone (10 μg/kg per day; n=8) and rapamycin alone (0.2 mg/kg per day; n=8). The dose of MBG is the same as we have previously reported.4, 5 The sixth group consisted of sham operated controls (n=8). All treatments were performed for 4 weeks with the use of osmotic minipumps (model 2004, ALZET; DURECT Corporation Cupertino, CA). Minipumps were inserted subcutaneously through a flank incision. Two minipumps were implanted for coadministration of rapamycin and MBG.
Blood Pressure, Cardiac Physiology, and Other In Vivo Measurements
Blood pressure (BP) was determined using the tail‐cuff method by IITC (Amplifier model 229, Monitor model 31, Test chamber Model 306; IITC Life Science, Woodland Hills, CA) at baseline, and once‐weekly for 4 weeks. Then, rats were euthanized and the heart weight and cardiac histology were determined. Plasma samples were stored at −80°C for biochemical analysis. Plasma MBG and creatinine were measured as we have previously described.6, 17
Isolation of Cardiac Fibroblasts and JEG‐3 Cell Experiments
Isolation of cardiac fibroblasts was carried out as previously reported by Brilla et al, with modifications.4, 18, 19, 20 Cardiac fibroblasts were grown to confluence in DMEM with 15% FBS and starved for 18 to 24 hours in medium containing 1% FBS before treatment. Cells were treated with rapamycin at a concentration of 0.1 and 0.01 μmol/L for 24 hours in media containing 1% FBS.21 Cells were also treated with either MBG alone at concentrations shown to induce procollagen production (1 and 100 nmol/L)19 or in combination with rapamycin. Cell lysates were prepared by washing the cells twice with ice‐cold PBS and incubating them on ice for 20 minutes with lysis buffer containing 10 mmol/L of Tris‐HCL (pH 7.5), 0.5% Nonidet P‐40, and protease and phosphatase inhibitors. Cell lysates were collected, vortexed for 30 seconds, and used immediately or stored at −80°C. Although we did not directly address the effects of cell proliferation on cardiac fibroblasts, in our experiments the cardiac fibroblasts were grown to confluence before serum starvation and treatment, and we did not visually note any significant morphological differences between rapamycin‐treated and untreated cells that might indicate any effects on proliferation. Furthermore, Poulalhon et al performed similar experiments in lung fibroblasts and did not note changes in proliferation at similar concentrations used.21
Human placental chorionic epithelial cells (JEG‐3) were purchased from a commercially available vendor (ATCC, Manassas, VA), cultured in 6‐well plates, and grown to confluence 80% to 85% in Eagle's minimum essential media (ATCC). Then, the 10% FBS media was replaced by 2.5% FBS media and rapamycin (1 μmol/L) was added into half the wells. Both vehicle‐treated control cells and rapamycin‐treated cells were sampled after 3 and 6 hours of incubation. Cultured media was collected for MBG measurements, and cells were collected for total protein measurement.14 MBG was extracted from the collected media using C18 columns (Waters, Milford, MA) as previously reported.17 Competitive immunoassays were performed using a monoclonal anti‐MBG antibody to determine the concentration of MBG in the samples also as previously reported.17
Protein Carbonylation
Whole‐cell lysates were prepared as aforementioned. Fifteen microgram of proteins from each sample were denatured with 6% SDS (final concentration), derivatized with 1× DNPH, and neutralized with neutralization buffer (30% of glycerol in 2 mol/L of Tris). Protein carbonylation levels were monitored by western blot with anti‐DNP antibody (Sigma D9656; Sigma‐Aldrich, St. Louis, MO). Ponceau S (Sigma P7170; Sigma‐Aldrich) staining of the membrane was used to verify uniform loading before immunoblotting with indicated antibodies.
Western Blot Analysis
Western blot analysis was performed on proteins from tissue homogenates and cell lysates as previously reported.4, 6, 18, 19 Goat anti‐type 1 collagen antibody (Southern Biotech, Birmingham, AL) was used to probe for collagen‐1, and secondary anti‐goat antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Rabbit anti‐phospho‐S6 antibody and total S‐6 antibody were purchased from Cell Signaling Technology (Danvers, MA). For detection, we used ECL and ECL plus purchased from Amersham Biosciences (Foster City, CA). Loading conditions were controlled using anti‐actin goat polyclonal antibody (Santa Cruz Biotechnology).
Histology
Left ventricle sections were immediately fixed in 4% formalin buffer solution (pH 7.2) for 18 hours, dehydrated in 70% ethanol, and then embedded in paraffin and cut with a microtome. Fast green staining with Sirius red (0.1%) was performed on left ventricular tissues as described previously.22 For quantitative morphometric analysis, 10 random images of Sirius red/Fast green stained slides were taken at ×20 magnification and electronically scanned into an RGB image which was analyzed using ImageJ software (ImageJ 1.36b; NIH, Bethesda, MD). The amount of fibrosis was then estimated with a macro by converting pixels of the image with substantially greater (>120%) red than green intensity to have the new grayscale amplitude=1, leaving other pixels as with amplitude=0. Representative photomicrographs were selected from the 10 random images taken from each group based on the quantified analysis. For confirmation of the histological findings, quantitative determination of collagen‐1 in left ventricular homogenates was performed using western blot.
Statistical Analysis
The results are presented as means±SEM. Data were analyzed using one‐way ANOVA and repeated‐measures ANOVA. If there was significant group difference shown by ANOVA, then post‐hoc confidence intervals for pair‐wise differences were conducted using the Tukey‐Kramer procedure to control for the family‐wise error rate when making all pair‐wise difference in mean comparisons. Other multiple comparisons were constructed based on the simultaneous inference procedures proposed by Hothorn et al.23 All analyses were performed in SAS (version 9.4; SAS Institute Inc., Cary, NC), R (version 3.0.; R Foundation for Statistical Computing, Vienna, Austria), and GraphPad Prism software (GraphPad Software Inc., San Diego, CA). A 2‐sided P value of less than 0.05 was considered to be statistically significant.
