

Abstract
Background
Stable plasma nitric oxide (NO) metabolites (NOM), composed predominantly of nitrate and nitrite, are attractive biomarkers of NO bioavailability. NOM levels integrate the influence of NO‐synthase‐derived NO production/metabolism, dietary intake of inorganic nitrate/nitrite, and clearance of NOM. Furthermore, nitrate and nitrite, the most abundant NOM, can be reduced to NO via the nitrate‐nitrite‐NO pathway.
Methods and Results
We compared serum NOM among subjects without heart failure (n=126), subjects with heart failure and preserved ejection fraction (HFpEF; n=43), and subjects with heart failure and reduced ejection fraction (HFrEF; n=32). LV mass and extracellular volume fraction were measured with cardiac MRI. Plasma NOM levels were measured after reduction to NO via reaction with vanadium (III)/hydrochloric acid. Subjects with HFpEF demonstrated significantly lower unadjusted levels of NOM (8.0 μmol/L; 95% CI 6.2–10.4 μmol/L; ANOVA P=0.013) than subjects without HF (12.0 μmol/L; 95% CI 10.4–13.9 μmol/L) or those with HFrEF (13.5 μmol/L; 95% CI 9.7–18.9 μmol/L). There were no significant differences in NOM between subjects with HFrEF and subjects without HF. In a multivariable model that adjusted for age, sex, race, diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate, HFpEF remained a predictor of lower NOM (β=−0.43; P=0.013). NOM did not correlate with LV mass, or LV diffuse fibrosis.
Conclusions
HFpEF, but not HFrEF, is associated with reduced plasma NOM, suggesting greater endothelial dysfunction, enhanced clearance, or deficient dietary ingestion of inorganic nitrate. Our findings may underlie the salutary effects of inorganic nitrate supplementation demonstrated in recent clinical trials in HFpEF.
Keywords: diastolic heart failure, heart failure, hypertrophy/remodeling, myocardial fibrosis, myocardial structure
Subject Categories: Heart Failure, Magnetic Resonance Imaging (MRI), Biomarkers, Fibrosis
Introduction
Nitric oxide (NO) is a key bioactive molecule for cardiovascular function. NO is synthesized by various NO synthase (NOS) isoforms, including neuronal, inducible, and endothelial isoforms. In addition, NO can also be produced via the nitrate‐nitrite‐NO pathway, which is increasingly recognized as an important source of NO in vivo.1, 2
The plasma concentration of stable NO metabolites (NOM) represents an attractive biomarker of NO bioavailability. Nitrate () and nitrite (), the most abundant circulating NOM species, are generated as byproducts of NOS‐derived NO3, 4, 5 as well from the ingestion of dietary inorganic nitrate and nitrite.1, 2, 6 Therefore, NOM levels integrate the influence of endogenous nitric oxide (NO) production, clearance, and dietary intake of NO precursors. Regardless of their source, plasma nitrate and nitrite constitute a circulating storage pool of NO precursors that can be readily converted to NO via the nitrate‐nitrite‐NO pathway.1, 2, 6, 7, 8 In this pathway nitrate undergoes a 2‐step reduction (to nitrite and then to NO) via the combined action of bacterial oxidoreductases and various nitrite reductases (predominantly deoxyhemoglobin and deoxymyoglobin).1, 2, 6, 7, 8
Recently, plasma NOM levels have been shown to independently correlate with electrocardiographic LV hypertrophy (LVH) in a Japanese population of normotensive men.9 However, whether NOM are related to the presence of heart failure (HF) is unknown. In particular, it is unknown whether low NOM levels are present in patients with HF with preserved ejection fraction (HFpEF), an epidemic condition in which abnormal phenotypes consistent with reduced NO bioavailability have been demonstrated.1, 10, 11, 12, 13, 14, 15 Furthermore, the relationship between NOM and LVH/remodeling in patients with and without HF requires further study. The previous report of a relationship between NOM and electrocardiographic LVH9 included normotensive men from Japan, a country in which dietary nitrate intake is high. Whether a relationship between LVH and NOM is present in Western populations with and without HF is unknown. Moreover, LVH is a complex process that may be driven by cellular hypertrophy, extracellular volume expansion due to diffuse interstitial fibrosis, or both. Cardiac MRI with T1 mapping before and after the injection of gadolinium contrast has emerged as a novel, highly precise method for the quantification of diffuse myocardial fibrosis.16
In this study, we aimed to (1) compare NOM levels among subjects with HFpEF, HF with reduced ejection fraction (HFrEF), and subjects without HF and (2) assess the relationship between NOM and LV remodeling (both macroscopic hypertrophy and diffuse interstitial myocardial fibrosis) in these populations.
