

Abstract
Background
The development of atherosclerosis is strongly linked to disorders of cholesterol metabolism. Matrix metalloproteinases (MMPs) are dysregulated in patients and animal models with atherosclerosis. Whether systemic MMP activity influences cholesterol metabolism is unknown.
Methods and Results
We examined MMP‐9–deficient (Mmp9 −/−) mice and found them to have abnormal lipid gene transcriptional responses to dietary cholesterol supplementation. As opposed to Mmp9 +/+ (wild‐type) mice, Mmp9 −/− mice failed to decrease the hepatic expression of sterol regulatory element binding protein 2 pathway genes, which control hepatic cholesterol biosynthesis and uptake. Furthermore, Mmp9 −/− mice failed to increase the expression of genes encoding the rate‐limiting enzymes in biliary cholesterol excretion (eg, Cyp7a and Cyp27a). In contrast, MMP‐9 deficiency did not impair intestinal cholesterol absorption, as shown by the 14C‐cholesterol and 3H‐sitostanol absorption assay. Similar to our earlier study on Mmp2 −/− mice, we observed that Mmp9 −/− mice had elevated plasma secreted phospholipase A2 activity. Pharmacological inhibition of systemic circulating secreted phospholipase A2 activity (with varespladib) partially normalized the hepatic transcriptional responses to dietary cholesterol in Mmp9 −/− mice. Functional studies with mice deficient in other MMPs suggested an important role for the MMP system, as a whole, in modulation of cholesterol metabolism.
Conclusions
Our results show that MMP‐9 modulates cholesterol metabolism, at least in part, through a novel MMP‐9–plasma secreted phospholipase A2 axis that affects the hepatic transcriptional responses to dietary cholesterol. Furthermore, the data suggest that dysregulation of the MMP system can result in metabolic disorder, which could lead to atherosclerosis and coronary heart disease.
Keywords: atherosclerosis, cholesterol, lipid metabolism, liver, matrix metalloproteinase, plasma phospholipase A2
Subject Categories: Coronary Artery Disease, Heart Failure
Introduction
The matrix metalloproteinase (MMP) system comprises 25 different zinc‐dependent and multifunctional endoproteases.1 Although collectively capable of cleaving extracellular matrix components, latent growth factors, cytokines, apolipoproteins, and cell membrane receptors, including the receptors for lipoproteins and metabolic hormones,1 MMPs are not typically viewed as important metabolic modulators.
We recently reported that the cardiohepatic metabolic phenotype in MMP‐2–deficient (Mmp2 −/−) mice can be largely explained by a novel heart–liver axis involving myocardial secretion of a unique phospholipase A2 (PLA2) called cardiac secreted PLA2 (sPLA2).2, 3 Purportedly, the MMP‐2–cardiac sPLA2 axis enables the heart to signal to the liver to satisfy its energy needs. In addition, the MMP‐2–cardiac sPLA2 system enhances the production of prostaglandin E2 in the heart, brain, and liver. Consequently, a heart‐centric mechanism using cardiac sPLA2 as a signal governed by MMP‐2 influences the metabolism of other noncardiac organs.2
Despite recent advances concerning the MMP‐2–cardiac sPLA2 axis, little is known about the metabolic functions of individual MMPs. In the current investigation, we investigated the potential contribution of MMP‐9 to the systemic modulation of metabolism using a murine model of global genetic loss of MMP‐9 (Mmp9 −/− mice). MMP‐9, also known as gelatinase B or 92‐kDa collagenase, shares remarkable structural similarities with MMP‐2 (and other MMPs) including the presence of a propeptide domain and a catalytic domain with a highly conserved Zn2+‐binding region; however, the tissue distribution of MMP‐9 differs from that of MMP‐2. MMP‐2, for example, is constitutively expressed in most tissues, whereas MMP‐9 expression is induced in response to inflammation.4 MMP‐9 activity contributes to atherosclerotic lesion progression,5, 6, 7, 8 pathological cardiovascular remodeling in left ventricular dilatation after myocardial infarction and aortic aneurysm formation, and complications of diabetes mellitus including nephropathy, cardiomyopathy, and retinopathy.9, 10 Moreover, obesity increases serum MMP‐9 levels in women.11, 12 MMP‐9–deficient and wild‐type (WT) littermate mice are similarly susceptible to obesity induced by a high fat diet (HFD) and exhibit normal adipose tissue development.11, 12 Little information, however, is available on the role of MMP‐9 in the regulation of lipid metabolism in the liver.
The liver responds to dietary cholesterol by decreasing hepatic cholesterol biosynthesis and increasing biliary cholesterol excretion.13, 14, 15, 16, 17 Disorders of cholesterol metabolism are strongly linked to the development of atherosclerosis.18, 19, 20 MMP‐9 activity is dysregulated in patients and experimental animal models with atherosclerosis or coronary artery disease.21, 22, 23
In this study, we showed that MMP‐9 modulates cholesterol metabolism, at least in part, through a novel MMP‐9–plasma sPLA2 axis that affects the hepatic transcriptional responses to dietary cholesterol. Functional studies with mice deficient in other MMPs further indicated that MMP‐9 (and the MMP system as a whole) strongly influences cholesterol homeostasis. We propose that dysregulation of the MMP system can result in metabolic disorders that could lead to atherosclerosis and coronary heart disease.
Materials and Methods
Reagents
Sterol regulatory element binding protein 2 (SREBP‐2) antibody was purchased from Abcam. Varespladib was from Selleck Chemicals. PNGase F was from Promega. Enhanced chemiluminescence immunoblotting detection reagent was from GE Healthcare. HRP‐conjugated antirabbit antibodies and the Bio‐Rad Protein Assay kit were from Bio‐Rad Laboratories.
Animals
All animal protocols and procedures were approved by the University of Alberta animal care committee and conducted in accordance with institutional guidelines issued by the Canada Council on Animal Care. Unless otherwise stated, WT mice aged 10 to 15 weeks were purchased from Charles River Laboratories (Wilmington, MA) or the Jackson Laboratory (Bar Harbor, ME) and compared against age‐ and sex‐matched Mmp7 −/− and Mmp9 −/− mice purchased from the Jackson Laboratory. Mmp2 −/− and Timp2 −/− mice were bred at the University of Alberta (Edmonton, Canada). All mice had the C57BL/6 background and were housed in the Health Sciences Laboratory Animal Services of the University of Alberta on a 12‐hour light/dark cycle. Mice were fed a chow diet ad libitum (PicoLab Rodent Diet 20; Lab Diet) unless otherwise specified. At the end of the experiments, mice were euthanized with 65 mg/kg of sodium pentobarbital, the blood was collected with EDTA‐coated syringes and tubes, and the organs were excised and snap‐frozen in liquid nitrogen. The experiments with the HFD were approved by the local ethics committee (KU Leuven, P06022). Experiments were performed in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The genetic background of the MMP‐9–deficient mice used in this subset of experiments was 50% CD1 and 50% Sv129 with corresponding WT littermates (Mmp9 +/+), as described elsewhere.12, 24 Male mice (aged 5 weeks) were kept in microisolation cages on a 12‐hour light/dark cycle and fed ad libitum with a standard fat diet (KM‐04‐k12, Muracon, containing 13% kcal as fat, with a caloric value of 10.9 kJ per gram; Carfil) or with an HFD (Harlan Teklad TD.88137; Envigo) containing 42% kcal as fat and a caloric value of 20.1 kJ per gram). All mice had ad libitum access to drinking water. Body weight and food intake were measured at weekly intervals. After 15 weeks, mice were anesthetized by intraperitoneal injection of 60 mg/kg sodium pentobarbital (Nembutal; Abbott Laboratories), following overnight fasting. Blood was collected via the retro‐orbital sinus with the addition of trisodium citrate (final concentration 0.01 mol/L), and plasma was stored at −80°C. Livers were removed, weighed, divided into portions, and snap‐frozen in liquid nitrogen for RNA extraction.
In Vivo Responses to Dietary Cholesterol, Fasting, and Fasting–Refeeding
The dietary regimens followed previously described protocols.25 In the cholesterol supplementation studies, the mice (aged 12–14 weeks) were fed chow supplemented with 0%, 0.15%, or 1.5% cholesterol for 2.5 or 6.5 days. In this study, the mice were not fasted before being euthanized. In the fasting and fasting–refeeding studies, mice (aged 10–22 weeks) were fasted for 16 hours or were fasted for 16 hours and then fed a “high carb” mouse diet (TD.88122; Envigo) for 4 hours, and then they were euthanized.
In Vivo Pharmacological Studies
To study the contribution of systemic sPLA2 to the lipid metabolic phenotype of MMP‐9 deficiency, mice were gavaged with the sPLA2 inhibitor varespladib (10 mg/kg per day). The varespladib stock was prepared in DMSO (Sigma Aldrich), as per the manufacturer's instructions, and diluted as required. Aqueous DMSO solution of the same concentration as in the varespladib working solution (equivalent to DMSO 2.6 μL/kg per day) was used in control experiments (vehicle). The mice were treated with or without varespladib for 2.5 days prior to cholesterol supplementation, and drug treatment was continued. Mice were then euthanized.
Metabolic Studies
Studies were conducted in metabolic cages at the Core Facility of the Cardiovascular Research Center, University of Alberta. Mice were individually housed in Oxymax/CLAMS metabolic chambers (Columbus Instruments) in which O2 consumption, CO2 production, food and water consumption, and movement (x and z) were measured over 2 days and 2 nights.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
RNA was extracted from tissues by homogenizing 30‐ to 50‐mg pieces of frozen tissue at 4°C in 1 mL of TRIzol reagent (Invitrogen) using the Bullet Blender (Next Advance). RNA was isolated from TRIzol according to the manufacturer's instructions, and cDNA was generated from RNA using random primers and Superscript II reverse transcriptase. RNA was quantitated in triplicate to obtain a value representative of the relevant tissue for each mouse. Expression analysis of genes was performed by TaqMan quantitative reverse transcriptase polymerase chain reaction using the ABI 7900 HT sequence detection system (Applied Biosystems). For data normalization, both Gapdh and Actb (to confirm interpretation of data relative to Gapdh) were used as internal standards at steady state. The quantitative reverse transcriptase polymerase chain reaction data are shown relative to Gapdh.
Protein Determinations
Total protein content was measured using the Bio‐Rad Protein Assay (Bio‐Rad Laboratories) or the Pierce BCA protein assay kit (Thermo Fisher Scientific), according to the manufacturers' instructions. Hepatic liver SREBP‐2 protein levels were assessed by immune blotting. Briefly, 15‐ to 25‐mg liver pieces were homogenized using the Bullet Blender at 4°C in a buffer of 5 mmol/L CaCl2, 150 mmol/L NaCl, 0.5 mmol/L, and 25 mmol/L Tris (pH 7.4) with a complete protease inhibitor (Roche). The homogenate was incubated for 1 hour at 37°C with 50 U alkaline phosphatase, NP‐40 was added to a concentration of 1%, and the samples were sonicated and then incubated for 3 hours at 37°C with PNGase (10 U/μL). The homogenate was diluted at 1:5 (vol/vol) with SDS‐PAGE loading buffer (15% SDS, 8 mol/L urea, 10% 2‐mercaptoethanol, 25% glycerol, 0.2 mol/L Tris [pH 6.8]), heated at 37°C for 20 minutes, and subjected to 10% SDS‐PAGE using the SE260 electrophoresis system (Hoefer). Following electrophoresis, proteins were transferred to a nitrocellulose membrane using the TE22 system (Hoefer). Proteins were visualized with Ponceau S acid stain, blocked in 5% bovine serum albumin in 20 mmol/L Tris and 150 mmol/L NaCl (pH 7.4) containing 0.1% Tween‐20, and probed overnight with primary antibodies to SREBP‐2. The membrane was then probed for 30 minutes with secondary antibodies and washed in 20 mmol/L Tris and 150 mmol/L NaCl (pH 7.4) containing 0.1% Tween‐20 to remove excess antibody. Immunoreactivity was revealed using enhanced chemiluminescence detection reagent.