Results
Effect of Rapamycin on MBG Production by JEG‐3 Cells
The chemical structure of MBG is shown in Figure 1A. Cultured human placental JEG‐3 cells were treated with 1 μmol/L of rapamycin to test the effects on MBG production. As presented in Figure 1B, rapamycin treatment significantly reduced MBG production compared to vehicle treated controls after 3 and 6 hours of treatment (84 vs 60 pmol/g of protein; P<0.01; and 243 vs 116 pmol/g of protein; P<0.01, correspondingly).
Figure 1.
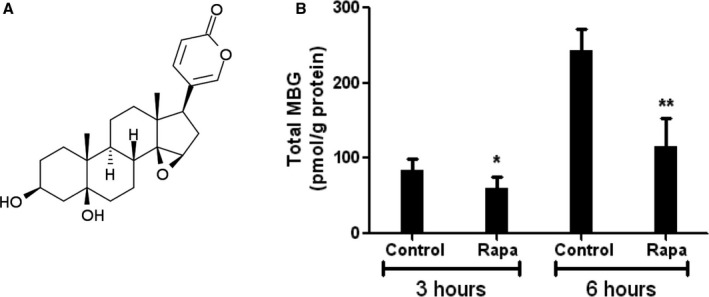
A, Chemical structure of marinobufagenin (MBG), 3β,5β‐dihydroxy‐14,15‐epoxy bufadienolide, PubChem CID: 11969465. B, MBG production in human placental chorionic epithelial cells (JEG‐3 cells) after incubation with 1 μmol/L of rapamycin (Rapa) for 3 and 6 hours. *P<0.05 versus control; **P<0.01 versus control.
Effect of Rapamycin on BP, MBG Levels
Rapamycin treatment alone demonstrated a slight elevation in systolic BP (123±3 vs 111±1 mm Hg; P=NS [not significant]), but did not significantly alter MBG levels compared to control animals (Table). PNx surgery substantially increased the heart weight/body weight ratio. PNx surgery and MBG infusion produced sustained hypertension throughout the duration of the experiment. PNx plasma MBG levels were similar to the MBG levels produced by MBG infusion alone (1025±60 vs 1092±57 pmol/L; P=NS). PNx surgery with rapamycin infusion showed a significant decrease in systolic BP by the third week (149±2 vs 166±1 mm Hg; P<0.01) compared to PNx alone, which persisted at 4 weeks when analyzed by t test (P<0.05), yet not by pair‐wise comparison. PNx surgery with rapamycin treatment also demonstrated a drastic decrease in MBG levels compared to PNx alone (373±46 vs 1025±60 pmol/L; P<0.01), although rapamycin treatment did not affect plasma MBG levels in normal animals. Coadministration of MBG with rapamycin did not attenuate systolic BP or plasma MBG levels. These data are summarized in Table.
Table 1.
Effects of Rapamycin (Rapa) on Physiological Measurements After PNx or Infusion of MBG
| Group | Sham (n=8) | Rapa (n=8) | PNx (n=10) | PNx+Rapa (n=6) | MBG (n=8) | MBG+Rapa (n=8) |
|---|---|---|---|---|---|---|
| Systolic BP, mm Hg | ||||||
| Baseline | 110±1 | 113±1 | 113±1 | 112±1 | 113±1 | 113±1 |
| Week 1 | 104±2 | 111±1 | 131±1a | 130±1 | 121±1a | 113±1c |
| Week 2 | 104±1 | 120±1 | 134±1a | 135±1 | 122±1a | 128±1c |
| Week 3 | 105±2 | 113±2 | 166±1a | 149±2b | 136±1a | 123±2c |
| Week 4 | 111±1 | 123±3 | 169±1a | 151±6b | 139±1a | 145±2 |
| Heart weight | ||||||
| BW, g | 529±0.01 | 533±0.02 | 425±0.02a | 368±0.04 | 493±0.01 | 528±0.01 |
| HW, g | 1.37±0.02 | 1.47±0.07 | 1.49±0.06 | 1.23±0.10b | 1.32±0.05 | 1.38±0.04 |
| HW/BW, ×103 | 2.60±0.06 | 2.77±0.08 | 3.57±0.20a | 3.42±0.24 | 2.73±0.08 | 2.63±0.05 |
| Plasma measurements | ||||||
| Creatinine, mg/dL | 0.39±0.06 | 0.48±0.03 | 0.55±0.04 | 0.57±0.09 | 0.31±0.03 | 0.51±0.11 |
| MBG, pmol/L | 377±53 | 439±42 | 1025±60a | 373±46b | 1092±57a | 1093±90 |
Data are presented as mean±SEM. For clarity purposes, statistical significance is designated for the following groups: sham versus PNx, sham versus MBG, PNx versus PNx+Rapa, and MBG versus MBG+Rapa. For complete pair‐wise differences, please refer to Figures S1 through S10. Sham refers to animals subject to sham surgery; PNx refers to partial nephrectomy; PNx+Rapa refers to PNx surgery and rapamycin infusion using minipumps; Rapa refers to rapamycin infusion using minipumps; MBG+Rapa refers to coadministration of MBG and rapamycin using minipumps; and MBG refers to MBG infusion using minipumps. BP indicates blood pressure; BW, body weight; HW, heart weight; MBG, marinobufagenin.