Methods
We prospectively enrolled a convenience sample of patients at the Corporal Michael J. Crescenz VA Medical Center. The protocol was approved by the Philadelphia VA Medical Center Institutional Review Board, and all subjects provided written informed consent.
HFrEF was defined as a symptomatic HF in the presence of a left ventricular ejection fraction (LVEF) <50%. HFpEF was defined as (1) NYHA Class II‐IV symptoms consistent with HF in the absence of significant aortic stenosis; (2) LV ejection fraction >50%; (3) a mitral E wave to annular e′ ratio >1417; or at least 2 of the following: (a) a mitral E wave to annular e′ ratio >8; (b) treatment with a loop diuretic for control of HF symptoms; (c) left atrial volume index >34 mL/m2 of body surface area (BSA); (d) NT‐pro B‐type natriuretic peptide level >200 pg/mL; and (e) LV mass index >149 g/m2 in men and 122 g/m2 in women (measured by cardiac MRI). Subjects without HF had an LVEF >50%, no significant valvular disease, and no symptoms and signs consistent with HF.
Key exclusion criteria were as follows: (1) claustrophobia; (2) presence of metallic objects or implanted medical devices in body; (3) more than mild aortic stenosis; (4) atrial fibrillation; (5) conditions that would make the study measurements less accurate or unreliable (ie, arrhythmia affecting cardiac gating, inability to perform an adequate breath hold for cardiac MRI acquisitions); (6) known infiltrative or hypertrophic cardiomyopathy or extracardiac amyloidosis or sarcoidosis.
Plasma NOM Measurements
Venous blood samples were obtained at the time of enrollment and stored at −80°C for batch analysis. NOM were measured as previously described.18 Briefly, samples were passed through a filter (AmiconUltra‐0.5 Centrifugal Filter Unit, EMD Millipore, Billerica, MA) to remove proteins with molecular weight >30 kDa. Samples were then injected into a custom‐made ice‐water‐cooled reaction chamber containing vanadium (III)/hydrochloric acid solution heated to 95°C. NO generated from the reduction of NOM was quantified by its gas‐phase chemiluminescence reaction with ozone (Nitric Oxide Analyzer, Sievers Instruments, Boulder, CO). Signal peaks (mV) were manually integrated, and the corresponding areas were used for the quantification of NOM concentrations. Authentic nitrate in the range of 0 to 50 μmol/L was injected into the system, and a 10‐point standard curve was constructed by plotting area against nitrate concentration. The detection limit of this assay was 1.6 μmol/L. The intraclass coefficient of variation of this method in our laboratory has been shown to be 0.96 (95% CI 0.92–0.98; P<0.001).
Measurement of LV Mass
Participants underwent a cardiac MRI examination to assess LV structure and function, using a 1.5‐Tesla (T) whole‐body MRI scanner (Avanto or Espree, Siemens, Malvern, PA) equipped with a phase‐array cardiac coil. LV volumes and ejection fraction (EF) were determined using balanced steady‐state free‐precession (SSFP) cine imaging. Typical parameters were as follows: TR=30.6 milliseconds; TE=1.3 milliseconds; phases=30; slice thickness=8 mm; matrix size=192×192; parallel image (IPAT) factor=2. LV short‐axis stack cine images were manually traced at end‐diastole and end‐systole using CMR42 software (Circle CVI, Calgary, AB, Canada). LV mass (LVM) was computed as the difference between epicardial and endocardial volumes, multiplied by myocardial density. LVM was normalized for body height in meters raised to the allometric power of 1.7.19
Assessment of Diffuse Myocardial Fibrosis
In a subset of participants (n=107), we assessed myocardial fibrosis with cardiac MRI. We used a modified Look‐Locker inversion recovery (MOLLI)20 sequence to assess T1 times prior to and following the intravenous administration of gadolinium contrast (gadopentetate dimeglumine, 0.15 mmol/kg or equivalent) in a midventricular short‐axis slice (Figure 1A). Scan parameters for the MOLLI protocol included field of view (FOV) ~340 mm; matrix size=144×192; slice thickness=6 mm; repetition time=24.9 milliseconds; echo time=1.18 milliseconds; flip angle=30. Myocardial T1 measurements were performed before and at several time points (~5, 10, 15, and 20‐40 minutes) post‐gadolinium administration. All available blood and myocardial T1 measurements were used to compute λ (myocardium‐blood partition coefficient) as the slope of the blood 1/T1 over the myocardial 1/T1 change, via linear regression21 (Figure 1B through 1D). The λ was used to compute the ECV as follows: ECV=λ×(1−hematocrit).
Figure 1.