In Vitro Assays of PLA2 Enzymatic Activity and Inhibitor Profiles
PLA2 activity was measured by 2 different methods. The Fernandez‐Patron laboratory used the commercial assay kit (Cayman) with diheptanoyl thio‐phosphatidylcholine as substrate, per the manufacturer's instructions, to measure PLA2 activity in tissue homogenates and plasma and to measure the ex vivo tissue release of sPLA2 activity.3 Confirmatory studies and extended characterization of sPLA2 biochemical properties were performed by the Lambeau laboratory using the highly sensitive [3H]‐oleic acid Escherichia coli membrane assay.26 Because E coli membranes are rich in phosphatidyl ethanolamine and do not contain phosphatidylcholine, the Cayman kit and E coli assay methods display different sensitivities. For comparative biochemical characterization, sPLA2 activity was assessed in the presence and absence of a panel of inhibitors of various enzyme classes. Briefly, samples were incubated for 15 minutes at room temperature prior to the assay in the presence of different inhibitors: dithiothreitol 10 mmol/L (reducing agent, incubation 30 minutes at 56°C; Euromedex), EDTA 40 mmol/L (a nonspecific inhibitor of Ca2+‐dependent PLA2; Euromedex), MJ33 30 μmol/L (PLA2 inhibitor; Santa Cruz Biotechnology), KH064 10 μmol/L (sPLA2 inhibitor; Sigma‐Aldrich), YM 26734 10 μmol/L (sPLA2 inhibitor; Tocris Bioscience), arachidonyl trifluoromethyl ketone (10 μmol/L (cytosolic PLA2 and calcium‐independent PLA2 inhibitor; Interchim), N‐(p‐amylcinnamoyl) anthranilic acid (100 μmol/L; PLA2 inhibitor, Calbiochem; EMD Millipore), bromoenol lactone (3 μmol/L; calcium‐independent PLA2 inhibitor; Interchim), or heparin 100 μg/mL (sPLA2 inhibitor; Sigma‐Aldrich).
Extraction of Lipids From Tissues
Lipids were extracted from tissues, as described previously.27 Tissue was homogenized in water (8–30 μL/mg) using the Bullet Blender. The homogenate or lysate (500–1000 μL) was mixed at a ratio of 3:4:8 aqueous homogenate/lysate:methanol:chloroform and vortexed for 1 minute, and then centrifuged for 15 minutes to separate phases. The bottom phase containing lipids was transferred into a new tube, dried under argon, and resuspended in 100 μL chloroform. The residue was used for lipid analysis.
Chromatographic Determinations of Lipid Content
Fast‐performance liquid chromatography analyses of plasma lipoproteins were performed at the Lipid and Lipid Metabolite Analysis Core Facility, part of the Women and Children's Health Research Institute and Faculty of Medicine and Dentistry at the University of Alberta. Plasma lipoprotein classes were resolved on a Superose 6 10/300 gel‐filtration fast‐performance liquid chromatography column run isocratically with 50 mmol/L NaCl buffer on a 1200 series high‐performance liquid chromatography system (Agilent Technologies). Cholesterol or triglycerides in lipoproteins were detected enzymatically by in‐line reaction at 37°C.
Thin layer chromatography plates were loaded with lipid extracts suspended in chloroform solution (10–40 μL) of sample. The sample was allowed to dry, and the plate was developed at room temperature for 40 minutes. Neutral lipids were separated using a diethyl ether–glacial acetic acid–n‐hexane developing solvent system, whereas phospholipids were separated in a chloroform–methanol–water solvent system, as described.28 Lipids were detected by exposure of the plate to iodine vapor.
14C‐Cholesterol and 3H‐Sitostanol Absorption Assay
Cholesterol absorption was measured using the fecal dual‐isotope ratio method.15 Briefly, mice were fasted overnight then intragastrically dosed with a mixture of 2 μCi (5,6‐3H)‐sitostanol (American Radiolabeled Chemicals Inc) and 1 μCi (4‐14C)‐cholesterol (New England Nuclear Corp). The mice were returned to fresh cages and refed, and stool was collected over the following 3 days. Aliquots of stool (1 g) were extracted with chloroform:methanol (2:1, vol/vol). The ratio of 14C to 3H was measured, and then the percentage of cholesterol absorption was determined, as described previously.29
Bile Acids Content in Stools
Fecal bile acid content was determined using a method described previously.30, 31, 32 Stools were collected daily over 3 consecutive days, after which the stool was dried, weighed, and ground into a fine powder. Bile acids were extracted from 1‐g aliquots of the sample, and total bile acid content was quantified using a commercial assay kit (BioVision).
Liver Function Analysis
Liver function was assessed by determining levels of alkaline phosphatases, alanine aminotransferases, and aspartate aminotransferases in plasma using standard laboratory assays.
Statistical Analysis
Unless otherwise indicated, the results are reported as mean±SEM. Data were analyzed with SigmaPlot 11 software (Systat Software) using 1‐way ANOVA or Student t test, as appropriate. ANOVA and repeated‐measures analysis was conducted to determine differences among groups in time‐course experiments. Differences between groups were analyzed with the nonparametric t test (Mann–Whitney) with statistically significant values of P<0.05.
Results
MMP‐9 Modulates Systemic Metabolism and Cholesterol Homeostasis
Chow‐fed Mmp9 −/− mice exhibited several metabolic abnormalities at baseline compared with age‐matched WT mice. In the Mmp9 −/− mice, locomotor activity and respiratory exchange ratio (a measure of energy substrate preference) were lower than in WT mice during the day, suggesting a relatively increased diurnal reliance on fatty acids as fuel; however, the respiratory exchange ratio during the night was higher than in WT mice, suggesting a switch to glucose oxidation. Oxygen consumption, energy expenditure (heat), body weight, and food intake were comparable for Mmp9 −/− and WT mice (Figure S1).
Analysis of hepatic lipids revealed further metabolic abnormalities in Mmp9 −/− mice including higher levels of triglycerides and cholesteryl esters in the liver of Mmp9 −/− mice compared with WT mice, particularly when refed after overnight fasting (Figure S2A). In Mmp9 −/− mice, plasma levels of triglycerides and cholesterol were higher in very low‐density lipoproteins and in high‐density lipoproteins than in WT mice (Figure 1A). The higher plasma cholesterol levels in Mmp9 −/− mice were not associated with abnormal cholesterol absorption, as demonstrated by the radioactive 14C‐cholesterol and 3H‐sitostanol absorption assay (Figure 1B). Cholesterol excretion in the stool was unchanged in MMP‐9–deficient mice (Figure S2B). There were no statistically significant changes in plasma bile acid content (mean levels were increased on average by 7.4% in Mmp9 −/− versus WT mice). The bile acid content, however, was higher in the stool of Mmp9 −/− mice fed regular chow than in the stool of WT mice, implying a role for MMP‐9 in the regulation of cholesterol excretion as bile acids (Figure 1C). When the chow was supplemented with 0.15% cholesterol, bile acid excretion remained high in Mmp9 −/− mice and was elevated in WT mice, as expected14 (Figure 1C).
Figure 1.
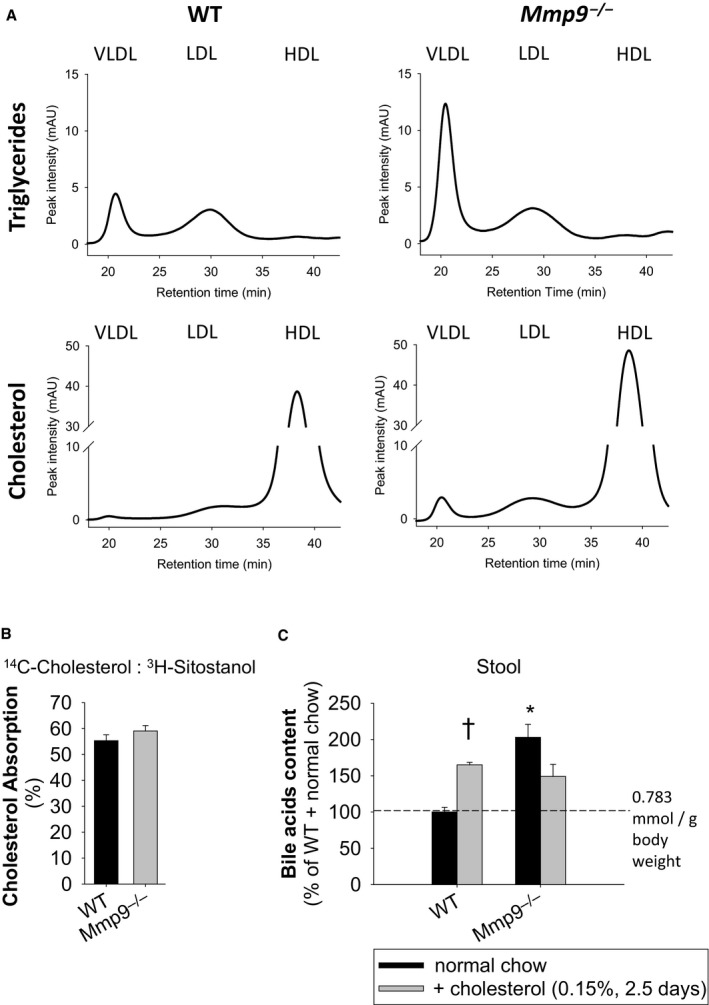
MMP‐9 deficiency is associated with abnormalities in lipid distribution and excretion. A, Triglyceride and cholesterol levels in lipoprotein fractions of plasma separated by FPLC. Traces correspond to pools of plasma from WT and Mmp9 −/− mice (n=4 per genotype). Semiquantitative assessment based on peak heights indicates that Mmp9 −/− mice have 2.7‐, 4.3‐, 1.5‐, and 1.2‐fold increases in VLDL triglycerides, VLDL cholesterol, LDL cholesterol, and HDL cholesterol, respectively (compared with WT mice). The same volume was injected onto the FPLC. B, Cholesterol absorption measured by radioactive 14C‐cholesterol and 3H‐sitostanol absorption assay (n=4 per genotype). C, Bile acid content in mouse stool in response to cholesterol (n=5 for WT with normal chow, n=5 for Mmp9 −/− with normal chow, n=4 for WT with cholesterol, n=5 for Mmp9 −/− with cholesterol). *P<0.05 vs WT. † P<0.05 vs normal chow, t test. FPLC indicates fast‐performance liquid chromatography; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MMP, matrix metalloproteinase; VLDL, very low‐density lipoprotein; WT, wild type.