P<0.01 versus sham.
P<0.01 versus PNx.
P<0.01 versus MBG.
Effect of Rapamycin on Cardiac Fibrosis
Cardiac fibrosis was assessed in the left ventricular myocardium by histological analysis (Sirius red with Fast green staining) and collagen 1 expression determined by western blot. Both PNx and MBG infusion resulted in substantial increases in collagen expression and cardiac scarring, whereas PNx with rapamycin infusion and coadministration of rapamycin with MBG significantly attenuated these effects (Figure 2A and 2B).
Figure 2.
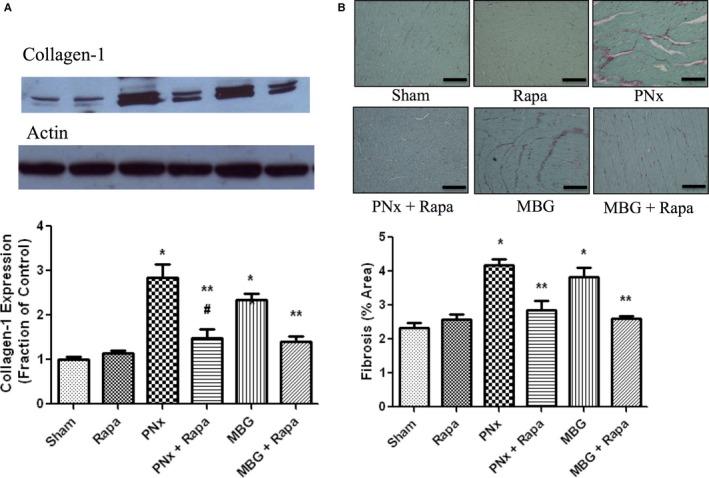
A, Representative (top) and quantitative analysis of collagen 1 (mean±SEM) western blots performed on cardiac tissue from the different groups. Actin was used as a loading control. B, Representative Sirius red and Fast green stained photomicrographs obtained from cardiac tissue derived from the different experimental groups. Scale bar=100 μmol/L. Amount of fibrosis expressed as mean±SEM measured using computer‐assisted morphometry, as we have previously described.5 Sham refers to animals subject to sham surgery (n=8); PNx refers to partial nephrectomy (n=10); PNx+Rapa refers to PNx surgery and rapamycin infusion using minipumps (n=6); Rapa refers to rapamycin infusion using minipumps (n=8); MBG+Rapa refers to coadministration of marinobufagenin (MBG) and rapamycin using minipumps (n=8); and MBG refers to MBG infusion using minipumps (n=8). *P<0.01 versus sham and Rapa; **P<0.01 versus PNx and MBG; # P<0.05 versus sham.
Effect of MBG and Rapamycin on Cardiac Fibroblast Phosphoribosomal S6, and Procollagen‐1 Protein Expression
Activation of the mTOR pathway results in phosphorylation of ribosomal S6, which is commonly used as an indicator of mTOR activation.9, 10, 12 Cultured cardiac fibroblasts treated with 1 and 100 nmol/L of MBG resulted in a significant increase in phosphorylation of ribosomal S6 protein expression determined by western blot compared to controls, indicating activation of the mTOR pathway (P<0.01; Figure 3). Treatment with rapamycin at 0.1 μmol/L alone and in combination with MBG at 1 and 100 nmol/L caused a significant reduction in phospho‐S6 expression compared to MBG treatment (P<0.01; Figure 3). Treatment with low‐dose rapamycin (0.01 μmol/L) in combination with MBG resulted in a less‐profound reduction in phospho‐S6 expression (Figure 3; P=NS). MBG treatment at 1 and 100 nmol/L resulted in a significant increase in procollagen‐1 protein expression compared to controls (both P<0.01; only 100 nmol/L data shown in Figure 4). Both the high‐ (0.1 μmol/L) and low‐dose (0.01 μmol/L) rapamycin treatments significantly attenuated MBG‐induced procollagen‐1 expression (P<0.01; Figure 4).
Figure 3.
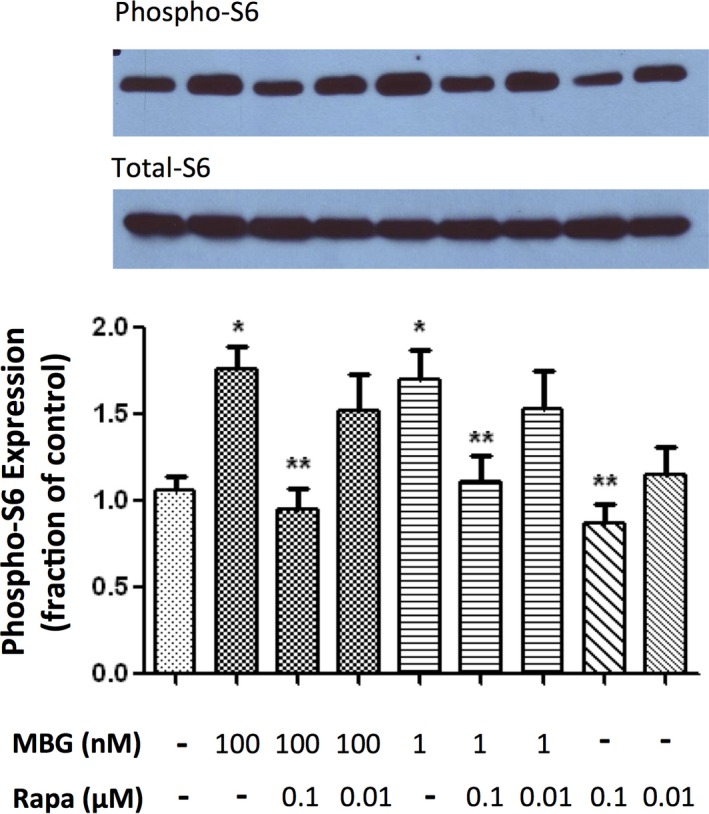
Representative (top) and quantitative analysis of phosphoribosomal S6 protein western blots derived from cardiac fibroblasts treated with marinobufagenin (MBG; 1 or 100 nmol/L), rapamycin (Rapa; 0.01 or 0.1 μmol/L), or in combination with the corresponding quantitative data shown as the mean±SEM of 5 experiments. Total ribosomal S6 protein was used as loading control. *P<0.01 versus control; **P<0.01 versus MBG 100 nmol/L, MBG 100 nmol/L+Rapa 0.01 μmol/L, MBG 1 nmol/L, and MBG 1 nmol/L+Rapa 0.01 μmol/L.