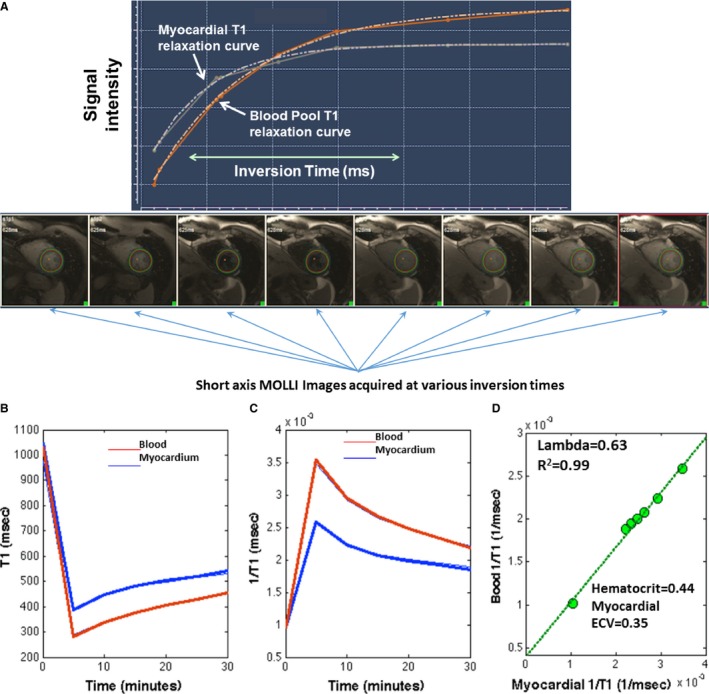
A, Myocardial T1 mapping using the modified Look‐Locker inversion recovery sequence (MOLLI). Example of MOLLI single‐slice T1 determinations performed at 8 different inversion times in a single breath hold. A region of interest (ROI) is defined manually in the images for the myocardium and for the blood pool. Then, the signal intensity in the images acquired at various inversion times is used to compute the time course of longitudinal relaxation (T1) for myocardium and blood. The myocardial T1 relaxation (white) and blood (orange) relaxation curves are shown. T1 for blood and myocardium is computed as the exponential time constant of the respective curves. B, MOLLI images are acquired before the administration of gadolinium‐based contrast (time “zero”) and at several time points after the administration of gadolinium (~5, 10 15, 20, and 30 minutes postinjection) in order to compute blood and myocardial T1 values at various time points for each subject (data shown correspond to 1 subject). Gadolinium administration reduces both myocardial and blood T1 (B) and thus increases T1 relaxivity (1/T1, C). Equilibrium is reached between blood and myocardium, such that when blood and myocardial T1 values are plotted against each other (D), a linear slope of the myocardial 1/T1 over the blood 1/T1 change (D) can be obtained with linear regression. This slope represents the gadolinium partition coefficient (lambda). Extracellular volume (ECV) is then computed as lambda×(1−hematocrit).
Statistical Methods
Continuous variables are presented as mean±SD unless otherwise stated. Categorical variables are presented as frequencies and percentages. Comparisons in NOM among subjects without HF, subjects with HFrEF, and subjects with HFpEF were performed with 1‐way analysis of variance (ANOVA), with post‐hoc pairwise comparisons performed with Bonferroni correction, after testing for homogeneity of variance with the Levine test. Linear regression was performed to determine the relationship between the presence of HFpEF or HFrEF and NOM, adjusting for multiple comorbidities. We also used linear regression to assess the association between NOM and measures of LV geometry (LV mass, end‐diastolic volume, myocardial ECV). NOM levels were log‐transformed because of their positively skewed distribution. All probability values are 2‐tailed. Statistical significance was defined α<0.05. Statistical analyses were performed using SPSS for Windows v22 (SPSS Inc, Chicago, IL).
Results
We included 201 subjects in this study, among whom 126 had no evidence of HF, 43 had HFpEF, and 32 had HFrEF. Baseline clinical characteristics for these groups are presented in Table 1. Subjects with HFrEF had lower body mass indexs, lower blood pressure, and significantly more coronary artery disease than control subjects and subjects with HFpEF. The prevalence of hypertension was high in all 3 groups, without significant differences among the groups.
Table 1.