Quantitative reverse transcriptase polymerase chain reaction analysis indicated no difference between Mmp9 −/− and WT mice in the expression of Nr1h3 (encoding liver X receptor α [LXR‐α], a major regulator of fatty acid synthesis and cholesterol excretion). Fasn (encodes fatty acid synthase) is an LXR‐α target and was also unaltered. Similarly unchanged were Srebf2 (encodes the transcription factor SREBP‐2) and SREBP‐2 target genes such as Hmgcr (encodes 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase, the rate‐limiting enzyme in the cholesterol and isoprenoid synthesis pathways) and Ldlr (encodes the LDL receptor, which is involved in clearance of low‐density lipoprotein from circulation). Paradoxically,2 LXR‐α target genes, the cholesterol transporters ATP‐binding cassette G5 and G8 (encoded by Abcg5 and Abcg8) and the SREBP‐2 target gene Pcsk9 (encodes proprotein convertase subtilisin/kexin type 9, a protein that binds and negatively regulates hepatic LDLR protein levels33) were all significantly higher at baseline in Mmp9 −/− mice than in WT mice (Figure 2).
Figure 2.
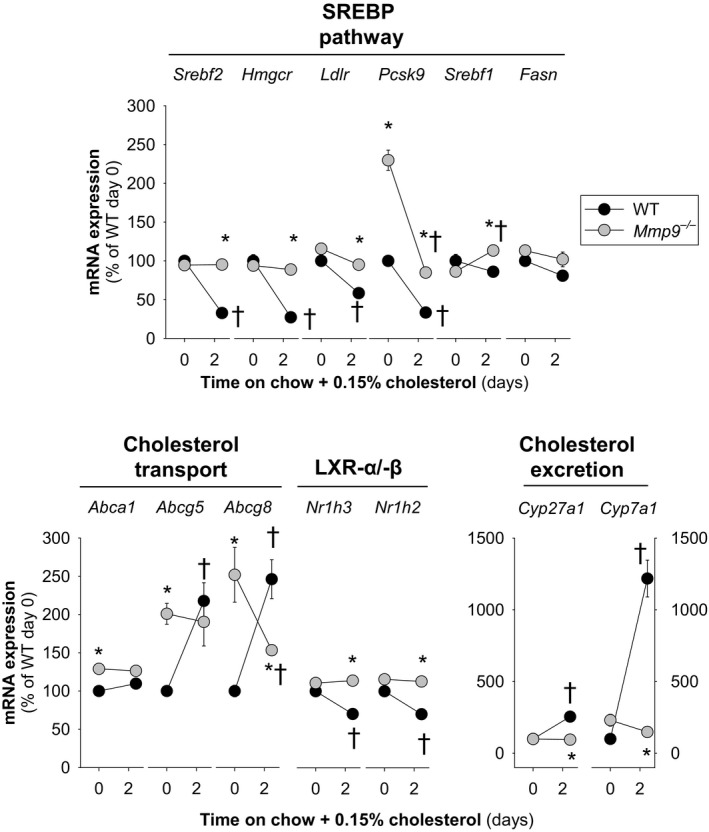
Hepatic transcriptional responses to dietary cholesterol supplementation. Mice were fed regular chow or chow supplemented with cholesterol (0.15%) for up to 2.5 days. Gene expression analysis was conducted at days 0 and 2.5 (n=4 to 5 mice per time point). *P<0.05 vs WT. † P<0.05 vs 0 days on cholesterol, 1‐way repeated‐measures ANOVA. Abca1 indicates ATP‐binding cassette sub‐family A member 1; Abcg5/Abcg8, ATP‐binding cassette sub‐family G member 5/8; Cyp27a1, sterol 27 hydroxylase; Cyp7a1, cholesterol 7 alpha hydroxylase; Fasn, fatty acid synthase; Hmgcr, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; Ldlr, low density lipoprotein receptor; Mmp, matrix metalloproteinase gene; Nr1h3/Nr1h2, liver X receptor α/β; Pcsk9, proprotein convertase subtilisin/kexin type 9; Srebf1, sterol regulatory element binding protein 1; Srebf2, gene for sterol regulatory element binding protein 2; SREBP, sterol regulatory element binding protein; WT, wild type.
Dietary cholesterol inhibits the SREBP‐2 pathway to decrease the hepatic synthesis and uptake of cholesterol. At the same time, oxysterols derived from dietary cholesterol activate LXR‐α signaling, which, in turn, increases both the synthesis of bile acids from hepatic cholesterol and the excretion of cholesterol into the bile.17, 34
As expected, WT mice fed chow supplemented with cholesterol exhibited time‐dependent (Figure 2) and concentration‐dependent (Figure 3) decreases in the expression of key genes involved in cholesterol synthesis and uptake: Srebf2, Hmgcr, Ldlr, and Pcsk9. The expression of Fatty acid synthase, a key enzyme in fatty acid biosynthesis, was concentration‐dependently downregulated by cholesterol. In addition, dietary cholesterol unexpectedly increased Abcg5 and Abcg8, caused a small decrease in Nr1h3 and Nh1h2 and strongly induced the expression of Cyp7a1 and Cyp27a, which encode the rate‐limiting enzymes in the classical (cholesterol 7α‐hydroxylase) and alternate (cholesterol 27α‐hydroxylase) bile acid biosynthesis pathways.
Figure 3.
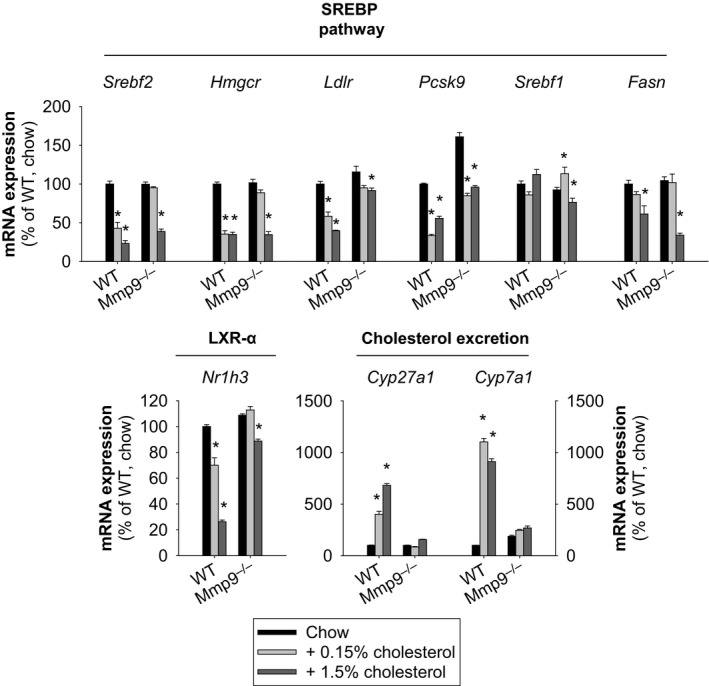
Impact of dietary cholesterol on hepatic transcriptional responses. Mice were fed either regular chow or chow supplemented with cholesterol (0.15% or 1.5%) for 2.5 days. Gene expression analysis was conducted at 0 and 2.5 days (n=4 to 5 mice per group [or treatment]). *P<0.05 vs normal chow for each genotype, all pairwise multiple comparisons vs control group (Holm–Sidak method), ANOVA. Cyp27a1 indicates sterol 27 hydroxylase; Cyp7a1, cholesterol 7 alpha hydroxylase; Fasn, fatty acid synthase; Hmgcr, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; Ldlr, low density lipoprotein receptor; LXR, liver X receptor; Mmp, matrix metalloproteinase gene; Nr1h3/LXR‐α, liver X receptor α; Pcsk9, proprotein convertase subtilisin/kexin type 9; Srebf1, sterol regulatory element binding protein 1; Srebf2, gene for sterol regulatory element binding protein 2; SREBP, sterol regulatory element binding protein; WT, wild type.
In contrast, the gene transcriptional responses to dietary cholesterol were markedly impaired in Mmp9 −/− mice (Figures 2 and 3). These findings identify MMP‐9 as a novel modulator of the hepatic transcriptional responses to dietary cholesterol.
The MMP‐9–Plasma sPLA2 Axis Modulates Hepatic Transcriptional Responses to Dietary Cholesterol
We recently found that in Mmp2 −/− mice, the heart secretes an as yet unidentified PLA2 (cardiac sPLA2), which circulates in plasma, acting as a signal that is governed by MMP‐2 and that modulates lipid metabolism in the liver.2, 3 In Mmp9 −/− mice, plasma sPLA2 activity was significantly higher than in WT plasma but otherwise was normal in the heart, as demonstrated by assaying the generation of free thiol from di‐heptanoyl‐thio‐phosphatidylcholine (substrate) (Figure 4A). The elevated sPLA2 activity in the Mmp9 −/− plasma was confirmed using a highly sensitive (3H)‐oleic acid radiolabeled E coli membranes assay (Figure 4B).
Figure 4.
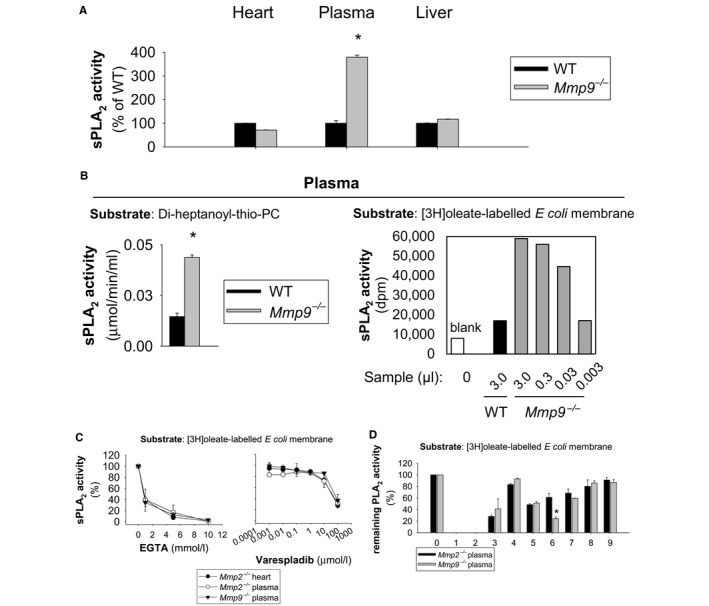
Plasma sPLA 2 activity is elevated by MMP‐9 deficiency. A, sPLA 2 activity in the plasma, liver, and heart of Mmp9 −/− mice (n=4 mice per genotype). *P<0.05 vs WT, t test. B, The elevated plasma PLA 2 activity in Mmp9 −/− mice (compared to WT) was confirmed using 2 unrelated assays: the Cayman sPLA 2 assay kit (substrate: di‐heptanoyl‐thio‐PC; n=4 per genotype; *P<0.05 vs WT, t test) and the 3H‐oleate E coli membrane assay (data are representative of technical duplicates for a pool of 5 mice per genotype). C, EGTA and varespladib inhibition profiles for the sPLA 2 from Mmp9 −/− plasma vs heart and plasma from Mmp2 −/− mice. The analysis was performed in duplicate using samples from pools of 4 mice per genotype using the 3H‐oleate E coli membrane assay. Similar results were obtained using the Cayman assay kit (data not shown). D, Profiling of PLA 2 activity inhibition demonstrates that the plasma sPLA 2 that is present in Mmp2 −/− and Mmp9 −/− mice is the same enzyme or very similar enzymes (pools of plasma: n=5 for Mmp2 −/− and n=5 for Mmp9 −/−). Data are representative of technical duplicates. For comparison, the activity of cardiac sPLA 2 from an Mmp2 −/− mouse (mouse “E” in figure S3 of Hernandez‐Anzaldo et al2) is presented. *P<0.05 vs Mmp2 −/− plasma, t test. The x‐axis values indicate (0) no inhibitor; (1) EDTA, inhibits Ca2+‐dependent PLA 2s; (2) dithiothreitol, sulfhydryl redox agent; (3) MJ33, active site‐directed PLA 2 inhibitor; (4) KH064, sPLA2 inhibitor; (5) YM 26734, sPLA 2 (PLAG2A, PLA2G5) inhibitor; (6) arachidonyl trifluoromethyl ketone, cytosolic PLA2 and iPLA 2 inhibitor; (7) N‐(p‐amylcinnamoyl) anthranilic acid, PLA 2 inhibitor; (8) bromoenol lactone, iPLA2 inhibitor; and (9) heparin, inhibits some sPLA 2s. AACOCF3 indicates arachidonyl trifluoromethyl ketone; ACA, N‐(p‐Amylcinnamoyl) anthranilic acid; BEL, bromoenol lactone; cPLA2, cytosolic phospholipase A2; DTT, dithiothreitol; E. coli, Escherichia coli; EDTA, ethylenediaminetetraacetic acid (divalent metal ion chelator); EGTA, ethylene glycol‐bis(?‐aminoethyl ether)‐tetraacetic acid (Ca2+ chelator); iPLA2, calcium‐independent phospholipase A2; MJ33, 1‐hexadecyl‐3‐trifluoroethylglycero‐sn‐2‐phosphomethanol; Mmp, matrix metalloproteinase gene; MMP, matrix metalloproteinase; MMP‐2, matrix metalloproteinase‐2; MMP‐9, matrix metalloproteinase‐9; PC, phosphatidylcholine; SH, sulfhydryl; sPLA2, secreted phospholipase A2; WT, wild type; YM 26734, 1,1?‐[5‐[3,4‐dihydro‐7‐hydroxy‐2‐(4‐hydroxyphenyl)‐2H‐1‐benzopyran‐4‐yl]‐2,4,6‐trihydroxy‐1,3‐phenylene]bis‐1‐dodecanone.