Figure 4.
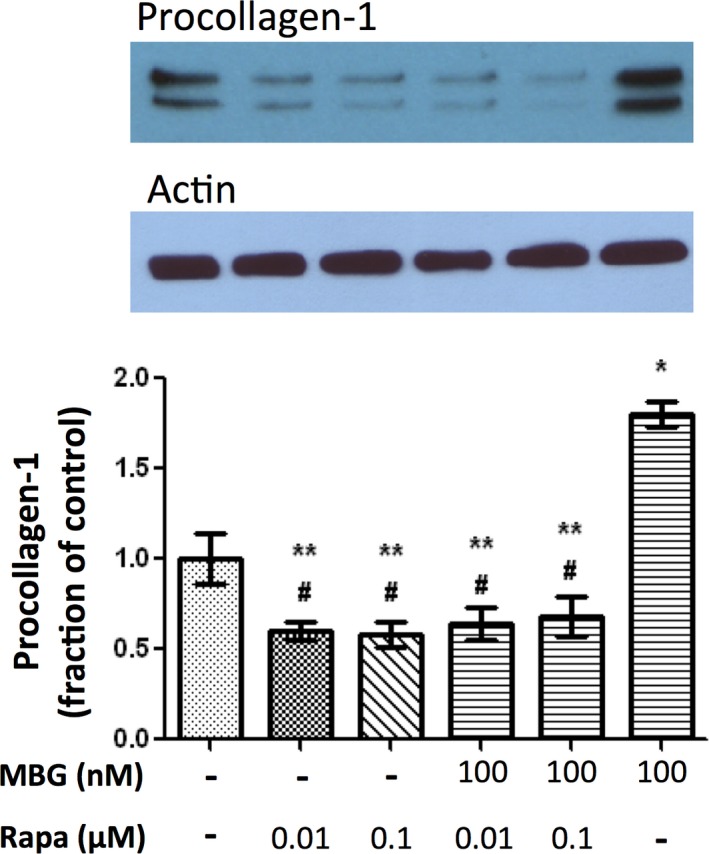
Representative (top) and quantitative analysis of procollagen‐1 western blots derived from cardiac fibroblasts treated with marinobufagenin (MBG) 100 nmol/L with or without rapamycin (Rapa; 0.01 or 0.1 μmol/L) with the corresponding quantitative data shown as the mean±SEM of 5 experiments. *P<0.01 versus control; # P<0.05 versus control; **P<0.01 versus MBG 100 nmol/L.
Effect of MBG and Rapamycin on Cardiac Fibroblast Oxidative Stress
MBG treatment (100 and 1 nmol/L) resulted in a significant increase in oxidative stress as measured by carbonylated protein expression compared to controls (P<0.01; Figure 5). Treatment with high‐dose rapamycin in combination with MBG at both 100 and 1 nmol/L significantly attenuated these effects (Figure 5). Treatment with low‐dose rapamycin significantly reduced oxidative stress in combination with MBG at 100 nmol/L compared to MBG 100 nmol/L alone (P<0.01, data not shown).
Figure 5.
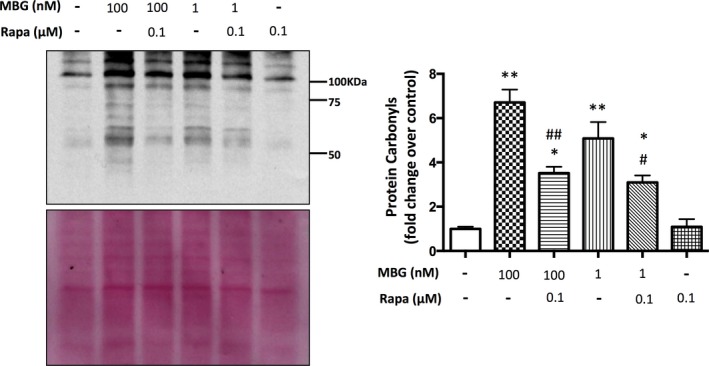
Representative and quantitative analysis of carbonylated protein western blots derived from cardiac fibroblasts treated with marinobufagenin (MBG; 100 or 1 nmol/L), rapamycin (Rapa; 0.1 μmol/L) or in combination with the corresponding quantitative data shown as the mean±SEM of 5 experiments. Ponceau S staining of the membrane was used to control for loading. A; *P=0.01 versus control; **P<0.01 versus control; ## P=0.01 versus MBG 100 nmol/L; # P<0.05 versus MBG 1‐nmol/L treatment.