Demographic, Clinical, and Laboratory Characteristics of the Study Population
| No HF | HFrEF | HFpEF | P Valuea | |
|---|---|---|---|---|
| n=126 | n=32 | n=43 | ||
| Age, y | 60.9, 12.7 | 65.2, 7.5 | 65.2, 10 | 0.038 |
| Male | 116 (92.1) | 31 (96.9) | 38 (88.4) | 0.40 |
| Race | 0.43 | |||
| White | 58 (46) | 13 (40.6) | 12 (27.9) | |
| Black | 61 (48.4) | 19 (59.4) | 30 (69.8) | |
| Other | 7 (5.5) | 0 (0) | 1 (2.3) | |
| Pro B‐type natriuretic peptide, pg/mL | 184.1, 200.6 | 3809.2, 4932.8 | 911.2, 1508 | <0.0001 |
| Hypertension | 100 (80) | 27 (84.4) | 39 (90.7) | 0.26 |
| Diabetes mellitus | 65 (52) | 15 (46.9) | 29 (67.4) | 0.14 |
| Current smoking | 27 (21.6) | 11 (34.4) | 9 (20.9) | 0.28 |
| Body mass index, kg/m2 | 31.7, 7.2 | 28.2, 6.3 | 35.1, 6.7 | <0.0001 |
| Systolic BP, mm Hg | 142.7, 17.4 | 139.5, 21.3 | 152.4, 18.8 | 0.004 |
| Diastolic BP, mm Hg | 83, 11.5 | 79.5, 10.4 | 85.4, 12.5 | 0.10 |
| LDL cholesterol, mg/dL | 97.9, 33.4 | 89.1, 41.1 | 97.9, 33.8 | 0.44 |
| HDL cholesterol, mg/dL | 42.2, 11.9 | 45.9, 12.2 | 42.9, 11.1 | 0.30 |
| eGFR, mL/min per 1.73 m2 | 89.1, 26.9 | 76.7, 22.8 | 76.7, 37.7 | 0.016 |
| ACE inhibitor use | 61 (48.8) | 26 (81.3) | 22 (51.2) | 0.004 |
| Angiotensin receptor blocker use | 5 (4) | 2 (6.3) | 10 (23.3) | <0.0001 |
| β‐Blocker use | 48 (38.4) | 29 (90.6) | 33 (76.7) | <0.0001 |
| Spironolactone | 3 (2.4) | 3 (9.4) | 1 (2.3) | 0.14 |
| Statin use | 76 (60.8) | 29 (90.6) | 31 (72.1) | 0.004 |
Numbers represent mean, SD or counts (%). ACE indicates angiotensin‐converting enzyme; BP, blood pressure; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEf, heart failure with reduced ejection fraction; LDL, low‐density lipoprotein; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association.
ANOVA P value for differences among the groups.
NOM Levels in Subjects With HFpEF, HFrEF, and no HF
NOM levels in subjects without HF versus those with HFrEF and HFpEF are shown in Figure 2. There was a significant difference between the groups (ANOVA P=0.013). Post‐hoc pairwise comparisons revealed that subjects with HFpEF exhibited significantly lower levels of NOM (8.0 μmol/L; 95% CI 6.2‐10.4 μmol/L) compared to (12.0 μmol/L; 95% CI 10.4‐13.9 μmol/L; P=0.025) or to those with HFrEF (13.5 μmol/L; 95% CI 9.7‐18.9 μmol/L; P=0.03). There were no significant differences in NOM between subjects with HFrEF and subjects without HF (P=1.00).
Figure 2.
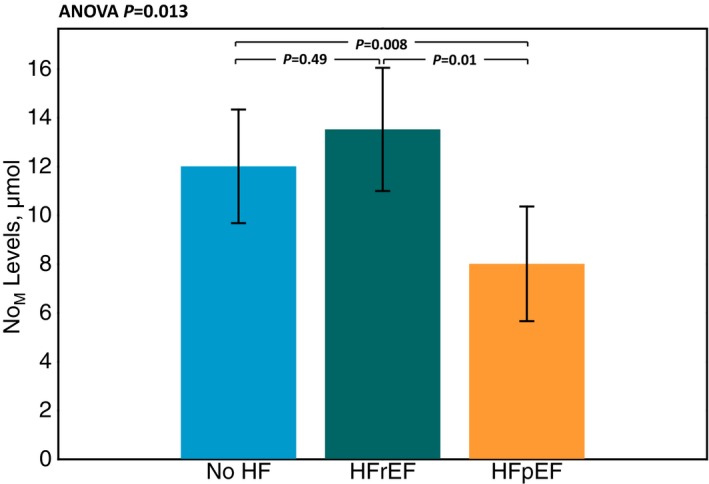
Comparison of metabolites (NOM) levels between subjects without HF, subjects with heart failure and preserved ejection fraction (HFpEF) and subjects with heart failure and reduced ejection fraction (HFrEF).
Multivariable Predictors of NOM Levels
Results of multivariable linear models in which HF status and various confounders were included as predictors of NOM levels are shown in Table 2.
Table 2.