Similar to the sPLA2 that is present in the plasma of Mmp2 −/− mice, the sPLA2 activity in Mmp9 −/− plasma was calcium dependent and inhibited by the broad‐spectrum sPLA2 inhibitor varespladib35 (Figure 4C). Furthermore, the plasma sPLA2 activities from Mmp2 −/− and Mmp9 −/− mice were inhibited to similar degrees by a panel of PLA2 inhibitors, suggesting that the plasma sPLA2 activities in Mmp2 −/− and Mmp9 −/− mice result from either the same or structurally homologous enzymes (Figure 4D).
Next, we screened for sPLA2 release from various mouse tissues. Only the spleen—not the heart, thymus, or bone marrow—exhibited an increased ex vivo release of sPLA2 in Mmp9 −/− mice relative to WT mice. Consequently, peripheral organs (eg, the spleen) may release sPLA2 into the circulation (Figure S3).
To determine whether the systemic circulating plasma sPLA2 activity mediated the hepatic transcriptional responses to dietary cholesterol in Mmp9 −/− mice, we administered the sPLA2 inhibitor varespladib (or vehicle) to Mmp9 −/− mice prior to dietary cholesterol supplementation (Figure 5). Administration of varespladib for 5 consecutive days fully normalized the levels of plasma sPLA2 activity in Mmp9 −/− mice (Figure 5A). In WT mice, varespladib did not affect the hepatic transcriptional responses to dietary cholesterol (Figure 5B and 5C). In Mmp9 −/− mice, varespladib partially normalized the hepatic transcriptional response to cholesterol for genes in the SREBP‐2 pathway (Figure 5D and Figure S4); however, varespladib failed to affect mRNA levels of the rate‐limiting enzymes in biliary cholesterol synthesis (Cyp7a1 and Cyp27a) (Figure 5D).
Figure 5.
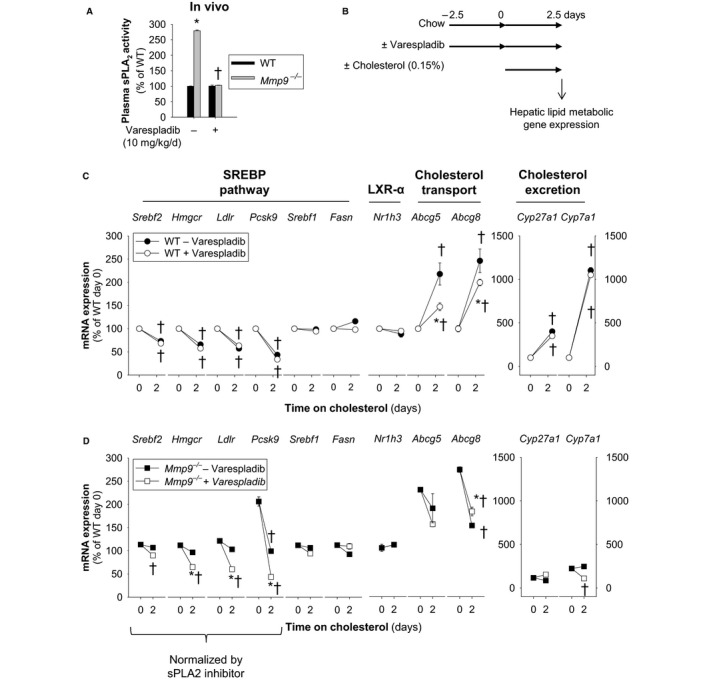
Circulating systemic sPLA 2 modulates hepatic transcriptional responses to dietary cholesterol supplementation. A, Plasma sPLA 2 activity ofMmp9 −/− mice administered the pan‐sPLA 2 inhibitor varespladib (10 mg/kg per day) or vehicle for 5 days (n=4 mice per group). *P<0.05 vs WT, t test. † P<0.05 vs untreated, t test. B, Study protocol for varespladib treatment prior to cholesterol supplementation. Mice were fed either regular chow or chow supplemented with 0.15% cholesterol for 2.5 days. Varespladib treatment (10 mg/kg per day for 5 days) started 2.5 days prior to commencement of cholesterol supplementation of the diet. C, Hepatic expression of lipid metabolic genes in WT mice administered varespladib (10 mg/kg per day; n=8 WT without varespladib and n=8 WT with varespladib mice, n=4 per time point). *P≤0.05 vs WT without varespladib at day 2.5. † P<0.05 vs day 0. All pairwise multiple comparisons vs control group (Holm–Sidak method), ANOVA. D, Hepatic expression of lipid‐metabolic genes in Mmp9 −/− mice administered varespladib (10 mg/kg per day; n=8 Mmp9 −/− without varespladib and n=8 Mmp9 −/− with varespladib, n=4 per time point). *P<0.05 vs Mmp9 −/− without varespladib at day 2.5. † P<0.05 vs day 0. All pairwise multiple comparisons vs control group (Holm–Sidak method), ANOVA. Abca1 indicates ATP‐binding cassette sub‐family A member 1; Abcg5/Abcg8, ATP‐binding cassette sub‐family G member 5/8; Cyp27a1, sterol 27 hydroxylase; Cyp7a1, cholesterol 7 alpha hydroxylase; Fasn, fatty acid synthase; Hmgcr, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; Ldlr, low density lipoprotein receptor; LXR, liver X receptor; Mmp, matrix metalloproteinase gene; MMP, matrix metalloproteinase; Nr1h3/Nr1h2, liver X receptor α/β; Pcsk9, proprotein convertase subtilisin/kexin type 9; sPLA2, secreted phospholipase A2; Srebf1, sterol regulatory element binding protein 1; Srebf2, gene for sterol regulatory element binding protein 2; SREBP, sterol regulatory element binding protein; WT, wild type.
Taken together, the data suggest that MMP‐9 deficiency results in a phenotype of relative hepatic insensitivity to dietary cholesterol mediated, at least in part, by systemic sPLA2 activity that is likely contributed by peripheral organs (Figure S5).
MMP‐9 Does Not Affect Liver Weight or Function in Mice Fed an HFD
Figures 3 and 6A indicate that either increasing dietary cholesterol from moderate (0.15%) to high (1.5%) over 2.5 days or extending cholesterol administration from 2.5 to 6.5 days resulted in the partial normalization of hepatic gene transcriptional responses of Mmp9 −/− mice, suggesting a finite contribution of MMP‐9 to regulation of cholesterol metabolism. We next compared the liver function of Mmp9 −/− and WT mice fed an HFD for 15 weeks (from 5 to 20 weeks of age) versus mice fed a standard fat diet. At 5 weeks of age, Mmp9 −/− and WT mice had similar body weights. The HFD markedly increased the levels of alkaline phosphatases and alanine and aspartate aminotransferases in both WT and Mmp9 −/− mice (versus standard fat diet); however, feeding the HFD did not differently influence plasma lipids,12 body weight, liver weight, or plasma levels of alkaline phosphatases or alanine and aspartate aminotransferases of Mmp9 −/− versus WT mice (Table).
Figure 6.
The MMP system modulates hepatic transcriptional responses to dietary cholesterol. A, Deficiency of MMP‐2, MMP‐7, or MMP‐9 is associated with abnormalities in the hepatic transcriptional responses to dietary cholesterol. Mice were fed either regular chow or chow supplemented with 0.15% cholesterol for 6.5 days. Gene expression analysis was conducted at 0, 2.5, and 6.5 days. Analysis involved 6 WT mice, 8Mmp2 −/− mice, 5 Mmp7 −/− mice, and 5 Mmp9 −/− mice. For clarity, the symbols indicating statistically significant differences were excluded. An expanded version of these analyses is presented in Figure S6. B, Proposed model for regulation of cholesterol homeostasis by systemic MMP activity. MMP deficiencies (due to genetic deletion or functional blockade) can alter the hepatic cholesterol metabolism. The mechanism by which MMPs act may or may not require the release of sPLA 2 activity from peripheral organs and is governed by tissue inhibitors of metalloproteinase. Cyp27a1 indicates sterol 27 hydroxylase; Cyp7a1, cholesterol 7 alpha hydroxylase; Fasn, fatty acid synthase; Hmgcr, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; Ldlr, low density lipoprotein receptor; Mmp, matrix metalloproteinase gene; MMP, matrix metalloproteinase; Nr1h3, liver X receptor α; Pcsk9, proprotein convertase subtilisin/kexin type 9; sPLA2, secreted phospholipase A2; Srebf1, sterol regulatory element binding protein 1; Srebf2, gene for sterol regulatory element binding protein 2; TIMP, tissue inhibitor of metalloproteinase; WT, wild type.
Table 1.