Discussion
The mTOR pathway has been shown to play a pivotal role in several different forms of renal disease.9 Treatment with rapamycin attenuates many of the physiological changes associated with a decrease in renal function, including interstitial fibrosis.9 Our current work demonstrates that rapamycin treatment, in the setting of experimental uremic cardiomyopathy, significantly reduces cardiac fibrosis. This is in support of a recent report with similar findings in a murine model of uremic cardiomyopathy.10 In the previously mentioned report, Siedlecki et al used a PNx mouse model to demonstrate that administration of rapamycin (2 mg/kg per day for 4 weeks) 4 weeks post‐PNx was able to reverse cardiac hypertrophy and fibrosis.10 Importantly, MBG was not measured in their study.10 In the current study, we demonstrated similar reductions in cardiac hypertrophy and fibrosis by administration of rapamycin concurrent with the PNx procedure. Furthermore, we demonstrate that rapamycin attenuates MBG production both in vitro (Figure 1B) and in vivo (Table) and that rapamycin is capable of attenuating MBG induced increases in both collagen formation (Figure 4) and protein carbonylation (Figure 5) in cardiac fibroblasts.
Recent work in animal models of renal disease has provided compelling evidence for the involvement of mTORC1 in the generation of fibrosis. In an animal model of unilateral obstructive nephropathy, as well as in fibroblasts, the profibrotic cytokine transforming growth factor beta (TGF‐β) was shown to activate mTORC1 acting through a phosphoinositide 3‐kinase pathway.24 In human fibroblasts, the mTOR pathway has been shown to regulate collagen type I production.25 Treatment with rapamycin has been shown to decrease TGF‐β, fibroblast proliferation, and renal fibrosis.24, 26, 27 In support of our results, rapamycin treatment significantly decreased cardiac fibrosis, as evaluated by trichrome staining, in a murine model of uremic cadiomyopathy.10 Similar results were reported using a transverse aortic constriction model.28
We have shown that MBG causes many of the pathophysiological changes associated with experimental uremic cardiomyopathy, including cardiac fibrosis,5 and that MBG induces cardiac fibroblasts to produce collagen.4 The transcription factor, Friend leukemia integration‐1 (Fli‐1), acts as a negative regulator of collagen production,29, 30 and activation of protein kinase C‐delta (PKC‐δ) can phosphorylate Fli‐1 to promote collagen synthesis.31 We have recently reported that MBG induces translocation of PKC‐δ, which phosphorylates Fli‐1 and leads to an increase in collagen synthesis.18 Signaling through PKC‐δ has been shown to activate the mTOR pathway.32
Progression of uremic cardiomyopathy begins with concentric hypertrophy, which results in decreases in ventricular systolic dimension and increased ejection fraction in the heart. It is known that this concentric hypertrophy cannot persist permanently because hypertrophy of individual myocytes occurs to such an extent that they then do not oxygenate well and cell death occurs, leading to pathological hypertrophy consisting of ventricular dilation and decreased ejection fraction.33 Interestingly, even mild degrees of chronic renal failure appear to confer a significant increase in cardiac disease.34 Using the currently proposed model of PNx in both mice and rats, we have shown that the concentric phase of hypertrophy occurs within 4 weeks,5, 35, 36 whereas pathological hypertrophy occurs within 8 weeks after the PNx procedure.35, 37 The current study concluded at 4 weeks, and although blood urea nitrogen measurements were not performed, we did measure cystatin C in the sham versus PNx rats, and this indicated levels that also increased significantly (1065±70 vs 2668±215 ng/mL; P<0.01).
The data summarized in Table indicate that rapamycin is capable of antagonizing the cardiac hypertrophy observed in PNx, and that this effect does not appear to be mediated by indirect effects on blood pressure. This is consistent with our observations that increased synthesis of cardiotonic steroids, such as MBG, plays a central role in mediating the cardiac growth effects observed in the PNx model,5, 6 and that these effects are independent of changes in BP given that antihypertensive therapy does not reduce cardiac hypertrophy and fibrosis post‐PNx.35 In the current study, we demonstrate that rapamycin antagonizes the synthesis of MBG both in vitro (JEG‐3 cells; Figure 1B) as well as in the in vivo PNx model. Given that rapamycin was not effective in antagonizing the cardiac growth effects or MBG levels observed with exogenous administration of MBG in vivo (Table), this underscores the notion that rapamycin mainly exerts its effects through blocking endogenous MBG production in this model. We did not observe any notable side effects in rats administered MBG. The dose of MBG used (10 μg/kg per day by subcutaneous minipump) did produce a modest decrease in body weight, although the heart weight/body weight ratio was also modestly, but not significantly, increased in the MBG group versus sham. Based on our in vitro data, we demonstrated that MBG is capable of inducing collagen formation in cardiac fibroblasts; thus, we expect that the increased fibrosis noted in MBG‐treated animals was the result of this increased collagen formation in vivo.
Treatment with rapamycin significantly reduced circulating MBG levels compared to PNx animals. In addition, treatment with rapamycin in JEG‐3 cells, which produce MBG, resulted in a 52% reduction in MBG levels after 6 hours of treatment. Cardiotonic steroid production is not sex specific, and we have previously shown that whereas partial nephrectomy caused the induction of smaller increases in blood pressure of female rats, both male and female rats are similarly susceptible to cardiac remodeling, hypertrophy, and fibrosis in this model, where cardiotonic steroids are elevated in both sexes.36 In addition to the adrenal cortex,4, 14, 38 the placenta is a known source of MBG synthesis, and our group and others have demonstrated the involvement of cardiotonic steroids, such as MBG, in the pathogenesis of preeclampsia.15, 17, 39, 40, 41 It was demonstrated that elevated plasma MBG levels in preeclampsia are responsible for BP increase and for the fibrosis development in placenta and umbilical arteries of preeclamptic patients versus controls with normal pregnancies, and immunoneutralization of MBG ex vivo results in improvement of vasorelaxation and reduces fibrosis and stiffening of umbilical arteries.2, 15, 17, 41, 42, 43, 44, 45 Accordingly, the JEG‐3 cell line has been commonly used to study production of cardiotonic steroids such as MBG in vitro, thus these experiments were performed as a proof of concept that rapamycin is capable of interrupting synthesis of cardiotonic steroids such as MBG.