HFpEF and HFrEF as Predictors of log‐NOM Levels in Adjusted Linear Regression Models
| Model 1 | Model 2 | Model 3 | ||||
|---|---|---|---|---|---|---|
| β±SE | P Value | β±SE | P Value | β±SE | P Value | |
| HFpEF | −0.42±0.16 | 0.009 | −0.43±0.17 | 0.013 | −0.41±0.19 | 0.028 |
| HFrEF | 0.1±0.18 | 0.573 | 0.03±0.19 | 0.887 | 0.04±0.20 | 0.83 |
Model 1: adjusted for age, sex, and race. Model 2: adjusted for variables in model 1 and further adjusted for the presence of diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate. Model 3: adjusted for variables in model 2 and further adjusted for ACE inhibitor, angiotensin receptor blocker, β‐blocker, spironolactone, statin, and long‐acting nitrate use. ACE indicates angiotensin‐converting enzyme; HFpEF, heart failure and preserved ejection fraction; HFrEF, heart failure and reduced ejection fraction; NOM, metabolites;
In a model that adjusted for age, sex, and race, HFpEF was a significant predictor of lower log‐NOM levels (β=−0.42; P=0.009). Similarly, in a model that further adjusted for the presence of diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate, HFpEF remained a significant predictor of lower log‐NOM (β=−0.43; P=0.013). With further adjustment for ACE inhibitor use, angiotensin receptor blocker use, β‐blocker use, spironolactone use, statin use, and long‐acting nitrate use, HFpEF remained a significant predictor of lower log‐NOM (β=−0.41; P=0.028). The presence of HFrEF was not a significant predictor of NOM levels in any of these multivariable models (P>0.05, Table 2).
Relationship Between NOM Levels and LV Mass
Results of multivariable linear models examining the predictors of LV mass index in the study population are shown in Table 3. In a model that included age, sex, race, log‐NOM, and the presence of HFpEF and HFrEF, both HFpEF (β=15.08 g/m1.7; P<0.0001) and HFrEF (β=15.06 g/m1.7; P<0.0001) were significantly associated with a greater LV mass index. However, NOM levels were not associated with LV mass index (β=0.16 g/m1.7; P=0.92). With incremental adjustment for the presence of diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate (Table 3), HFpEF and HFrEF remained significant predictors of LV mass index, whereas NOM was not (Table 3).
Table 3.
HFpEF, HFrEF, and log‐NOM as Predictors of LV Mass Index and Myocardial Extracellular Volume (ECV) in Linear Regression Models
| Model 1 | Model 2 | |||
|---|---|---|---|---|
| β±SE | P Value | β±SE | P Value | |
| LV mass index | ||||
| HFpEF | 15.08±3.61 | <0.0001 | 12.58±3.59 | 0.001 |
| HFrEF | 15.06±3.7 | <0.0001 | 17.77±3.67 | <0.0001 |
| Log‐NOM | 0.16±1.6 | 0.92 | 0.88±1.57 | 0.58 |
| Myocardial ECV | ||||
| HFpEF | 1.87±1.52 | 0.223 | 3.31±1.58 | 0.039 |
| HFrEF | 5.01±1.55 | 0.002 | 4.44±1.67 | 0.009 |
| Log‐NOM | 0.3±0.71 | 0.676 | 0.43±0.72 | 0.553 |
Model 1: adjusted for age, sex, and race. Model 2: model 1, with further adjustment for the presence of diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate. HFpEF indicates heart failure and preserved ejection fraction; HFrEF, heart failure and reduced ejection fraction; LV, left ventricular; NOM, metabolites;
Relationship Between NOM Levels and Myocardial ECV
Results of multivariable linear models examining the predictors of LV mass index in the study population are shown in Table 3. In unadjusted analyses ECV was significantly different among the groups (ANOVA P=0.004). This was due to a significantly greater ECV in HFrEF compared to participants without HF (P=0.004). In analyses that adjusted for age, sex, race, presence of diabetes mellitus, body mass index, current smoking, systolic blood pressure, and glomerular filtration rate, both HFpEF (β=3.31; P=0.039) and HFrEF (β=4.44; P=0.009) were significantly associated with a greater ECV. However, NOM levels were not associated with ECV (β=0.43; P=0.55).
Effect Modification
We found no significant effect modification by either HFpEF or HFrEF status on the relationship between NOM and either LV mass index or ECV (all P>0.05).
Discussion
In this study we demonstrate that HFpEF, but not HFrEF, is associated with reduced plasma NOM, suggesting greater endothelial dysfunction, enhanced clearance, or deficient ingestion or metabolism of dietary inorganic nitrate. These findings support the concept of reduced NO bioavailability in HFpEF and may underlie the recently demonstrated salutary effects of inorganic nitrate supplementation observed in recent randomized trials in HFpEF.1, 2, 18, 22 We also assessed the relationship between NOM and LV remodeling. In contrast to a previous report that reported a relationship between electrocardiographic LVH and NOM levels in a Japanese normotensive population,9 we did not observe such a relationship with LV mass measured with cardiac MRI in the current study, which included a Western population. Furthermore, we did not find a relationship between NOM and diffuse myocardial fibrosis.