Effect of MMP‐9 Deficiency on Body and Liver Weight and Plasma Hepatic Enzyme Activities
| Variable | SFD | HFD | ||
|---|---|---|---|---|
| Mmp9 +/+ | Mmp9 −/− | Mmp9 +/+ | Mmp9 −/− | |
| Experiments, no. | 7 | 7 | 7 | 11 |
| Body weight start, g | 21±1.2 | 19±1.0 | 20±1.0 | 17±1.4 |
| Body weight end, g | 34±1.8 | 33±1.6 | 39±1.3 | 38±2.2 |
| Liver weight, mg | 1153±135 | 963±55 | 1343±60 | 1533±215a |
| AP, U/L | 39±0.9 | 37±1.8 | 246±51a | 221±31a |
| AST, U/L | 24±2.8 | 28±4.5 | 219±54a | 302±45a |
| ALT, U/L | 14.0±1.8 | 13±1.8 | 156±46a | 272±56a |
Male mice were fed an SFD or an HFD for 15 weeks. Data are shown as mean±SEM of the number of experiments in each group. AP indicates alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; HFD, high fat diet; MMP, matrix metalloproteinase; SFD, standard fat diet.
P<0.05 vs SFD. Diet composition is described in Materials and Methods.
The MMP System Modulates Cholesterol Metabolism
We recently discovered numerous differences in expression of lipid metabolic genes between WT and MMP‐2–deficient mice including an upregulation of genes in the SREBP‐2 pathway2, 32, 33 and impaired hepatic transcriptional responses to dietary cholesterol.2
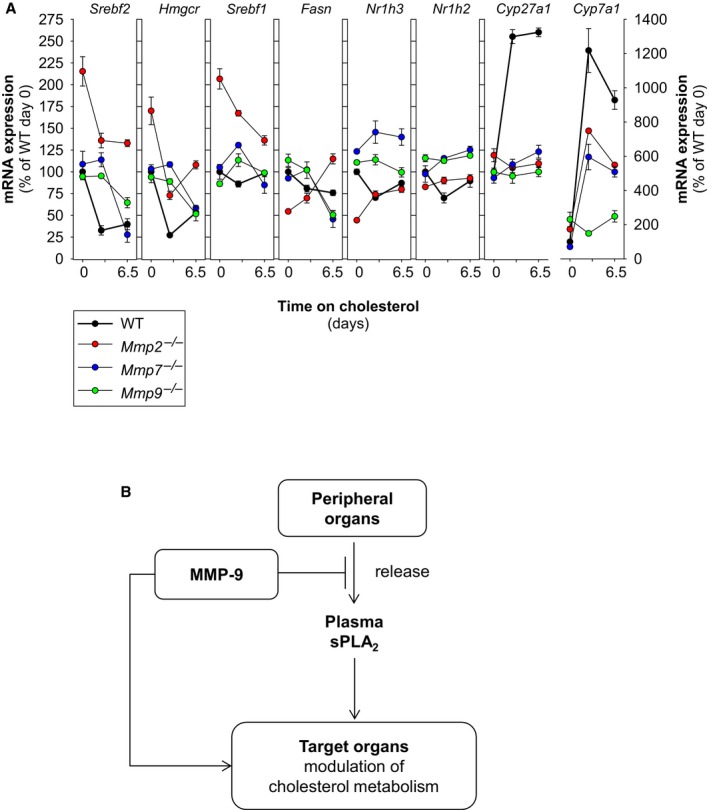
We further compared the hepatic gene transcriptional responses to dietary cholesterol in mice deficient in MMP‐22 and in MMP‐9. Compared with WT mice, mice deficient in either MMP‐2 or MMP‐9 exhibited abnormal regulation of genes involved in cholesterol metabolism (Figure 6A); however, the transcriptional responses to dietary cholesterol of MMP‐2–deficient mice were, generally, not the same as those of MMP‐9–deficient mice. Chow‐fed MMP‐2–deficient mice, for example, exhibited higher Srebf2 mRNA expression than MMP‐9–deficient and WT mice (Figure 6A and reference2). When the chow was supplemented with cholesterol for 2.5 days, the Srebf2 mRNA levels of MMP‐2–deficient mice dropped but remained above those of WT mice. Chow‐fed MMP‐9–deficient mice exhibited similar Srebf2 mRNA expression as WT mice but had a delayed response to dietary cholesterol supplementation. The response to cholesterol of Fasn was also different in MMP‐2–deficient and MMP‐9–deficient mice. Deficiency of either MMP‐2 or MMP‐9 markedly impaired the response of Cyp27a1 and Cyp7a1 to dietary cholesterol supplementation, whereas WT mice had a dramatic response.
Taken together, our results suggest that individual MMPs can modulate the hepatic transcriptional responses to dietary cholesterol in an MMP‐specific fashion. This notion was supported by analysis of mice deficient in MMP‐7, a “minimal” MMP produced in Kupffer cells and hepatocytes.36 MMP‐7 contains a conserved catalytic domain but lacks the fibronectin repeats and hemopexin domain present in MMP‐2 and MMP‐9.4 Deficiency of MMP‐7 induced hepatic insensitivity to dietary cholesterol that resembled the phenotype of MMP‐9–deficient mice rather than that of MMP‐2 deficient mice (Figure 6A).
Systemic MMP activity is regulated by tissue inhibitors of metalloproteinase (TIMPs) that form MMP/TIMP complexes37 that can affect MMP activation, activity, and binding to interaction partners and substrates.38 TIMP‐2 is both an activator of MMP‐2 and a broad‐spectrum MMP inhibitor in a concentration‐dependent fashion.38, 39, 40 TIMP‐2–deficient mice have an increased susceptibility to HFD‐induced obesity.41 Figure S6 shows that Timp2 −/− mice exhibit abnormalities in both hepatic lipid gene expression and transcriptional responses to dietary cholesterol resembling those of both WT and MMP‐deficient mice. Timp2 −/− mice, for example, exhibited decreased mRNA levels of Srebf2 and Hmgcr when fed chow, and their response to dietary cholesterol supplementation was different from those of Mmp2 −/−, Mmp7 −/−, Mmp9 −/−, and WT mice (Figure S6).
Additional studies confirmed that MMP expression affects mRNA expression of multiple lipid metabolic genes and their transcriptional responses to dietary cholesterol in the cardiohepatic circuit (Figure S7).
Discussion
Our results identify MMP‐9 as a new modulator of cholesterol metabolism. Furthermore, the results suggest that dysregulation of MMP‐9 activity (and the MMP system as a whole) can alter the hepatic transcriptional responses to dietary cholesterol (Figure 6B), resulting in metabolic disorder that could lead to atherosclerosis and coronary heart disease.
Our study also suggests a novel endocrine mechanism that modulates the sensitivity of the liver to cholesterol: the production and secretion of a unique sPLA2 with plasma levels that could reflect a cumulative release from multiple peripheral organs. Similar to MMP‐2,2 MMP‐9 acts as a negative regulator of sPLA2 release from the source tissue into the circulation. Our current enzymological data suggest that the plasma sPLA2 activity that is increased in MMP‐9– and MMP‐2–deficient plasma corresponds to 1 enzyme (or to homologous enzymes). Proteomic studies are under way in our laboratories to identify the relevant variants of sPLA2.
It is noteworthy that in MMP‐2–deficient mice, the heart is a major source of plasma sPLA2 activity2, whereas in MMP‐9–deficient mice, the spleen is a likely source of plasma sPLA2. The reason for this apparent MMP‐dependent tissue specificity warrants further investigation, and a possible mechanism has been postulated.42 This MMP‐9–plasma sPLA2 axis profoundly influences transcription responses of lipid metabolic genes to dietary cholesterol in the liver, particularly those of enzymes in the SREBP‐2 pathway, which is a major regulator of hepatic cholesterol biosynthesis and uptake.43, 44 Although intestinal cholesterol absorption is normal in Mmp9 −/− mice, plasma cholesterol and bile acid excretion are elevated. Most intriguingly, the mRNA levels of Cyp7a1 and Cyp27a1—the rate‐limiting enzymes in the classical and alternative bile acid biosynthesis pathways,13, 14 respectively—are significantly insensitive to dietary cholesterol supplementation. These findings clearly identify MMP‐9 as an important modulator of cholesterol homeostasis and suggest the existence of MMP‐regulated pathways of bile acid synthesis and cholesterol excretion. This latter notion is supported by earlier research linking PLA2 activity to gallstone formation,45, 46, 47 although these earlier studies do not invoke regulation of either PLA2 activity or biliary cholesterol synthesis by MMPs. Similarly, PLA2 activity has been linked previously to remodeling of lipoproteins and cardiovascular disease,48, 49, 50, 51 although these earlier studies focused on group II PLA2 (PLA2G2A; which is not made by C57BL mice) and lipoprotein lipase, PLA2s that do not fit the activity or inhibition profile of the plasma sPLA2 activity that is elevated in mice deficient in either MMP‐9 (as shown in this paper) or MMP‐2.2 Previous research has attributed roles in cholesterol metabolism to several sPLA2 family members including PLA2G1B, PLA2G2E, PLA2G5, PLA2G10, and PLA2G12B (the latter is not catalytically active).52, 53, 54, 55, 56, 57 These earlier findings are consistent with our current results.
It appears that MMP‐9 affects cholesterol metabolism in ways that we could not predict. We did not expect, for example, that the mRNA levels of ABCG5/8 would be higher in the liver of Mmp9 −/− compared with WT mice fed chow or that CYP7 and CYP27 gene expression would remain almost unchanged when Mmp9 −/− mice were fed cholesterol. These findings are very interesting. Notably, it has been reported previously that PLA2G10 influences gene expression of ABCG5/8 and LXR‐α.56 In the case of the former gene, PLA2G10 activity works in a manner opposite to that of MMP‐9 deficiency, whereas in the case of the LXR‐α, PLA2G10 works in a manner similar to MMP‐2‐deficiency.2 Consequently, it appears that the convergence of MMP‐ and PLA2‐dependent pathways can strongly affect lipid‐metabolic gene transcription in somewhat unintuitive ways.
Our operating hypothesis is that sPLA2 release into plasma is globally regulated by proinflammatory cytokines and MMPs in an MMP‐ and tissue‐specific fashion.2 Downstream hydrolysis of target phospholipids by the sPLA2 might influence liver metabolism and affect cholesterol homeostasis. Importantly, not all lipid‐metabolic abnormalities associated with MMP‐9 deficiency were normalized by systemic sPLA2 inhibition using varespladib; therefore, other MMP‐9–dependent but sPLA2 activity–independent mechanisms are likely at play. A putative mechanism is the MMP‐9 proteolytic processing of cell membrane lipoprotein receptors and lipid transporters such as CD36.58 Furthermore, MMPs bind to lipoprotein and cargo receptors.59, 60 The LDLR‐related protein 1 (LRP1) is a member of the LDLR superfamily, also known as CD91.59 LRP1 is an endocytic receptor for multiple extracellular ligands including apolipoprotein E–containing lipoproteins such as chylomicron remnants (which carry dietary cholesterol and other lipids to the liver) and plasma MMPs such as MMP‐2, MMP‐9, and MMP‐13, including complexes of these MMPs with TIMPs. LRP1‐mediated endocytosis removes MMPs from the circulation.59, 60 The hemopexin domain of MMP‐9, which is conserved in MMP‐2 but absent in MMP‐7, contains the high‐affinity binding site required for LRP‐mediated endocytosis of MMP‐9. The endocytosis of MMP‐9 results in catabolism of MMP‐9 and thus may play a major role in modulating remodeling of the extracellular matrix by regulating extracellular proteinase activity. Conversely, LRP1‐mediated endocytosis of MMPs reduces cell surface levels of LRP1. Consequently, MMPs compete with apolipoprotein E–containing lipoproteins such as chylomicron remnants and intermediate‐density LDL for LRP‐mediated endocytosis, providing mechanisms through which the MMP system can influence systemic lipid metabolism. Further research is warranted to establish the precise mechanisms by which MMPs regulate cholesterol metabolism.