Endogenous cardiotonic steroids have been postulated to be synthesized from the classic steroidogenesis pathway through cholesterol side‐chain cleavage and pregnenolone precursors.2 Though this theory is still upheld for other cardiotonic steroids, such as ouabain, there have been controversial results with regard to MBG production.2 In toads, the biosynthesis of MBG occurs through the bile acid pathway form cholanic acids.46 Rapamycin acts as a competitive inhibitor of CYP27A1, a key rate‐limiting enzyme of the bile acid pathway. Our data provide preliminary evidence indicating that the drastic reduction in MBG levels in both PNx animals and JEG‐3 cells, treated with rapamycin, may be attributed to competitive inhibition of CYP27A1. Our results are supported by the work of Fedorova et al, which demonstrated that post‐transcriptional silencing of CYP27A1 in human JEG‐3 cells and rat adrenocortical cells significantly reduced bile acid production and MBG.14 In addition, the investigators report that Dahl‐S rats, following a high‐salt diet, demonstrated that a significant elevation in circulating MBG levels was accompanied by upregulation of adrenocortical CYP27A1 mRNA and increase in CYP27A1 protein levels.14 Thus, in the setting of experimental uremic cardiomyopathy, rapamycin may have a dual effect of both inhibiting cardiac fibrosis and reducing MBG production. Interestingly, rapamycin did not reverse the hypertensive response in rats treated with MBG (Table) in the presence of a pronounced effect on collagen abundance (Figure 2). MBG initiates ionic and signaling pathways by binding to Na/K‐ATPase.2 The former pathway participates in BP regulation through increased contractility of vascular smooth muscle cells, and the latter pathway initiates profibrotic signaling. Thus, rapamycin may block the profibrotic effect of MBG downstream in the setting of exogenous MBG in the circulation without affecting ionic signaling.2
Importantly, we did see a significant reduction in cardiac fibrosis with combined MBG infusion and rapamycin treatment, although we did not observe substantial effects on plasma MBG levels with coadministration of rapamycin. In the current study, we demonstrated that MBG‐induced oxidative stress is significantly attenuated by treatment with rapamycin in cardiac fibroblasts (Figure 5). Although the bulk of our in vitro work would suggest that rapamycin causes both reductions in MBG synthesis as well as interferes with the cardiotonic steroid signaling process at a postreceptor level (ie, distal to the Na/K‐ATPase), it is still unclear where this interference occurs, and whether higher doses of rapamycin would overcome the effects of continuous MBG infusion.
In conclusion, the mTOR pathway has been implicated in the generation of renal fibrosis during renal failure. Our results suggest that treatment with rapamycin has a dual effect of both blocking the mTOR pathway and reducing MBG levels resulting in attenuation of cardiac fibrosis. Treatment with rapamycin may provide a novel therapy for reducing MBG levels and cardiac fibrosis in the setting of uremic cardiomyopathy.
Sources of Funding
Haller has received support from the American Heart Association (National and Ohio Valley Affilate) (Grant No.: 13POST16860035). Tian is supported by the National Institutes of Health (NIH; Grant No.: HL‐105649). Kennedy is supported by an American Heart Association Scientist Development Grant (12SDG12050473), the David and Helen Boone Foundation Research Fund, and an Early Career Development Award from the Central Society for Clinical and Translational Research. Cooper has received support from the National Heart, Lung and Blood Institute (NHLBI), NIH (5U01HL071556). Shapiro has received support from the NIH (1R01HL109015‐01) and NHLBI, NIH (5U01HL071556). Zijian Xie has received support from the NIH (1R01HL109015‐01). Fedorova and Bagrov are supported by the Intramural Research Program, National Institute on Aging, NIH.
Disclosures
None.
Supporting information
Figure S1. Pair‐wise comparisons of baseline systolic blood pressure with a 95% family‐wise confidence level.
Figure S2. Pair‐wise comparisons of week 1 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S3. Pair‐wise comparisons of week 2 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S4. Pair‐wise comparisons of week 3 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S5. Pair‐wise comparisons of week 4 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S6. Pair‐wise comparisons of body weight with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S7. Pair‐wise comparisons of heart weight with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S8. Pair‐wise comparisons of heart weight/body weight ratio with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S9. Pair‐wise comparisons of creatinine with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S10. Pair‐wise comparisons of marinobufagenin (MBG) with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Acknowledgments
The authors would like to gratefully acknowledge the expert assistance of Dr Tian Chen (University of Toledo, Department of Mathematics and Statistics) for her assistance in performing the statistical analysis.