Circulating NOM represents an attractive integrated biomarker of NO bioavailability. Several studies have demonstrated that plasma NOM levels are influenced by endogenous NO production by NOS.3, 4 However, plasma NOM levels can also be substantially increased via the ingestion of dietary nitrate.1, 2, 18 Nitrate and nitrite, the main NOM, undergo enterosalivary cycles and are ultimately excreted by the kidneys.1 Therefore, circulating NOM integrate the influence of endogenous NOS‐derived NO production, dietary intake of NO precursors, and clearance of NO metabolites. Interestingly, HFpEF remained associated with reduced NOM levels after adjustment for estimated glomerular filtration rate, suggesting that renal clearance is unlikely to account for the observed difference.
Overall, our findings support the concept of reduced NO bioavailability in HFpEF, as suggested by recent studies.23, 24 Reduced NO bioavailability has been demonstrated in myocardial tissue of patients with HFpEF compared to those with HFrEF,25, 26 with reduced nitrate/nitrite content within myocardial homogenates of HFpEF patients.26 Similarly, increased oxidative stress, which tends to reduce NO bioavailability, has been demonstrated in HFpEF participants as compared to those with HFrEF.25, 26 In addition to reduced NOS‐derived endogenous NO production, it is also possible that the reduced NOM levels observed in HFpEF are the result of deficient ingestion or bioavailability of dietary inorganic nitrate and nitrite. It seems unlikely, however, that HFpEF patients have dietary intake patterns different from both HFrEF and control subjects, although this will need to be properly assessed in future studies.
It should be noted that, regardless of their source, plasma nitrate and nitrite (the most abundant circulating NOM species) constitute a physiologic circulating storage pool of NO precursors, which can be readily converted to NO via the nitrate‐nitrite‐NO pathway.1, 2, 6, 7, 8 This conversion is particularly effective in the presence of hypoxia and acidosis, as occurs in skeletal muscle during exercise.1, 2 Interestingly, a reduced vasodilatory reserve with exercise has been demonstrated in HFpEF, and inorganic nitrate supplementation led to an improvement in this vasodilatory reserve and increased oxygen consumption in this patient population.18 Our findings are therefore consistent and may underlie the positive effects of inorganic nitrate administration in recent randomized trials in patients with HFpEF. These trials have shown that inorganic nitrate supplementation with either a single dose of 12.9 mmol or sustained administration of 6 mmol/day for 1 week increases NOM levels and improves exercise capacity in HFpEF.1, 2, 18, 22 Of note, these findings are in contrast to attempts at increasing endogenous NO signaling in this patient population using a phosphodiesterase inhibitor to prevent the breakdown of cyclic GMP, the second messenger of NO.27 The latter findings have been attributed to low endogenous NO bioavailability. The low levels of NOM observed in our study, which suggest reduced NO bioavailability, are consistent with this hypothesis.
Interestingly, NOM were not reduced in HFrEF compared to subjects without HF. The reasons behind these observations are unclear and need to be assessed in future studies. Activation of inducible nitric oxide synthase (iNOS), leading to higher NO and NOM levels, which has been reported in HFrEF, may partially explain this finding.28, 29
An additional novel contribution of our study is the assessment of the relationship between plasma NOM and LV remodeling. We examined both macroscopic LVH (myocardial mass) and interstitial myocardial fibrosis using state‐of‐the‐art MRI methods. We found that patients with HFpEF and, particularly, HFrEF exhibited greater myocardial fibrosis, which is consistent with other recent studies. However, in contrast to a previous study that demonstrated a relationship between ECG LVH and NOM levels in a Japanese population of normotensive men,9 we did not find any relationship between NOM levels and either LV mass or ECV (a measure of interstitial myocardial fibrosis). It is likely that multiple pathways are involved in the development of LVH and fibrosis, which may confound the association between NOM and LV remodeling. It is also possible that high NOM levels are “protective” against LV remodeling only in the presence of high dietary nitrate ingestion, as seen in Japanese diets. The previous study from Japan reported a median NOM level of 34.1 (IQR 22.6, 52.9) μmol/L, which is higher than the levels found in the present study; however, this comparison should be taken with caution, given potential differences in absolute NOM levels due to different methods of measurement.
Our study should be interpreted in the context of its strengths and limitations. Strengths of our study include the inclusion of patients with and without HF, appropriate adjudication of HFpEF versus HFrEF, and the use of gold‐standard noninvasive assessments of LV mass and diffuse myocardial fibrosis. Our study also has limitations. The sample size was limited, and some tests may have failed to reach significance due to limited statistical power. Dietary intake was not standardized at the time of our serum acquisition, and therefore, we cannot account for differences in inorganic nitrate/nitrite ingestion among the groups. Our population was a convenience clinical sample, which may not fully represent population‐based trends. This is particularly true for the smaller subsample of subjects who underwent ECV measurements; our findings about ECV should be interpreted with caution and confirmed in future studies. Our study does provide inference about causality or reasons behind the reduced levels of NOM in HFpEF. Further studies to assess the mechanisms leading to reduced levels of NOM in HFpEF are needed. Similarly, further studies of interventions to increase NOM levels (such as nitrate supplementation) are under way. Finally, we acknowledge that a measure of endothelial function (such as flow‐mediated dilation) would have strengthened the study.