Some of the effects of MMP‐9 deficiency were obliterated when the cholesterol level of the diet was sufficiently high (eg, by supplementation of the chow diet with 1.5% cholesterol or by prolonged administration of an HFD). These data suggest that the intracellular cholesterol‐sensing mechanisms remain functional in MMP‐9 deficiency. Furthermore, the individual contribution of MMP‐9 to cholesterol homeostasis, although significant, may be relatively small. Most interestingly, similar to MMP‐9–deficient mice, the lack of any of MMP‐2, MMP‐7, or TIMP‐2 resulted in a significant perturbation of hepatic transcriptional responses to dietary cholesterol. These observations demonstrate a potentially important role of MMP‐9 (and the MMP/TIMP system as a whole) in the modulation of cholesterol metabolism. As such, the current research substantially expands previous reports by our groups2, 3, 11, 12, 61, 62, 63, 64, 65 and other investigators23, 41, 66 and points to important metabolic actions of MMPs.
In most in vitro and in vivo studies of cholesterol metabolism, alterations in MMPs and TIMPs are not normally monitored; therefore, detection of the modulatory effects of individual MMPs and TIMPs on cholesterol metabolism has likely been precluded. These effects could be profoundly important if ≥1 MMP or TIMP was altered, genetically or pharmacologically, and should not be neglected.
Insufficiencies in hepatic cholesterol uptake tend to elevate plasma cholesterol favoring extrahepatic cholesterol deposition and the development of atherosclerotic plaque at sites of vascular injury, a process in which the role of MMPs (including MMP‐9) is well documented but poorly understood.21, 22, 67, 68 Restoring the sensitivity of the liver to cholesterol is crucial for managing hypercholesterolemia and atherosclerosis.18, 69, 70, 71, 72 Our data suggest that cholesterol homeostasis could be targeted through broad‐spectrum pharmacological manipulation of the proteolytic activity of MMPs. The efficacy of therapeutic strategies directed at decreasing plasma cholesterol would be enhanced if MMP‐2, MMP‐7, and MMP‐9 levels were kept normal. Whether elevating these MMPs above normal levels would increase hepatic cholesterol sensitivity warrants further investigation and could be a novel therapeutic target for restoring sensitivity of the liver to cholesterol.
In the current study, we could not detect statistically significant changes for liver weight or plasma aspartate and alanine aminotransferase concentrations between Mmp9 −/− and Mmp9 +/+ mice on an HFD or a standard fat diet; however, these observations do not exclude a role for MMP‐9 in inflammation. Like MMP‐2, MMP‐9 has been implicated in hepatitis and liver fibrosis.73, 74, 75, 76, 77, 78, 79 In an experimental murine colitis model, Mmp9 −/− mice show an attenuated intestinal permeability and a lower degree of intestinal inflammation.80 In addition, leukocyte recruitment resulting in the induction of proinflammatory cytokine expression was markedly impaired in the liver of Mmp9 −/− animals after hepatic ischemia–reperfusion injury.81 Future studies should investigate the expression of proinflammatory markers and immune cell infiltration in the livers of Mmp9 −/− versus Mmp9 +/+ mice.
Further research is also warranted before the current findings can be extrapolated to human physiology. Most of our knowledge about the inflammatory response has been derived from studies using mice; how closely mice mimic the inflammatory response in humans remains a matter of debate.82, 83 In summary, this study and our previous reports1 identify MMPs as new modulators of lipid metabolism. We propose that the dysregulation of the MMP system can contribute to the development of metabolic disorders that could, ultimately, lead to atherosclerosis and coronary heart disease.
Sources of Funding
This study was supported by operating grants from the Canadian Institutes of Health Research (CIHR) and Natural Sciences and Engineering Research Council of Canada (to Fernandez‐Patron). HPLC analysis of lipids was performed at the Faculty of Medicine and Dentistry Lipid Analysis Core that receives partial funding from the Women and Children Health Research Institute. The Center for Molecular and Vascular Biology is supported by the “Programma Financiering KU Leuven” (Project PF/10/014).
Disclosures
None.
Supporting information
Figure S1. Systemic metabolic profile of MMP‐9 deficient mice. A, Oxygen consumption. B, Carbon dioxide production. C, Heat/energy expenditure (normalized to body weight). D through F, Locomotor activity. G, Body weight. H, Total food consumption. The studies were conducted in metabolic cages (n=7 WT mice, n=5 Mmp9 −/− mice). *P≤0.05 vs WT. MMP indicates matrix metalloproteinase; WT, wild type.
Figure S2. Supplemental quantitative analysis of lipids. A, Esterified lipids were elevated in livers from Mmp9 −/− mice compared with livers from WT mice, particularly when the mice were fasted and refed a high‐carbohydrate diet. In contrast to Mmp9 −/− mice, Mmp7 −/− mice had a different hepatic lipid profile (n=4 mice per group, except n=3 for Mmp7 −/− fasted and refed). † P≤0.05 vs fasted for each genotype.*P≤0.05 vs WT fasted and refed. B, Cholesterol excretion was unchanged in Mmp9 −/− mice (n=4 mice per genotype). WT indicates wild type.
Figure S3. Spleen is a likely source of plasma sPLA2 in MMP‐9–deficient mice. sPLA2 activity was analyzed in duplicate using pools of the indicated tissues (n=3 WT and n=4 Mmp9 −/− mice). *P<0.05 vs WT. MMP indicates matrix metalloproteinase; sPLA2, secretory phospholipase A2; WT, wild type.
Figure S4. SREBP‐2 expression in response to dietary choloesterol and varespladib. Western blot (left) showing amount of hepatic SREBP‐2 protein in Mmp9 −/− mice in response to varespladib and dietary cholesterol supplementation. The experiment involved 4 to 5 mice per group (or treatment). For analysis, livers were pooled and homogenized, and the fraction containing nuclei was subjected to Western blot analysis with SREBP‐2 antibodies. Quantitative analysis (right) for 2 independent preparations and Western blots. *P≤0.05 vs untreated. SREBP‐2 indicates sterol regulatory element binding protein 2.
Figure S5. Proposed model. MMP‐9 regulates cholesterol metabolism through PLA2‐dependent and ‐independent mechanisms. Important elements are peripheral organs (eg, the spleen) acting as a source of plasma sPLA2 activity and MMP‐9 (inhibitor of sPLA2 release from peripheral organs). Once in the circulation, sPLA2 acts on plasma lipoproteins or target organs (eg, the liver) to release lipid mediators from phospholipids that ultimately influence cholesterol metabolism. Furthermore, the direct action of MMP‐9 in the liver may influence hepatic cholesterol through as yet unclear PLA2‐independent pathways. MMP indicates matrix metalloproteinase; PLA2, phospholipase A2; sPLA2, secretory phospholipase A2.
Figure S6. Supplement to Figure 7A containing the quantitative analysis of hepatic transcriptional responses to dietary cholesterol for the indicated genes and genotypes (n=6 WT mice, n=8 Mmp2 −/− mice, n=5 Mmp7 −/− mice, n=5 Mmp9 −/− mice, n=5 Timp2 −/− mice). *P≤0.05 vs WT. † P≤0.05 vs 0 days on cholesterol. WT indicates wild type.
Figure S7. Extended quantitative analysis of the relative mRNA expression of cardiac and hepatic lipid‐metabolic and metalloproteinase genes and their response to dietary supplementation with 0.15% cholesterol for 2.5 days in mice deficient in one of several MMPs (n=6 WT mice, n=8 Mmp2 −/− mice, n=5 Mmp7 −/− mice, n=5 Mmp9 −/− mice). Slots without bars: Gene expression was not determined. MMP indicates matrix metalloproteinase; WT, wild type.
Acknowledgments
The authors thank L. Frederix for expert technical assistance.
(J Am Heart Assoc. 2016;5:e004228 doi: 10.1161/JAHA.116.004228)
Note
Contributor Information
Roger H. Lijnen, Email: roger.lijnen@med.kuleuven.be.
Carlos Fernandez‐Patron, Email: cf2@ualberta.ca.