(J Am Heart Assoc. 2016;5:e004106 doi: 10.1161/JAHA.116.004106)
References
- 1. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154–2169. [DOI] [PubMed] [Google Scholar]
- 2. Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61:9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolmakova EV, Haller ST, Kennedy DJ, Isachkina AN, Budny GV, Frolova EV, Piecha G, Nikitina ER, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids in chronic renal failure. Nephrol Dial Transplant. 2011;26:2912–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49:215–224. [DOI] [PubMed] [Google Scholar]
- 5. Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. [DOI] [PubMed] [Google Scholar]
- 6. Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens. 2012;25:690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hall MN. mTOR‐what does it do? Transplant Proc. 2008;40:S5–S8. [DOI] [PubMed] [Google Scholar]
- 8. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument‐Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. [DOI] [PubMed] [Google Scholar]
- 9. Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493–2502. [DOI] [PubMed] [Google Scholar]
- 10. Siedlecki AM, Jin X, Muslin AJ. Uremic cardiac hypertrophy is reversed by rapamycin but not by lowering of blood pressure. Kidney Int. 2009;75:800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J, Ren J, Liu X, Jiang L, He W, Yuan W, Yang J, Dai C. Rictor/mTORC2 signaling mediates TGFbeta1‐induced fibroblast activation and kidney fibrosis. Kidney Int. 2015;88:515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu C, Bai Y, Chen Y, Wang Y, Sottejeau Y, Liu L, Li X, Lingrel JB, Malhotra D, Cooper CJ, Shapiro JI, Xie ZJ, Tian J. Reduction of Na/K‐ATPase potentiates marinobufagenin‐induced cardiac dysfunction and myocyte apoptosis. J Biol Chem. 2012;287:16390–16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gueguen Y, Ferrari L, Souidi M, Batt AM, Lutton C, Siest G, Visvikis S. Compared effect of immunosuppressive drugs cyclosporine A and rapamycin on cholesterol homeostasis key enzymes CYP27A1 and HMG‐CoA reductase. Basic Clin Pharmacol Toxicol. 2007;100:392–397. [DOI] [PubMed] [Google Scholar]
- 14. Fedorova OV, Zernetkina VI, Shilova VY, Grigorova YN, Juhasz O, Wei W, Marshall CA, Lakatta EG, Bagrov AY. Synthesis of an endogenous steroidal Na pump inhibitor marinobufagenin, implicated in human cardiovascular diseases, is initiated by CYP27A1 via bile acid pathway. Circ Cardiovasc Genet. 2015;8:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fedorova OV, Tapilskaya NI, Bzhelyansky AM, Frolova EV, Nikitina ER, Reznik VA, Kashkin VA, Bagrov AY. Interaction of Digibind with endogenous cardiotonic steroids from preeclamptic placentae. J Hypertens. 2010;28:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inman SR, Davis NA, Olson KM, Lukaszek VA, McKinley MR, Seminerio JL. Rapamycin preserves renal function compared with cyclosporine A after ischemia/reperfusion injury. Urology. 2003;62:750–754. [DOI] [PubMed] [Google Scholar]
- 17. Fedorova OV, Simbirtsev AS, Kolodkin NI, Kotov AY, Agalakova NI, Kashkin VA, Tapilskaya NI, Bzhelyansky A, Reznik VA, Frolova EV, Nikitina ER, Budny GV, Longo DL, Lakatta EG, Bagrov AY. Monoclonal antibody to an endogenous bufadienolide, marinobufagenin, reverses preeclampsia‐induced Na/K‐ATPase inhibition and lowers blood pressure in NaCl‐sensitive hypertension. J Hypertens. 2008;26:2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elkareh J, Periyasamy SM, Shidyak A, Vetteth S, Schroeder J, Raju V, Hariri IM, El‐Okdi N, Gupta S, Fedorova L, Liu J, Fedorova OV, Kahaleh MB, Xie Z, Malhotra D, Watson DK, Bagrov AY, Shapiro JI. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli‐1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol. 2009;296:F1219–F1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tian J, Shidyak A, Periyasamy SM, Haller S, Taleb M, El‐Okdi N, Elkareh J, Gupta S, Gohara S, Fedorova OV, Cooper CJ, Xie Z, Malhotra D, Bagrov AY, Shapiro JI. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension. 2009;54:1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol. 1994;26:809–820. [DOI] [PubMed] [Google Scholar]
- 21. Poulalhon N, Farge D, Roos N, Tacheau C, Neuzillet C, Michel L, Mauviel A, Verrecchia F. Modulation of collagen and MMP‐1 gene expression in fibroblasts by the immunosuppressive drug rapamycin. A direct role as an antifibrotic agent? J Biol Chem. 2006;281:33045–33052. [DOI] [PubMed] [Google Scholar]
- 22. Lopez‐De Leon A, Rojkind M. A simple micromethod for collagen and total protein determination in formalin‐fixed paraffin‐embedded sections. J Histochem Cytochem. 1985;33:737–743. [DOI] [PubMed] [Google Scholar]
- 23. Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50:346–363. [DOI] [PubMed] [Google Scholar]
- 24. Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF‐beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol. 2010;298:F142–F149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shegogue D, Trojanowska M. Mammalian target of rapamycin positively regulates collagen type I production via a phosphatidylinositol 3‐kinase‐independent pathway. J Biol Chem. 2004;279:23166–23175. [DOI] [PubMed] [Google Scholar]
- 26. Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lloberas N, Cruzado JM, Franquesa M, Herrero‐Fresneda I, Torras J, Alperovich G, Rama I, Vidal A, Grinyo JM. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006;17:1395–1404. [DOI] [PubMed] [Google Scholar]
- 28. Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD, Dart A, Du XJ. Inhibition of mTOR reduces chronic pressure‐overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663–1670. [DOI] [PubMed] [Google Scholar]