In conclusion, we report, for the first time, a reduction in plasma levels of NOM in HFpEF, which may suggest either reduced endogenous synthesis of NO, increased clearance of NOM, or decreased dietary intake of inorganic nitrate/nitrite. We did not observe a relationship between NOM and either LV or diffuse myocardial fibrosis measured with cardiac MRI. Our findings regarding reduced NOM levels in HFpEF may underlie the therapeutic effects of inorganic nitrate supplementation in HFpEF demonstrated in recent trials.
Sources of Funding
This study was supported by NIH grants R56HL‐124073‐01A1 (Chirinos), R01 HL 121510‐01A1 (Chirinos), RO1 HL054926 (Ischiropoulos), and 5‐R21‐AG‐043802‐02 (Chirinos). Zamani is funded by the Translational Medicine and Therapeutics of the University of Pennsylvania (5UL1TR000003‐09 from the National Center for Research Resources), 5‐T32‐HL007843‐17, and 1‐K23‐HL‐130551‐01. The project described was supported by Grant Number UL1RR024134 from the National Center for Research Resources and by Grant Number UL1TR000003 from the National Center for Advancing Translational Sciences, National Institutes of Health; the content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosures
Chirinos has received consulting fees from Bristol‐Myers Squibb, OPKO Healthcare, Fukuda Denshi, Microsoft Research, Merck, and Vital Labs. Chirinos received research grants from National Institutes of Health, American College of Radiology Network, Fukuda Denshi, Bristol‐Myers Squibb, Microsoft Research, and CVRx Inc, and device loans from Atcor Medical. He is named as inventor in a University of Pennsylvania patent application for the use of inorganic nitrates/nitrites for the treatment of Heart Failure and Preserved Ejection Fraction. Cappola has received funding from BG medicine and Abbott Diagnostics. Margulies has received research grants from Juventis Therapeutics, Celladon Corporation, Thoratec Corporation, Innolign Biomedical, LLC, and Merck, Inc and has been a consultant/Advisory Board member for NovoNordisk (unpaid), Janssen, Merck, Pfizer, Ridgetop Research, and AstraZeneca. Townsend received consulting fees from Medtronic, Fukuda Denshi, and Relypsa. Other authors have no conflict of interest.
(J Am Heart Assoc. 2016;5:e004133 doi: 10.1161/JAHA.116.004133)
References
- 1. Chirinos JA, Zamani P. The nitrate‐nitrite‐NO pathway and its implications for heart failure and preserved ejection fraction. Curr Heart Fail Rep. 2016;13:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vanderpool R, Gladwin MT. Harnessing the nitrate‐nitrite‐nitric oxide pathway for therapy of heart failure with preserved ejection fraction. Circulation. 2015;131:334–336. [DOI] [PubMed] [Google Scholar]
- 3. Kelm M, Preik‐Steinhoff H, Preik M, Strauer BE. Serum nitrite sensitively reflects endothelial NO formation in human forearm vasculature: evidence for biochemical assessment of the endothelial L‐arginine‐NO pathway. Cardiovasc Res. 1999;41:765–772. [DOI] [PubMed] [Google Scholar]
- 4. Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA. 2001;98:12814–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lauer T, Heiss C, Balzer J, Kehmeier E, Mangold S, Leyendecker T, Rottler J, Meyer C, Merx MW, Kelm M, Rassaf T. Age‐dependent endothelial dysfunction is associated with failure to increase plasma nitrite in response to exercise. Basic Res Cardiol. 2008;103:291–297. [DOI] [PubMed] [Google Scholar]
- 6. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate‐nitrite‐nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. [DOI] [PubMed] [Google Scholar]
- 7. Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia‐reperfusion damage. Proc Natl Acad Sci USA. 2004;101:13683–13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme‐independent formation of nitric oxide in biological tissues. Nat Med. 1995;1:804–809. [DOI] [PubMed] [Google Scholar]
- 9. Kamezaki F, Tsutsui M, Takahashi M, Sonoda S, Kubo T, Fujino Y, Adachi T, Abe H, Takeuchi M, Mayumi T, Otsuji Y. Plasma levels of nitric oxide metabolites are markedly reduced in normotensive men with electrocardiographically determined left ventricular hypertrophy. Hypertension. 2014;64:516–522. [DOI] [PubMed] [Google Scholar]
- 10. Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD, Redfield MM. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010;56:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hornig B, Maier V, Drexler H. Physical training improves endothelial function in patients with chronic heart failure. Circulation. 1996;93:210–214. [DOI] [PubMed] [Google Scholar]