References
- 1. Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. [DOI] [PubMed] [Google Scholar]
- 2. Hernandez‐Anzaldo S, Berry E, Brglez V, Leung D, Yun TJ, Lee JS, Filep JG, Kassiri Z, Cheong C, Lambeau G, Lehner R, Fernandez‐Patron C. Identification of a novel heart‐liver axis: matrix metalloproteinase‐2 negatively regulates cardiac secreted phospholipase A2 to modulate lipid metabolism and inflammation in the liver. J Am Heart Assoc. 2015;4:e002553 doi: 10.1161/JAHA.115.002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berry E, Hernandez‐Anzaldo S, Ghomashchi F, Lehner R, Murakami M, Gelb MH, Kassiri Z, Wang X, Fernandez‐Patron C. Matrix metalloproteinase‐2 negatively regulates cardiac secreted phospholipase A2 to modulate inflammation and fever. J Am Heart Assoc. 2015;4:e001868 doi: 10.1161/JAHA.115.001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silvello D, Narvaes LB, Albuquerque LC, Forgiarini LF, Meurer L, Martinelli NC, Andrades ME, Clausell N, dos Santos KG, Rohde LE. Serum levels and polymorphisms of matrix metalloproteinases (MMPs) in carotid artery atherosclerosis: higher MMP‐9 levels are associated with plaque vulnerability. Biomarkers. 2014;19:49–55. [DOI] [PubMed] [Google Scholar]
- 6. Wagsater D, Zhu C, Bjorkegren J, Skogsberg J, Eriksson P. MMP‐2 and MMP‐9 are prominent matrix metalloproteinases during atherosclerosis development in the Ldlr(‐/‐)Apob(100/100) mouse. Int J Mol Med. 2011;28:247–253. [DOI] [PubMed] [Google Scholar]
- 7. Ii H, Hontani N, Toshida I, Oka M, Sato T, Akiba S. Group IVA phospholipase A2‐associated production of MMP‐9 in macrophages and formation of atherosclerotic lesions. Biol Pharm Bull. 2008;31:363–368. [DOI] [PubMed] [Google Scholar]
- 8. Fiotti N, Altamura N, Fisicaro M, Carraro N, Uxa L, Grassi G, Torelli L, Gobbato R, Guarnieri G, Baxter BT, Giansante C. MMP‐9 microsatellite polymorphism and susceptibility to carotid arteries atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1330–1336. [DOI] [PubMed] [Google Scholar]
- 9. Zile MR, Desantis SM, Baicu CF, Stroud RE, Thompson SB, McClure CD, Mehurg SM, Spinale FG. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail. 2011;4:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ikonomidis JS, Barbour JR, Amani Z, Stroud RE, Herron AR, McClister DM Jr, Camens SE, Lindsey ML, Mukherjee R, Spinale FG. Effects of deletion of the matrix metalloproteinase 9 gene on development of murine thoracic aortic aneurysms. Circulation. 2005;112:I242–I248. [DOI] [PubMed] [Google Scholar]
- 11. Bauters D, Van Hul M, Lijnen HR. Gelatinase B (MMP‐9) gene silencing does not affect murine preadipocyte differentiation. Adipocyte. 2014;3:50–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Hul M, Piccard H, Lijnen HR. Gelatinase B (MMP‐9) deficiency does not affect murine adipose tissue development. Thromb Haemost. 2010;104:165–171. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest. 2012;122:1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. [DOI] [PubMed] [Google Scholar]
- 15. Yu L, Gupta S, Xu F, Liverman AD, Moschetta A, Mangelsdorf DJ, Repa JJ, Hobbs HH, Cohen JC. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem. 2005;280:8742–8747. [DOI] [PubMed] [Google Scholar]
- 16. Repa JJ, Mangelsdorf DJ. Nuclear receptor regulation of cholesterol and bile acid metabolism. Curr Opin Biotechnol. 1999;10:557–563. [DOI] [PubMed] [Google Scholar]
- 17. Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. [DOI] [PubMed] [Google Scholar]
- 18. Goldstein MR, Mascitelli L, Pezzetta F. Cholesterol, statins, and mortality. Lancet. 2008;371:1161; author reply 1162‐1163. [DOI] [PubMed] [Google Scholar]
- 19. Brown MS, Goldstein JL. Koch's postulates for cholesterol. Cell. 1992;71:187–188. [DOI] [PubMed] [Google Scholar]
- 20. Brown MS, Goldstein JL. Lowering plasma cholesterol by raising LDL receptors. N Engl J Med. 1981;305:515–517. [DOI] [PubMed] [Google Scholar]
- 21. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase‐9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beaudeux JL, Giral P, Bruckert E, Foglietti MJ, Chapman MJ. Matrix metalloproteinases, inflammation and atherosclerosis: therapeutic perspectives. Clin Chem Lab Med. 2004;42:121–131. [DOI] [PubMed] [Google Scholar]
- 24. Itoh T, Tanioka M, Matsuda H, Nishimoto H, Yoshioka T, Suzuki R, Uehira M. Experimental metastasis is suppressed in MMP‐9‐deficient mice. Clin Exp Metastasis. 1999;17:177–181. [DOI] [PubMed] [Google Scholar]
- 25. Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, Liang G. Overexpression of insig‐1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin‐stimulated lipogenesis. J Clin Invest. 2004;113:1168–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rouault M, Bollinger JG, Lazdunski M, Gelb MH, Lambeau G. Novel mammalian group XII secreted phospholipase A2 lacking enzymatic activity. Biochemistry. 2003;42:11494–11503. [DOI] [PubMed] [Google Scholar]
- 27. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 28. Krell K, Hashim SA. Measurement of serum triglycerides by thin‐layer chromatography and infrared spectrophotometry. J Lipid Res. 1963;4:407–412. [PubMed] [Google Scholar]
- 29. Turley SD, Herndon MW, Dietschy JM. Reevaluation and application of the dual‐isotope plasma ratio method for the measurement of intestinal cholesterol absorption in the hamster. J Lipid Res. 1994;35:328–339. [PubMed] [Google Scholar]
- 30. Daggy BP, O'Connell NC, Jerdack GR, Stinson BA, Setchell KD. Additive hypocholesterolemic effect of psyllium and cholestyramine in the hamster: influence on fecal sterol and bile acid profiles. J Lipid Res. 1997;38:491–502. [PubMed] [Google Scholar]
- 31. Turley SD, Daggy BP, Dietschy JM. Effect of feeding psyllium and cholestyramine in combination on low density lipoprotein metabolism and fecal bile acid excretion in hamsters with dietary‐induced hypercholesterolemia. J Cardiovasc Pharmacol. 1996;27:71–79. [DOI] [PubMed] [Google Scholar]
- 32. Suckling KE, Benson GM, Bond B, Gee A, Glen A, Haynes C, Jackson B. Cholesterol lowering and bile acid excretion in the hamster with cholestyramine treatment. Atherosclerosis. 1991;89:183–190. [DOI] [PubMed] [Google Scholar]
- 33. Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(suppl):S172–S177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch‐like control of SREBP‐2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oslund RC, Cermak N, Gelb MH. Highly specific and broadly potent inhibitors of mammalian secreted phospholipases A2. J Med Chem. 2008;51:4708–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang CC, Chuang JH, Chou MH, Wu CL, Chen CM, Wang CC, Chen YS, Chen CL, Tai MH. Matrilysin (MMP‐7) is a major matrix metalloproteinase upregulated in biliary atresia‐associated liver fibrosis. Mod Pathol. 2005;18:941–950. [DOI] [PubMed] [Google Scholar]
- 37. Bode W, Fernandez‐Catalan C, Grams F, Gomis‐Ruth FX, Nagase H, Tschesche H, Maskos K. Insights into MMP‐TIMP interactions. Ann N Y Acad Sci. 1999;878:73–91. [DOI] [PubMed] [Google Scholar]
- 38. Strongin AY, Marmer BL, Grant GA, Goldberg GI. Plasma membrane‐dependent activation of the 72‐kDa type IV collagenase is prevented by complex formation with TIMP‐2. J Biol Chem. 1993;268:14033–14039. [PubMed] [Google Scholar]
- 39. Shiryaev SA, Remacle AG, Golubkov VS, Ingvarsen S, Porse A, Behrendt N, Cieplak P, Strongin AY. A monoclonal antibody interferes with TIMP‐2 binding and incapacitates the MMP‐2‐activating function of multifunctional, pro‐tumorigenic MMP‐14/MT1‐MMP. Oncogenesis. 2013;2:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chernov AV, Sounni NE, Remacle AG, Strongin AY. Epigenetic control of the invasion‐promoting MT1‐MMP/MMP‐2/TIMP‐2 axis in cancer cells. J Biol Chem. 2009;284:12727–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jaworski DM, Sideleva O, Stradecki HM, Langlois GD, Habibovic A, Satish B, Tharp WG, Lausier J, Larock K, Jetton TL, Peshavaria M, Pratley RE. Sexually dimorphic diet‐induced insulin resistance in obese tissue inhibitor of metalloproteinase‐2 (TIMP‐2)‐deficient mice. Endocrinology. 2011;152:1300–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernandez‐Patron C, Leung D. Emergence of a metalloproteinase/phospholipase A2 axis of systemic inflammation. Metalloproteinases Med. 2015;2:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab. 2008;19:65–73. [DOI] [PubMed] [Google Scholar]
- 44. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell. 1997;89:331–340. [DOI] [PubMed] [Google Scholar]
- 45. Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, van der Giet M, Tietge UJ. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J Lipid Res. 2010;51:743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sunami Y, Tazuma S, Chayama K. Is a role of phospholipase A(2) in cholesterol gallstone formation phospholipid species‐dependent? Biochim Biophys Acta. 2001;1532:51–59. [DOI] [PubMed] [Google Scholar]
- 47. Hattori Y, Tazuma S, Yamashita G, Ochi H, Sunami Y, Nishioka T, Hyogo H, Yasumiba S, Kajihara T, Nakai K, Tsuboi K, Asamoto Y, Sakomoto M, Kajiyama G. Role of phospholipase A2 in cholesterol gallstone formation is associated with biliary phospholipid species selection at the site of hepatic excretion: indirect evidence. Dig Dis Sci. 2000;45:1413–1421. [DOI] [PubMed] [Google Scholar]
- 48. Holmes MV, Simon T, Exeter HJ, Folkersen L, Asselbergs FW, Guardiola M, Cooper JA, Palmen J, Hubacek JA, Carruthers KF, Horne BD, Brunisholz KD, Mega JL, van Iperen EP, Li M, Leusink M, Trompet S, Verschuren JJ, Hovingh GK, Dehghan A, Nelson CP, Kotti S, Danchin N, Scholz M, Haase CL, Rothenbacher D, Swerdlow DI, Kuchenbaecker KB, Staines‐Urias E, Goel A, van‘t Hooft F, Gertow K, de Faire U, Panayiotou AG, Tremoli E, Baldassarre D, Veglia F, Holdt LM, Beutner F, Gansevoort RT, Navis GJ, Mateo Leach I, Breitling LP, Brenner H, Thiery J, Dallmeier D, Franco‐Cereceda A, Boer JM, Stephens JW, Hofker MH, Tedgui A, Hofman A, Uitterlinden AG, Adamkova V, Pitha J, Onland‐Moret NC, Cramer MJ, Nathoe HM, Spiering W, Klungel OH, Kumari M, Whincup PH, Morrow DA, Braund PS, Hall AS, Olsson AG, Doevendans PA, Trip MD, Tobin MD, Hamsten A, Watkins H, Koenig W, Nicolaides AN, Teupser D, Day IN, Carlquist JF, Gaunt TR, Ford I, Sattar N, Tsimikas S, Schwartz GG, Lawlor DA, Morris RW, Sandhu MS, Poledne R, Maitland‐van der Zee AH, Khaw KT, Keating BJ, van der Harst P, Price JF, Mehta SR, Yusuf S, Witteman JC, Franco OH, Jukema JW, de Knijff P, Tybjaerg‐Hansen A, Rader DJ, Farrall M, Samani NJ, Kivimaki M, Fox KA, Humphries SE, Anderson JL, Boekholdt SM, Palmer TM, Eriksson P, Pare G, Hingorani AD, Sabatine MS, Mallat Z, Casas JP, Talmud PJ. Secretory phospholipase A(2)‐IIA and cardiovascular disease: a Mendelian randomization study. J Am Coll Cardiol. 2013;62:1966–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ferguson JF, Hinkle CC, Mehta NN, Bagheri R, Derohannessian SL, Shah R, Mucksavage MI, Bradfield JP, Hakonarson H, Wang X, Master SR, Rader DJ, Li M, Reilly MP. Translational studies of lipoprotein‐associated phospholipase A(2) in inflammation and atherosclerosis. J Am Coll Cardiol. 2012;59:764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tietge UJ, Maugeais C, Lund‐Katz S, Grass D, deBeer FC, Rader DJ. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA‐I in response to inflammation in human apoA‐I transgenic mice. Arterioscler Thromb Vasc Biol. 2002;22:1213–1218. [DOI] [PubMed] [Google Scholar]
- 51. Tietge UJ, Maugeais C, Cain W, Grass D, Glick JM, de Beer FC, Rader DJ. Overexpression of secretory phospholipase A(2) causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A‐I. J Biol Chem. 2000;275:10077–10084. [DOI] [PubMed] [Google Scholar]