- 29. Czuwara‐Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli‐1 inhibits collagen type I production in dermal fibroblasts via an Sp1‐dependent pathway. J Biol Chem. 2001;276:20839–20848. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54:2271–2279. [DOI] [PubMed] [Google Scholar]
- 31. Jinnin M, Ihn H, Yamane K, Mimura Y, Asano Y, Tamaki K. Alpha2(I) collagen gene regulation by protein kinase C signaling in human dermal fibroblasts. Nucleic Acids Res. 2005;33:1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minhajuddin M, Bijli KM, Fazal F, Sassano A, Nakayama KI, Hay N, Platanias LC, Rahman A. Protein kinase C‐delta and phosphatidylinositol 3‐kinase/Akt activate mammalian target of rapamycin to modulate NF‐kappaB activation and intercellular adhesion molecule‐1 (ICAM‐1) expression in endothelial cells. J Biol Chem. 2009;284:4052–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Di Lullo L, Gorini A, Russo D, Santoboni A, Ronco C. Left ventricular hypertrophy in chronic kidney disease patients: from pathophysiology to treatment. Cardiorenal Med. 2015;5:254–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henry RM, Kostense PJ, Bos G, Dekker JM, Nijpels G, Heine RJ, Bouter LM, Stehouwer CD. Mild renal insufficiency is associated with increased cardiovascular mortality: the Hoorn Study. Kidney Int. 2002;62:1402–1407. [DOI] [PubMed] [Google Scholar]
- 35. Kennedy DJ, Elkareh J, Shidyak A, Shapiro AP, Smaili S, Mutgi K, Gupta S, Tian J, Morgan E, Khouri S, Cooper CJ, Periyasamy SM, Xie Z, Malhotra D, Fedorova OV, Bagrov AY, Shapiro JI. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–F454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drummond CA, Buddny G, Haller ST, Liu J, Yan Y, Xie Z, Malhotra D, Shapiro JI, Tian J. Gender differences in the development of uremic cardiomyopathy following partial nephrectomy: role of progesterone. J Hypertens (Los Angel). 2013;2:doi: 10.4172/2167‐1095.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drummond CA, Sayed M, Evans KL, Shi H, Wang X, Haller ST, Liu J, Cooper CJ, Xie Z, Shapiro JI, Tian J. Reduction of Na/K‐ATPase affects cardiac remodeling and increases c‐kit cell abundance in partial nephrectomized mice. Am J Physiol Heart Circ Physiol. 2014;306:H1631–H1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dmitrieva RI, Bagrov AY, Lalli E, Sassone‐Corsi P, Stocco DM, Doris PA. Mammalian bufadienolide is synthesized from cholesterol in the adrenal cortex by a pathway that is independent of cholesterol side‐chain cleavage. Hypertension. 2000;36:442–448. [DOI] [PubMed] [Google Scholar]
- 39. Fedorova OV, Kolodkin NI, Agalakova NI, Namikas AR, Bzhelyansky A, St‐Louis J, Lakatta EG, Bagrov AY. Antibody to marinobufagenin lowers blood pressure in pregnant rats on a high NaCl intake. J Hypertens. 2005;23:835–842. [DOI] [PubMed] [Google Scholar]
- 40. Goodlin RC. Antidigoxin antibodies in eclampsia. N Engl J Med. 1988;318:518–519. [DOI] [PubMed] [Google Scholar]
- 41. Ishkaraeva‐Yakovleva VV, Fedorova OV, Solodovnikova NG, Frolova EV, Bzhelyansky AM, Emelyanov IV, Adair CD, Zazerskaya IE, Bagrov AY. DigiFab interacts with endogenous cardiotonic steroids and reverses preeclampsia‐induced Na/K‐ATPase inhibition. Reprod Sci. 2012;19:1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nikitina ER, Mikhailov AV, Nikandrova ES, Frolova EV, Fadeev AV, Shman VV, Shilova VY, Tapilskaya NI, Shapiro JI, Fedorova OV, Bagrov AY. In preeclampsia endogenous cardiotonic steroids induce vascular fibrosis and impair relaxation of umbilical arteries. J Hypertens. 2011;29:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Averina IV, Tapilskaya NI, Reznik VA, Frolova EV, Fedorova OV, Lakatta EG, Bagrov AY. Endogenous Na/K‐ATPase inhibitors in patients with preeclampsia. Cell Mol Biol. 2006;52:19–23. [PubMed] [Google Scholar]
- 44. Vu HV, Ianosi‐Irimie MR, Pridjian CA, Whitbred JM, Durst JM, Bagrov AY, Fedorova OV, Pridjian G, Puschett JB. Involvement of marinobufagenin in a rat model of human preeclampsia. Am J Nephrol. 2005;25:520–528. [DOI] [PubMed] [Google Scholar]
- 45. Lopatin DA, Ailamazian EK, Dmitrieva RI, Shpen VM, Fedorova OV, Doris PA, Bagrov AY. Circulating bufodienolide and cardenolide sodium pump inhibitors in preeclampsia. J Hypertens. 1999;17:1179–1187. [DOI] [PubMed] [Google Scholar]
- 46. Chen C, Osuch MV. Biosynthesis of bufadienolides–3‐beta‐hydroxycholanates as precursors in Bufo marinus bufadienolides synthesis. Biochem Pharmacol. 1969;18:1797–1802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pair‐wise comparisons of baseline systolic blood pressure with a 95% family‐wise confidence level.
Figure S2. Pair‐wise comparisons of week 1 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S3. Pair‐wise comparisons of week 2 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S4. Pair‐wise comparisons of week 3 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S5. Pair‐wise comparisons of week 4 systolic blood pressure with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S6. Pair‐wise comparisons of body weight with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S7. Pair‐wise comparisons of heart weight with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S8. Pair‐wise comparisons of heart weight/body weight ratio with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S9. Pair‐wise comparisons of creatinine with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.
Figure S10. Pair‐wise comparisons of marinobufagenin (MBG) with a 95% family‐wise confidence level. Red lines indicate statistical significance at P<0.01.