- 12. Hundley WG, Bayram E, Hamilton CA, Hamilton EA, Morgan TM, Darty SN, Stewart KP, Link KM, Herrington DM, Kitzman DW. Leg flow‐mediated arterial dilation in elderly patients with heart failure and normal left ventricular ejection fraction. Am J Physiol Heart Circ Physiol. 2007;292:H1427–H1434. [DOI] [PubMed] [Google Scholar]
- 13. Katz SD, Schwarz M, Yuen J, LeJemtel TH. Impaired acetylcholine‐mediated vasodilation in patients with congestive heart failure. Role of endothelium‐derived vasodilating and vasoconstricting factors. Circulation. 1993;88:55–61. [DOI] [PubMed] [Google Scholar]
- 14. Katz SD, Krum H, Khan T, Knecht M. Exercise‐induced vasodilation in forearm circulation of normal subjects and patients with congestive heart failure: role of endothelium‐derived nitric oxide. J Am Coll Cardiol. 1996;28:585–590. [DOI] [PubMed] [Google Scholar]
- 15. Drexler H, Hayoz D, Munzel T, Hornig B, Just H, Brunner HR, Zelis R. Endothelial function in chronic congestive heart failure. Am J Cardiol. 1992;69:1596–1601. [DOI] [PubMed] [Google Scholar]
- 16. Kellman P, Hansen MS. T1‐mapping in the heart: accuracy and precision. J Cardiovasc Magn Reson. 2014;16:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagueh SF, Smiseth OA, Appleton CP, Byrd BF 3rd, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, Marino P, Oh JK, Popescu BA, Waggoner AD. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2016;29:277–314. [DOI] [PubMed] [Google Scholar]
- 18. Zamani P, Rawat D, Shiva‐Kumar P, Geraci S, Bhuva R, Konda P, Doulias PT, Ischiropoulos H, Townsend RR, Margulies KB, Cappola TP, Poole DC, Chirinos JA. Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation. 2015;131:371–380; discussion 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chirinos JA, Segers P, De Buyzere ML, Kronmal RA, Raja MW, De Bacquer D, Claessens T, Gillebert TC, St John‐Sutton M, Rietzschel ER. Left ventricular mass: allometric scaling, normative values, effect of obesity, and prognostic performance. Hypertension. 2010;56:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Messroghli DR, Greiser A, Frohlich M, Dietz R, Schulz‐Menger J. Optimization and validation of a fully‐integrated pulse sequence for modified look‐locker inversion‐recovery (MOLLI) T1 mapping of the heart. J Magn Reson Imaging. 2007;26:1081–1086. [DOI] [PubMed] [Google Scholar]
- 21. Liu CY, Liu YC, Wu C, Armstrong A, Volpe GJ, van der Geest RJ, Liu Y, Hundley WG, Gomes AS, Liu S, Nacif M, Bluemke DA, Lima JA. Evaluation of age‐related interstitial myocardial fibrosis with cardiac magnetic resonance contrast‐enhanced T1 mapping: MESA (Multi‐Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2013;62:1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eggebeen J, Kim‐Shapiro DB, Haykowsky M, Morgan TM, Basu S, Brubaker P, Rejeski J, Kitzman DW. One week of daily dosing with beetroot juice improves submaximal endurance and blood pressure in older patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2016;4:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrari R, Bohm M, Cleland JG, Paulus WJ, Pieske B, Rapezzi C, Tavazzi L. Heart failure with preserved ejection fraction: uncertainties and dilemmas. Eur J Heart Fail. 2015;17:665–671. [DOI] [PubMed] [Google Scholar]
- 24. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 25. van Heerebeek L, Hamdani N, Falcao‐Pires I, Leite‐Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–839. [DOI] [PubMed] [Google Scholar]
- 26. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite‐Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4:312–324. [DOI] [PubMed] [Google Scholar]
- 27. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishibashi Y, Shimada T, Sakane T, Takahashi N, Sugamori T, Ohhata S, Inoue S, Katoh H, Sano K, Murakami Y, Hashimoto M. Contribution of endogenous nitric oxide to basal vasomotor tone of peripheral vessels and plasma B‐type natriuretic peptide levels in patients with congestive heart failure. J Am Coll Cardiol. 2000;36:1605–1611. [DOI] [PubMed] [Google Scholar]
- 29. Winlaw DS, Smythe GA, Keogh AM, Schyvens CG, Spratt PM, Macdonald PS. Increased nitric oxide production in heart failure. Lancet. 1994;344:373–374. [DOI] [PubMed] [Google Scholar]