- 52. Pucer A, Brglez V, Payre C, Pungercar J, Lambeau G, Petan T. Group X secreted phospholipase A(2) induces lipid droplet formation and prolongs breast cancer cell survival. Mol Cancer. 2013;12:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang B, Fan P, Shimoji E, Itabe H, Miura S, Uehara Y, Matsunaga A, Saku K. Modulating effects of cholesterol feeding and simvastatin treatment on platelet‐activating factor acetylhydrolase activity and lysophosphatidylcholine concentration. Atherosclerosis. 2006;186:291–301. [DOI] [PubMed] [Google Scholar]
- 54. Shridas P, Zahoor L, Forrest KJ, Layne JD, Webb NR. Group X secretory phospholipase A2 regulates insulin secretion through a cyclooxygenase‐2‐dependent mechanism. J Biol Chem. 2014;289:27410–27417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boyanovsky BB, Bailey W, Dixon L, Shridas P, Webb NR. Group V secretory phospholipase A2 enhances the progression of angiotensin II‐induced abdominal aortic aneurysms but confers protection against angiotensin II‐induced cardiac fibrosis in apoE‐deficient mice. Am J Pathol. 2012;181:1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shridas P, Bailey WM, Gizard F, Oslund RC, Gelb MH, Bruemmer D, Webb NR. Group X secretory phospholipase A2 negatively regulates ABCA1 and ABCG1 expression and cholesterol efflux in macrophages. Arterioscler Thromb Vasc Biol. 2010;30:2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boyanovsky BB, Li X, Shridas P, Sunkara M, Morris AJ, Webb NR. Bioactive products generated by group V sPLA(2) hydrolysis of LDL activate macrophages to secrete pro‐inflammatory cytokines. Cytokine. 2010;50:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeLeon‐Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML. CD36 is a matrix metalloproteinase‐9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet. 2016;9:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mantuano E, Inoue G, Li X, Takahashi K, Gaultier A, Gonias SL, Campana WM. The hemopexin domain of matrix metalloproteinase‐9 activates cell signaling and promotes migration of schwann cells by binding to low‐density lipoprotein receptor‐related protein. J Neurosci. 2008;28:11571–11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van den Steen PE, Van Aelst I, Hvidberg V, Piccard H, Fiten P, Jacobsen C, Moestrup SK, Fry S, Royle L, Wormald MR, Wallis R, Rudd PM, Dwek RA, Opdenakker G. The hemopexin and O‐glycosylated domains tune gelatinase B/MMP‐9 bioavailability via inhibition and binding to cargo receptors. J Biol Chem. 2006;281:18626–18637. [DOI] [PubMed] [Google Scholar]
- 61. Bauters D, Scroyen I, Van Hul M, Lijnen HR. Gelatinase A (MMP‐2) promotes murine adipogenesis. Biochim Biophys Acta. 2015;1850:1449–1456. [DOI] [PubMed] [Google Scholar]
- 62. Van Hul M, Bauters D, Himmelreich U, Kindt N, Noppen B, Vanhove M, Lijnen HR. Effect of gelatinase inhibition on adipogenesis and adipose tissue development. Clin Exp Pharmacol Physiol. 2012;39:49–56. [DOI] [PubMed] [Google Scholar]
- 63. Lijnen HR, Demeulemeester D, Van Hoef B, Collen D, Maquoi E. Deficiency of tissue inhibitor of matrix metalloproteinase‐1 (TIMP‐1) impairs nutritionally induced obesity in mice. Thromb Haemost. 2003;89:249–255. [PubMed] [Google Scholar]
- 64. Wang X, Berry E, Hernandez‐Anzaldo S, Takawale A, Kassiri Z, Fernandez‐Patron C. Matrix metalloproteinase‐2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element‐binding protein‐2/3‐hydroxy‐3‐methylglutaryl‐CoA reductase pathway in the heart. Hypertension. 2015;65:882–888. [DOI] [PubMed] [Google Scholar]
- 65. Wang X, Berry E, Hernandez‐Anzaldo S, Sun D, Adijiang A, Li L, Zhang D, Fernandez‐Patron C. MMP‐2 inhibits PCSK9‐induced degradation of the LDL receptor in Hepa1‐c1c7 cells. FEBS Lett. 2015;589:490–496. [DOI] [PubMed] [Google Scholar]
- 66. Federici M, Hribal ML, Menghini R, Kanno H, Marchetti V, Porzio O, Sunnarborg SW, Rizza S, Serino M, Cunsolo V, Lauro D, Mauriello A, Smookler DS, Sbraccia P, Sesti G, Lee DC, Khokha R, Accili D, Lauro R. Timp3 deficiency in insulin receptor‐haploinsufficient mice promotes diabetes and vascular inflammation via increased TNF‐alpha. J Clin Invest. 2005;115:3494–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schonbeck U, Mach F, Libby P. Generation of biologically active IL‐1 beta by matrix metalloproteinases: a novel caspase‐1‐independent pathway of IL‐1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 68. Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma contain PPARgamma: differentiation‐dependent peroxisomal proliferator‐activated receptor gamma(PPARgamma) expression and reduction of MMP‐9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vogel RA. PCSK9 inhibition: the next statin? J Am Coll Cardiol. 2012;59:2354–2355. [DOI] [PubMed] [Google Scholar]
- 70. Porter KE, Turner NA. Statins and myocardial remodelling: cell and molecular pathways. Expert Rev Mol Med. 2011;13:e22. [DOI] [PubMed] [Google Scholar]
- 71. Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho‐associated coiled‐coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009;119:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Adhyaru BB, Jacobson TA. New cholesterol guidelines for the management of atherosclerotic cardiovascular disease risk a comparison of the 2013 American College of Cardiology/American Heart Association Cholesterol Guidelines with the 2014 National Lipid Association Recommendations for Patient‐Centered Management of Dyslipidemia. Endocrinol Metab Clin North Am. 2016;45:17–37. [DOI] [PubMed] [Google Scholar]
- 73. Abdel‐Latif MS. Plasma levels of matrix metalloproteinase (MMP)‐2, MMP‐9 and tumor necrosis factor‐alpha in chronic hepatitis C virus patients. Open Microbiol J. 2015;9:136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Akca G, Tuncbilek S, Sepici‐Dincel A. Association between matrix metalloproteinase (MMP)‐2, MMP‐9 and total antioxidant status of patients with asymptomatic hepatitis C virus infection. Lett Appl Microbiol. 2013;57:436–442. [DOI] [PubMed] [Google Scholar]
- 75. Nunez O, Fernandez‐Martinez A, Majano PL, Apolinario A, Gomez‐Gonzalo M, Benedicto I, Lopez‐Cabrera M, Bosca L, Clemente G, Garcia‐Monzon C, Martin‐Sanz P. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut. 2004;53:1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chung TW, Kim JR, Suh JI, Lee YC, Chang YC, Chung TH, Kim CH. Correlation between plasma levels of matrix metalloproteinase (MMP)‐9/MMP‐2 ratio and alpha‐fetoproteins in chronic hepatitis carrying hepatitis B virus. J Gastroenterol Hepatol. 2004;19:565–571. [DOI] [PubMed] [Google Scholar]
- 77. Chung TW, Moon SK, Lee YC, Kim JG, Ko JH, Kim CH. Enhanced expression of matrix metalloproteinase‐9 by hepatitis B virus infection in liver cells. Arch Biochem Biophys. 2002;408:147–154. [DOI] [PubMed] [Google Scholar]
- 78. Lichtinghagen R, Michels D, Haberkorn CI, Arndt B, Bahr M, Flemming P, Manns MP, Boeker KH. Matrix metalloproteinase (MMP)‐2, MMP‐7, and tissue inhibitor of metalloproteinase‐1 are closely related to the fibroproliferative process in the liver during chronic hepatitis C. J Hepatol. 2001;34:239–247. [DOI] [PubMed] [Google Scholar]
- 79. Lichtinghagen R, Huegel O, Seifert T, Haberkorn CI, Michels D, Flemming P, Bahr M, Boeker KH. Expression of matrix metalloproteinase‐2 and ‐9 and their inhibitors in peripheral blood cells of patients with chronic hepatitis C. Clin Chem. 2000;46:183–192. [PubMed] [Google Scholar]
- 80. Nighot P, Al‐Sadi R, Rawat M, Guo S, Watterson DM, Ma T. Matrix metalloproteinase 9‐induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am J Physiol Gastrointest Liver Physiol. 2015;309:G988–G997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase‐9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47:186–198. [DOI] [PubMed] [Google Scholar]
- 82. Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2015;112:1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald‐Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller‐Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van Hul M, Bauters D, Lijnen RH. Differential effects of a gelatinase inhibitor on adipocyte differentiation and adipose tissue development. Clin Exp Pharmacol Physiol. 2013;40:689–697. [DOI] [PubMed] [Google Scholar]
- 85. Van Hul M, Lijnen HR. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. J Thromb Haemost. 2008;6:1198–1206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Systemic metabolic profile of MMP‐9 deficient mice. A, Oxygen consumption. B, Carbon dioxide production. C, Heat/energy expenditure (normalized to body weight). D through F, Locomotor activity. G, Body weight. H, Total food consumption. The studies were conducted in metabolic cages (n=7 WT mice, n=5 Mmp9 −/− mice). *P≤0.05 vs WT. MMP indicates matrix metalloproteinase; WT, wild type.
Figure S2. Supplemental quantitative analysis of lipids. A, Esterified lipids were elevated in livers from Mmp9 −/− mice compared with livers from WT mice, particularly when the mice were fasted and refed a high‐carbohydrate diet. In contrast to Mmp9 −/− mice, Mmp7 −/− mice had a different hepatic lipid profile (n=4 mice per group, except n=3 for Mmp7 −/− fasted and refed). † P≤0.05 vs fasted for each genotype.*P≤0.05 vs WT fasted and refed. B, Cholesterol excretion was unchanged in Mmp9 −/− mice (n=4 mice per genotype). WT indicates wild type.
Figure S3. Spleen is a likely source of plasma sPLA2 in MMP‐9–deficient mice. sPLA2 activity was analyzed in duplicate using pools of the indicated tissues (n=3 WT and n=4 Mmp9 −/− mice). *P<0.05 vs WT. MMP indicates matrix metalloproteinase; sPLA2, secretory phospholipase A2; WT, wild type.
Figure S4. SREBP‐2 expression in response to dietary choloesterol and varespladib. Western blot (left) showing amount of hepatic SREBP‐2 protein in Mmp9 −/− mice in response to varespladib and dietary cholesterol supplementation. The experiment involved 4 to 5 mice per group (or treatment). For analysis, livers were pooled and homogenized, and the fraction containing nuclei was subjected to Western blot analysis with SREBP‐2 antibodies. Quantitative analysis (right) for 2 independent preparations and Western blots. *P≤0.05 vs untreated. SREBP‐2 indicates sterol regulatory element binding protein 2.
Figure S5. Proposed model. MMP‐9 regulates cholesterol metabolism through PLA2‐dependent and ‐independent mechanisms. Important elements are peripheral organs (eg, the spleen) acting as a source of plasma sPLA2 activity and MMP‐9 (inhibitor of sPLA2 release from peripheral organs). Once in the circulation, sPLA2 acts on plasma lipoproteins or target organs (eg, the liver) to release lipid mediators from phospholipids that ultimately influence cholesterol metabolism. Furthermore, the direct action of MMP‐9 in the liver may influence hepatic cholesterol through as yet unclear PLA2‐independent pathways. MMP indicates matrix metalloproteinase; PLA2, phospholipase A2; sPLA2, secretory phospholipase A2.
Figure S6. Supplement to Figure 7A containing the quantitative analysis of hepatic transcriptional responses to dietary cholesterol for the indicated genes and genotypes (n=6 WT mice, n=8 Mmp2 −/− mice, n=5 Mmp7 −/− mice, n=5 Mmp9 −/− mice, n=5 Timp2 −/− mice). *P≤0.05 vs WT. † P≤0.05 vs 0 days on cholesterol. WT indicates wild type.
Figure S7. Extended quantitative analysis of the relative mRNA expression of cardiac and hepatic lipid‐metabolic and metalloproteinase genes and their response to dietary supplementation with 0.15% cholesterol for 2.5 days in mice deficient in one of several MMPs (n=6 WT mice, n=8 Mmp2 −/− mice, n=5 Mmp7 −/− mice, n=5 Mmp9 −/− mice). Slots without bars: Gene expression was not determined. MMP indicates matrix metalloproteinase; WT, wild type.